

Background: Some retinoid X receptor (RXR) agonists have potential as cancer drugs.
Results: Structures of RXR in complex with two different agonists show similar folds. Dynamics analysis reveals unique ligand-induced dynamics in helices 3, 11, and 12.
Conclusion: Two networks of interactions that connect RXR agonists to coactivator binding are defined.
Significance: Recognition of common conformational changes and distinguishing dynamics of RXR-selective agonists is necessary for advances in drug design.
Keywords: Nuclear Receptors, Protein Dynamics, Retinoid, Structural Biology, X-ray Crystallography, 9-cis-UAB30, RXR Receptor, Agonists, Targretin
Abstract
Retinoid X receptors (RXRs) are obligate partners for several other nuclear receptors, and they play a key role in several signaling processes. Despite being a promiscuous heterodimer partner, this nuclear receptor is a target of therapeutic intervention through activation using selective RXR agonists (rexinoids). Agonist binding to RXR initiates a large conformational change in the receptor that allows for coactivator recruitment to its surface and enhanced transcription. Here we reveal the structural and dynamical changes produced when a coactivator peptide binds to the human RXRα ligand binding domain containing two clinically relevant rexinoids, Targretin and 9-cis-UAB30. Our results show that the structural changes are very similar for each rexinoid and similar to those for the pan-agonist 9-cis-retinoic acid. The four structural changes involve key residues on helix 3, helix 4, and helix 11 that move from a solvent-exposed environment to one that interacts extensively with helix 12. Hydrogen-deuterium exchange mass spectrometry reveals that the dynamics of helices 3, 11, and 12 are significantly decreased when the two rexinoids are bound to the receptor. When the pan-agonist 9-cis-retinoic acid is bound to the receptor, only the dynamics of helices 3 and 11 are reduced. The four structural changes are conserved in all x-ray structures of the RXR ligand-binding domain in the presence of agonist and coactivator peptide. They serve as hallmarks for how RXR changes conformation and dynamics in the presence of agonist and coactivator to initiate signaling.
Introduction
Nuclear receptor (NR)3 proteins are ligand-inducible transcription factors that up-regulate target genes. Ligand binding causes a major structural rearrangement of the ligand-binding domain (LBD) that exposes/buries key residues on the LBD surface for coactivator binding. Many coactivator proteins contain amphipathic helices with an LXXLL motif that bind the LBD to enhance gene expression. Corepressor proteins compete for this binding site and down-regulate target gene expression. Thus, NR signaling depends on two factors: the presence/absence on the NR agonist and the abundance of coactivators relative to corepressors. Understanding how NR agonists induce conformational changes to generate a surface on the LBD to recruit coactivator proteins and activate transcription is a central question remaining in NR signaling.
Vitamin A acids (retinoids) bind and activate two classes of NR proteins: the retinoic acid receptors (RARs) and retinoid X receptors (RXRs). When RXR was discovered, it was designated an orphan NR because its endogenous ligand was not identified (1). In 1992, 9cRA (Fig. 1) was proposed to be the high affinity ligand for RXR (2, 3). 9cRA bound to RXRs in the nanomolar range (Kd ∼10 nm) and activated gene expression 40 times more potently than all-trans-retinoic acid, the high affinity ligand for RAR (2). 9cRA also binds/activates RAR, making it a pan-agonist for both RAR and RXR signaling pathways (4, 5). Moras and co-workers (6) solved the x-ray structure of 9cRA bound to hRXRα-LBD. When compared with their structure of apo-hRXRα-LBD homodimer (7), they reported that Helix 12 (H12) and other residues at the carboxyl end of the LBD significantly changed confirmation moving from an extended position into the folded active conformation when agonist was bound (mouse trap model). However, HDX MS and NMR analyses of the agonist-bound RXRα-LBD without coactivator peptides firmly demonstrate that H12 remains dynamic rather than static as predicted in the mousetrap model (8–10). It has been demonstrated that HDX MS provides a better method for comparing how agonists that bind the same ligand binding pocket (LBP) in an NR produce changes in protein dynamics for proteins that undergo small conformational changes (8, 11).
FIGURE 1.

9cRA, Targretin, and 9cUAB30 prevent oncogenic transformation in epithelial cells. A, the structures and numbering schemes of 9cRA, Targretin, and 9cUAB30. B, percentage of oncogenic transformation of epithelial cells treated with 9cRA, Targretin, or 9cUAB30 relative to DMSO controls. Top right, KLF4-ER tamoxifen-induced oncogenic transformation; bottom right, ErbB2-ER tamoxifen-induced oncogenic transformation. Error bars, S.D.
Recently, we solved the x-ray crystal structure of hRXRα-LBD bound to 9cRA with a coactivator peptide, glucocorticoid receptor-interacting protein-1 (GRIP-1). These studies revealed the structural and dynamical rearrangements of H12 and other residues associated with coactivator binding to the holo-RXRα-LBD (12). In this structure, we observed four tertiary structural changes that connected the retinoid in the LBP to H12 residues. In each of these changes, residues moved from a solvent-exposed environment to one that interacts with residues on H12. Our HDX MS results revealed reduced dynamics in the protein backbone, which correlated with each of the four tertiary structural changes in the LBD. In fact, the changes in the crystal structures induced by coactivator binding were subtle compared with the significant reduction in dynamics of the peptides associated with these residues, which reside one layer away from the coactivator binding site and point toward the LBP of RXR. These studies provide a hypothesis for how 9cRA remolds the surface of the LBD to create a hydrophobic cleft to promote coactivator binding.
Based on these results, we were interested to explore if the structural and dynamical changes identified for coactivator binding to hRXRα-LBD containing the pan-agonist 9cRA are also present for hRXRα-LBD containing selective RXR agonists. There are numerous examples of RXR agonists that selectively enhance signaling through RXRs independent of RAR signaling. Targretin was discovered by the Dawson and Pfahl groups (SR-11247) as a potent rexinoid (13), and this rexinoid was translated to the clinic by Ligand Pharmaceuticals (14). Targretin shares little structural similarity with 9cRA (Fig. 1), but it does preserve the overall shape and polarity of 9cRA. Numerous studies have shown that Targretin is as effective as 9cRA in cancer prevention and therapy models (15–18). Human clinical trials revealed that Targretin was better tolerated than 9cRA and did not produce many of the classic retinoic acid-associated toxicities (except for hyperlipidemia) (19, 20).
