

Background: Phospholipase D (PLD) and phosphatidic acid regulate fundamental cellular processes that contribute to cancer cell proliferation and survival.
Results: Inhibition of PLD2 decreases activation of the pro-survival kinase Akt leading to cell death through inhibition of autophagic flux.
Conclusion: PLD2 promotes autophagy through regulation of Akt in glioblastoma cells.
Significance: PLD presents a novel opportunity for Akt inhibition without directly targeting the kinase.
Keywords: Akt, Autophagy, Cell Signaling, Glioblastoma, Phosphatidic Acid, Phospholipase D
Abstract
The lack of innovative drug targets for glioblastoma multiforme (GBM) limits patient survival to approximately 1 year following diagnosis. The pro-survival kinase Akt provides an ideal target for the treatment of GBM as Akt signaling is frequently activated in this cancer type. However, the central role of Akt in physiological processes limits its potential as a therapeutic target. In this report, we show that the lipid-metabolizing enzyme phospholipase D (PLD) is a novel regulator of Akt in GBM. Studies using a combination of small molecule PLD inhibitors and siRNA knockdowns establish phosphatidic acid, the product of the PLD reaction, as an essential component for the membrane recruitment and activation of Akt. Inhibition of PLD enzymatic activity and subsequent Akt activation decreases GBM cell viability by specifically inhibiting autophagic flux. We propose a mechanism whereby phosphorylation of beclin1 by Akt prevents binding of Rubicon (RUN domain cysteine-rich domain containing beclin1-interacting protein), an interaction known to inhibit autophagic flux. These findings provide a novel framework through which Akt inhibition can be achieved without directly targeting the kinase.
Introduction
Glioblastoma multiforme (GBM),2 the most common and aggressive glioma, is a highly lethal type of brain tumor with poor patient prognosis. Despite advances in imaging and neurosurgery over the past 30 years, GBM remains one of the most difficult tumors to manage with a median survival time of ∼14 months following diagnosis (1). Treatment options are limited and invasive, typically including a combination of surgical resection, radiotherapy, and adjuvant chemotherapy (2). The less invasive small molecule therapies for GBM have been met with limited success due, in part, to the poor blood-brain barrier penetrance of current chemotherapeutics that limits their access to GBM tumors. Moreover, the aggressive nature of GBM, its genetic variability, and antineoplastic drug resistance further curbs the effectiveness of small molecule inhibitors to treat the disease. The combination of these clinical obstacles contributes to the comparatively short survival times and patient death rate (3). Thus, the field requires a more thorough understanding of GBM signaling and metabolic pathways to develop novel treatment options.
Among the most frequently deregulated pathways in GBM are components of the phosphoinositide 3-kinase (PI3K)/Akt pathway (4). Activation of PI3K either by cell-surface receptor stimulation or constitutively activating mutations results in phosphatidylinositol 3,4,5-trisphosphate (PIP3) production and subsequently initiates signaling cascades by recruiting a variety of molecules containing lipid-binding domains to membranes (5). The serine/threonine kinase Akt was identified as the eukaryotic homolog of the retroviral oncogene protein v-Akt, which becomes activated following PI3K generation of PIP3 (6, 7). Akt mediates a variety of intracellular functions critical to oncogenic processes, including cell growth, proliferation, metabolism, and survival (8). Mutations that result in PI3K activation, such as constitutive growth factor receptor activation (9) or inactivation of phosphatase and tensin homolog (PTEN) (10), the lipid phosphatase that hydrolyzes PIP3, are common in GBM. Although small molecule inhibitors of the PI3K/Akt pathway hold promise in clinical trials for GBM (11), global inhibition of the Akt isoenzymes results in side effects that limit their clinical potential (12).
In addition to PIP3, other lipids are known to mediate intracellular signaling events that are required for oncogenic processes. Phospholipase D (PLD) enzymes hydrolyze membrane phospholipids, such as phosphatidylcholine (PC), to generate phosphatidic acid (PtdOH), an important lipid second messenger. Multiple cancer types, including breast, gastric, and renal cancers, show elevated PLD activity compared with normal tissue (reviewed in Selvy et al. (13)). In keeping with these observations, cells overexpressing PLD demonstrate increased anchorage-independent growth (14), invasiveness (15), and tumorigenesis in nude mice (16). Mechanistically, PLD and PtdOH regulate cytoskeletal rearrangement (17), angiogenesis (18), and expression of matrix metalloproteases (15), which are all requirements for invasion and metastasis. PLD also participates in a multitude of intracellular signaling pathways critical for cell survival, including the mitogen-activated protein kinase pathways (16, 19, 20), the mammalian target of rapamycin (mTOR) pathway (21), and nonreceptor tyrosine kinase pathways such as focal adhesion kinase (22) and Src kinase (23). The development of small molecule PLD inhibitors that decrease cancer cell invasiveness (24), along with the development of PLD knock-out mice that show no overt negative phenotypes (25, 26), makes PLD a promising therapeutic target.
Recent reports have suggested a possible relationship between PLD and Akt involving both direct (27, 28) and indirect (29) mechanisms. Interestingly, PLD from Neisseria gonorrhoeae regulates human Akt kinase activity upon infection of cervical epithelial cells (30). In this report, we investigate the regulation of Akt by human PLD and demonstrate a novel mechanism by which PtdOH activates Akt and mediates survival signaling in GBM cells. By targeting PLD, we explore novel treatment options for regulating Akt kinase activity for the treatment of human brain cancers.
EXPERIMENTAL PROCEDURES
Cell Culture
U87MG and U118MG cells (ATCC) and HEK293-TREx (Invitrogen) were maintained in DMEM (Invitrogen) + 10% FBS (Atlanta Biologicals) + 1% penicillin/streptomycin (Invitrogen). myrAkt1-U87MG cells were maintained in DMEM + 10% tetracycline-free FBS (Atlanta Biologicals) + 1% penicillin/streptomycin. CD133+ glioma stem cells were cultured as described previously (31). Stem cells were maintained in neurobasal media containing glutamine, B27, sodium pyruvate (all from Invitrogen), 20 ng/ml fibroblast growth factor, and epidermal growth factor (PeproTech). All human cells were maintained at 37 °C in a humidified incubator with 5% CO2. Sf21 insect cells were obtained from Orbigen and maintained in Grace's media (Invitrogen) supplemented with lactalbumin hydrolysate, yeastolate, sodium bicarbonate, and 10% FBS. Sf21 cells were maintained at 27 °C.
Plasmids and Baculovirus Production
The following plasmids were obtained from Addgene: pcDNA3 T7 Akt1 (William Sellers (32), plasmid 9003), pcDNA3 myr HA Akt1 (William Sellers (32), plasmid 1036), ptfLC3 (Tamotsu Yoshimori (33), plasmid 21074), and pcDNA4 beclin1-HA (Qing Zhong (34), plasmid 24399). FLAG-PLD1 and PLD2 were created by PCR amplification of the PLD open reading frames (PLD1 cDNA was obtained from Open Biosystems MGC collection, clone 6068382, and PLD2 cDNA was a generous gift from Dr. David Lambeth at Emory University) using forward primers containing FLAG epitope sequence and ligating into pcDNA5/TO (Invitrogen). To create the protein A-Tev-Strep-tagged PLD2 construct (PtS- PLD2), the PtS tag from p31-N-PtS (a kind gift from Dr. Yisong Wang (35)) was shuttled into pcDNA5/TO to create PtS-pcDNA5/TO, and the PLD2 ORF was subsequently ligated 3′ of the PtS ORF into PtS-pcDNA5 to create a PLD2 construct with an N-terminal PtS tag. To create the PtS-PLD2 baculovirus, the PtS-PLD2 ORF was ligated into pENTR1A (Invitrogen). After LR recombination into pDEST8 (Invitrogen), baculovirus was produced according to the manufacturer's instructions. A bacterial expression vector for the PtS tag was created by amplification of the PtS tag from PtS-pcDNA5/TO and ligated into pET16b (EMD Millipore). For His6-Akt1 baculovirus production, the Akt1 ORF was amplified from pcDNA3 myr HA Akt1 and ligated into pENTR3C (Invitrogen). pENTR3C was LR-recombined into pDEST10 (Invitrogen) to generate a His6-Akt1 construct, and baculovirus was produced according to the manufacturer's instructions.
Transfection and RNAi
For protein expression, cells were transfected using FuGENE 6 (Roche Applied Science) according to the manufacturer's instructions. All siRNA was obtained from Dharmacon as a pool of 4 oligonucleotide targeting sequences per relevant target (ON-TARGETplus). Cells were transfected according to the manufacturer's instructions using the Dharmafect 1 reagent and a final concentration of 100 nm siRNA.
Endogenous PLD Activity Assays
PLD activity assays were performed essentially as described previously (36). Following experimental treatments, cells were treated with 0.3% deuterated n-butanol (n-butanol-d10) for 30 min prior to phospholipid extraction and quantification of PLD-generated phosphatidylbutanol species. To generate concentration-response curves, U87MG cells were serum-starved for ∼24 h. Cells were treated with the indicated concentration of PLD inhibitor for 15 min prior to addition of n-butanol-d10 and phospholipid extraction.
Viability Assays
Cells were seeded into clear-bottom, black-walled 96-well tissue culture plates and allowed to adhere overnight to achieve ∼60% confluence the following day. Cells were serum-starved in the presence of indicated inhibitors overnight. Viability was measured by addition of the WST-1 reagent (Roche Applied Science) and reading absorbance at 450 nm. Time of WST-1 incubation was cell line-dependent and ranged from 30 min to 2 h.
To measure viability following RNAi treatment, cells were seeded in 60-mm tissue culture plates at 540,000 cells/plate and transfected with 100 nm siRNA. The next day, cells were split into 96-well plates, allowed to adhere overnight, and serum-starved in the presence of inhibitors the following day. Viability was assessed ∼72 h post siRNA transfection
Anchorage-independent Growth Assays
Base layers of 0.5 ml of 0.7% agarose (SeaKem GTG-agarose, Cambrex BioScience, Rockland, ME) containing complete neurobasal growth media were prepared in 12-well tissue culture plates. PLD inhibitors or DMSO vehicle controls were incorporated into the base layers. A 0.5-ml overlayer of 0.35% agarose containing CD133+ cells (5.0 × 103) in compete growth medium with PLD inhibitors was applied. Each condition was plated in triplicate wells. Plates were incubated at 37 °C in a humidified atmosphere of 5% CO2 in air. Cells were fed every 2–3 days with complete growth media plus PLD inhibitor. Colony formation progressed for 8 weeks. Crystal violet (0.005%) was used to stain colonies. Large colonies (>50 cells) were scored at ×10 magnification with an inverted phase microscope using an average of four random fields per sample.
