

Abstract
Background
Many microRNAs (miRNAs) are downregulated in proliferative vascular disease. Thus, upregulation of these miRNAs has become a major focus of research activity. However, there is a critical barrier in gene therapy to upregulate some miRNAs such as miR‐145 and miR‐143 because of their significant downregulation by the unclear endogenous mechanisms under disease conditions. The purpose of this study was to determine the molecular mechanisms responsible for their downregulation and to overcome the therapeutic barrier.
Methods and Results
In cultured proliferative rat vascular smooth muscle cells (VSMCs) in vitro and in diseased rat and mouse arteries in vivo, we have identified that the impairment of pri‐miR‐145 into pre‐miR‐145 is the critical step related to the downregulation of miR‐145, in which the PI3‐kinase/Akt/p53 pathway is involved. We further identified that the flank sequences of pri‐miR‐145 are the critical structural components responsible for the aberrant miR‐145 expression. Switching of the flank sequence of downregulated miR‐145 and miR‐143 to the flank sequence of miR‐31 confers resistance to their downregulation. The genetically engineered miR‐145 (smart miR‐145) restored the downregulated miR‐145 in proliferative rat VSMCs and in rat carotid arteries with balloon injury and mouse atherosclerotic aortas and demonstrated much better therapeutic effects on the abnormal growth of VSMCs, expression of its target gene, KLF5 expression, VSMC marker gene expression, and vascular neointimal growth.
Conclusions
The flank sequences of miR‐145 and miR‐143 play a critical role in their aberrant expression in VSMCs and vascular walls. The genetically engineered “smart” miRNAs based on their flank sequences may have broadly therapeutic applications for many vascular diseases.
Keywords: cell growth, gene expression, microRNAs, smart microRNAs, vascular disease
Introduction
MicroRNAs (miRNAs) are a class of endogenous, small, noncoding RNAs that negatively regulate >30% of genes in a cell via degradation or translational inhibition of their target mRNAs.1–2 There are 3 key steps in miRNA biogenesis. First, miRNA is initially transcribed in the nucleus to form a large pri‐miRNA. The structure of pri‐miRNA contains 1 loop, 2 arms, and 2 strands of flank sequence. Then, the pri‐miRNA is processed in the nucleus into a 70‐nucleotide pre‐miRNA. After that, the pre‐miRNA enters the cytoplasm to form an 18‐ to 24‐nucleotide (nt) mature miRNA. Functionally, an individual miRNA is able to regulate the expression of its multiple target genes. It is therefore not surprising that miRNAs are involved in the regulation of all major cellular functions and may play important roles in the pathogenesis of many human diseases.3–5 Indeed, many miRNAs are aberrantly downregulated in proliferative human diseases such as cancer and cardiovascular disease.
Atherosclerotic vascular diseases such as atherosclerosis and restenosis are still the major cause of morbidity and a major cost in our healthcare system. Recent studies from us6–9 and other groups10–13 have demonstrated that miRNAs may play important roles in the pathogenesis of atherosclerotic vascular diseases. Among the dysregulated miRNAs in diseased vascular walls, vascular smooth muscle cell (VSMC)–enriched miR‐145 has received special attention because it is the most abundant miRNA in normal VSMCs and vascular walls and its expression is downregulated in dedifferentiated, proliferative VSMCs and in diseased arteries.6–7,6–12 Both pharmacological and genetic evidence suggests that miR‐145 is a critical controller for VSMC phenotype and vascular neointimal growth. Indeed, spontaneous huge neointimal lesion formation was displayed in arteries from miR‐145‐knockout mice without any additional injuries.11 Thus, miRNAs such as miR‐145 could be promising novel therapeutic targets for vascular diseases such as atherosclerosis.
Upregulation of the aberrantly downregulated miRNAs may represent novel therapeutics. However, our recent unpublished data revealed that there is a critical barrier in gene therapy to upregulate these miRNAs such as miR‐145 and miR‐143 because of the downregulation of these exogenous miRNA genes by the endogenous mechanisms under disease conditions. Here we demonstrate that the flank sequences of pri‐miRNAs such as pri‐miR‐145 are critical for the downregulation of these miRNAs in proliferative VSMCs and in diseased vascular walls. Modification or replacement of the flank sequences of these miRNAs is able to create the “smart” miRNA genes that could avoid their downregulation by the endogenous mechanisms. These smart miRNAs may have broadly therapeutic applications for proliferative diseases such as vascular disease.
Methods
Cell Culture
VSMCs and vascular endothelial cells were obtained from the aortas of male Sprague‐Dawley rats (5 weeks old) by using an enzymatic dissociation method as described previously.6–8 VSMCs were cultured with DMEM containing 10% fetal bovine serum. For all experiments, rat VSMCs and vascular endothelial cells from passages 3 to 6 were used. Human alveolar adenocarcinoma cell line A549 cells (American Type Culture Collection) and human embryonic kidney cells (HEK 293A; Invitrogen) were cultured in DMEM with 10% fetal bovine serum.
RNA Isolation and qRT‐PCR
RNA levels were determined by qRT‐PCR.6–9 Briefly, RNA from VSMCs, vascular endothelial cells, A549 cells, HEK 293A cells, rat carotid arteries, and mouse aortas was isolated with TRIzol (Invitrogen; Carlsbad, CA). qRT‐PCR for miRNA was performed on cDNA generated from 100 ng of total RNA using TaqMan MiRNA Reverse Transcription and TaqMan MiRNA assays (Applied Biosystems). qRT‐PCR for p53, pri‐miR‐145, pre‐miR‐145, pri‐miR‐143, or pre‐miR‐143 was performed on cDNA generated from 200 ng of total RNA using the protocol of a qRT‐PCR mRNA Detection Kit (Roche). Amplification and detection of specific products were performed with a Roche Lightcycler 480 Detection System. As an internal control, U6 was used for miRNA template normalization and GADPH was used for other template normalizations. Pri‐miRNA and pre‐miRNA was distinguished on the basis of primer designs and isolation methods as described previously.14–15 In brief, for detection of pre‐miRNA, we used a small RNA‐enriched fraction isolated with an miRNeasy Mini Kit and mirVana miRNA Isolation Kit and performed a PCR reaction by using the primer set for stem loop (Applied Biosystems). For detection of pri‐miRNA, we use total RNA and performed a PCR reaction using the primers of the franking region in pre‐miRNA (Applied Biosystems). The sequences of the primers used are shown in Table. Fluorescent signals were normalized to an internal reference, and the threshold cycle (Ct) was set within the exponential phase of the PCR. Relative gene expression was calculated by comparing cycle times for each target PCR. The target PCR Ct values were normalized by subtracting the U6 or GADPH Ct value, which provided the ΔCt value. Relative expression between treatments was then calculated using the following equation: relative gene expression=2−(ΔCt sample−ΔCt control).
Table 1.