To achieve even higher RXR selectivity, many groups, including our own, have designed tissue-selective rexinoids that can be administered for cancer prevention without exhibiting dose-limiting lipid toxicities. 9cUAB30 uses a tetralone ring to replace the trimethylcyclohexenyl ring of 9cRA (Fig. 1). Although 9cUAB30 is slightly less potent than Targretin or 9cRA, it displays a higher degree of RXR selectivity. 9cUAB30 has also been shown to be effective in cancer prevention models (21–25). 9cUAB30 is a tissue-selective RXR agonist; it does not act as an agonist in liver to activate lipid biosynthesis although it has agonist capabilities in cancer cells (26). A Phase I human clinical trial has shown that 9cUAB30 is well tolerated in human volunteers without lipid toxicity (27).
In this study, we compare the structural and dynamical changes in hRXRα-LBD homodimers due to binding of the coactivator peptide GRIP-1 when either Targretin or 9cUAB30 is present. We were particularly interested in observing if coactivator binding induces similar structural and dynamical changes in the hRXRα-LBD homodimers bound to a rexinoid as we observed for homodimers bound with the pan-agonist 9cRA. Is there a common set of structural and dynamical changes characteristic of potent RXR agonists, and which changes (if any) are dependent on the type of rexinoid present in the LBD (rexinoid-specific changes)?
EXPERIMENTAL PROCEDURES
Materials
9cUAB30 was synthesized at the University of Alabama at Birmingham according to previous methods (21, 24). Targretin was provided by Dr. Clinton Grubbs at UAB. A 13-mer peptide derived from residues 686–698 of GRIP-1 was synthesized by AnaSpec Inc. (KHKILHRLLQDSS) with a molecular mass of 1575.9 Da. The structures and purity of the rexinoids and peptide were confirmed by NMR and LC-MS. The concentration of peptide was determined by 1H NMR added to a known concentration of tryptophan as reported previously (12).
Oncogenic Transformation Assay
The oncogenic transformation was performed as described previously by Jiang et al. (28). Cells infected with KLF4-ER were given DMEM supplemented with 4-hydroxytamoxifen (0.3 μm; Calbiochem) and 9cRA, 9cUAB30, Targretin, or vehicle (DMSO) every other day performed in duplicate for each concentration. Three weeks postinfection, transformed foci were fixed, stained, and counted and compared against the DMSO control.
Protein Expression and Purification
The protein expression and purification of hRXRα-LBD (Thr223–Thr462) was accomplished according to Egea and Moras (29) and Xia et al. (12). Briefly, the His6-tagged hRXRα-LBD fusion protein was expressed in BL21-(DE3) Escherichia coli bacteria (Invitrogen), which was grown in Luria broth (LB) medium at 20 °C. Protein expression was induced with 1 mm isopropyl-β-d-thiogalactopyranoside. Cells were lysed using a French press (1500 p.s.i.) and then centrifuged at 25,000 rpm for 30 min. The His6-tagged hRXRα-LBD was eluted from a nickel-chelating column (GE Healthcare) using a 20 mm Tris (pH 8.0) buffer containing 300 mm imidazole and 500 mm NaCl. The eluted hRXRα-LBD fractions were dialyzed in a 10 mm Tris buffer (pH 8) containing 50 mm NaCl, 0.5 mm EDTA, and 2 mm DTT. The His6 tag was hydrolyzed using α-thrombin (Novagen, Madison, WI) at 4 °C. The hRXRα-LBD homodimers were separated at 4 °C from tetramers using a HiLoad Superdex 75 gel filtration column (GE Healthcare) with a 1.0 ml/min flow rate. SDS-PAGE and MALDI mass spectrometry were used to establish a purity of >97% and mass of the monomers (m/z = 26,433.1 Da). Native PAGE confirmed that the isolated fractions were hRXRα-LBD homodimers.
Crystallization of hRXRα-LBD Homodimers Bound to 9cUAB30/Targretin and to GRIP-1
Manipulation of 9cUAB30 and Targretin was performed under dimmed red light due to light sensitivity of the compounds. Crystallization of hRXRα-LBD·9cUAB30·GRIP-1 and hRXRα-LBD·9cRA·GRIP-1 homodimer complexes was similar to the method described previously (12) and those reported by Moras and co-workers (30). The crystals of hRXRα-LBD homodimers with 9cUAB30 were obtained in a reservoir solution containing 5–10% PEG 4000, 2–5% glycerol, 0.1 m BisTris at pH 7.0. The crystals of the hRXRα-LBD containing Targretin and GRIP-1 formed more slowly than those for 9cUAB30. An additional microseeding step was applied by transferring tiny crystals of the complex into a new crystallization reservoir. X-ray quality crystals of the hRXRα-LBD·Targretin·GRIP-1 complex were obtained at 5–15% PEG 4000, 6–12% glycerol, 0.1 m BisTris at pH 7.0. Both crystals had a P4(3)2(1)2 space group, which was the same as the crystals of the hRXRα-LBD complex with 9cRA and GRIP-1 reported in our previous paper (12).
Data Collection, Structure Determination, and Refinement
Diffraction data of these two crystals were collected using the Rigaku R-AXIS IV+ at the Center for Biophysical Sciences and Engineering at the University of Alabama at Birmingham or an R-AXIS IV++ at BioCryst. Inc. Data collection, structure determination, and refinement strategy were described in detail previously (12). A summary of the diffraction data and crystal structure refinement statistics is given in Table 1. The contacts between hRXRα-LBD and rexinoid (either 9cUAB30 or Targretin) were determined by ligand-protein contacts (31). Contacts between GRIP-1 and hRXRα-LBD were determined by contacts of structural units (31). VOIDOO (32) and MAPMAN (33) programs were used for determining the LBP.
TABLE 1.