Immunoblotting
Lysates were prepared by incubating cell pellets in lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 0.5% Nonidet P-40, 40 mm β-glycerophosphate, 20 mm sodium pyrophosphate, 1 mm Na3VO4, 2 mm EDTA, 2 mm EGTA, 5 mm NaF, 1 mm DTT, and complete protease inhibitor mixture from Roche Applied Science) for 30 min at 4 °C. Protein concentrations were measured using the Bio-Rad protein assay reagent. pan-Akt, Akt-S473, Akt-T308, beclin1, p62, Atg7, p70S6K, p70S6K-T389 antibodies were from Cell Signaling; GAPDH and PLD1 antibodies were from Santa Cruz Biotechnology; β-actin and FLAG (M2) antibodies were from Sigma; pan-cadherin and Rubicon antibodies were from Abcam; and PLD2, HA tag, strep tag and LC3 antibodies were from Abgent, Covance, Qiagen, and Novus, respectively. Bands were quantified using the gel analyzer function of ImageJ (National Institutes of Health).
Protein Purification
PtS-PLD2 infected Sf21 cells were harvested and collected by centrifugation at 500 × g for 5 min. Cells were lysed by sonication in lysis buffer (50 mm Tris, pH 8.0, 500 mm NaCl, 0.5% Nonidet P-40, 2.5 mm EDTA, and 50 μg/ml avidin, 1 mm DTT, complete protease inhibitor tablet (Roche Applied Science), and 1 mm PMSF added immediately prior to sonication). Lysate was cleared by centrifugation at 12,000 × g for 10 min. Cleared lysate was incubated with strep-tactin affinity resin (IBA) overnight at 4 °C. Beads were washed three times with wash buffer (lysis buffer with 0.01% Nonidet P-40). PtS-PLD2 was batch-eluted by incubation of beads with 5 mm desthiobiotin (Sigma) in wash buffer for 10 min, centrifugation at 1,000 × g, and collecting supernatants containing soluble PtS-PLD2. For protein-protein interaction studies, PtS-PLD2 eluates were dialyzed (5,000 MWCO, Invitrogen) overnight against wash buffer to remove desthiobiotin.
The PtS protein, used as a control for protein interaction experiments, was produced by transforming BL21 Escherichia coli (Agilent) with the pET16b-PtS plasmid. Bacteria were grown at 37 °C until the A600 reached 0.7. At that point, protein expression was induced by adding 100 μm isopropyl 1-thio-β-d-galactopyranoside and growing bacteria overnight at 18 °C. Bacteria were lysed by incubating in lysis buffer (30 mm sodium phosphate buffer, pH 7.4, 500 mm NaCl, complete protease inhibitor mixture, and 1 mg/ml lysozyme) for 30 min followed by sonication. Lysates were clarified by centrifugation at 14,500 × g for 30 min at 4 °C and then loaded onto a 1-ml Hi-Trap chelating column (GE Healthcare). The column was washed until the A280 returned to base line, and nonspecific proteins were eluted using a 40 mm imidazole step gradient. Once the A280 returned to base line, PtS was eluted in a linear imidazole gradient from 40–500 mm. Eluates were pooled and loaded onto a 120-ml Sephadex 75 gel filtration column (GE Healthcare), previously equilibrated with 50 mm Tris, pH 7.4, 0.5 mm EGTA, 150 mm NaCl, and 2 mm DTT. Fractions containing PtS protein were collected and pooled for use in binding assays.
N-terminal Akt PH domain His6-GST fusion proteins were produced by PCR amplifying the first 123 amino acids of Akt1 (37), ligating into pBG105 (Vanderbilt Structural Biology Core), and transforming BL21 E. coli. Bacteria were grown at 37 °C until A600 reached 0.7. At that point, protein expression was induced by adding 250 μm isopropyl 1-thio-β-d-galactopyranoside and growing bacteria overnight at 27 °C. Bacteria were lysed by incubating in lysis buffer (50 mm Tris buffer, pH 7.5, 150 mm NaCl, 1 mm EGTA, 1 mm EDTA, 1 mm Na3VO4, 10 mm β-glycerophosphate, 50 mm NaF, 5 mm DTT, complete protease inhibitor mixture (Roche Applied Science), and 1 mg/ml lysozyme) for 30 min followed by sonication. Lysates were clarified by centrifugation at 14,500 × g for 30 min at 4 °C and then applied to glutathione-agarose (Sigma) for 2 h at 4 °C. Resin was washed twice with lysis buffer and twice with wash buffer (50 mm Tris, pH 7.5, 300 mm NaCl, 0.1 mm EGTA, and 5 mm DTT) and eluted by incubating in wash buffer containing 10 mm reduced glutathione. His6-Akt1 was purified from Sf21 cells essentially as described previously (38).
Immunoprecipitation and in Vitro Protein-Protein Interaction Assays
Cells were resuspended in lysis buffer (see under “Immunoblotting”) and lysed with three freeze/thaw cycles in dry ice/ethanol, drawing lysate through a 25-gauge syringe needle between cycles. Clarified (10,000 × g for 10 min) lysates were pre-cleared with protein G-agarose (Millipore), and immunoprecipitating antibodies were incubated with lysate overnight. Complexes were captured using protein G-agarose, washed three times in lysis buffer, then eluted by boiling in 2× SDS-PAGE loading buffer.
For in vitro protein-protein interaction assays, 25 nm PtS-PLD2 or PtS tag was incubated with 50 nm His6-Akt1 for 2 h in 50 mm Tris, pH 8.0, 0.01% Nonidet P-40, 150 mm NaCl, 0.5 mm EDTA, and 50 μg/ml avidin. Where indicated, 10 μm inhibitor was included in the reaction mixture. PtS-tagged proteins were captured by incubation with streptactin resin for 2 h. Resin was washed three times and proteins eluted by boiling in 2× SDS-PAGE loading buffer
Protein-Lipid Binding
The general procedure for measuring Akt binding to lipid spots on nitrocellulose membranes has been described previously (39). The membrane containing various classes of lipids was obtained from Avanti Polar Lipids and contained 2 μg of the indicated lipid per spot. For vesicle competition assays, vesicles were prepared by drying lipids under N2 gas and resuspending in 50 mm Tris, pH 7.4, 150 mm NaCl, and 2 mm EGTA. Lipids were vortexed and sonicated until in solution. The vesicles were composed of either 100% 32:0 PC or 95% PC + 5% 32:0 PtdOH (mol %). Recombinant Akt was incubated for 2 h with 200 μm bulk vesicles and then incubated with nitrocellulose membranes overnight. Lipid-bound Akt was determined using a total Akt antibody and chemiluminescence.
Exogenous PtdOH Rescue
Lipids were obtained as chloroform solutions from Avanti Polar Lipids. Lipids were dried under N2 gas in glass Pyrex tubes. Dried lipid film was vortexed in DMEM + 0.25 mg/ml fatty acid-free BSA and then sonicated for 10 min. The sonicated lipid mixture was further diluted in the DMEM/BSA mixture to a final concentration of 1 mm, and cells were treated for the duration of time indicated in the text.
Membrane Isolation from U87MG Cells
U87MG cells were seeded on 150-mm tissue culture plates at 2.6 × 106 cells/plate (four plates per condition) and allowed to adhere overnight. The following day, cells were washed and media replaced with DMEM plus indicated inhibitor or vehicle, and cells were treated for 6 h. Cells were washed twice in 1× PBS and then scraped in homogenization buffer (20 mm HEPES, pH 7.4, 1 mm EDTA, 250 mm sucrose). Cells were pelleted by centrifugation at 1,000 × g for 5 min at 4 °C. Cell pellets were resuspended in homogenization buffer containing complete protease inhibitor mixture (Roche Applied Science), 10 mm β-glycerophosphate, 1 mm sodium pyrophosphate, 1 mm Na3VO4, and 5 mm NaF and lysed by nitrogen cavitation (1,000 p.s.i. for 5 min, 4 °C). Lysate was collected dropwise then centrifuged at 2,000 × g for 10 min to pellet unbroken cells, nuclei, and heavy debris. The supernatant was subsequently centrifuged at 100,000 × g for 60 min. The supernatant was saved as the cytosolic fraction and the 100,000 × g pellet was washed once by resuspension in lysis buffer then centrifuged again under the same conditions.
A stock iodixanol (Optiprep, Sigma) gradient solution was prepared by diluting the 60% iodixanol solution from the manufacturer in dilution buffer (120 mm HEPES, pH 7.4, 250 mm sucrose, 6 mm EDTA, 60 mm β-glycerophosphate, 6 mm Na3VO4, and 60 mm sodium pyrophosphate) in a ratio of 5 parts 60% iodixanol to 1 part dilution buffer to create a 50% iodixanol working solution. 2.5, 10, 17.5, 25, and 30% iodixanol solutions were prepared by mixing the appropriate ratios of 50% iodixanol with homogenization buffer. Washed membranes from the second 100,000 × g spin were resuspended in 30% iodixanol (∼500 μl) and added to 11-ml polycarbonate ultracentrifuge tubes (Beckman Coulter). Equal volumes of 25, 17.5, 10, and 2.5% iodixanol were layered on top of the membrane suspension, and tubes were centrifuged for 3.5 h at 165,000 × g in a swinging bucket rotor. 1-ml fractions were collected from the bottom by introducing a small hole with a 25-gauge syringe needle and collecting droplets. Samples were then boiled in 6× SDS-PAGE loading buffer prior to immunoblotting. Membranes predominantly banded at the 10/17.5% iodixanol interface.
Immunofluorescence
U87MG-tfLC3 cells were seeded on glass coverslips in 6-well plates in complete media and allowed to adhere overnight. The following day, cells were washed and treated with inhibitors in serum-free DMEM for 24 h. Cells were fixed for 15 min in 2% paraformaldehyde followed by washing in PBS. Coverslips were removed and mounted onto glass slides in Vectashield mounting media containing DAPI (Vector Laboratories). GFP/RFP images were acquired using Nikon A1R laser scanning confocal microscope equipped with a Plan Apo VC 60 × 1.4 N.A. and 40× oil immersion lens.
Statistical Analysis
Statistical analyses used for each figure are listed in the figure legend. Graphs of PLD activity and cell viability are representative from at least three experiments. Quantified immunoblots represent pooled data from at least three independent experiments unless otherwise noted in the text.