Sequences of the Primers Used in the Study
| Primer Name | Sequence (5′ to 3′) |
|---|---|
| GAPDH FP | AAGCTCACTGGCATGGCCTT |
| GAPDH RP | CGGCATGTCAGATCCACAAC |
| U6 FP | CTCGCTTCGGCAGCACA |
| U6 RP | AACGCTTCACGAATTTGCGT |
| Pri‐miR‐145 FP | GGCACTGCTGAAGGCATCTC |
| Pri‐miR‐145 RP | CTGTTAAGCCATGACCTCAAGAAC |
| Pre‐miR‐145 FP | GTCCAGTTTTCCCAGGAATC |
| Pre‐miR‐145 RP | AGAACAGTATTTCCAGGAAT |
| Pri‐miR‐143 FP | TCTGAGATGAAGCACTGTAG |
| Pri‐miR‐143 RP | TGTTACGGATGGCATAAGA |
| p53 FP | GAGGATTCACAGTCGGATA |
| p53 RP | ATCATCTGGAGGAAGAAGTT |
| Ad‐miR‐145 FP | TGAATTCAGAAGTGAGGTGCATATAGCACC |
| Ad‐miR‐145 RP | TAAGCTTCTCCGATGCACCTCCTCCTC |
| Ad‐miR‐143 FP | TGGTACCGACAAAGGACACGAAGATGGACG |
| Ad‐miR‐143 RP | TTGATATCTGTTACGGATGGCATAAGAGTG |
| miR‐145:40 FP | TTGAATTCTCTCTCTCTCCCACCTTGTCC |
| miR‐145:40 RP | TTGGATCCTCTCTCTCTCCCACCTTGACC |
| miR‐145:90 FP | TTGAATTCAGAGAACTGCTGGTCCCTAGG |
| miR‐145:90 RP | TTGGATCCGGAGACAGATCCAGCTGCTAAGC |
| miR‐145:180 FP | TTGAATTCGGAAGGAGGGTGTATACAGCG |
| miR‐145:180 RP | TTGGATCCGAACCGCCTCCTCCTCCTCCCTACT |
Adenovirus and Plasmid Generation
The adenoviruses expressing miR‐145 (Ad‐miR‐145), genetically engineered smart miR‐145 (Ad‐smart‐miR‐145), miR‐143 (Ad‐miR‐143), genetically engineered smart miR‐143 (Ad‐smart‐miR‐143), genetically engineered miR‐145‐21 (Ad‐miR‐145‐21), miR‐31 (Ad‐miR‐31), genetically engineered miR‐miR‐31 (Ad‐modified miR‐31), p53 (Ad‐p53), adenovirus expressing dominate negative Akt (Ad‐dn‐Akt), control empty adenovirus (Ad‐CMV), and control adenoviruses expressing GFP (Ad‐GFP) were generated using ViraPower Adenoviral Gateway Expression system (Invitrogen, Carlsbad, CA) according to the manufacturer's protocols as described previously.2 For example, to produce Ad‐miR‐145, a fragment containing the precursor miR‐145 was amplified with its primers (Rno miR‐145 FP and Rno miR‐145 RP) from rat genomic DNA and inserted into pENTR‐3C vectors (Invitrogen, Carlsbad, CA) at EcoRI and XhoI sites. The construct named pENTR‐miR‐145 was sequenced to confirm the DNA sequences. By Cre recombinase, the fragment was excised from the pENTR‐miR‐145 donor vector and was inserted into the pAd/CMV/V5‐DEST Gateway receptor vector, which was named pAd‐miR‐145. To produce recombinant adenoviruses with Lipofectamine 2000 (Invitrogen, Carlsbad, CA), the plasmid pAd‐miR‐145 was digested by Pac and transfected into low‐passage HEK 293A cells.
Ad‐smart‐miR‐145 was a genetically engineered miR‐145 in which the 110‐nt‐long flank sequences of pri‐miR‐145 were switched to the 110‐nt flank sequences of pri‐miR‐31. Ad‐smart‐miR‐143 was a genetically engineered miR‐143 in which the 110‐nt flank sequences of pri‐miR‐143 were switched to the 110‐nt‐long flank sequences of pri‐miR‐31. Ad‐miR‐145‐21 was a genetically engineered miR‐145 in which the 110‐nt‐long flank sequences of pri‐miR‐145 were switched to the 110‐nt flank sequences of pri‐miR‐21. Ad‐modified miR‐31 was a genetically engineered miR‐31 in which the 110‐nt‐long flank sequences of pri‐miR‐31 were switched to the 110 flank sequences of pri‐miR‐145. The resulting adenoviruses were further amplified by infection of HEK 293A cells and purified by cesium chloride gradient ultracentrifugation. The titers of purified adenoviruses were determined by using Adeno‐X Rapid Titer Kit (Clontech, Mountain View, CA).
In addition, plasmids expressing miR‐145 vectors with different lengths of the flank sequences (40, 90, or 180 nt from both the 3′ end and the 5′ end)—pDNR‐miR‐145:40, pDNR‐miR‐145:90, and pDNR‐miR‐145:180—were synthesized by Integrated DNA Technology.
Oligonucleotide Transfection and Adenovirus Infection in Cultured VSMCs
Oligonucleotide transfection was performed as described in our previous studies.6–9 Briefly, cells were transfected using a transfection reagent (Qiagen, Valencia, CA) 24 hours after seeding into the wells. Transfection complexes were prepared according to the manufacturer's instructions. p53 Gene knockdown was performed using siRNA p53 (50 nmol/L; Invitrogen, Grand Island, NY). The transfection medium was replaced 4 hours posttransfection by the regular culture medium. Vehicle and scramble controls (Ambion, Inc) were applied. For miR‐145 or miR‐143 overexpression, Ad‐miR‐145, Ad‐smart‐miR‐145, Ad‐miR‐143, Ad‐smart‐miR‐143, miR‐145‐21 miR‐145:40, miR‐145:110, or miR‐145:160 was added to the culture medium at 50 multiplicities of infection (MOI) or indicated MOI. Ad‐p53 (MOI) was used to upregulate the expression of p53. Ad‐GFP or adenovirus containing an empty CMV promoter (Ad‐CMV) was used as the adenovirus controls. Forty‐eight or 72 hours later, the cells were harvested for RNA or protein isolation, respectively.
Western Blot Analysis
Proteins were isolated from cultured VSMCS, and the protein levels were determined by Western blot analysis. Briefly, equal amounts of protein were subjected to SDS‐PAGE. Standard Western blot analysis was conducted using p53 (1:1000 dilution; Cell Signaling) and phosphor‐Akt (p‐Akt, ser473, 1:1000 dilution; Cell Signaling). GADPH antibody (1:5000 dilution; Cell Signaling) was used as a loading control.
Kinase Inhibition
In cultured VSMCs, PI3‐kinase, JNK, ERK, and p38 were inhibited by their inhibitors, LY294002 (20 μmol/L, PI3‐kinase inhibitor), SP600125 (20 μmol/L, JNK inhibitor), PD98059 (20 μmol/L, ERK inhibitor), and SB203580 (10 μmol/L, p38 inhibitor). To inhibit PI3‐kinase in rat and mouse arteries in vivo, wortmannin (30 μg/kg per day IP) was used for 3 days.
Cell Model of Proliferation
VSMCs grown to 30% to 50% confluence were made quiescent by incubation with 0.1% fetal bovine serum for 48 hours. Then, platelet‐derived growth factor (PDGF; 10 ng/mL; Cell Signaling) was added to the culture medium to induce cell proliferation. VSMC proliferation was determined by cell counting and cell proliferation assay via an MTT kit (Roche).
Rat Carotid Artery Balloon Injury Model
Carotid artery balloon injury was induced in male Sprague‐Dawley rats (230 to 300 g) as described in our previous studies.6–8 Rats were anesthetized with ketamine (80 mg/kg)/xylazine (5 mg/kg). Under a dissecting microscope, the right common carotid artery was exposed through a midline cervical incision. A 2F Fogarty catheter (Baxter Edwards) was introduced via an arteriotomy in the external carotid artery, and then the catheter was advanced to the proximal edge of the omohyoid muscle. To produce carotid artery injury, we inflated the balloon with saline and withdrew it 3 times from just under the proximal edge of the omohyoid muscle to the carotid bifurcation. After balloon injury, solutions (100 μL) of Ad‐miR‐145 (1×109 pfu/mL), Ad‐smart–miR‐145 (1×109 pfu/mL), high‐dose Ad‐miR‐145 (5×109 pfu/mL), or control empty virus (Ad‐CMV, 1×109 pfu/mL) or Ad‐GFP (5×109 pfu/mL) were infused into the injured segment of the common carotid artery for 30 minutes. The external carotid artery was then permanently ligated with a 6‐0 silk suture, and blood flow in the common carotid artery was restored. All protocols were approved by the Institutional Animal Care and Use Committee at UMDNJ and were consistent with the Guide for the Care and Use of Laboratory Animals (NIH publication 85‐23, revised 1985).