Crystallographic data collection and refinement statistics for hRXRα-LBD complexes
| 9cUAB30 | Targretin | |
|---|---|---|
| Data collection | ||
| Unit cell parameters (Å) | a = 66.04, b = 66.04, c = 111.16 | a = 68.15, b = 68.15, c = 106.59 |
| Unit cell parameters (degrees) | α = 90, β = 90, γ = 90 | α = 90, β = 90, γ = 90 |
| Space group | P43212 | P43212 |
| Resolution (last shell) (Å) | 29.53–2.00 (2.07–2.00) | 29.30–2.10 (2.18–2.10) |
| No. of total/unique reflections | 93,556/17,180 | 124,225/15,301 |
| Rmerge (last shell) (%) | 0.048 (0.322) | 0.078 (0.350) |
| Redundancy (last shell) | 5.45 (4.47) | 8.12 (7.73) |
| Completeness (last shell) (%) | 99.5 (99.1) | 100.0 (99.7) |
| I/σ (last shell) | 17.3 (4.6) | 14.1 (4.5) |
| Refinement | ||
| No. of residues | 223 | 223 |
| No. of protein atoms | 1789 | 1789 |
| No. of water molecules | 115 | 71 |
| No. of ligand atoms | 22 | 26 |
| Rcryst (%) | 23.95 | 24.62 |
| Rfree (%) | 28.31 | 28.87 |
| Root mean square deviation deviation from ideality | ||
| Bond length (Å) | 0.0068 | 0.0091 |
| Bond angles (degrees) | 1.089 | 1.162 |
| Ramachandran plot, residues in | ||
| Most favored regions (%) | 94.98 | 95.89 |
| Additional allowed regions (%) | 3.65 | 3.20 |
| Generously allowed regions (%) | 1.37 | 0.91 |
| Average B factor (Å2) | 37.33 | 43.04 |
| Non-hydrogen protein atoms (Å2) | 37.23 | 43.31 |
| Non-hydrogen ligand atoms (Å2) | 36.73 | 30.19 |
| Water molecules (Å2) | 38.96 | 40.92 |
Isothermal Titration Calorimetry of GRIP-1 Binding to Holo-hRXRα-LBD
A VP-Isothermal titration calorimeter (Microcal, Piscataway, NJ) was used to measure binding of the GRIP-1 coactivator peptide to holo-hRXRα-LBD homodimers as described previously (12). Each titration experiment consisted of 30 injections of 8 μl of GRIP-1 peptide (0.04–0.12 mm) into the sample cell containing 1.34 ml of hRXRα-LBD homodimers (0.05 mm) in 10 mm Tris buffer (pH 8.0) containing 50 mm NaCl, 0.5 mm EDTA, and 2 mm DTT. 9cUAB30 and Targretin were dissolved in DMSO and added at a ratio of 2:1 (retinoid/protein). Both retinoid solution and hRXRα-LBD solution were degassed at least 15 min before the retinoids were added to the protein solution. The isothermal calorimetry (ITC) data were processed using the ORIGIN 7 software. The titration curves were fit to a single site binding model by a non-linear least squares method. The ITC experiments were performed at 20, 25, and 30 °C, and the heat capacity change was found by linear regression of the corrected enthalpies versus temperature.
Automated Hydrogen Deuterium Exchange Experiments
Solution phase H/D exchange experiments were performed using an automated LEAP Technologies Twin PAL HTS autosampler (LEAP Technologies, Carrboro, NC) (8). In short, 40 pmol of hRXR-LBD homodimer in the presence of 10 times the concentration of ligand or vehicle (DMSO) and GRIP-1 coactivator peptide (>95% saturation) were dispensed into a mixing tray and diluted with 16 μl of deuterated buffer or protonated buffer for control experiments. Following on-exchange periods (4 °C), the entire sample was picked up and dispensed into 30 μl of quench buffer (3 m urea, 1% TFA, 50 mm TCEP, 1 °C). Samples were taken at 10, 30, 60, 300, 900, and 3600 s and performed in triplicate. Samples were digested for 2.5 min on an in-house prepared pepsin column (1 °C). Sample loading, digestion, and desalting were driven with an isocratic HPLC pump at 200 μl min−1. Peptic peptides were then loaded onto a desalting C8 phase trap column (3.5 μm; Agilent), followed by the analytical column (50 mm × 2.1, 5 μm; Thermo Scientific). Eluent was injected into the instruments at 50 μl min−1, and spectra were acquired over the scan range of 200–2000 m/z. Each spectrum was the sum of two microscans. HDX MS raw files were processed and visualized by use of the HD Desktop software (34). Differential HDX perturbation values were calculated by taking the averaged differences in deuterium incorporation for five on-exchange time points from the deuterium incorporation of the reference, apo-hRXRα-LBD·GRIP-1. HDX perturbations greater than ±5% were considered significant as described previously (8, 12). For identification of discriminatory dynamics of individual HDX MS peptides between the three agonists, changes in the measured percentage of deuterium incorporation (%D) relative to the apo control were compared at the 30 s or 30 min time points. Statistically significant differences were determined by use of a two-tailed t test as described previously (11).
Peptide Sequencing
Proteolytic digestion prior to mass spectromic analysis allowed regional monitoring of deuterium incorporation. Identification of hRXRα-LBD peptides was achieved by use of positive ion electrospray ionization LC tandem mass spectrometry (MS/MS) on a hybrid linear quadrupole ion trap 7 Tesla Fourier transform ion cyclotron resonance mass spectrometer (Thermo Fisher Scientific). Pilot experiments optimized protein digestion conditions, and peptides were identified by data-dependent tandem LC MS/MS as described previously (35).