RESULTS
Phospholipase D2 Activity Is Required for Glioma Viability Following Serum Withdrawal
Multiple cancer types require PLD and its product, PtdOH, for sustained survival under stress conditions (20). Serum withdrawal, a known stimulus of PLD activity in multiple cancer cell lines (40), is frequently used to simulate the harsh growth environments encountered by neoplastic cells prior to vascularization and restoration of nutrient supply within the tumor mass. Viability is compromised when normal cells are cultured in serum-depleted conditions. Cells with elevated PI3K/Akt activity, however, continue to proliferate under these harsh culture conditions (41). To investigate the role of PLD in GBM survival, we measured PLD activity following serum withdrawal in the PTEN-null U87MG GBM cell line. Cells were grown overnight in complete growth media (DMEM with FBS) before growth in media lacking FBS for times ranging from 1 to 24 h. Serum withdrawal resulted in a time-dependent increase in PLD activity with the most robust activation observed after 16 h, and longer durations of serum withdrawal did not further increase PLD activity (Fig. 1A). By contrast, we did not observe increased PLD activity in the nontumorigenic HEK293 line under the same conditions (Fig. 1A), suggesting a cancer line-specific PLD response. Serum deprivation leading to PLD activation in U87MG cells is consistent with published reports on other cancer cell lines showing similar trends (40).
FIGURE 1.
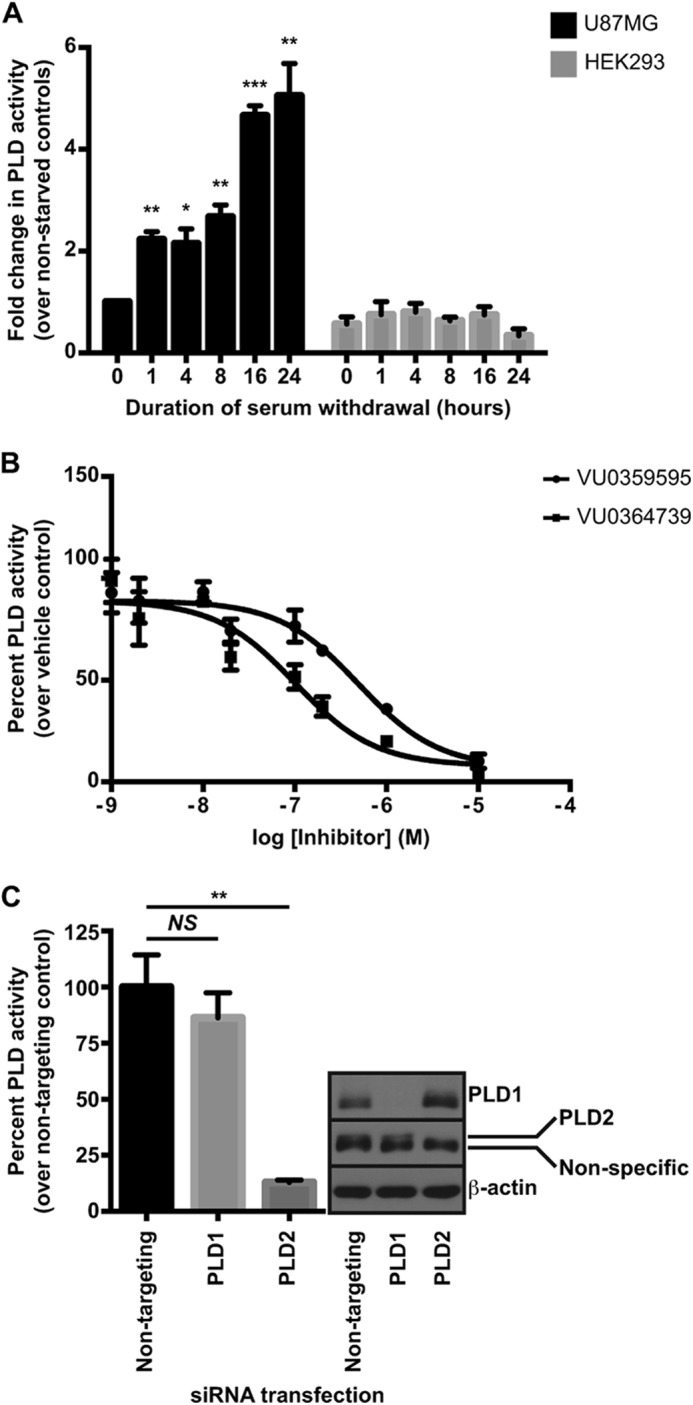
Serum withdrawal stimulates PLD2 activity in GBM cells. A, cells were seeded ∼24 h prior to washing and incubation in serum-free media for the indicated length of time. n-Butanol (n-butanol-d10) was added 30 min prior to glycerophospholipid extraction and subsequent phosphatidylbutanol quantification. B, U87MG cells were seeded as in A, and serum was withdrawn for 24 h. Cells were pretreated with inhibitors 30 min prior to measurement of PLD activity. Data are presented as the percent activity remaining after PLD inhibitor treatment relative to control. C, U87MG cells were transfected with siRNA targeting either PLD1 or PLD2 for 48 h prior to a 24-h serum starvation before measuring PLD activity. Note the PLD2 antibody recognizes a nonspecific band of similar molecular weight to PLD2, and the band of interest is directly above the nonspecific band. *, p < 0.5; **, p < 0.01; ***, p < 0.005, NS, not significant, unpaired Student's t test. Error bars, mean ± S.E.
Two isoforms of PLD have been identified, PLD1 (42) and PLD2 (43), and each demonstrates distinct regulatory properties. PLD1 is quiescent under normal growth conditions and requires stimulation by proteins, including small GTPases such as Arf (44), whereas PLD2 displays higher basal activity and is generally unresponsive to activators of PLD1 (43). To better understand the role of each isoform in this stress pathway, we explored which PLD isoform was preferentially activated following serum withdrawal using both pharmacological and genetic tools. In the first approach, U87MG cells were serum-deprived overnight and then treated with various concentrations of previously reported, isoform-preferring PLD inhibitors. VU0359595 is a 1,700-fold PLD1-preferring compound (45), and VU0364739 is a 75-fold PLD2-preferring compound (46), as determined with cell-based assays designed to measure activity of individual PLD isoforms. In the U87MG cells, which express both PLD1 and PLD2, VU0359595 and VU0364739 attenuated PLD activity following serum withdrawal with IC50 values of ∼500 and 100 nm, respectively (Fig. 1B). The 5-fold greater potency of the PLD2-preferring compound suggests that the PLD2 isoform is responsible for the vast majority of PLD activity in these cells following serum withdrawal, although the PLD1 isoform may partially compensate following acute inhibitor treatment. To further explore the contribution of individual isoforms to the total PLD activity, we utilized isoform-specific siRNA to knock down either PLD1 or PLD2 and to measure PLD activity following overnight serum withdrawal. Silencing of PLD2, but not PLD1, resulted in a significant decrease in PLD activity (Fig. 1C), further implicating PLD2 as the predominant isoform in the serum withdrawal response.
To determine whether PLD activity was required for viability in U87MG cells following serum withdrawal, we measured cell viability following overnight treatment with various concentrations of PLD inhibitors. U87MG cell viability decreased in a concentration-dependent manner (Fig. 2A), consistent with concentrations needed to completely ablate PLD activity (Fig. 1B), suggesting that complete suppression of PLD activity compromises viability in these cells. By contrast, treatment of HEK293 cells with PLD inhibitors resulted in significantly less cell death when compared with U87MG cells (Fig. 2B), further implicating PLD as necessary for cancer cell survival.
FIGURE 2.

PLD activity is required for cell viability and anchorage-independent growth in GBM cells. A, cells were treated with indicated concentration of PLD inhibitor for 24 h in serum-free media. Following inhibitor treatment, viability was measured using the WST-1 reagent. B, U87MG or HEK293 cells were grown in complete growth media for 24 h. Cells were then treated for 24 h with 10 μm VU0364739 or 20 μm VU0359595 in serum-free DMEM. Viability was measured using the WST-1 reagent. ***, p < 0.001 two-way analysis of variance (ANOVA) with Sidek's post hoc test to compare viability between each cell line within each inhibitor treatment group. C, CD133+ glioma stem cells were seeded into 96-well plates in media containing growth supplements and laminin to facilitate adhesion. 24 h after seeding, cells were treated with 10 μm VU0364739 or 20 μm VU0359595 in neurobasal media without supplements for an additional 24 h. Viability was measured as in A. D, anchorage-independent growth of glioma stem cell clone 4302 was assessed using soft agar colony formation. Growth media were replaced every 2–3 days with 10 μm PLD inhibitor or vehicle. Colonies were allowed to form for 8 weeks. Large colonies were scored after visualization with Crystal Violet. C and D, **, p < 0.01; ***, p < 0.005, unpaired Student's t test. Error bars, mean ± S.E.
Although U87MG cells are a well characterized GBM line, we wanted to extend our study on PLD to a more disease-relevant model, namely cells isolated from biopsies of primary human GBMs. Glioma stem cells can be isolated from patient tumors by sorting for surface expression of the CD133 antigen. These stem cells are tumorigenic and phenocopy the patient's original tumor when injected into immunocompromised mice (47). Two glioma stem cell clones (31), derived from individual patients, both showed reduced viability following PLD inhibitor treatment under growth factor starvation (Fig. 2C). Anchorage-independent growth, the most important measure of tumorigenicity (48), was then assessed in these stem cells. Following PLD inhibitor treatment, GBM stem cells formed significantly fewer colonies than vehicle control samples in soft agar, even in the presence of growth factor supplements (Fig. 2D). Together, these results demonstrate that PLD activity is required for proliferation and survival in glioma cells.