Mouse Atherosclerosis Model
Atherosclerosis was induced in the aortas of 8‐week‐old ApoE‐knockout male mice on a C57BL/6 background (Jackson Laboratories) by a Western diet containing 21% fat, 0.15% cholesterol, and 19.5% casein for 20 weeks as described previously.16 Atherosclerotic lesions were confirmed by oil red O staining using en face preparation of whole aortas and hematoxylin‐eosin staining of cross‐sections of proximal aortas (aortic sinus, ascending aorta, arch, and thoracic aorta) as shown in our previous publications.17–18 To upregulate the miR‐145 in mouse aortas, 50‐μL solutions of Ad‐miR‐145 (5×1010 pfu/mL) or control Ad‐GFP (5×1010 pfu/mL) were injected via the tail vein. One week after injection, RNA was isolated from these mouse aortas to determine the levels of miR‐145.
Morphometric Analysis for Neointimal Lesion Formation
Morphometric analysis via computerized image analysis system (NIS Elements BR 3.0) was performed in sections stained with Masson's trichrome staining as described.6–9 In brief, 6 sections (5 μm thick) sectioned at equally spaced intervals of injured carotid arteries were used. The intimal‐to‐medial area ratio of each section was calculated. The average intimal‐to‐medial area ratio of the 6 sections was used as the intimal‐to‐medial area ratio of each animal.
Statistics
All data are presented as mean±standard error. For relative gene expression, the mean value of the vehicle control group is defined as 100% or 1. Two‐tailed unpaired Student t tests and ANOVAs were used for statistical evaluation of the data. SPSS 17.0 was used for data analysis. A P<0.05 was considered significant. As some sample sizes per group were relatively small, the results were further verified by the Wilcoxon rank sum or the Kruskal–Wallis test.
Results
PI3‐Kinase/Akt/p53 Is the Critical Signaling Pathway Related to Downregulation of miR‐145 in Proliferative VSMCs in Balloon‐Injured Arteries or in Atherosclerotic Arteries
PI3‐kinase, JNK, ERK, and p38 are important signaling molecules involved in PDGF and balloon‐injury‐induced gene expression, VSMC proliferation, and vascular neointimal growth.19–21 To determine the key signaling pathways related to the regulation of miR‐145 expression, we examined the roles of these kinase pathways in miR‐145 expression by using their specific inhibitors. In cultured VSMCs, PI3‐kinase, JNK, ERK, and p38 were inhibited by their inhibitors, LY294002 (20 μmol/L, PI3‐kinase inhibitor), SP600125 (20 μmol/L, JNK inhibitor), PD98059 (20 μmol/L, ERK inhibitor), and SB203580 (10 μmol/L, p38 inhibitor). Then VSMCs were treated with PDGF (10 ng/mL) for 24 hours, and the expression of miR‐145 was determined by qRT‐PCR. VSMCs without PDGF treatment were used as the vehicle control. As shown in Figure 1A, PDGF‐induced downregulation of miR‐145 expression was inhibited by LY294002 in cultured VSMCs. However, no significant effect of SP600125, PD98059, and SB203580 on the expression of miR‐145 was demonstrated in PDGF‐treated VSMCs. To inhibit PI3‐kinase in rat carotid arteries in vivo, wortmannin (30 μg/kg per day IP) was used for 3 days after angioplasty. As shown in Figure 1B, the downregulation of miR‐145 induced by balloon injury was also significantly blocked by inhibition of PI3‐kinase. The successful inhibition of PI3‐kinase via LY294002 and wortmannin was confirmed by the decreased p‐Akt via Western blot analysis (Figure 1C). The successful inhibition of JNK, ERK, and p38 was also confirmed by the decreased phospho‐JNK, phospho‐ERK, and phospho‐p38 (data not shown). Moreover, the downregulated miR‐145 in atherosclerotic aortas from ApoE‐knockout mice was restored by PI3K inhibition (wortmannin, 30 μg/kg per day, ip, for 3 days) (Figure 1D). It is well established that Akt is the downstream molecule of PI3‐kinase. We thus determined the role of Akt in the downregulation of miR‐145. As shown in Figure 1E, PDGF‐induced downregulation of miR‐145 was successfully inhibited by the adenovirus‐expressing dominant negative Akt (Ad‐dn‐Akt).
Figure 1.

PI3‐kinase/Akt/p53 is the critical signaling pathway related to the downregulation of miR‐145 in proliferative VSMCs and in balloon‐injured or atherosclerotic arteries. A, PI3‐kinase inhibitor blocked PDGF‐induced downregulation of miR‐145 in cultured VSMCs. In cultured VSMCs, PI3‐kinase, JNK, ERK, and p38 were inhibited by their inhibitors: LY294002 (20 μmol/L, PI3‐kinase inhibitor), SP600125 (20 μmol/L, JNK inhibitor), PD98059 (20 μmol/L, ERK inhibitor), and SB203580 (10 μmol/L, p38 inhibitor). Then, VSMCs were treated with PDGF (10 ng/mL) for 24 hours, and the expression of miR‐145 was determined by qRT‐PCR. n=6; *P<0.05 compared with vehicle group; #P<0.05 compared with that in PDGF‐treated VSMCs without kinase inhibitor. B, Inhibition of miR‐145 downregulation by wortmannin (30 μg/kg per day IP) in balloon‐injured rat carotid arteries. n=5; *P<0.05 compared with that in balloon‐injured arteries treated with vehicle (injury). C, Successful inhibition of PI3‐kinase via LY294002 or wortmannin was confirmed by decreased p‐Akt via Western blot analysis. D, Downregulation of miR‐145 in atherosclerotic aortas (AS) of ApoE‐knockout mice was inhibited by PI3‐kinase inhibitor wortmannin (30 μg/kg per day, IP). n=3; *P<0.05 compared with that in AS treated with vehicle. E, Downregulation of miR‐145 in PDGF‐treated VSMCs was inhibited by Ad‐dn‐Akt (50 MOI). n=3; *P<0.05 compared with that in Ad‐GFP and PDGF‐treated cells. F, p53 Inducer doxorubicin (Doxo, 1 μg/mL) increased the expression of miR‐145 in VSMCs treated with PDGF (10 ng/mL). n=6; *P<0.05 compared with the PDGF‐treated group without Doxo. G, Knockdown of p53 expression at protein levels by siRNA p53 (50 nmol/L). n=6; *P<0.05 compared with the scramble‐treated group. H, Representative Western blots of p53 protein in VSMCs treated with vehicle, siRNA control (scramble, 50 nmol/L), or siRNA p53 (50 nmol/L). I, siRNA p53 (50 nmol/L) decreased the expression of miR‐145 in VSMCs. n=6; *P<0.05 compared with the scramble‐treated group. J, Overexpression of p53 expression at protein levels by Ad‐p53 (50 MOI). n=6; *P<0.05 compared with the Ad‐GFP‐treated group. K, Representative Western blots of p53 protein in VSMCs treated with Ad‐GFP (50 MOI) or Ad‐p53 (50 MOI). L, Ad‐p53 (50 nmol/L) increased the expression of miR‐145 in VSMCs. n=6; *P<0.05 compared with the Ad‐GFP‐treated group. M, PI3‐kinase inhibitor LY294002 (20 μmol/L) increased the expression of p53 in VSMCs treated with PDGF (10 ng/mL). n=3; *P<0.05 compared with the vehicle‐treated group. N, Representative Western blots of p53 in vehicle or LY294002‐treated VSMCs with PDGF (10 ng/mL). Ad‐dn‐Akt indicates adenovirus expressing dominate negative Akt; Ad‐GFP, adenoviruses expressing GFP; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; MOI, multiplicity of infection; PDGF, platelet‐derived growth factor; qRT‐PCR, quantitative real‐time polymerase chain reaction; VSMCs, vascular smooth muscle cells.