RESULTS
In Vitro Analysis of RXRα Agonist Anticancer Properties
Previous reports have shown that 9cRA, Targretin, and 9cUAB30 are potent agonists of RXRα transcription (2, 14, 21). Here we compared each RXR agonist for its capacity to inhibit oncogenic transformation of rat kidney epithelial (RK3E) cells. In this assay, oncogenic transformed RK3E epithelial cells form dense foci surrounded by contact-inhibited parental cells in the absence of retinoid (28, 36–42). For this study, the RK3E cells were transduced with retrovirus encoding a conditional, 4-hydroxytamoxifen-inducible Krüppel-like factor 4 (KLF4) allele (KLF4-ER) (43), ErbB2, or empty vector as the control. When the cells were dosed with three different concentrations of retinoid every other day for 3 weeks, formation of foci was significantly decreased in either ErbB2- or KLF4-ER-infected RK3E cells relative to DMSO controls in a dose-dependent manner (Fig. 1). Targretin was more effective than 9cUAB30 at the highest dose (75% versus 59%), which correlated with its potency as an RXR agonist. The pan-agonist 9cRA was the most effective retinoid tested (95%), but this pan-agonist can inhibit oncogenesis by activating both RAR and RXR signaling pathways.
Binding Affinity of GRIP-1 with hRXRα-LBD Containing 9cUAB30 or Targretin
ITC was used to measure the thermodynamics of GRIP-1 binding to hRXRα-LBD containing either 9cUAB30 or Targretin at 20, 25, and 30 °C (Table 2). As observed for 9cRA complexes in our previous analysis (12), the binding stoichiometry was nearly 1:1 (coactivator peptide/LBD monomer unit). The free energy of GRIP-1 binding to the hRXRα-LBD complexes containing rexinoid was driven strongly by a large negative enthalpy change, and it is opposed by entropy although the coactivator peptide contains the LXXLL motif (Table 2). The magnitude of this thermodynamic signature was very similar for homodimers containing rexinoid (9cUAB30 or Targretin) or pan-agonist (9cRA) in its LBP (Table 2) (12). Using the temperature dependence of the enthalpy change for binding, the heat capacity changes for GRIP-1 binding to the hRXRα-LBD homodimers containing rexinoid were determined. The heat capacity changes were very similar for Targretin and 9cRA complexes (−401 ± 18 cal mol−1 K−1 and −413 ± 25 cal mol−1 K−1), and it was slightly smaller for 9cUAB30 complex (−356 ± 29 cal mol−1 K−1).
TABLE 2.
Summary of ITC measurements of GRIP-1 to hRXRα-LBD·retinoid complexes
| Temperature | Ka | ΔH | −TΔS | ΔG | n | ΔCp |
|---|---|---|---|---|---|---|
| °C | μm−1 | kcal/mol | kcal/mol | kcal/mol | cal mol−1 K−1 | |
| Targretin | ||||||
| 20 | 1.80 ± 0.07 | −7.18 ± 0.03 | −1.21 ± 0.06 | −8.39 ± 0.03 | 0.96 | −413 ± 25 |
| 25 | 1.59 ± 0.04 | −9.46 ± 0.02 | 0.84 ± 0.04 | −8.62 ± 0.02 | 0.96 | |
| 30 | 1.25 ± 0.04 | −11.31 ± 0.03 | 2.86 ± 0.06 | −8.45 ± 0.03 | 0.97 | |
| 9cUAB30 | ||||||
| 20 | 1.62 ± 0.06 | −7.12 ± 0.02 | −1.21 ± 0.04 | −8.33 ± 0.02 | 1.06 | −356 ± 29 |
| 25 | 1.33 ± 0.04 | −8.88 ± 0.03 | 0.52 ± 0.06 | −8.36 ± 0.03 | 1.05 | |
| 30 | 0.95 ± 0.02 | −10.68 ± 0.02 | 2.39 ± 0.04 | −8.29 ± 0.02 | 1.05 | |
| 9cRA | ||||||
| 20 | 2.15 ± 0.07 | −6.78 ± 0.02 | −1.71 ± 0.04 | −8.49 ± 0.02 | 0.98 | −401 ± 18 |
| 25 | 1.81 ± 0.06 | −9.15 ± 0.03 | 0.61 ± 0.06 | −8.54 ± 0.03 | 0.92 | |
| 30 | 1.38 ± 0.03 | −11.13 ± 0.03 | 2.61 ± 0.06 | −8.52 ± 0.03 | 0.95 | |
Comparisons of the X-ray Crystal Structures of hRXRα-LBD Homodimers Bound to RXR Agonists and GRIP-1
To explore the structures of this NR with different rexinoids and a coactivator peptide, we crystallized hRXRα-LBD homodimers bound with the GRIP-1 coactivator peptide and either 9cUAB30 or Targretin. Both crystal structures belonged to the P4(3)2(1)2 space group, and each asymmetric unit contained two monomers with GRIP-1 bound to each monomer. The structures were determined to 2.0 Å resolution for the hRXRα-LBD·9cUAB30·GRIP-1 (4K4J) complex and 2.25 Å resolution for the hRXRα-LBD·Targretin·GRIP-1 complex (4K6I) by use of the molecular replacement method (Table 1). Each structure had identical two-dimensional topology (Fig. 2A). This LBD formed a three-layer helical sandwich with H1 and H3 on one side and H6, H7, H10, H11, and H12 on the other (Fig. 2). The x-ray structures of hRXRα-LBD bound with either Targretin or 9cUAB30 and GRIP-1 were overlaid onto the structure of hRXRα-LBD·9cRA·GRIP-1 (12). The overlay showed that the hRXRα-LBD backbone atoms of these three crystal structures were very similar (Fig. 2B). The root mean square deviation was 0.137 Å for the 229 backbone residues of hRXRα-LBD·9cUAB30 (4K4J versus 3OAP), and it was 0.210 Å for hRXRα-LBD·Targretin (4K6I versus 3OAP).
FIGURE 2.
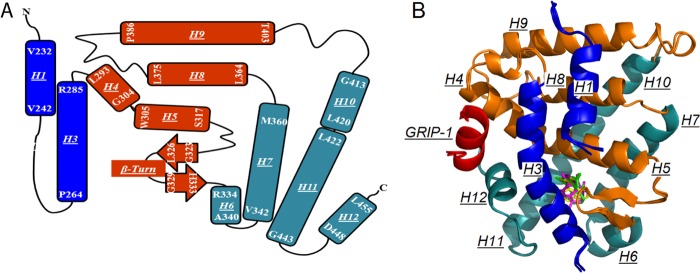
X-ray crystal structures of hRXRα-LBD bound to either Targretin, 9cUAB30, or 9cRA and GRIP-1. A, topology map of holo-hRXRα-LBD showing a three-sandwich helical fold. H4, H5, H8, H9, and the β-sheet (orange) are sandwiched by H1, H2 (missing), and H3 (blue) at one side and H6, H7, H10, and H11 (dark cyan) at the other side. B, overlay of three hRXRα-LBD·GRIP-1·agonist structures. Helices are rendered in the same colors as in A; GRIP-1 is shown in red.