PLD2 Is Required for Akt Activation in GBM Cells
After establishing a requirement for PLD2 in glioma cell viability, we wanted to determine the mechanism by which PLD2 regulates survival signaling. The PI3K/Akt pathway is frequently up-regulated in cancer and promotes survival by inhibiting apoptotic processes and by regulating metabolism and nutrient utilization (8). Additionally, extracellular pathogens are known to engage the Akt pathway upon infection, and bacterial PLD from N. gonorrhoeae was demonstrated to interact with and activate human Akt upon infection of human cervical epithelial cells (30). We hypothesized there could be an interaction or regulation between human PLD and Akt. To determine whether human PLD2 regulated Akt activation in PTEN-null glioma lines, we measured Akt phosphorylation following treatment with PLD inhibitors under various growth conditions. Under the canonical Akt activation sequence, PI3K generates PIP3, which serves as a membrane recruitment signal for Akt (7, 49). Membrane-bound Akt is subsequently activated via a phosphorylation dependent mechanism whereby 3-phosphoinositide-dependent kinase 1 phosphorylates Akt at threonine 308 in the activation loop, and other kinases such as the mammalian target of rapamycin complex 2 (mTORC2) phosphorylate Akt in its hydrophobic motif at serine 473 (50, 51). Cells were treated overnight with PLD inhibitors in either serum-free DMEM, DMEM + 10% FBS, or DMEM followed by stimulation for 10 min the following day with 20% FBS. Because the PLD1- and PLD2-preferring inhibitors are chemically unique compounds that have few structural similarities, using either inhibitor individually at concentrations high enough to inhibit both isozymes (Fig. 1B) without causing substantial cell death (Fig. 2A) allowed us to minimize possible off-target effects associated with an individual compound. Inhibition of PLD in the PTEN-null U87MG (Fig. 3, A and B) and U118MG (Fig. 3, C and D) cell lines resulted in decreased levels of activated Akt under serum-depleted conditions as assessed by phosphorylation of threonine 308 and serine 473. Akt phosphorylation was less affected by PLD inhibition when cells were cultured normally or when stimulated with 20% FBS. These results strongly suggest that PLD regulates Akt activation predominantly under stressful, serum-depleted conditions. Additionally, these results suggest that the PLD inhibitors do not inhibit upstream kinases or Akt directly because growth factor signaling to Akt remains unperturbed. By contrast, PLD inhibitors do not reduce phosphorylated Akt in the nontumorigenic HEK293 cell line under any condition (Fig. 3, E and F), suggesting a cell type-specific regulation of Akt by PLD. We next knocked down either PLD1 or PLD2 to dismiss any off-target effects of PLD inhibitors and also to further link a specific PLD isoform to this process. Transfection of U87MG cells with PLD2, but not PLD1, siRNA resulted in a significant decrease in phosphorylated Akt at both threonine 308 and serine 473 (Fig. 3, G and H). Taken together, the data demonstrate that PLD regulates Akt activation under serum-depleted conditions and that regulation is due to the PLD2 isoform.
FIGURE 3.

Akt activation requires PLD activity in GBM cells. U87MG (A and B), U118MG (C and D), or HEK293 (E and F) cells were incubated overnight in the presence of 10 μm VU0359595 or 5 μm VU0364739 in either DMEM (starved) or DMEM + 10% FBS (normal). Another set of cells was starved overnight and then stimulated the following day with 20% FBS for 10 min (stimulated) for comparison. Akt activation was assessed by immunoblotting for phosphorylation at Thr-308 and Ser-473. Blots were quantified by calculating the ratio of phospho-Akt at Thr-308 to total Akt using densitometry. Ser-473 was not quantified because no qualitative differences were seen between Ser-473 and Thr-308. *, p < 0.05; **, p < 0.01; ***, p < 0.005. ANOVA with Dunnett's post hoc test comparing the PLD inhibitor treatment to vehicle within the growth condition. G, U87MG cells were transfected with either PLD1 or PLD2 siRNA for 48 h prior to overnight serum starvation and immunoblotting. H, quantification of phospho-Akt (Thr-308 and Ser-473) following PLD siRNA treatment in U87MG cells. Data represent the fold change in phospho-Akt relative to the nontargeting siRNA controls and are averages from three independent experiments. *, p < 0.05, paired Student's t test. Error bars, mean ± S.E. NS, not significant.
Phosphatidic Acid Regulates Akt Activation
To determine the mechanism by which PLD2 regulates Akt activation following serum withdrawal in GBM cells, we initially investigated a potential protein-protein interaction as was demonstrated with PLD from N. gonorrhoeae (30). Although we detected a specific interaction of Akt with PLD2, but not PLD1, in cell lysates (Fig. 4, A and B) and with recombinant, purified proteins (Fig. 4, C and D), PLD inhibitors did not disrupt the PLD2-Akt complex formation (Fig. 4E). These data suggest that the small molecule PLD inhibitors are not mediating Akt regulation by disrupting the PLD2-Akt protein complex.
FIGURE 4.

Akt and PLD2 form a direct protein complex. A, U87MG cells were transfected with vector or FLAG-tagged PLD1 or PLD2 for 48 h. FLAG-PLD1 or PLD2 was immunoprecipitated (IP), and binding of endogenous Akt was assessed by immunoblotting FLAG-PLD complexes for Akt. B, U87MG cells were lysed and endogenous Akt immunoprecipitated overnight using a pan-specific Akt antibody. Nonspecific proteins were immunoprecipitated using normal rabbit IgG. Note, ∼0.5% of the material used for IP was loaded into the lysate lanes for A and B. C, Coomassie Brilliant Blue-stained gel of PLD2 and Akt to demonstrate protein purity. Numbers indicate molecular weights in kilodaltons. D, purified Akt was incubated with either PLD2 or protein A-tev-Strep (PtS) tag for 2 h, and complexes were captured using affinity resin. Bound Akt was determined by immunoblotting for Akt to demonstrate a direct protein-protein interaction between PLD2 and Akt. E, same procedure as D except 10 μm VU0359595, VU0364739, or Akt inhibitor MK2206 was included in the reaction mixture.
Because PLD inhibitors did not disrupt the PLD2-Akt protein complex, we investigated a potential regulation of Akt by PtdOH, the catalytic product of PLD. To confirm that the decrease in Akt phosphorylation following PLD inhibitor treatment or siRNA knockdown was due to the decrease in PtdOH production, we attempted to rescue Akt phosphorylation by co-treatment of U87MG cells with PLD inhibitors and exogenously added PtdOH. Our laboratory recently published a detailed lipidomic characterization of PtdOH species generated by PLD in an astrocytoma cell line (52). Based upon this analysis and others, we attempted to rescue Akt phosphorylation using 36:2 PtdOH, a species generated by PLD. We observed complete rescue of Akt phosphorylation with exogenously added PtdOH (Fig. 5, A and B), suggesting that decreased Akt phosphorylation following PLD inhibitor treatment was due to decreased production of PtdOH by PLD.
FIGURE 5.

Akt recruitment to membranes is enhanced by binding to PtdOH. A, U87MG cells were treated overnight with 10 μm VU0359595 or 5 μm VU0364739 in DMEM containing 0.25 mg/ml fatty acid-free BSA. Where indicated, cells were co-treated with 1 mm PtdOH or BSA control, and Akt phosphorylation was assessed by immunoblotting Thr-308 and Ser-473. B, quantification of PtdOH rescue of Akt phosphorylation following PLD inhibitor treatment. Fold changes in phospho-Akt (Thr-308) were determined relative to the vehicle-treated BSA control. Data were analyzed by repeated measures ANOVA across all conditions and post hoc paired t tests in indicated conditions. *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, 5 nm recombinant Akt was incubated overnight with nitrocellulose membranes containing 2 μg of the indicated lipid. Bound Akt was measured using an Akt-specific antibody. D, recombinant Akt (3 nm) was incubated with 200 μm bulk lipid vesicles for 1 h and then the Akt/vesicle mixtures were incubated overnight with nitrocellulose spotted with PtdOH or PIP3. Bound Akt was determined using an Akt-specific antibody. E, protein-lipid overlay measuring wild-type or R25C PIP3 binding-deficient mutant Akt PH domain (3 nm) binding to PtdOH or PIP3. F and G, U87MG cells were treated for 6 h with 10 μm PLD inhibitor in the absence (F) or presence (G) of 1 mm 36:2 PtdOH in DMEM + 0.25 mg/ml fatty acid-free BSA and separated into cytosol and membranes for immunoblotting. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and pan-cadherin were used as cytosolic and membrane markers, respectively. Error bars, mean ± S.E. WCL, whole cell lysate; NS, not significant. PE, phosphatidylethanolamine; PC, phosphatidylcholine; PG, phosphatidylglycerol; PS, phosphatidylserine; PI, phosphatidylinositol.
Several studies have recently suggested that Akt binds other phospholipids in addition to phosphoinositide, including phosphatidylserine (53) and PtdOH (54). To compare the relative affinity of Akt for various phospholipids, we measured Akt binding using a commercially available protein-lipid overlay assay (39) in which phospholipids are spotted onto nitrocellulose, and binding of recombinant protein is detected immunologically. In agreement with other studies, recombinant Akt bound PtdOH and with higher affinity than other phospholipids (Fig. 5C).
Phosphatidic Acid Enhances Akt Binding to PIP3 and Subsequent Membrane Recruitment
To understand the mechanism by which PtdOH regulates Akt activation, we first determined whether PtdOH and PIP3 share a binding site on Akt. Recombinant Akt was incubated with lipid vesicles composed of PC alone or PC with PtdOH. Akt binding to PtdOH and PIP3 was then assessed using a protein-lipid overlay assay. When preincubated with vesicles containing PtdOH, binding of Akt to PtdOH on nitrocellulose was diminished, and this condition served as an internal control for the experiment (Fig. 5D). Intriguingly, binding of Akt to PIP3 was strongly enhanced by preincubation with PtdOH-containing vesicles (Fig. 5D). Because PIP3 is known to bind the Akt PH domain based on published crystal structures (37), we investigated whether the PH domain also mediates the interaction with PtdOH. GST-Akt PH domain fusion proteins were purified from E. coli, and lipid binding was again assessed using a protein-lipid overlay. We purified the wild-type (WT) Akt PH domain and an Akt PH domain mutant deficient in PIP3 binding, R25C (37), to determine whether perturbing PIP3 binding would also alter PtdOH binding. The WT PH domain of Akt was sufficient to bind PtdOH, and disruption of PIP3 binding with the R25C mutant had no effect on PtdOH binding (Fig. 5E). These results suggest that PtdOH binds a distinct site on the PH domain of Akt and that the binding of PtdOH acts cooperatively to increase the affinity of Akt for PIP3.
Because PIP3 recruits Akt to membranes (6, 7) and PtdOH increased Akt binding to PIP3, we investigated the possibility that PLD-generated PtdOH regulates membrane localization of Akt. Membranes from serum-starved U87MG cells treated with vehicle or PLD inhibitors were prepared by flotation through a discontinuous iodixanol gradient. Under control conditions, Akt was present in both cytosolic and membrane fractions, and as we hypothesized, inhibition of PLD decreased the levels of both total and phosphorylated Akt in the membrane fraction (Fig. 5F). Akt membrane recruitment was less sensitive to PLD inhibition in the presence of serum, and Akt was not detected in the membrane fractions of serum-starved HEK293 cells except under conditions where film was extremely overexposed (data not shown), consistent with constitutive activation of Akt in PTEN-null GBM cells. To confirm that PLD inhibitors decreased Akt membrane localization in a PtdOH-dependent manner, we co-treated U87MG cells with PLD inhibitors and the PLD product PtdOH before preparing membranes. Interestingly, co-treatment of cells with PtdOH not only rescued Akt membrane localization but PtdOH treatment resulted in a dramatic relocalization of cytosolic Akt to the membrane fraction (Fig. 5G). These data implicate PLD-generated PtdOH as a crucial mediator of Akt membrane recruitment in GBM cells.