There are 2 important downstream transcription factors involved in PI3‐kinase/Akt‐mediated gene suppression: Forkhead and p53. Because transient transfection experiments indicated that Forkhead had no significant effect on miR‐145, we then tested whether p53 was involved in miR‐145 expression. In this experiment, VSMCs were treated with PDGF (10 ng/mLl) for 24 hours with or without p53 inducer doxorubicin (1 μg/mL). As shown in Figure 1F, PDGF‐mediated downregulation of miR‐145 was partially inhibited by p53 activation induced by doxorubicin. To further confirm the role of p53 in the expression of miR‐145, both gain‐of‐function and loss‐of‐function approaches were applied. The expression of p53 was knocked down by its siRNA (siRNA p53; Figure 1G and 1H) and was overexpressed by adenovirus expressing p53 (Ad‐p53; Figure 1I and 1J). Interestingly, the expression of miR‐145 in VSMCs was inhibited by siRNA p53 (Figure 1K), but was increased by overexpression of p53 (Figure 1L). In addition, the effect of the PI3‐kinase/Akt pathway on the expression of p53 in VSMCs was verified by Western blot, in which its expression was upregulated by PI3 inhibitor LY294002 (20 μmol/L; Figure 1M and 1N). The results suggested that the downstream signaling molecule of PI3‐kinase/Akt, p53, is indeed an important modulator that controls the expression of miR‐145 in proliferative VSMCs.
Exogenous miR‐145 Gene‐Mediated Increase in miR‐145 Expression Is Markedly Inhibited in Proliferative VSMCs and in Diseased Vessels Induced by Balloon Injury or Atherosclerosis
As shown in Figure 2A, the expression of miR‐145 in cultured VSMCs without growth stimulus was significantly increased by adenovirus expressing miR‐145 (Ad‐miR‐145, 50 MOI). However, when the cells were treated with PDGF (10 ng/mL), the increase in miR‐145 expression by Ad‐miR‐145 was markedly inhibited in these PDGF‐treated proliferative cells. No difference in transfection efficiency of Ad‐miR‐145 was found between PDGF‐treated cells and vehicle‐treated cells under the culture condition of 50 MOI (data not shown). Although miR‐145 expression was easily upregulated by Ad‐miR‐145 in normal mouse aortas (Figure 2B) and uninjured rat carotid arteries (Figure 2C), it failed to efficiently upregulate the expression of miR‐145 to the normal level in atherosclerotic aortas from ApoE‐knockout mice and in balloon‐injured rat carotid arteries (Figure 2B and 2C). If we used the levels of miR‐145 in normal untreated VSMCs or in normal mouse and rat arteries as the basal levels, the increase in miR‐145 expression by Ad‐miR‐145 in PDGF‐simulated VSMCs or in diseased arteries was much smaller compared with that induced by Ad‐miR‐145 in the normal controls (Figure 2D). It should be noted that the failure to upregulate the downregulated miRNAs because of endogenous downregulatory mechanisms in the proliferative cells and in diseased tissues was not limited to miR‐145. For example, miR‐143, another downregulated miRNA in proliferative diseases, also failed to be upregulated efficiently by the adenovirus expressing exogenous miR‐143 (Ad‐miR‐143) in proliferative cells (Figure 3).
Figure 2.
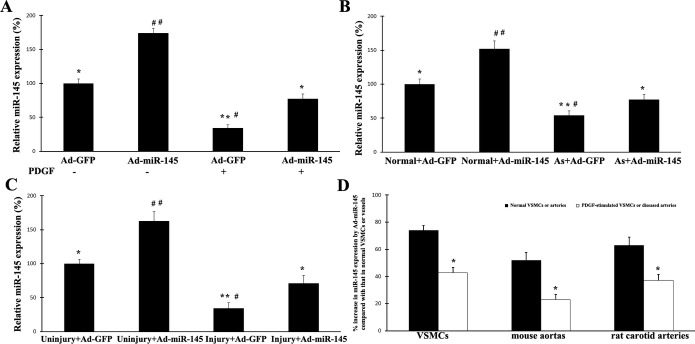
Effects of exogenous miR‐145 on the expression of miR‐145 in untreated VSMCs, proliferative VSMCs, and diseased vessels induced by balloon injury and atherosclerosis: A, Expression of miR‐145 in cultured VSMCs without growth stimulus was significantly increased by Ad‐miR‐145 (50 MOI). However, when the cells were treated with PDGF (10 ng/mL), the increase in miR‐145 expression by Ad‐miR‐145 was markedly inhibited in these proliferative cells. n=6; *P<0.01 and **P<0.001 compared with that in Ad‐miR‐145‐treated cells without PDGF; #P<0.05 and ##P<0.001 compared with that in Ad‐miR‐145‐treated cells with PDGF. B, Expression of miR‐145 in normal mouse aortas was significantly increased by Ad‐miR‐145 (1×109 pfu/mL). However, Ad‐miR‐145 failed to efficiently overexpress miR‐145 to a normal level in atherosclerotic aortas from ApoE‐knockout mice. n=5; *P<0.01 and **P<0.001 compared with that in normal aortas with Ad‐miR‐145 treatment; #P<0.05 and ##P<0.001 compared with that in atherosclerotic aortas with Ad‐miR‐145 treatment. C, Expression of miR‐145 in normal uninjured rat carotid arteries was significantly increased by Ad‐miR‐145. However, Ad‐miR‐145 failed to efficiently overexpress miR‐145 to a normal level in rat carotid arteries after balloon injury. n=5; *P<0.01 and **P<0.001 compared with that in uninjured rat carotid arteries with Ad‐miR‐145 treatment; #P<0.05 and ##P<0.001 compared with that in injured rat carotid arteries with Ad‐miR‐145 treatment. D, Effects of exogenous miR‐145 on the expression of miR‐145 in untreated VSMCs, PDGF‐treated VSMCs, normal vessels, and diseased vessels induced by balloon injury and atherosclerosis. Levels of vmiR‐145 in normal untreated VSMCs or in normal mouse and rat arteries were used as basal levels. The increase in miR‐145 expression by Ad‐miR‐145 (50 MOI) in PDGF‐simulated VSMCs or in diseased mouse and rat arteries was much smaller compared with that induced by Ad‐miR‐145 in the normal controls. n=6; *P<0.05 compared with that in the normal controls. Ad‐GFP indicates adenoviruses expressing GFP; MOI, multiplicity of infection; PDGF, platelet‐derived growth factor; VSM‐HMC, smooth muscle myosin heavy chain; SMCs, vascular smooth muscle cells.
Figure 3.
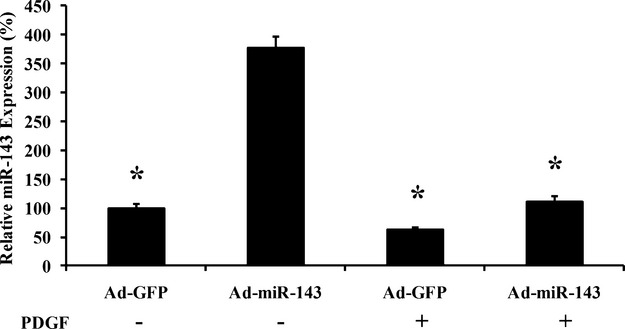
Effects of Ad‐miR‐143 on the expression of miR‐143 in cultured VSMCs treated with vehicle or PDGF. n=5; *P<0.05 compared with that in Ad‐miR‐143‐treated cells without PDGF. Ad‐GFP indicates adenoviruses expressing GFP; PDGF, platelet‐derived growth factor; VSMCs, vascular smooth muscle cells.