GRIP-1·Holo-hRXRα·LBD Contacts
GRIP-1 coactivator peptide contains 13 residues (686KHKILHRLLQDSS698), including one LXXLL motif. In both structures, GRIP-1 adopted a two-turn amphipathic helix (Fig. 3A) and was positioned in the coactivator binding site of hRXRα-LBD formed by H3, H4, and H12 (12, 30). The helical conformation of GRIP-1 and the interactions of the coactivator peptide with hRXRα-LBD were nearly identical in these two structures (Fig. 3B). The hydrophobic surface of the amphipathic peptide (Ile689, Leu690, Leu693, and Leu694) interacted with a hydrophobic binding pocket on the surface of the LBD (Fig. 3B). There were subtle differences in the manner in which GRIP-1 interacted with the surface of the hRXRα-LBD (Fig. 3B). When Targretin occupied the LBP, the hydrophobic contact between Ile689 of GRIP-1 and Thr449 (H12) of hRXRα-LBD was smaller (4.8 and 17.5 Å2) compared with the same interaction when 9cUAB30 (3.5 and 45.5 Å2) or 9cRA (3.6 and 46.0 Å2) was present. The sec-butyl group of Ile689 changed conformations in the Targretin-bound structure. Additionally, Lys686 of GRIP-1 interacted with Glu456 and Ala457 (C terminus) of the hRXRα-LBD when Targretin was present, but this interaction was weaker when 9cRA or 9cUAB30 was bound (Fig. 3C). Last, the imidazole group of His691 of GRIP-1 shifted slightly toward the carboxylate group of Asp295 (H4) when Targretin was bound to the LBD. The contact surface area between His691 in GRIP-1 and H4 residues (Leu294, Asp295, Val298) was 20% less when Targretin was bound (56.7 Å2) relative to when 9cUAB30 (71.9 Å2) or 9cRA (81.4 Å2) was bound.
FIGURE 3.
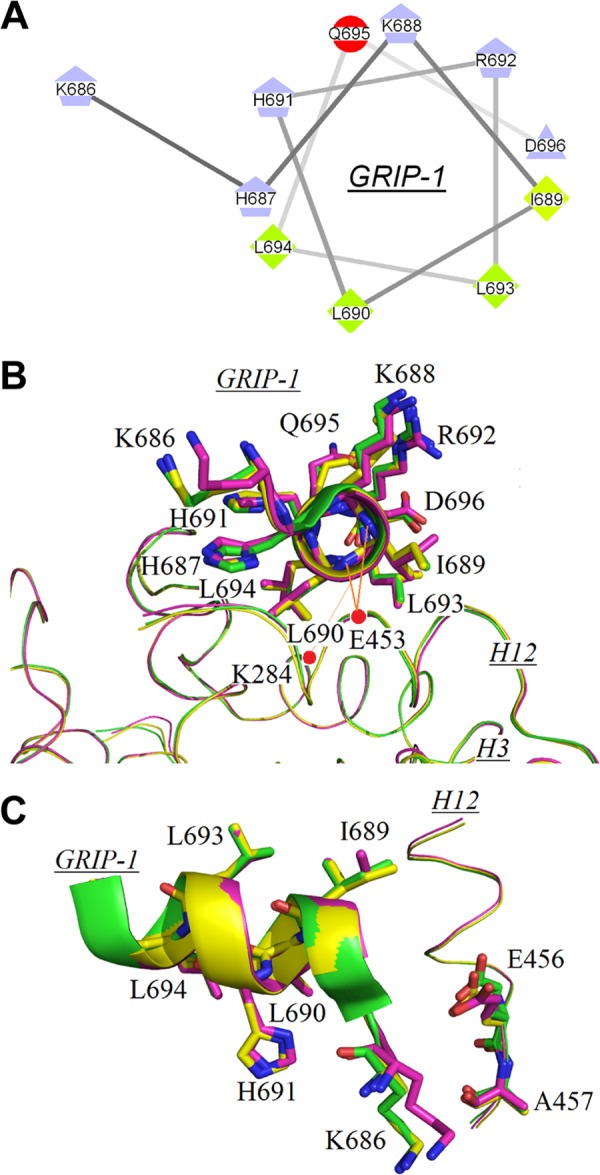
Interactions of the coactivator peptide GRIP-1 with surface residues on holo-hRXRα-LBD. A, helical wheel view of GRIP-1 bound to holo-hRXRα-LBD illustrates its amphipathic character. B, overlay of GRIP-1 in holo-hRXRα-LBD structures containing 9cRA (green), 9cUAB30 (yellow), and Targretin (magenta). Two charge clamps between GRIP-1 and hRXRα-LBD are indicated by red dashed lines. C, side view of GRIP-1 binding to LBD showing relative positions of Glu456 and Ala457 on H12 of hRXRα-LBD to Lys686 on GRIP-1. The color code is the same as in B.
Structural Changes Induced by GRIP-1 Binding
The structural changes induced by GRIP-1 binding to hRXRα-LBD when either Targretin or 9cUAB30 was bound to the LBD were compared with those of hRXRα-LBD·9cRA (Protein Data Bank code 1FBY) and to hRXRα-LBD·9cRA·GRIP-1 (3OAP). In the two crystal structures containing rexinoid and GRIP-1, Phe277 formed a π-hydrogen bond with Phe450 (Fig. 4A). This interaction was found in 3OAP but absent in 1FBY. Phe277 (H3) and Phe450 (H12) also interacted with two hydrophobic residues of GRIP-1 (Ile689 and Leu693). The carboxylate group of Asp273 (H3) formed hydrogen bonds to the backbone amide nitrogens of Phe450 and Thr449, which are both on H12 (Fig. 4A). H3 contains three nonpolar residues (Ile268, Ala271, and Ala272), which form the LBP and are on the opposite side of the H3 from Asp273 (Fig. 4A). These interactions are displayed by blue dashed lines in Fig. 4A. These interactions provide a molecular bridge between the coactivator peptide binding sites and carboxylate end of the retinoid.