PLD2 Inhibition Induces Autophagy-dependent Cell Death
After establishing a requirement for PLD2-generated PtdOH in the activation of Akt in U87MG cells, we next determined the mechanism of cell death following inhibition of the PLD2-Akt pathway by first measuring markers for apoptosis in U87MG cells. PLD inhibition only modestly induced caspase-3/7 cleavage relative to a well characterized apoptotic stimulus, staurosporine (55), and treatment of U87MG cells with a pan-caspase inhibitor failed to rescue PLD inhibitor-induced cell death (data not shown). Together, these results suggest that PLD inhibition predominantly leads to nonapoptotic cell death in this GBM line.
In addition to apoptosis, cells undergo another type of programmed cell death requiring autophagy (56), a process known to be stimulated by nutrient or growth factor deprivation (57). Autophagy is a multistep process involving formation of double-membrane autophagosomes that engulf cytosolic components and deliver cargo to lysosomes for digestion and nutrient recycling (58). To determine whether autophagy was perturbed following PLD or Akt inhibitor (MK2206) (59) treatment, we measured the expression levels of the well characterized autophagy markers microtubule-associated protein 1A/1B-LC3 and p62. Autophagosome number is frequently assessed by measuring conversion of cytosolic LC3-I to the membrane-associated, lipidated LC3-II, which is readily measured as a faster migrating species of LC3 during SDS-PAGE (60). The other marker, p62, is an LC3 and ubiquitin-binding protein involved in the regulation of protein aggregates and is degraded by autophagy (61). Induction of autophagy and successful degradation of autophagosomes would thus be accompanied by a decrease in p62 levels. Overnight treatment of U87MG cells with PLD or Akt inhibitors robustly induced LC3-II conversion and also increased p62 levels (Fig. 6, A and B). These results suggest an increased number of autophagosomes resulting from a deficiency in autophagosome turnover as is often observed under conditions where autophagy is defective (62). In the presence of PtdOH, PLD inhibitors failed to increase autophagy markers, which further validates the specificity of our inhibitors (Fig. 6, C–E). LC3-II and p62 levels also increased in other glioma cell lines, including the CD133+ glioma stem cells (Fig. 6F) and U118MG cells (data not shown) following PLD inhibitor treatment.
FIGURE 6.

PLD inhibitors induce autophagy-dependent cell death in GBM. A, immunoblot of autophagy markers from U87MG cells treated overnight with 10 μm VU0359595, 5 μm VU0364739, or 10 μm MK2206 in serum-free DMEM. B, quantification of LC3-II and p62 increases in U87MG cells following PLD or Akt inhibition. Fold change in immunoreactivity relative to vehicle control. *, p < 0.05; **, p < 0.01, ANOVA with Dunnett's post hoc test comparing inhibitor treatment to vehicle control. C, immunoblot of autophagy markers from U87MG cells treated overnight with PLD inhibitors with or without 1 mm 36:2 PtdOH in DMEM + 0.25 mg/ml fatty acid-free BSA. D and E, quantification of LC3-II and p62 from C. *, p < 0.05; **, p < 0.01, ANOVA with Dunnett's post hoc test comparing inhibitor treatment to vehicle control within the PtdOH treatment conditions. F, immunoblot of autophagy markers from CD133+ glioma stem cells treated overnight with 10 μm VU0359595 or 5 μm VU0364739 in neurobasal media without growth supplements. G, comparison of autophagy marker expression in U87MG or HEK293-TREx cells treated overnight with 10 μm VU0359595, 5 μm VU0364739, or 10 μm MK2206 in serum-free DMEM. H, viability assay from U87MG cells treated with siRNA targeting Atg7. Cells were treated overnight with PLD or Akt inhibitors in serum-free DMEM before measuring viability with the WST-1 reagent. *, p < 0.05; **, p < 0.01, two-way ANOVA with post hoc Sidak multiple comparison test on Atg7 siRNA effect within inhibitor treatment groups. I, immunoblot of LC3 and Atg7 for experiment in H. Error bars, mean ± S.E. N/T, nontargeting siRNA.
To determine whether the effects on autophagy following PLD inhibition were cell type-specific, we compared LC3/p62 levels between U87MG cells and HEK293 cells. PLD and Akt inhibitors increased LC3-II conversion in both cell types (Fig. 6G). However, the levels of LC3 and p62 under basal conditions were much higher in the U87MG cells, suggesting that glioma cells utilize autophagy more so than other cell types, rendering them particularly sensitive to compounds that perturb autophagy.
To confirm that U87MG cells were undergoing autophagy-dependent cell death, we measured the ability of PLD inhibitors to decrease viability when machinery required for autophagosome formation was perturbed by siRNA knockdown. Atg7 is a ubiquitination E1-like enzyme required for autophagosome formation (63). Knockdown of Atg7 significantly increased viability (Fig. 6H) and decreased LC3-II conversion (Fig. 6I) following PLD inhibition in U87MG cells. These results strongly suggest that GBM cell death resulting from PLD inhibition is predominantly through an autophagy-dependent mechanism.
PLD and Akt Inhibition Reduces Autophagic Flux
The increased conversion of LC3-II and increased expression of p62 after inhibitor treatments in glioma cells suggest that autophagic flux requires PLD and Akt activity. To conclusively determine that flux, rather than autophagosome initiation, is regulated by PLD, we measured LC3-II conversion in the presence of the lysosomal proton pump inhibitor bafilomycin A1, which prevents autophagosome fusion to lysosomes and inhibits degradation of autophagosomes. Thus, bafilomycin A1 is commonly used to discriminate the effects of a compound on autophagy initiation versus flux by assessing LC3-II levels in the presence of a test compound after clamping degradation of autophagosomes (64). Bafilomycin A1, PLD, and Akt inhibitor treatment increased LC3-II levels relative to vehicle control (Fig. 7, A and B). However, no additional accumulation of LC3-II was measured when PLD or Akt inhibitors were added in the presence of bafilomycin A1, confirming that PLD and Akt were controlling degradation of autophagosomes.
FIGURE 7.

PLD inhibition decreases autophagic flux. A, immunoblot of LC3 from U87MG cells treated with 10 nm bafilomycin A1 for 30 min followed by treatment with 10 μm VU0359595, 5 μm VU0364739, or 10 μm MK2206 for 6 h in serum-free DMEM before cell harvest. B, quantification of LC3-II conversion in the presence of bafilomycin A1 and PLD/Akt inhibitors. Data are presented as the ratio of band intensity for LC3-II relative to actin. *, p < 0.05; ***, p < 0.001, two-way ANOVA with Tukey's post hoc test on PLD/Akt inhibitor effects versus vehicle control; NS, no significant impact of PLD inhibitors in conditions with bafilomycin A1. C, representative images of U87MG stable cells expressing a GFP/RFP-LC3 tandem-fluorescent tag. Cells were treated overnight in serum-free DMEM with 10 μm VU0359595, 5 μm VU0364739, 10 μm MK2206, or 10 nm bafilomycin A1 and then fixed and imaged using confocal microscopy. Error bars, mean ± S.E.
To further demonstrate decreased degradation of autophagosomes following PLD2/Akt inhibition, we generated a stable U87MG cell line to express a tandem-fluorescent LC3 reporter used to assess autophagosome maturation (33). This reporter system consists of an RFP and GFP fused to LC3. As autophagosome numbers increase, either due to increased autophagy initiation or decreased degradation, fluorescence intensity increases as LC3 clusters on autophagosome membranes. Unlike RFP, GFP is quenched by low pH, and LC3 present in lysosomes should predominantly emit an RFP signal. Under situations where autophagosome degradation is perturbed, the GFP and RFP signals co-localize because autophagosomes do not fuse to acidic lysosomes. The tandem-fluorescent LC3 reporter U87MG cells were treated with PLD or Akt inhibitors overnight, fixed, and imaged for GFP and RFP signals. Under vehicle-treated conditions, numbers of LC3 puncta were low and predominantly visualized with the pH-insensitive RFP tag, indicative of functional autophagy. However, PLD or Akt inhibitor treatments induced a robust relocalization of cytosolic LC3-I to large fluorescent puncta, and when merged, the GFP/RFP signals highly co-localized, indicating a perturbation in the ability of the cell to effectively degrade and process autophagosomes (Fig. 7C).
After establishing that PLD and Akt promote autophagic flux, we sought to understand the molecular mechanism. The mammalian target of rapamycin (mTOR) pathway suppresses autophagy under nutrient-rich conditions, and PLD has been implicated as an upstream positive regulator of mTOR (65). Although we measured diminution of mTOR activity with Akt inhibition, we measured little to no change in mTOR effector phosphorylation status with PLD inhibition (Fig. 8A), suggesting the mTOR pathway was not mediating the effects of PLD inhibitors on autophagy and also suggesting that PLD2 and mTOR signaling are uncoupled in the U87MG cell line. Because mTOR regulation did not explain the effects of PLD inhibition on autophagy, we investigated other Akt substrates. Recently, Akt was shown to phosphorylate beclin1 and promote autophagy (66). Beclin1 is a component of the core autophagy complex (67) and exists in multiple protein complexes during progressive stages of autophagy (68). Autophagosome maturation and subsequent degradation is, in part, regulated by the interaction of beclin1 with Rubicon (69, 70), which is believed to negatively impact autophagosome maturation. We hypothesized that the phosphorylation of beclin1 by Akt might inhibit the interaction with Rubicon, and either PLD2 or Akt inhibition would thereby enhance the interaction. As expected, The PLD2 inhibitor VU0364739 and Akt inhibitor MK2206 increased the amount of Rubicon that co-immunoprecipitated with beclin1 from U87MG cells (Fig. 8, B and C). To address whether the interaction of Rubicon with beclin1 was mediated by Akt phosphorylation, we mutated the two putative Akt phosphorylation residues on beclin1, serine 234 and serine 295 (66), and assessed Rubicon binding. Previous studies identified serine 295 as the predominant Akt phosphorylation site on beclin1 (66). Alanine mutation of serine 295, but not 234, increased Rubicon binding to beclin1 compared with wild-type controls (Fig. 8B). PLD and Akt inhibition failed to increase binding of Rubicon to the S295A mutant of beclin1, supporting the model that Akt activity enhances autophagic flux by preventing binding of Rubicon to beclin1 (Fig. 8, B and C).
FIGURE 8.

PLD and Akt promote autophagic flux by dissociating Rubicon from beclin1. A, U87MG cells were treated overnight with 10 μm VU0359595, 5 μm VU0364739, 10 μm MK2206, or 1 μm mTOR inhibitor Torin1. Cells were immunoblotted for total and phosphorylated p70S6K1. B, U87MG cells were transfected with HA-tagged wild type or mutant beclin1 for 48 h. Cells were treated with 10 μm VU0364739 or MK2206 for 6 h in serum-free DMEM prior to cell harvest and immunoprecipitation (IP) of HA-beclin1 with an HA antibody. Immunoprecipitates were probed for co-IP of endogenous Rubicon. C, quantification of the increased binding of Rubicon to beclin1 following PLD or Akt inhibition. Band intensities of Rubicon and beclin1 were determined, and the ratio of Rubicon to beclin1 was calculated for each sample. Fold changes in this ratio were calculated by comparing inhibitor conditions to the vehicle-treated conditions within the wild-type or S295A beclin1 groups (n = 4). **, p < 0.01, ANOVA with Dunnett's post hoc test. Error bars, mean ± S.E.