Pri‐miR‐145 to Pre‐miR‐145 Is the Critical Step of miR‐145 Biogenesis Responsible for the Downregulation of miR‐145 in Proliferative VSMCs and in Diseased Vascular Walls in Which p53 Is Involved
To determine which step is responsible for the downregulation of miR‐145 in proliferative VSMCs and in diseased vascular walls, pri‐miR‐145, pre‐miR‐145, and mature miR‐145 were determined by qRT‐PCR. As shown in Figure 4A and 4B, there was only a mild decrease in pri‐miR‐145 expression in both PDGF‐stimulated proliferative VSMCs and in balloon‐injured rat carotid arteries. However, a huge decrease in pre‐miR‐145 expression was found in these cells and vessels. The result suggested that pri‐miR‐145 to pre‐miR‐145 could be the critical step for the downregulation of miR‐145. To further confirm it, 2 strategies were applied in cultured VSMCs treated with PDGF. First, Ad‐miR‐145 (50 MOI) was used. Second, pre‐miR‐145 (10 nmol/L) was applied. As shown in Figure 4C, pri‐miR‐145 could be increased by >70‐fold via Ad‐miR‐145. The failure in the response of pre‐miR‐145 and mature miR‐145 suggested that miR‐145 biogenesis was blocked in the second step (pri‐miR‐145 to pre‐miR‐145). In contrast, a >5‐fold increase was found in miR‐145 expression in VSMCs treated with pre‐miR‐145 (Figure 4D), suggesting that the biogenesis of miR‐145 from pre‐miR‐145 to mature miR‐145 was still intact. To determine the potential role of p53 in the impaired process of pri‐miR‐145 to pre‐miR‐145 in PDGF‐stimulated VSMCs, we quantified pri‐miR‐145, pre‐miR‐145, and mature miR‐145 in VSMCs with (Ad‐p53, 50 nmol/L) or without (Ad‐GFP) overexpression of p53. As shown in Figure 4E, the process of pri‐miR‐145 to pre‐miR‐145, as well as the product of mature miR‐145, was significantly increased in p53‐overexpressed cells. In addition, pri‐miR‐145 was also mildly increased in Ad‐p53‐treated cells, suggesting that p53 may also have a weak effect on the transcription of pri‐miR‐145.
Figure 4.

Pri‐miR‐145 to pre‐miR‐145 is the critical step of miR‐145 biogenesis responsible for the downregulation of miR‐145 in proliferative VSMCs and in diseased vascular walls. A, Expression of pri‐miR‐145, pre‐miR‐145, and mature miR‐145 in VSMCs with or without PDGF treatment (10 ng/mL). n=5; *P<0.05 and **P<0.001 compared with that in VSMCs without PDGF (vehicle). B, Expression of pri‐miR‐145, pre‐miR‐145, and mature miR‐145 in normal uninjured rat carotid arteries and in arteries with balloon injury. n=5; *P<0.05 and **P<0.001 compared with that in uninjured arteries. C, Expression of pri‐miR‐145, pre‐miR‐145, and mature miR‐145 in PDGF‐stimulated VSMCs treated with Ad‐GFP or Ad‐miR‐145 (50 MOI). n=5; *P<0.05 and **P<0.001 compared with that in the Ad‐GFP‐treated group. D, pre‐miR‐145 (10 nmol/L) increased the expression of mature miR‐145. n=5; **P<0.001 compared with that in control oligo‐treated group. E, Effect of p53 overexpression on the levels of pri‐miR‐145, pre‐miR‐145, and mature miR‐145 in PDGF‐stimulated (10 ng/mL) VSMCs. n=6; *P<0.05 and **P<0.01 compared with that in the Ad‐GFP‐treated group. Ad‐GFP indicates adenoviruses expressing GFP; MOI, multiplicity of infection; PDGF, platelet‐derived growth factor; VSMCs, vascular smooth muscle cells.
Flank Sequences of Pri‐miR‐145 Are the Critical Structure Components Responsible for Downregulation of miR‐145 in Proliferative VSMCs and in Diseased Vascular Walls
As pri‐miR‐145 to pre‐miR‐145 is the critical step in control of the expression of miR‐145, we hypothesized that the flank sequences of pri‐miR‐145 should be critical genomic sequences for control of the expression of miR‐145. To test this hypothesis, we generated plasmids expressing miR‐145 vectors with different lengths of the flank sequence (40, 90, or 180 nt)—pDNR‐miR‐145:40, pDNR‐miR‐145:90, and pDNR‐miR‐145:180—and transfected them into HEK 293A cells. In HEK 293 cells, the transfection efficacy of these plasmids is about 93% with lipofectamine 2000 without any difference among the different groups (data not shown). As shown in Figure 5A, the expression of mature miR‐145 in these miR‐145‐treated cells was different. The result suggested that the flank sequence of pri‐miR‐145 indeed plays an important role in control of the expression of miR‐145.
Figure 5.

Generation of the smart‐miR‐145. A, Effects of plasmids expressing miR‐145 vectors with different lengths of the flank sequences (40, 90, or 180 nt)—pDNR‐miR‐145:40, pDNR‐miR‐145:90, and pDNR‐miR‐145:180—on the expression of miR‐145 in HEK 293A cells. n=5; *P<0.05 compared with pDNR‐miR‐145:40. B, Design of the smart miR‐145 in which the 110‐nt‐long flank sequences of pri‐miR‐145 were switched to the 110‐nt‐long flank sequences of pri‐miR‐31. C, Ad‐smart‐miR‐145 could avoid of the endogenous downregulatory mechanisms in proliferative VSMCs treated with PDGF. n=6; *P<0.05 compared with the Ad‐miR‐145 group. D, Effects of Ad‐smart‐miR‐145 on the expression of miR‐145 in balloon‐injured rat carotid arteries. n=5; *P<0.05 compared with the Ad‐GFP group. E. Switching of the 110‐nt‐long flank of miR‐31 to the flank sequences of miR‐145 (Ad‐modified‐miR‐31) impaired the expression of miR‐31 in VSMCs treated with PDGF (10 ng/mL). n=6; *P<0.05 compared with the Ad‐miR‐31 group. F, Effects of Ad‐miR‐145 (50 MOI) and Ad‐smart‐miR‐145 (50 MOI) on the levels of pri‐miR‐145, pre‐miR‐145, and mature miR‐145 in PDGF‐treated (10 ng/mL) VSMCs. n=6; *P<0.05 compared with the Ad‐miR‐145 group. Ad‐GFP indicates adenoviruses expressing GFP; CMV, control empty adenovirus; MOI, multiplicity of infection; PDGF, platelet‐derived growth factor; VSMCs, vascular smooth muscle cells.
Switching of Flank Sequences of miR‐145 to Flank Sequences of miR‐31 Can Create a Smart miR‐145
We have found that miR‐31, in contrast to miR‐145, is increased in proliferative VSMCs, in diseased vessels, and in other diseased tissues such as cancer. In the current study, we identified that the flank sequence of pri‐miR‐145 is related to control of the expression of miR‐145 (Figure 5A). These results let us generate a hypothesis: switching of the flank sequences of miR‐145 to the flank sequences of miR‐31 may create the smart miR‐145 that could avoid the endogenous downregulatory mechanisms of miR‐145 in proliferative VSMCs and in diseased vessels. To test our hypothesis, we switched the 110‐nt flank sequences of miR‐145 to the 110‐nt flank sequences of miR‐31 and named it the “smart” miR‐145 (smart‐miR‐145; Figure 5B). We then determined the effect of adenovirus expressing smart miR‐145 (Ad‐smart‐miR‐145) on the expression of miR‐145 in PDGF‐treated VSMCs (Figure 5C). We found that the Ad‐smart‐miR‐145 could indeed avoid the endogenous downregulatory mechanisms in proliferative VSMCs treated with PDGF. Moreover, smart‐miR‐145 could also avoid the endogenous downregulatory mechanisms in diseased rat arteries after balloon injury (Figure 5D). To unequivocally prove that impaired upregulation of miR‐145 by normal Ad‐miR‐145 is related to its flanking sequences, we also replaced the flanking sequences of miR‐31 with the flanking sequences from miR‐145 to create Ad‐modified‐miR‐31 and transfected it into PDGF‐treated VSMCs. As shown in Figure 5E, the expression of miR‐31 in Ad‐modified‐miR‐31‐infected cells was lower than that in VSMCs infected with normal Ad‐miR‐31.