FIGURE 4.
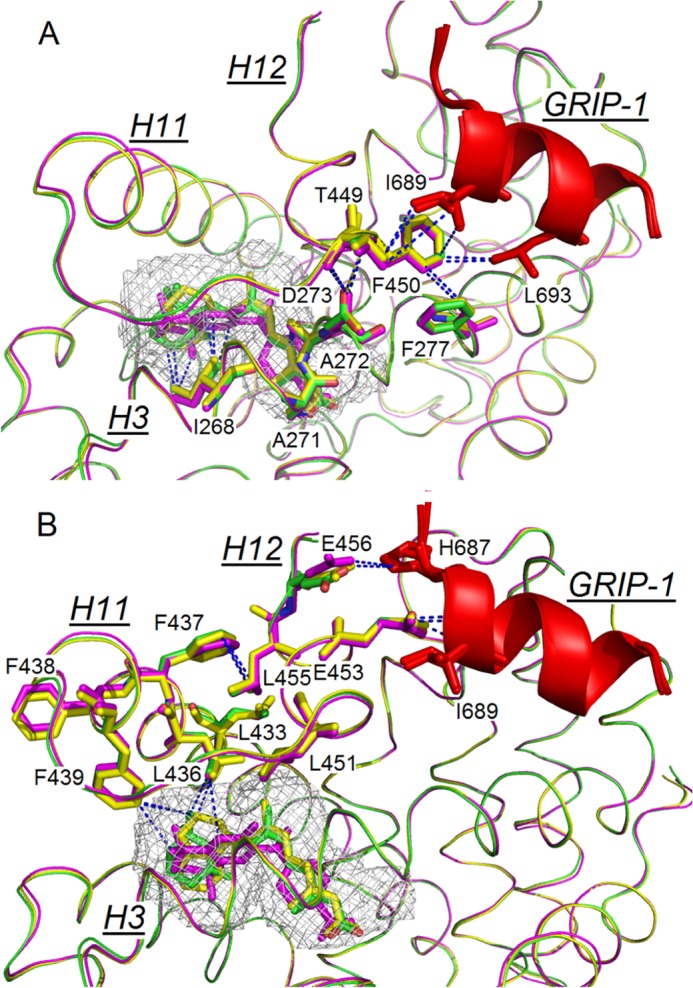
Networks of interacting residues between the GRIP-1 (red) and H3, H11, and H12 of the holo-hRXRα-LBD containing 9cRA (green), 9cUAB30 (yellow), and Targretin (magenta). Blue dashed lines link interacting residues. A, network between retinoid ring, H3 residues, H12 residues, and GRIP-1. B, network between retinoid ring, H11 residues, H12 residues, and GRIP-1.
A second molecular bridge was identified that connects the coactivator peptide to the nonpolar retinoid rings. At the surface of NR, two glutamates on H12 (Glu453/Glu456), which were solvent-exposed in 1FYB, interacted with GRIP-1. Glu453 (H12) formed ionic interactions with the amides on the N terminus of GRIP-1 and is part of the charge clamp (44) (Figs. 3B and 4B). Glu456 (H12) also contributed to this charge clamp by interacting with the side chain of His687 on GRIP-1 (Fig. 4B). The guanidinium group of the highly conserved Arg302 on H4 (45) formed strong hydrogen bonds with the backbone amides of Glu453 and Glu456 (∼3.2 Å). These interactions were identical to what was found previously (12). GRIP-1 binding reoriented Phe437 (H11) inward so that it was now capable of interacting with Leu455 of H12 (Glu453 and Glu456 are on the opposite face of H12 from Leu455). Phe437 (H11) changed conformations and pointed toward H12 so that the phenyl side chain is within van der Waals contact with the iso-butyl group of Leu455 (H12). Leu455 (H12) is one of four leucine residues (with Leu451 (H12), Leu436 (H11), and Leu433 (H11)) that form a hydrophobic core between H11 and H12. A leucine hydrophobic core is one layer deeper from the surface of coactivator binding (Fig. 4B). Phe439, Leu436, and Leu451 make direct contacts to the retinoid rings of 9cRA, Targretin, and 9cUAB30.
Differences in the Ligand-Protein Contacts of Targretin/9cUAB30/9cRA Bound to hRXRα-LBD
We next examined the conformation of the retinoids in the LBP. Electron density maps clearly showed the presence of 9cUAB30 and Targretin in the LBP of hRXRα-LBD. Both retinoids adopted nonplanar, L-shaped conformations that fit well into the LBP (Fig. 5A). For 9cUAB30, the C8–C9 bond (C7-C8-C9-C10 torsional angle 121°) was twisted in a right-handed screw sense (Fig. 5C). For Targretin, the twist occurred about the C3–C11 bond (C2-C3-C11-C12 torsional angle −85°). In contrast, 9cRA twisted about the C6–C7 bond linking the polyene chain to the trimethylcylcohexenyl ring (C5-C6-C7-C8 torsional angle −14°). In each case, the twisted conformation of the retinoid relieved steric repulsion between methyl groups. Each of three achiral retinoids adopted a right-handed screw sense in the LBP when viewed from the retinoid ring toward the carboxylate group down the long axis of the rexinoid.
FIGURE 5.

Conformation of rexinoids and 9cRA in the ligand binding pocket of holo-hRXRα-LBD. A, overlay of 9cRA (green), 9cUAB30 (yellow), and Targretin (magenta) in the ligand binding pocket. Shown are electron density maps (2Fo − Fc) for the retinoid binding pocket of hRXRα-LBD. B, 9cRA; C, 9cUAB30; D, Targretin. To the right of each electron density map, a space-filling rendering of each retinoid is given with one of the other retinoids overlaid.