Functional Rescue of Akt Restores Autophagic Flux and Viability in Glioma Cells Following PLD Inhibition
To confirm that the effects on autophagy and cell death following PLD inhibition in glioma cells were due to the regulation of Akt by PLD2, we developed a stable U87MG cell line expressing a constitutively active form of Akt under the transcriptional control of the tetracycline repressor protein. This Akt construct contains the myristoylation sequence from Src kinase (71) and is constitutively membrane-associated and active. If PtdOH serves to enhance membrane docking of Akt, then PLD inhibition should not decrease phosphorylation of myristoylated Akt (myrAkt1) because this construct bypasses lipid recruitment signals for membrane association. As expected, PLD inhibitors failed to reduce levels of phosphorylated Akt in myrAkt1 U87MG cells (Fig. 9A). Unlike PLD inhibitors, Torin1, an ATP-site mTOR inhibitor (72), decreased phosphorylation of myrAkt1, demonstrating that mTORC2 activity is still required for myrAkt1 phosphorylation and that inhibition of PLD activity does not decrease mTORC2 activity in this cell line (Fig. 9B). This result is consistent with PLD regulating Akt by enhancing membrane recruitment rather than regulating kinases or phosphatases that modulate phosphorylation of threonine 308 and serine 473.
FIGURE 9.

Restoration of Akt function rescues cell viability following PLD inhibitor treatment. A, immunoblot of phosphorylated Akt from parental or myrAkt1-expressing U87MG cells treated overnight with 10 μm VU0359595 or 5 μm VU0364739 in serum-free DMEM. B, parental or myrAkt1 U87MG cells were treated with the indicated inhibitors as in A overnight before blotting for phosphorylated and total Akt. C, immunoblot of LC3 from parental or myrAkt1 U87MG following a 6-h treatment with PLD or Akt inhibitors in serum-free DMEM. D, quantification of LC3-II conversion in parental and myrAkt1 U87MG cells following PLD and Akt inhibition. Fold changes were determined by calculating the ratio of LC3-II in inhibitor-treated samples to vehicle-treated samples within each cell line. *, p < 0.05; ***, p < 0.005, two-way ANOVA with Tukey's post hoc test. E, MyrAkt1-U87MG cells were seeded and treated as in Fig. 8B. IP, immunoprecipitation. F, quantification of Rubicon binding to beclin1 in myrAkt1 U87MG cells. Binding was quantified as in Fig. 8C; *, p < 0.05 using a paired Student's t test (n = 4). G, WST-1 viability assay with parental or myrAkt1 U87MG cells treated for 24 h with 20 μm VU0359595 or 10 μm VU0364739 in serum-free DMEM. Data are presented as the viability remaining following inhibitor treatment compared with the vehicle control within each cell type. ***, p < 0.005, two-way ANOVA with Sidek's post hoc test. Error bars, mean ± S.E.
Because phosphorylation of myrAkt1 was resistant to PLD inhibition, we next determined whether autophagic flux was restored following myrAkt1 expression in PLD inhibitor-treated cells. Expression of myrAkt1 produced a modest increase in the basal level of LC3-II versus the parental U87MG line (Fig. 9C). However, the fold induction of LC3-II due to PLD inhibitor treatment versus vehicle control was significantly less than in the parental U87MG line (Fig. 9, C and D), suggesting that the decrease in autophagic flux was due to inactivation of Akt via a PLD-dependent mechanism. Mechanistically, expression of myrAkt1 should prevent the increased binding of Rubicon to beclin1 following treatment with PLD inhibitors. Treatment of myrAkt1 U87MG cells with Akt inhibitor MK2206, but not VU0364739, increased beclin1 binding to Rubicon even in the presence of myrAkt1 (Fig. 9, E and F), supporting the proposed mechanism that PLD2 inhibition results in the inactivation of Akt, which promotes the Rubicon-beclin1 interaction and inhibits autophagic flux.
Finally, to confirm that the decrease in viability following PLD inhibition was due to inhibition of Akt, we measured U87MG cell viability in the parental and myrAkt1 lines. Restoration of Akt function significantly increased viability and protected the GBM cells from PLD inhibitor-induced cell death (Fig. 9G). Taken together, these data suggest that PLD activity is required for full Akt activation in GBM cells and that when inhibited the cells undergo autophagic death.
DISCUSSION
Although cancer research has improved the lives and long term survival of many patients over recent decades, the prognoses and treatment options for patients with GBM remain grim. Treatment of drug-resistant GBM demands new understanding of the survival mechanisms used to sustain GBM growth and viability under stress conditions. The insight that most GBM are resistant to apoptotic stimuli has prompted investigators to examine autophagy as a survival mechanism that may be exploited for drug treatment (73). The chemotherapeutic drugs most successful in treating GBM, such as temozolomide, induce autophagy (74). Drugs that interfere with autophagic flux, such as the anti-malarial drug chloroquine, have shown promise in clinical trials for potentiating cell death induced by conventional GBM chemotherapies (75). Therefore, identification of novel autophagy drug targets is clinically relevant for GBM.
In this report, we identified PLD, specifically the PLD2 isoform, as a regulator of autophagy and cell survival in gliomas through its regulation of Akt kinase (Fig. 10). Phosphorylation of Akt, under serum-depleted conditions, requires PLD2-generated PtdOH for recruitment to membranes (Figs. 3 and 5). The phosphorylation status of a constitutively membrane-associated Akt, myrAkt, is unperturbed by PLD inhibition (Fig. 9, A and B), suggesting that PLD2 functions to regulate Akt activation by membrane recruitment as opposed to modulating the activities of upstream kinases such as the mTORC2 complex, as has been suggested in other cell lines (18, 29). Additionally, we report that PLD2 and Akt co-immunoprecipitate from U87MG cell lysates and form a direct protein-protein interaction (Fig. 4). Several Akt-interacting proteins have been identified and mediate a variety of effects, including modulation of kinase activity and enhancing the ability of upstream kinases to activate Akt by rendering Akt a better kinase substrate (76). An interesting hypothesis emerges in which PLD2-generated PtdOH recruits Akt to membranes and allows PLD2 to directly interact with Akt and regulate kinase activity or activation by upstream kinases, and this is the subject of ongoing investigations.
FIGURE 10.
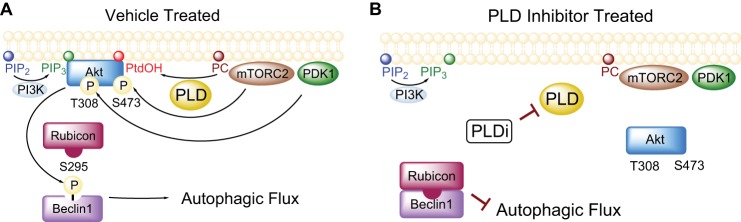
Mechanism of Akt and autophagy regulation by PLD. A, in the absence of inhibitors, PLD generates PtdOH and recruits Akt to the membrane allowing for phosphorylation of beclin1 by Akt at serine 295 and disruption of the beclin1-Rubicon complex and promotion of autophagic flux. B, PLD inhibitors reduce PtdOH production and subsequent Akt membrane recruitment. The inactivation of Akt results in reduced phosphorylation of beclin1 at serine 295 and formation of the beclin1-Rubicon complex.
The coupling of PtdOH to Akt activation is apparent in GBM lines but not other cells such as the nontumorigenic HEK293 line (Fig. 3). The GBM cell lines used in this study, U87MG and U118MG, are both PTEN-null and have higher basal levels of Akt relative to other cell lines. Studies are emerging that suggest Akt-lipid binding profiles may be altered by post-translational modifications and could account for the differences we observe between various cell lines. For example, Mahajan et al. (54) reported that phosphorylation at tyrosine 176 increases the affinity of Akt for PtdOH. In our study, PtdOH appears to increase the affinity of Akt for PIP3 by binding a distinct site in the Akt PH domain, suggesting that PLD and PI3K might either work together or independently to activate Akt (Fig. 5). Crystal structures of the phox homology domain from the NADPH oxidase protein p47Phox revealed a PtdOH binding pocket distinct from the well characterized phosphoinositide-binding site (77). Similar to our findings with Akt, occupation of the PtdOH binding pocket on p47Phox dramatically increased binding affinity for phosphoinositide-containing membranes (77). These findings open the possibility that other lipid-binding proteins are subject to dual regulation by PtdOH and phosphoinositides and expose the potential for alternative therapy strategies targeting one or both pathways as PLD and PI3K may function together to fine-tune subcellular localization and regulate specific effector pathways. In tandem, selective inhibitors might reduce the effective concentrations and thereby minimize undesired side effects.
In our studies, PLD inhibition did not induce global changes in the phosphorylation status of known Akt effectors such as glycogen synthase kinase 3 (GSK3) or proline-rich Akt substrate of 40 kDa (PRAS40) (data not shown). However, the loss of glioma viability following PLD inhibition appears to be mediated by the inactivation of Akt (Fig. 9) and subsequent inhibition of autophagic flux. Modulation of autophagy at either the initiation or degradation steps appear to have varying outcomes on cell viability when GBM cells are treated with compounds that induce autophagy. For example, GBM cell death induced by temozolomide (74) or imatinib (78) is potentiated by inhibition of autophagosome degradation with bafilomycin A1. Paradoxically, chemical or genetic inhibition of autophagosome initiation protects GBM cells against temozolomide- (74) or imatinib (78)-induced cell death. In this study and others (79), PLD inhibition decreased autophagic flux and inhibition of autophagy initiation via siRNA knockdown of Atg7-protected U87MG cells against PLD inhibitor-induced cell death (Fig. 6, H and I). Autophagosomes are known to carry damaged organelles or toxic aggregates that must be successfully cleared for cell survival. Therefore, the accumulation of this toxic cargo that cannot be degraded following inhibition of autophagic flux by PLD inhibition or bafilomycin A1 treatment results in cell death, and decreasing the number of these toxic autophagosomes by reducing autophagy initiation appears to be cytoprotective under certain circumstances.
Akt kinase has emerged as a regulator of autophagy, and chemical inhibitors or genetic silencing can modulate autophagy in GBM cells (80), although the mechanisms have not been fully elucidated. Here, we have characterized a novel function for the Akt-mediated phosphorylation of beclin1. Phosphorylation of beclin1 by Akt appears to prevent binding to Rubicon (Fig. 8, B and C), an interaction that is known to inhibit autophagosome maturation (69, 70). Further elucidation of novel Akt substrates in autophagy pathways will potentially shed light on other molecular mechanisms underlying the regulation of autophagy by PLD2 and Akt.