To further understand the process of miR‐145 expression induced by Ad‐smart‐miR‐145, we determined the levels of pri‐miR‐145, pre‐miR‐145, and mature miR‐145 in PDGF‐treated VSMCs after transfection with Ad‐smart‐miR‐145 or Ad‐miR‐145 (50 MOI). As shown in Figure 5F, the accumulation of pri‐miR‐145 in Ad‐smart‐miR‐145‐treated cells was significantly decreased compared with that in Ad‐miR‐145‐treated VSMCs. Accordingly, the expression of pre‐miR‐145 and mature miR‐145 was increased in Ad‐smart‐miR‐145‐treated VSMCs compared with that in Ad‐miR‐145‐treated cells (Figure 5F).
Therapeutic Effects of Smart miR‐145 on Its Target Gene, Krüppel‐Like Zinc‐Finger Transcription Factor 5 (KLF5), Expression, VSMC Marker Genes, VSMC Proliferation In Vitro, and Vascular Neointimal Growth In Vivo
In our recent study, we have demonstrated that miR‐145 is able to increase the expression of differentiation marker genes such as SM alpha‐actin, calponin, and SM‐MHC and inhibit cell proliferation via its direct target gene, KLF5, in PDGF‐stimulated VSMCs.7 We thus compared the effects of the original miR‐145 (Ad‐miR‐145) and the smart miR‐145 (Ad‐smart‐miR‐145) on the expression of KLF5 in PDGF‐treated VSMCs. As shown in Figure 6A and 6B, both were able to decrease the expression of KLF5. However, the inhibitory effect on the expression of KLF5 in Ad‐smart‐miR‐145‐treated cells was stronger compared with that in Ad‐miR‐145‐treated cells. To compare the therapeutic effects of Ad‐miR‐145 and Ad‐smart‐miR‐145, VSMC proliferation and VSMC differentiation marker genes were determined in PDGF‐treated VSMCs. We found that the inhibitory effect of smart miR‐145 on cell proliferation was significantly increased compared with that of original miR‐145, as demonstrated by cell counting (Figure 6C) and the cell proliferation assay via MTT (Figure 6D). In addition, the expression of the differentiation marker genes in smart miR‐145‐treated VSMCs was significantly higher than that in original miR‐145‐treated VSMCs at both mRNA (Figure 6E) and protein (Figure 6F and 6G) levels.
Figure 6.
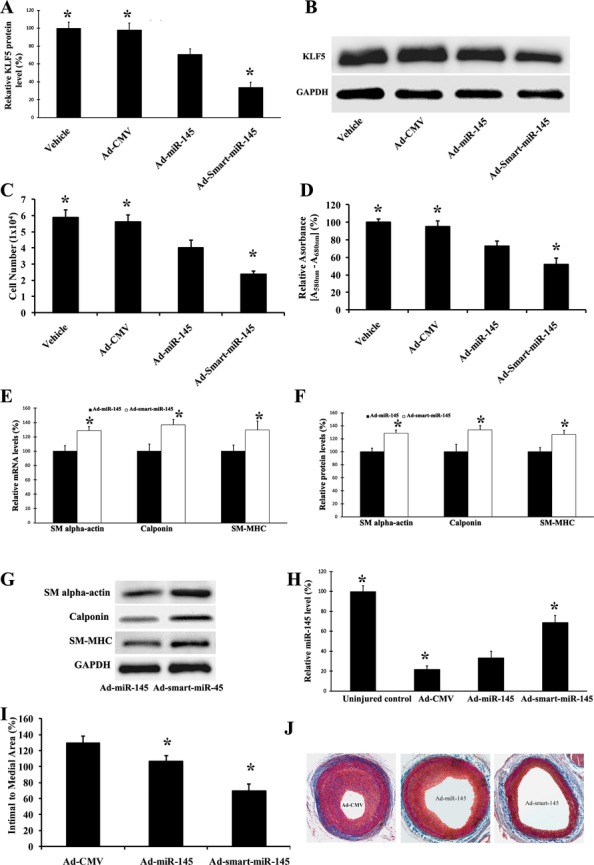
Therapeutic effects of smart miR‐145 on expression of its target, KLF5, VSMC proliferation, and VSMC marker genes in vitro in PDGF‐treated cells, and on vascular neointimal growth in vivo. A, Effects of Ad‐miR‐145 (50 MOI) and Ad‐smart‐miR‐145 (50 MOI) on the expression of KLF5 in VSMCs. n=5; *P<0.05 compared with the Ad‐miR‐145 group. B, Representative Western blots of KLF5 protein in VSMCs treated with vehicle, control virus, Ad‐GFP (50 MOI), Ad‐miR‐145 (50 MOI), or Ad‐smart‐miR‐145 (50 MOI). C, The inhibitory effect of Ad‐smart‐miR‐145 on cell proliferation was increased compared with that of Ad‐miR‐145 as demonstrated by cell counting. n=5; *P<0.05 compared with the Ad‐miR‐145 group. D, The inhibitory effect of Ad‐smart‐miR‐145 on cell proliferation was increased compared with that of Ad‐miR‐145 as demonstrated by the MTT assay. n=5; *P<0.05 compared with the Ad‐miR‐145 group. E, Upregulatory effects of Ad‐smart‐miR‐145 on the expression of VSMC marker genes, SM alpha‐actin, calponin, and SM‐MHC, was enhanced at the mRNA level compared with those of Ad‐miR‐145. n=5; *P<0.05 compared with the Ad‐miR‐145 group. F, Upregulatory effects of Ad‐smart‐miR‐145 on the expression of VSMC marker genes, SM alpha‐actin, calponin, and SM‐MHC, was enhanced at the protein level compared with those of Ad‐miR‐145. n=5; *P<0.05 compared with Ad‐miR‐145 group. G, Representative Western blots of SM alpha‐actin, calponin, and SM‐MHC in VSMCs treated with Ad‐miR‐145 (50 MOI), or Ad‐smart‐miR‐145 (50 MOI). H, Effects of Ad‐miR‐145 and Ad‐smart‐miR‐145 (1×109 pfu/mL) on the expression of miR‐145 in balloon‐injured rat carotid arteries. n=5; *P<0.05 compared with the Ad‐miR‐145 group. I, Effects of Ad‐miR‐145 and Ad‐smart‐miR‐145 (1×109 pfu/mL) on neointimal growth in rat carotid arteries 14 days after balloon injury. n=6; *P<0.05 compared with the Ad‐miR‐145 group. J, Representative images of rat carotid arteries treated with control virus (Ad‐CMV), Ad‐miR‐145, and Ad‐smart‐miR‐145. Ad‐GFP indicates adenoviruses expressing GFP; KLF5, Krüppel‐like zinc‐finger transcription factor 5; MOI, multiplicity of infection; PDGF, platelet‐derived growth factor; SM‐HMC, smooth muscle myosin heavy chain; VSMCs, vascular smooth muscle cells.
In our recent report, we identified that Ad‐miR‐145 at a large dose (100 μL, 5×109 pfu/mL) is able to inhibit vascular neointimal growth in rat carotid arteries after balloon injury.7 To compare the therapeutic effects of the original miR‐145 and smart miR‐145 in vivo, a low dose of Ad‐miR‐145 (100 μL, 1×109 pfu/mL) or Ad‐smart‐miR‐145 (100 μL, 1×109 pfu/mL) was applied by the local delivery model in rat carotid arteries after balloon injury.7 As expected, a mild increase in miR‐145 expression was found in the Ad‐miR‐145‐treated vessels (Figure 6H). Obviously, Ad‐smart‐miR‐145 at the same dose had given an additional increase in the expression of miR‐145 in these balloon‐injured vessels, although the level was still lower than that in the uninjured artery control (Figure 6F). Interestingly, the inhibitory effect of Ad‐smart‐miR‐145 on neointimal growth was significantly increased compared with that of Ad‐miR‐145 at the same dose (Figure 6I). Representative images of rat carotid arteries treated with control virus (Ad‐CMV), Ad‐miR‐145, and Ad‐smart‐miR‐145 are displayed in Figure 6J.