The LBP of hRXRα-LBD is formed by residues contributed from H1, H3, H5, H7, H9, H11, H12, and the short two-turn β-sheet (6). The LBP is comprised of mainly hydrophobic residues and a few hydrophilic ones centered on the two-turn β-sheet and H5. The LBP of hRXRα-LBD bound to 9cRA and coactivator peptide GRIP-1 was formed by 34 residues, and its volume was 493 Å3 (12). 9cRA (300 Da) occupied 371 Å3 of the pocket volume (75% occupancy). In the structures reported here for the two rexinoids bound to hRXRα homodimers and GRIP-1, the LBPs were both slightly smaller (442 Å3 for 9cUAB30; 456 Å3 for Targretin) than that found for 9cRA. Thus, Targretin and 9cUAB30 filled this volume more than 9cRA (84% occupancy for Targretin; 78% occupancy for 9cUAB30).
In the 9cRA or rexinoid structures, the LBPs were formed by the same 24 residues (Fig. 5, compare B, C, and D). The elongated pockets (∼16.8 Å) were lined with the 16 hydrophobic residues. Four H3 residues (Ile268, Cys269, Ala271, and Ala272) and four H7 residues (Val342, Ile345, Phe346, and Val349) formed the lower part of the LBP (Fig. 4B). Four H5 residues (Trp305, Asn306, Leu309, and Phe313) and four H11 residues (Cys432, His435, Leu436, Phe439) formed the other side of the pocket. The total contact surface area of the ligand-protein contacts between this portion of 9cUAB30 and H3 and H5 residues was very similar. Although 9cUAB30 contains a structurally different ring from 9cRA, the total surface area of the contacts was similar (12). The overlaid conformations of space-filled renderings of these two agonists were very similar (Fig. 5B).
The structure of Targretin in the LBP was more highly twisted than either 9cRA or 9cUAB30. When a space-filled overlay was made, C27 of Targretin occupied space in the LBP that 9cUAB30 or 9cRA did not (Fig. 5, C and D). The C24, C26, and C27 methyl groups of Targretin interacted more strongly with residues on H3 and H7 than 9cRA or 9cUAB30. The total surface area for the contacts between the ring carbons on Targretin and H3 and H7 residues increased by about 30 and 20 Å2, respectively, over those found for the other two agonists. In contrast to H3 and H7, the interactions between Targretin and the residues on H11 decreased by 20 Å2 relative to what was observed in 9cRA. The single structural change in the Targretin LBP versus that of 9cRA occurs at Cys432. In the Targretin structure, the thiol group was rotated by about 116° and now points toward the C4 methyl group (4.6 Å away). Changes also occurred closer to the middle of the structure. The methylene C21 of Targretin formed a new contact to Ile310 on H5 (17 Å2; 4.0 Å), which was a protein residue that does not interact directly to 9cRA or 9cUAB30.
HDX MS Analysis of hRXRα-LBD Complexes
Several differential HDX MS studies have shown that the dynamics of hRXRα-LBD change when agonist binds (8, 10, 12, 46). We also observed that GRIP-1 reduced deuterium incorporation of apo-hRXRα-LBD in the absence of agonist (12). Here we performed differential HDX MS analysis using apo-hRXRα-LBD·GRIP-1 as the reference control to address the question of whether rexinoid binding produced distinct hRXRα-LBD HDX exchange rates in the presence of GRIP-1. Ninety-seven peptides comprising 99% of hRXRα-LBD (<2 ppm error) were followed by HDX MS (supplemental Table S1). The %D was calculated for each peptide at each time point in triplicate by comparison with a no deuterium control (11). Overall changes in %D were examined across all time points relative to the apo-hRXRα-LBD-GRIP-1 reference. Additionally, individual HDX MS peptides were examined at the 30 s and/or 30 min time points to identify statistically significant differences in individual peptide %D between the hRXRα agonists.
The addition of rexinoids to the NR LBD complex in the presence of GRIP-1 reduced deuterium incorporation in peptides from H2, H3, β-sheet-H6, H7, H11, and H12. For peptides spanning H2, the β-sheet-H6, and H7, the change in %D was negative, and the extent of reduction was the same regardless of which rexinoid was bound to the homodimer. For example, the change in %D for the β-sheet peptide Leu326–Gly343 was −8 ± 1% at 30 s regardless of agonist bound to the hRXRα-LBD (Fig. 6). Results for all overlapping H2 and H7 peptides followed this same pattern (supplemental Table S1). For the majority of LBD HDX MS peptides, Targretin and 9cUAB30 binding mimicked the changes in %D of 9cRA binding in the presence of GRIP-1. The list of all changes in %D is provided in supplemental Table S1.
FIGURE 6.

HDX MS deuterium incorporation plots for hRXRα-LBD H3, H4, H11, and H12 peptic peptides (sequence indicated at the top). A, %D is plotted versus a log time scale for each peptide. The rexinoids (Targretin and 9cUAB30) show distinct changes in %D compared with the pan-agonist 9cRA in the H3, H11, and H12 regions but not in H4. Shown is HDX MS analysis of hRXRα-LBD in the presence of retinoid and GRIP-1 (9cRA (green squares), 9cUAB30 (yellow triangles), and Targretin (magenta diamonds)) relative to the controls of the digested LBD without retinoid but with GRIP-1 (blue circles). B, comparison of individual time points (30 or 900 s) for the same four HDX MS peptides shows that the changes in %D for the H3, H11, and H12 peptides when the two rexinoids are bound (9cUAB30 (yellow) and Targretin (magenta)) are statistically significant relative to the same peptide when 9cRA (green) is bound to the hRXRα-LBD. Error bars, S.D.
For 15 (of 97) peptides, the overall reduction in %D was 10% or more. These HDX MS peptides encompassed three regions within the hRXRα-LBD and show statistically significant dynamics upon binding of Targretin, 9cUAB30, or the pan-agonist 9cRA. Four of the peptides included H3 residues, 10 peptides spanned the longer H11 and a C-terminal adjacent loop, and a single peptide included H12 residues. Fig. 6 shows representative %D versus time comparative HDX MS plots from these 15 peptides, including the H3 peptide Ala271–Ala278, H11 peptide Pro423–Phe438, and H12 peptide Phe450–Met454 when bound to each ligand. Significantly, the overall reduction in %D was different for each of the agonists in these three areas, with the two rexinoids showing more reduction in %D for H3 (Targretin, −46%; 9cUAB30, −50%), H11 (Targretin, −32%; 9cUAB30, −28%), and H12 (Targretin, −11%; 9cUAB30, −12%) compared with 9cRA (H3, −26%; H11, −14%; H12, 1%). The overlapping peptides for H3 and H11 showed the same trend (supplemental Table S1). Fig. 6 also includes an H4 peptide as an example of an HDX MS peptide that does not change in %D in the presence of ligand, whether it is the two rexinoids or the pan-agonist 9cRA.