Our results support PLD as a novel drug target for GBM therapy. By regulating specific functions of Akt, such as autophagy (Fig. 10), the side effects of global Akt inhibition (12, 81) are potentially avoided by targeting PLD2. Knock-out mice for PLD1 and PLD2 (25, 26) are viable and show no overt phenotypes, suggesting that small molecule inhibition of PLD may circumvent toxic side effects seen with conventional chemotherapies. Although we characterized the molecular mechanisms of cell survival in a model system, U87MG cells, PLD inhibitors decreased autophagic flux, reduced viability, and reduced anchorage-independent growth in two glioma stem cell lines isolated from human biopsies (Figs. 2C and 6F), which are notoriously resistant to conventional chemotherapies (3). Therefore, the development of PLD inhibitors as a stand-alone or combination therapy has exciting potential for GBM treatment.
Acknowledgments
We thank Sarah Scott for conducting anchorage-independent growth assays; Pavlina Ivanova, Steve Milne, and David Myers for assistance with mass spectrometry analysis of lipid products; Michelle Armstrong for assistance with editing and figures; Jialiang Wang in the department of neurosurgery for providing CD133-expressing stem cells from primary human gliomas; Kacie Sims and Walt Shaw at Avanti Polar Lipids for providing the Lipid Snoopers®; and Shawn Goodwin at the Morphology Core at Meharry Medical College.
This work was supported, in whole or in part, by National Institutes of Health Grant P01-ES013125 from NIEHS and Grant U54 MH084659. This work was also supported by the McDonnell Foundation for Brain Cancer Research, a Lai Sulin scholarship (to R. C. B.), and a Bixler-Johnson-Mayes Endowed Chair (to H. A. B.).
This article was selected as a Paper of the Week.
- GBM
- glioblastoma multiforme
- PIP3
- phosphatidylinositol 3,4,5-trisphosphate
- PTEN
- phosphatase and tensin homolog
- PLD
- phospholipase D
- PC
- phosphatidylcholine
- PtdOH
- phosphatidic acid
- mTOR
- mammalian target of rapamycin
- mTORC2
- mTOR complex 2
- PH
- pleckstrin homology
- PtS
- protein A-Tev-Strep tag
- LC3
- microtubule-associated protein 1A/1B-light chain
- Atg7
- autophagy-related protein 7
- RFP
- red fluorescent protein
- Rubicon
- RUN-domain cysteine rich domain containing, beclin1-interacting protein
- myrAkt1
- myristoylated Akt1
- ANOVA
- analysis of variance.
REFERENCES
- 1. Stupp R., Mason W. P., van den Bent M. J., Weller M., Fisher B., Taphoorn M. J., Belanger K., Brandes A. A., Marosi C., Bogdahn U., Curschmann J., Janzer R. C., Ludwin S. K., Gorlia T., Allgeier A., Lacombe D., Cairncross J. G., Eisenhauer E., Mirimanoff R. O. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 [DOI] [PubMed] [Google Scholar]
- 2. DeAngelis L. M. (2001) Brain tumors. N. Engl. J. Med. 344, 114–123 [DOI] [PubMed] [Google Scholar]
- 3. Eramo A., Ricci-Vitiani L., Zeuner A., Pallini R., Lotti F., Sette G., Pilozzi E., Larocca L. M., Peschle C., De Maria R. (2006) Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 13, 1238–1241 [DOI] [PubMed] [Google Scholar]
- 4. Cheng C. K., Fan Q. W., Weiss W. A. (2009) PI3K signaling in glioma-animal models and therapeutic challenges. Brain Pathol. 19, 112–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cantley L. C. (2002) The phosphoinositide 3-kinase pathway. Science 296, 1655–1657 [DOI] [PubMed] [Google Scholar]
- 6. Bellacosa A., Testa J. R., Staal S. P., Tsichlis P. N. (1991) A retroviral oncogene, Akt, encoding a serine-threonine kinase containing an SH2-like region. Science 254, 274–277 [DOI] [PubMed] [Google Scholar]
- 7. Franke T. F., Yang S. I., Chan T. O., Datta K., Kazlauskas A. (1995) The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 81, 727–736 [DOI] [PubMed] [Google Scholar]
- 8. Manning B. D., Cantley L. C. (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Libermann T. A., Nusbaum H. R., Razon N., Kris R., Lax I. (1985) Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313, 144–147 [DOI] [PubMed] [Google Scholar]
- 10. Haas-Kogan D., Shalev N., Wong M., Mills G., Yount G., Stokoe D. (1998) Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr. Biol. 8, 1195–1198 [DOI] [PubMed] [Google Scholar]
- 11. Furnari F. B., Fenton T., Bachoo R. M., Mukasa A., Stommel J. M., Stegh A., Hahn W. C., Ligon K. L., Louis D. N., Brennan C., Chin L., DePinho R. A., Cavenee W. K. (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Gene Dev. 21, 2683–2710 [DOI] [PubMed] [Google Scholar]
- 12. Yap T. A., Yan L., Patnaik A., Fearen I., Olmos D., Papadopoulos K., Baird R. D., Delgado L., Taylor A., Lupinacci L., Riisnaes R., Pope L. L., Heaton S. P., Thomas G., Garrett M. D., Sullivan D. M., de Bono J. S., Tolcher A. W. (2011) First-in-man clinical trial of the oral pan-AKT Inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 29, 4688–4695 [DOI] [PubMed] [Google Scholar]
- 13. Selvy P. E., Lavieri R. R., Lindsley C. W., Brown H. A. (2011) Phospholipase D: enzymology, functionality, and chemical modulation. Chem. Rev. 111, 6064–6119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Min D. S. (2001) Neoplastic transformation and tumorigenesis associated with overexpression of phospholipase D isozymes in cultured murine fibroblasts. Carcinogenesis 22, 1641–1647 [DOI] [PubMed] [Google Scholar]
- 15. Park M. H., Ahn B. H., Hong Y. K., Min do S. (2009) Overexpression of phospholipase D enhances matrix metalloproteinase-2 expression and glioma cell invasion via protein kinase C and protein kinase A/NF-κB/Sp1-mediated signaling pathways. Carcinogenesis 30, 356–365 [DOI] [PubMed] [Google Scholar]
- 16. Buchanan F. G., McReynolds M., Couvillon A., Kam Y., Holla V. R., Dubois R. N., Exton J. H. (2005) Requirement of phospholipase D1 activity in H-RasV12-induced transformation. Proc. Natl. Acad. Sci. U.S.A. 102, 1638–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Henkels K. M., Boivin G. P., Dudley E. S., Berberich S. J., Gomez-Cambronero J. (2013) Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model. Oncogene 10.1038/onc.2013.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen Q., Hongu T., Sato T., Zhang Y., Ali W., Cavallo J. A., van der Velden A., Tian H., Di Paolo G., Nieswandt B., Kanaho Y., Frohman M. A. (2012) Key roles for the lipid signaling enzyme phospholipase D1 in the tumor microenvironment during tumor angiogenesis and metastasis. Sci. Signal. 5, ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rizzo M. A., Shome K., Vasudevan C., Stolz D. B., Sung T. C., Frohman M. A., Watkins S. C., Romero G. (1999) Phospholipase D and its product, phosphatidic acid, mediate agonist-dependent raf-1 translocation to the plasma membrane and the activation of the mitogen-activated protein kinase pathway. J. Biol. Chem. 274, 1131–1139 [DOI] [PubMed] [Google Scholar]
- 20. Foster D. A., Xu L. (2003) Phospholipase D in cell proliferation and cancer. Mol. Cancer Res. 1, 789–800 [PubMed] [Google Scholar]
- 21. Fang Y., Vilella-Bach M., Bachmann R. (2001) Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294, 1942–1945 [DOI] [PubMed] [Google Scholar]
- 22. Knoepp S. M., Chahal M. S., Xie Y., Zhang Z., Brauner D. J., Hallman M. A., Robinson S. A., Han S., Imai M., Tomlinson S., Meier K. E. (2008) Effects of active and inactive phospholipase D2 on signal transduction, adhesion, migration, invasion, and metastasis in EL4 lymphoma cells. Mol. Pharmacol. 74, 574–584 [DOI] [PubMed] [Google Scholar]
- 23. Ahn B. H., Kim S. Y., Kim E. H., Choi K. S., Kwon T. K., Lee Y. H., Chang J. S., Kim M. S., Jo Y. H., Min D. S. (2003) Transmodulation between phospholipase D and c-Src enhances cell proliferation. Mol. Cell. Biol. 23, 3103–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scott S. A., Selvy P. E., Buck J. R., Cho H. P., Criswell T. L., Thomas A. L., Armstrong M. D., Arteaga C. L., Lindsley C. W., Brown H. A. (2009) Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat. Chem. Biol. 5, 108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elvers M., Stegner D., Hagedorn I., Kleinschnitz C., Braun A., Kuijpers M. E., Boesl M., Chen Q., Heemskerk J. W., Stoll G., Frohman M. A., Nieswandt B. (2010) Impaired αIIbβ3 integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci. Signal. 3, ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oliveira T. G., Chan R. B., Tian H., Laredo M., Shui G., Staniszewski A., Zhang H., Wang L., Kim T. W., Duff K. E., Wenk M. R., Arancio O., Di Paolo G. (2010) Phospholipase D2 ablation ameliorates Alzheimer's disease-linked synaptic dysfunction and cognitive deficits. J. Neurosci. 30, 16419–16428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Di Fulvio M., Frondorf K., Gomez-Cambronero J. (2008) Mutation of Y179 on phospholipase D2 (PLD2) up-regulates DNA synthesis in a PI3K-and Akt-dependent manner. Cell. Signal. 20, 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patel S., Djerdjouri B., Raoul-Des-Essarts Y., Dang P. M., El-Benna J., Périanin A. (2010) Protein kinase B (AKT) mediates phospholipase D activation via ERK1/2 and promotes respiratory burst parameters in formylpeptide-stimulated neutrophil-like HL-60 cells. J. Biol. Chem. 285, 32055–32063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Toschi A., Lee E., Xu L., Garcia A., Gadir N., Foster D. A. (2009) Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol. Cell. Biol. 29, 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Edwards J. L., Apicella M. A. (2006) Neisseria gonorrhoeae PLD directly interacts with Akt kinase upon infection of primary, human, cervical epithelial cells. Cell Microbiol. 8, 1253–1271 [DOI] [PubMed] [Google Scholar]
- 31. Wang J., Wakeman T. P., Lathia J. D., Hjelmeland A. B., Wang X. F., White R. R., Rich J. N., Sullenger B. A. (2010) Notch promotes radioresistance of glioma stem cells. Stem Cells 28, 17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramaswamy S., Nakamura N., Vazquez F., Batt D. B., Perera S., Roberts T. M., Sellers W. R. (1999) Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 2110–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kimura S., Noda T., Yoshimori T. (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 [DOI] [PubMed] [Google Scholar]