Discussion
Recent studies have revealed that miRNAs have strong biological functions that may affect almost every aspect of biology and biomedicine.3–5 It is well established that multiple miRNAs are aberrantly downregulated in many human diseases and are active participators in the pathogenesis of these diseases. Thus, upregulation of these downregulated miRNAs could be the promising novel therapeutic approach for human diseases including vascular disease.
In the current study, we have identified that there is a barrier to upregulating some miRNAs under disease conditions, because some endogenous mechanisms are able to downregulate the exogenous miRNAs. Indeed, although adenovirus‐mediated gene transfer could successfully upregulate miRNAs such as miR‐145 in normal vascular cells and in normal vessels, it failed to efficiently upregulate the downregulated miRNA in PDGF‐stimulated proliferative VSMCs and in diseased vessels with neointimal growth, as described in this study. This phenomenon is not limited to miR‐145, because a similar response was found in the expression of miR‐143 (Figure 3). The results suggest that some endogenous mechanisms in proliferative VSMCs and diseased arteries cannot only inhibit endogenous miR‐145/miR‐143 expression but also block the expression of exogenous miR‐145/miR‐143 by virus vectors. Uncovering the endogenous mechanisms responsible for the downregulation of these miRNAs and overcoming them are critical for miRNA‐based therapeutics in many diseases.
To overcome the critical barrier in miRNA‐145‐based therapy, we first determined the potential molecular mechanisms for its downregulation in proliferative VSMCs and in diseased arteries. It is well established that although the environmental cues affecting gene expression of VSMCs and vascular neointimal growth such as growth factors, reactive oxygen species, cell–cell contacts, and extracellular matrix components are divergent, the divergent cues converge into less redundant late nuclear events affecting gene expression and cell growth. Intercellular kinases such as PI3‐kinase, JNK, ERK, and p38 are thought to transduce these divergent extracellular signals to the nucleus.19–21 We therefore hypothesized that these kinases might be involved in the downregulation of miR‐145 in proliferative VSMCs and in diseased vessels. In the current study, we found that the downregulation in PDGF‐induced proliferative VSMCs was completely inhibited by inhibition of PI3‐kinase. However, no significant effects were found on the expression of miR‐145 in VSMCs treated with inhibitor of JNK, ERK, or p38. The results clearly showed that PI3‐kinase is the critical upstream signaling molecule responsible for miR‐145 downregulation in proliferative VSMCs. To further verify the role of PI3‐kinase in the regulation of miR‐145 expression in vascular walls in vivo, the rat carotid artery balloon‐injury model and the atherosclerotic mouse model were applied. The results demonstrated that the downregulation of miR‐145 in these diseased vessels was significantly inhibited by inhibition of PI3‐kinase. Thus, PI3‐kinase is also the critical signaling molecule in the control of expression of miR‐145 in the vascular walls in vivo. Akt is the well‐established downstream signal molecule of PI3‐kinase. Akt then could achieve its gene regulatory functions via its 2 major critical downstream transcription factors, Forkhead and p53. In the current study, we identified that PI3‐kinase/Akt/p53 is the critical signaling pathway responsible for the downregulation of miR‐145 in proliferative VSMCs and in balloon‐injured or atherosclerotic arteries.
PI3‐kinase/Akt/p53 might affect the expression of miR‐145 via any of the 3 key steps in its biogenesis. In this study, we have identified that pri‐miR‐145 to pre‐miR‐145 may be the critical step of miR‐145 biogenesis responsible for the downregulation of miR‐145 in proliferative VSMCs and in diseased vascular walls, which is regulated by PI3‐kinase/Akt/p53. Indeed, p53 activation or overexpression increased the expression of pre‐miR‐145, as well as mature miR‐145. In addition, pri‐miR‐145 was also mildly increased in Ad‐p53‐treated cells, suggesting that p53 may also have a weak effect on the transcription of pri‐miR‐145. The potential critical role of p53 in miR‐145 processing was also demonstrated by recent studies from other groups.15,22–23
In the current study, we found that the process of miR‐145 from pre‐miR‐145 to mature miR‐145 was still intact in proliferative VSMCs and in diseased vessels. Thus, the 2 arms and the loop structure of pri‐miR‐145 may be unrelated to control of the expression of miR‐145. We then hypothesized that the flank sequences of pri‐miR‐145 should be critical genomic components for control of the expression of miR‐145. Indeed, we identified that modification of the flank sequences of miR‐145 had a strong effect on the expression of miR‐145.
In contrast to miRNAs such as miR‐145, some miRNAs including miR‐31 were upregulated in proliferative VSMCs and in diseased vessels. These findings allowed us to generate a hypothesis: switching of the flank sequences of miR‐145 to the flank sequences of miR‐31 may create a novel genetically engineered smart miR‐145 that might be able to avoid the endogenous downregulatory mechanisms of miR‐145 in proliferative VSMCs and in diseased vessels. Our results have clearly demonstrated that smart miR‐145 is indeed able to avoid the endogenous downregulatory mechanisms and efficiently upregulate the expression of miR‐145 both in PDGF‐stimulated proliferative VSMCs and in diseased vessels compared with the original miR‐145. To further understand the process of miR‐145 expression induced by Ad‐smart‐miR‐145, we determined the levels of pri‐miR‐145, pre‐miR‐145, and mature miR‐145 in PDGF‐treated VSMCs after transfection with Ad‐smart‐miR‐145 or Ad‐miR‐145. The result revealed that the accumulation of pri‐miR‐145 in Ad‐smart‐miR‐145‐treated cells was significantly decreased, compared with that in Ad‐miR‐145‐treated VSMCs. Accordingly, the expression of pre‐miR‐145 and mature miR‐145 was increased in the Ad‐smart‐miR‐145‐treated groups.
It should be noted that creating the smart miRNAs by switching the flank sequences of the downregulated miRNAs in proliferative cells to the flank sequences of miR‐31 is not limited to miR‐145. For example, switching the flank sequences of the miR‐143 to the flank sequences of miR‐31 was able to generate smart miR‐143 (Figure 7A). Ad‐smart‐miR‐143 was able to avoid the endogenous downregulatory mechanisms of miR‐143 in proliferative VSMCs (Figure 7B). However, the new flank sequences in a smart miRNA cannot be freely selected from an upregulated miRNA in proliferative disease. For example, if we used the flank sequences of miR‐21, another upregulated miRNA in proliferative diseases,6,24 to replace the flank sequences of miR‐145, the newly created miR‐145, miR‐145‐21, was not a “smart” miR‐145. It could not efficiently upregulate the expression of miR‐145 in proliferative VSMCs, although the mechanism is unclear (Figure 8). Moreover, the importance of the flanking segments in pri‐miR‐145 processing was also demonstrated by adenovirus expressing the modified miR‐31, in which the flanking sequences of pri‐miR‐31 were replaced with the flanking sequences from pri‐miR‐145. As shown in Figure 5E, the expression of miR‐31 in Ad‐modified‐miR‐31‐infected cells was lower than that in VSMCs infected with normal Ad‐miR‐31.
Figure 7.

Generation of smart‐miR‐143. A, Design of smart miR‐143. B, Effects of Ad‐smart miR‐143 on the expression of miR‐143 in cultured VSMCs treated with vehicle or PDGF. n=5; *P<0.05 compared with that in Ad‐miR‐145‐treated cells. Ad‐GFP indicates adenoviruses expressing GFP; PDGF, platelet‐derived growth factor; VSMCs, vascular smooth muscle cells.
Figure 8.
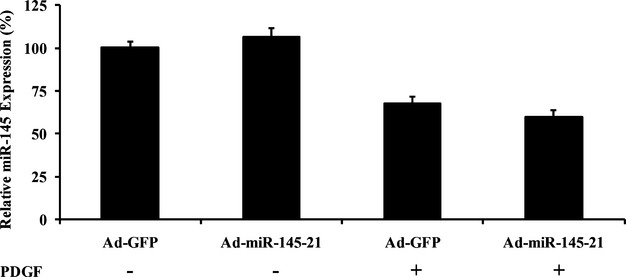
Effects of Ad‐miR‐145‐21 on the expression of miR‐145 in cultured VSMCs treated with vehicle or PDGF. Ad‐GFP indicates adenoviruses expressing GFP; PDGF, platelet‐derived growth factor; VSMCs, vascular smooth muscle cells.