- 34. Sun Q., Fan W., Chen K., Ding X., Chen S., Zhong Q. (2008) Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U.S.A. 105, 19211–19216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Giannone R., McDonald W. H., Hurst G., Huang Y., Wu J., Liu Y., Wang Y. (2007) Dual-tagging system for the affinity purification of mammalian protein complexes. BioTechniques 43, 296–302 [DOI] [PubMed] [Google Scholar]
- 36. Brown H. A., Henage L. G., Preininger A. M., Xiang Y., Exton J. H. (2007) Biochemical analysis of phospholipase D. Methods Enzymol. 434, 49–87 [DOI] [PubMed] [Google Scholar]
- 37. Thomas C. C., Deak M., Alessi D. R., van Aalten D. M. (2002) High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr. Biol. 12, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 38. Kumar C. C., Diao R., Yin Z., Liu Y., Samatar A. A. (2001) Expression, purification, characterization and homology modeling of active Akt/PKB, a key enzyme involved in cell survival signaling. Biochim. Biophys. Acta 1526, 257–268 [DOI] [PubMed] [Google Scholar]
- 39. Dowler S., Kular G., Alessi D. R. (2002) Protein lipid overlay assay. Sci. STKE 2002, pl6. [DOI] [PubMed] [Google Scholar]
- 40. Zheng Y., Rodrik V., Toschi A., Shi M., Hui L., Shen Y., Foster D. A. (2006) Phospholipase D couples survival and migration signals in stress response of human cancer cells. J. Biol. Chem. 281, 15862–15868 [DOI] [PubMed] [Google Scholar]
- 41. Sun H., Lesche R., Li D. M., Liliental J., Zhang H., Gao J., Gavrilova N., Mueller B., Liu X., Wu H. (1999) PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 6199–6204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hammond S. M., Altshuller Y. M., Sung T. C. (1995) Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J. Biol. Chem. 270, 29640–29643 [DOI] [PubMed] [Google Scholar]
- 43. Colley W. C., Sung T. C., Roll R., Jenco J., Hammond S. M., Altshuller Y., Bar-Sagi D., Morris A. J., Frohman M. A. (1997) Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr. Biol. 7, 191–201 [DOI] [PubMed] [Google Scholar]
- 44. Brown H. A., Gutowski S., Moomaw C. R., Slaughter C., Sternweis P. C. (1993) ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell 75, 1137–1144 [DOI] [PubMed] [Google Scholar]
- 45. Lewis J. A., Scott S. A., Lavieri R., Buck J. R., Selvy P. E., Stoops S. L., Armstrong M. D., Brown H. A., Lindsley C. W. (2009) Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg. Med. Chem. Lett. 19, 1916–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lavieri R. R., Scott S. A., Selvy P. E., Kim K., Jadhav S., Morrison R. D., Daniels J. S., Brown H. A., Lindsley C. W. (2010) Design, synthesis, and biological evaluation of halogenated N-(2-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)benzamides: discovery of an isoform-selective small molecule phospholipase D2 inhibitor. J. Med. Chem. 53, 6706–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Singh S. K., Hawkins C., Clarke I. D., Squire J. A., Bayani J., Hide T., Henkelman R. M., Cusimano M. D., Dirks P. B. (2004) Identification of human brain tumour initiating cells. Nature Cell Biol. 432, 396–401 [DOI] [PubMed] [Google Scholar]
- 48. Shin S. I., Freedman V. H., Risser R., Pollack R. (1975) Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc. Natl. Acad. Sci. U.S.A. 72, 4435–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. James S. R., Downes C. P., Gigg R., Grove S. J., Holmes A. B., Alessi D. R. (1996) Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem. J. 315, 709–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alessi D. R., James S. R., Downes C. P., Holmes A. B. (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B. Curr. Biol. 7, 261–269 [DOI] [PubMed] [Google Scholar]
- 51. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 52. Scott S. A., Xiang Y., Mathews T. P., Cho H. P., Myers D. S., Armstrong M. D., Tallman K. A., O'Reilly M. C., Lindsley C. W., Brown H. A. (2013) Regulation of phospholipase D activity and phosphatidic acid production after purinergic (P2Y6) receptor stimulation. J. Biol. Chem. 288, 20477–20487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huang B. X., Akbar M., Kevala K., Kim H. Y. (2011) Phosphatidylserine is a critical modulator for Akt activation. J. Cell Biol. 192, 979–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mahajan K., Coppola D., Challa S., Fang B., Chen Y. A., Zhu W., Lopez A. S., Koomen J., Engelman R. W., Rivera C., Muraoka-Cook R. S., Cheng J. Q., Schönbrunn E., Sebti S. M., Earp H. S., Mahajan N. P. (2010) Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PLoS One 5, e9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bertrand R., Solary E., O'Connor P., Kohn K. W., Pommier Y. (1994) Induction of a common pathway of apoptosis by staurosporine. Exp. Cell Res. 211, 314–321 [DOI] [PubMed] [Google Scholar]
- 56. Tsujimoto Y., Shimizu S. (2005) Another way to die: autophagic programmed cell death. Cell Death Differ. 12, 1528–1534 [DOI] [PubMed] [Google Scholar]
- 57. Kroemer G., Mariño G., Levine B. (2010) Autophagy and the integrated stress response. Mol. Cell 40, 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ravikumar B., Futter M., Jahreiss L., Korolchuk V. I., Lichtenberg M., Luo S., Massey D. C., Menzies F. M., Narayanan U., Renna M., Jimenez-Sanchez M., Sarkar S., Underwood B., Winslow A., Rubinsztein D. C. (2009) Mammalian macroautophagy at a glance. J. Cell Sci. 122, 1707–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hirai H., Sootome H., Nakatsuru Y., Miyama K., Taguchi S., Tsujioka K., Ueno Y., Hatch H., Majumder P. K., Pan B.-S., Kotani H. (2010) MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 9, 1956–1967 [DOI] [PubMed] [Google Scholar]
- 60. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Komatsu M., Waguri S., Koike M., Sou Y. S., Ueno T., Hara T., Mizushima N., Iwata J., Ezaki J., Murata S., Hamazaki J., Nishito Y., Iemura S., Natsume T., Yanagawa T., Uwayama J., Warabi E., Yoshida H., Ishii T., Kobayashi A., Yamamoto M., Yue Z., Uchiyama Y., Kominami E., Tanaka K. (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163 [DOI] [PubMed] [Google Scholar]
- 62. Wang Q. J., Ding Y., Kohtz D. S., Kohtz S., Mizushima N., Cristea I. M., Rout M. P., Chait B. T., Zhong Y., Heintz N., Yue Z. (2006) Induction of autophagy in axonal dystrophy and degeneration. J. Neurosci. 26, 8057–8068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., Chiba T. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yamamoto A., Tagawa Y., Yoshimori T., Moriyama Y., Masaki R., Tashiro Y. (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 23, 33–42 [DOI] [PubMed] [Google Scholar]
- 65. Foster D. A. (2009) Phosphatidic acid signaling to mTOR: signals for the survival of human cancer cells. Biochim. Biophys. Acta 1791, 949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang R. C., Wei Y., An Z., Zou Z., Xiao G., Bhagat G., White M., Reichelt J., Levine B. (2012) Akt-mediated regulation of autophagy and tumorigenesis through beclin 1 phosphorylation. Science 338, 956–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liang X. H., Jackson S., Seaman M., Brown K., Kempkes B. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 [DOI] [PubMed] [Google Scholar]
- 68. Kihara A. (2001) Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2, 330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Matsunaga K., Saitoh T., Tabata K., Omori H., Satoh T., Kurotori N., Maejima I., Shirahama-Noda K., Ichimura T., Isobe T., Akira S., Noda T., Yoshimori T. (2009) Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11, 385–396 [DOI] [PubMed] [Google Scholar]
- 70. Zhong Y., Wang Q. J., Li X., Yan Y., Backer J. M., Chait B. T., Heintz N., Yue Z. (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 11, 468–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kohn A. D., Summers S. A., Birnbaum M. J., Roth R. A. (1996) Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 271, 31372–31378 [DOI] [PubMed] [Google Scholar]
- 72. Thoreen C. C., Kang S. A., Chang J. W., Liu Q., Zhang J., Gao Y., Reichling L. J., Sim T., Sabatini D. M., Gray N. S. (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284, 8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lefranc F., Facchini V., Kiss R. (2007) Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist 12, 1395–1403 [DOI] [PubMed] [Google Scholar]
- 74. Kanzawa T., Germano I. M., Komata T., Ito H., Kondo Y., Kondo S. (2004) Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 11, 448–457 [DOI] [PubMed] [Google Scholar]
- 75. Sotelo J., Briceño E., López-González M. A. (2006) Adding chloroquine to conventional treatment for glioblastoma multiforme. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 144, 337–343 [DOI] [PubMed] [Google Scholar]
- 76. Du K., Tsichlis P. N. (2005) Regulation of the Akt kinase by interacting proteins. Oncogene 24, 7401–7409 [DOI] [PubMed] [Google Scholar]
- 77. Karathanassis D., Stahelin R. V., Bravo J., Perisic O., Pacold C. M., Cho W., Williams R. L. (2002) Binding of the PX domain of p47phox to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J. 21, 5057–5068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shingu T., Fujiwara K., Bögler O., Akiyama Y., Moritake K., Shinojima N., Tamada Y., Yokoyama T., Kondo S. (2009) Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int. J. Cancer 124, 1060–1071 [DOI] [PubMed] [Google Scholar]
- 79. Dall'Armi C., Hurtado-Lorenzo A., Tian H., Morel E., Nezu A., Chan R. B., Yu W. H., Robinson K. S., Yeku O., Small S. A., Duff K., Frohman M. A., Wenk M. R., Yamamoto A., Di Paolo G. (2010) The phospholipase D1 pathway modulates macroautophagy. Nat. Commun. 1, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Degtyarev M., De Mazière A., Orr C., Lin J., Lee B. B., Tien J. Y., Prior W. W., van Dijk S., Wu H., Gray D. C., Davis D. P., Stern H. M., Murray L. J., Hoeflich K. P., Klumperman J., Friedman L. S., Lin K. (2008) Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol. 183, 101–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Carlson R. H. (2013) AKT inhibitor activity confirmed by hyperglycemia “On-target effect.” Oncology Times 35, 46–47 [Google Scholar]