Finally, the therapeutic effects of the smart miR‐145 on the expression of its target gene, KLF5, VSMC proliferation and VSMC maker gene expression in vitro, and vascular neointimal growth in vivo were determined and compared with those of the original miR‐145. Indeed, smart miR‐145 has a much better therapeutic effect both in vitro and in vivo. It should be noted that the improved therapeutic effects of the Ad‐smart miR‐145 is not related to its transfection efficiency, because we did not find a significant difference in transfection efficiency between Ad‐smart miR‐145 and Ad‐miR‐145. Although in this study we used proliferative vascular disease and proliferative VSMCs as examples, downregulation of the exogenous miRNA genes by endogenous mechanisms might be a general biological phenomenon in many proliferative human diseases. For example, in cancer cells, the responses of Ad‐miR‐145 and Ad‐smart‐miR‐145 were also different (Figure 9).
Figure 9.

Effects of Ad‐miR‐145 and Ad‐smart‐miR‐145 on the expression of miR‐145 in cultured human alveolar adenocarcinoma cell line A549 cells. n=5; *P<0.05 compared with that in Ad‐miR‐145‐treated cells.
We have found that the flank sequences of pri‐miR‐145 are critical genomic components for the downregulation of miR‐145 expression in proliferative VSMCs and in diseased arteries in which the PI3‐kinase/Akt/p53 pathway is involved; however, a major limitation of current study is that the detailed molecular mechanisms about how the flank sequences are regulated are still unclear. It is well established that miRNA processing from pri‐miRNA is a complex process in which many proteins such as Drosha, DGCR8, SMAD proteins, and p53 are involved, as described in some recent studies.14–15,14–23,14–27 The detailed regulatory mechanisms of these proteins on the flank sequences of pri‐miR‐145 in VSMCs and vascular walls need to be defined in future studies. Another limitation is that the sample sizes of the experiment groups are relatively very small, which might have affected the explanation of the experimental results.
In summary, in the current study we uncovered the molecular mechanisms of the downregulation of miR‐145 in proliferative VSMCs and in vascular disease and created the first genetically engineered smart miRNA in the treatment of vascular diseases. The study is also important for its transformative potential to other miRNAs and other human diseases. Thus, the smart miRNAs may have broadly therapeutic applications for many human diseases.
Sources of Funding
This work was supported by 3 NIH grants, HL095707, HL109656, NR013876, and a grant from the American Heart Association, 09GRNT2250567 (to C. Zhang). The research is also partially supported by RNA Bioscience.
Disclosures
C.Z., Y.C., J.Y., and X.L. are named as coinventors on a US patent application pertaining to Therapeutic and Diagnostic MIRNA Products (Patent Application #61476974).
References
- 1.Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003; 113:673-676 [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281-297 [DOI] [PubMed] [Google Scholar]
- 3.Zhang C. Novel functions for small RNA molecules. Curr Opin Mol Ther. 2009; 11:541-651 [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards JK, Pasqualini R, Arap W, Calin GA. MicroRNAs and ultraconserved genes as diagnostic markers and therapeutic targets in cancer and cardiovascular diseases. J Cardiovasc Transl Res. 2010; 3:271-279 [DOI] [PubMed] [Google Scholar]
- 5.Port JD, Sucharov C. Role of microRNAs in cardiovascular disease: therapeutic challenges and potentials. J Cardiovasc Pharmacol. 2010; 56:444-453 [DOI] [PubMed] [Google Scholar]
- 6.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense‐mediated depletion reveal an essential role of microRNA in vascular neointimal lesion formation. Circ Res. 2007; 100:1579-1588 [DOI] [PubMed] [Google Scholar]
- 7.Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA‐145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009; 105:158-166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR‐222 and miR‐221 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009; 104:476-487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin Y, Liu X, Cheng Y, Yang J, Huo Y, Zhang C. Involvement of microRNAs in hydrogen peroxide‐mediated gene regulation and cellular injury response in vascular smooth muscle cells. J Biol Chem. 2009; 284:7903-7913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR‐145 and miR‐143 regulate smooth muscle cell fate and plasticity. Nature. 2009; 460:705-710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boettger T, Beetz N, Kostin S, Schneider J, Krüger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009; 119:2634-2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G. The knockout of miR‐143 and ‐145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009; 16:1590-1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, Hristov M, Köppel T, Jahantigh MN, Lutgens E, Wang S, Olson EN, Schober A, Weber C. Delivery of microRNA‐126 by apoptotic bodies induces CXCL12‐dependent vascular protection. Sci Signal. 2009; 2:ra81. [DOI] [PubMed] [Google Scholar]
- 14.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA‐mediated microRNA maturation. Nature. 2008; 454:56-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009; 460:529-533 [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Zhang W, Zhu C, Bucher C, Blazar BR, Zhang C, Chen JF, Linden J, Wu C, Huo Y. Inactivation of the adenosine A2A receptor protects apolipoprotein E‐deficient mice from atherosclerosis. Arterioscler Thromb Vasc Biol. 2009; 29:1046-1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Y, Zhang C, Nair U, Du X, Yoo TJ. The morphological and functional alterations of the cochlea in apolipoprotein E deficient mice. Hear Res. 2005; 208:54-67 [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Zhang W, Tang R, Hebbel RP, Kowalska MA, Zhang C, Marth JD, Fukuda M, Zhu C, Huo Y. Core2 1‐6‐N‐glucosaminyltransferase‐I deficiency protects injured arteries from neointima formation in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol. 2009; 29:1053-1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Y, Cheng L, Hochleitner BW, Xu Q. Activation of mitogen‐activated protein kinases (ERK/JNK) and AP‐1 transcription factor in rat carotid arteries after balloon injury. Arterioscler Thromb Vasc Biol. 1997; 17:2808-2816 [DOI] [PubMed] [Google Scholar]
- 20.Mii S, Khalil RA, Morgan KG, Ware JA, Kent KC. Mitogen‐activated protein kinase and proliferation of human vascular smooth muscle cells. Am J Physiol. 1996; 270:H142-H150 [DOI] [PubMed] [Google Scholar]
- 21.Wang AB, Li HL, Zhang R, She ZG, Chen HZ, Huang Y, Liu DP, Liang CC. A20 attenuates vascular smooth muscle cell proliferation and migration through blocking PI3k/Akt singling in vitro and in vivo. J Biomed Sci. 2007; 14:357-371 [DOI] [PubMed] [Google Scholar]
- 22.Suzuki HI, Miyazono K. p53 actions on microRNA expression and maturation pathway. Methods Mol Biol. 2013; 962:165-181 [DOI] [PubMed] [Google Scholar]
- 23.Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY. p53 represses c‐Myc through induction of the tumor suppressor miR‐145. Proc Natl Acad Sci USA. 2009; 106:3207-3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng Y, Zhu P, Yang J, Liu X, Dong S, Wang X, Chun B, Zhuang J, Zhang C. Ischaemic preconditioning‐regulated miR‐21 protects heart against ischaemia/reperfusion injury via anti‐apoptosis through its target PDCD4. Cardiovasc Res. 2010; 87:431-439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeom KH, Lee Y, Han J, Suh MR, Kim VN. Characterization of DGCR8/Pasha, the essential cofactor for Drosha in primary miRNA processing. Nucleic Acids Res. 2006; 34:4622-4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.La Rocca G, Shi B, Sepp‐Lorenzino L, Baserga R. Expression of micro‐RNA‐145 is regulated by a highly conserved genomic sequence 3′ to the pre‐miR. J Cell Physiol. 2011; 226:602-607 [DOI] [PubMed] [Google Scholar]
- 27.Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha‐DGCR8 complex. Cell. 2006; 125:887-901 [DOI] [PubMed] [Google Scholar]