To determine if these comparative HDX MS differences for individual peptides were significant, we examined individual time points for each data set relative to their matched controls of unbound ligand. Chalmers et al. (11) have demonstrated that comparison of statistically significant differences in individual peptides %D at a single time point can be used to delineate between NR agonists based on their %D signatures. Fig. 6B provides this comparison for the four peptides in A. The change in %D for in H3, H11, and H12 when bound to either rexinoid showed statistically significant differences (p < 0.0001) compared with the same peptides when 9cRA was bound to the hRXRα-LBD in the presence of GRIP-1. There was no significant difference in the changes in %D for the H4 peptide between the three ligands as was the case for the majority of the observed HDX MS peptides.
The dynamical changes from all comparative HDX MS peptides with >5% change in %D were painted onto one monomer of the holo-hRXRα-LBD structures containing GRIP-1 (Fig. 7). (GRIP-1 is colored red and viewed along its helical axis in Fig. 7). The regions of change in %D for the two rexinoids mimic those found with 9cRA in the presence of GRIP-1 (12). However, H12 is stabilized by coactivator binding to hRXRα-LBD only when a rexinoid was present. GRIP-1 binding also stabilized H3 and the C terminus of H11. The largest effects were present when Targretin was present in the retinoid binding site. When 9cUAB30 was bound, the stabilization was slightly less than observed for Targretin but significantly larger than that when 9cRA was present.
FIGURE 7.
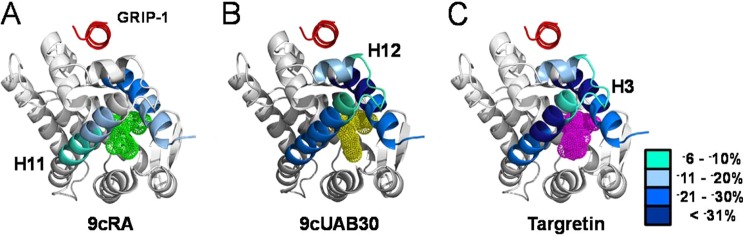
HDX MS analyses mapped onto hRXRα-LBD·GRIP-1·agonist structures. A, 9cRA; B, 9cUAB30; C, Targretin. Regions that show <5% difference in %D incorporation between agonist structures are gray. Regions that show >5% difference in %D are colored according to the heat map. Significant changes occur in H3, H11, and H12 regions and distinguish the two rexinoids from the pan-agonist 9cRA.
We and others have shown that an increase in %D was observed for the H12 peptide when the hRXRα-LBD had bound retinoid but lacked coactivator peptide (10, 12, 47). This indicated that the dynamics of H12 residues increased for holo-hRXRα-LBD containing 9cRA over apo-hRXRα-LBD. Based on these previous results, we evaluated HDX MS of the holo-hRXRα-LBD complexes with the two rexinoids without coactivator peptide and used apo-hRXRα-LBD as the reference control. In the absence of GRIP-1, H12 peptides also showed a positive HDX perturbation (ranging from 6 to 13%) when hRXRα-LBD was bound with rexinoid only. The %D for H3 and H11 peptides was reduced relative to the reference control. However, the extent of decrease in %D was dependent on retinoid compared with the analysis for this region when GRIP-1 was present. For example, the H3 Ala271–Ala278 peptide displayed a −52% perturbation when 9cRA was bound to homodimers and a −30% perturbation when 9cUAB30 was bound. Likewise, for the H11 peptide Pro423–Phe438, the greatest HDX perturbations were found when homodimers were bound to Targretin (−27%) or 9cRA (−24%), but this reduction was smaller when 9cUAB30 was bound (−18%). Because H11 contains 21 residues, there were overlapping peptides that spanned smaller sections of this α-helix. Based on the peptide analysis of overlapping peptides, we pinpointed the C-terminal end of H11 (Ile428–Phe438) as the sequence whose HDX perturbations were dependent on agonist. A complete listing the HDX perturbations of hRXRα-LBD peptides is provided in supplemental Table S2.
DISCUSSION
We have demonstrated that 9cUAB30 compared well with Targretin in its capacity to prevent mammary cancer (21–25). The in vitro studies presented here also show how similar each rexinoid is in blocking ErbB2-oncogenic transformation, consistent with in vivo studies. At the molecular level, each rexinoid recruits the coactivator peptide GRIP-1 to the surface of its LBD in 1:1 stoichiometry. The thermodynamics for the formation of the coactivator complex with this NR are the same regardless of which rexinoid is bound to the LBD. We investigated the structures and dynamics of GRIP-1 binding to the holo-hRXRα-LBD complex with the rexinoid Targretin or 9cUAB30. Through our analysis of these two structures and our previously reported 9cRA structure, we identify four structural changes that occur when GRIP-1 binds to the holo-hRXRα-LBD·rexinoid complex that appear to be hallmarks of hRXRα-LBD agonist plus coactivator active conformations. These changes are as follows: 1) the carboxylate group of Asp273 (H3) forms a strong interaction with Thr449 and Phe450 of H12; 2) the phenyl ring of Phe277 (H3) forms a π-hydrogen bond with Phe450 (H12); 3) the guanidinium group of Arg302 (H4) forms ionic interactions with Glu453 and Glu456 of H12; and 4) H11 changes its helical axis by 10°, and Phe437 (H11) moves from a solvent-exposed environment to one that interacts with hydrophobic side chain carbon atoms on Leu455 on H12. These structural changes are very similar to those identified for this NR bound to 9cRA (18). Our comparative HDX MS analysis of hRXRα-LBD dynamics clearly show that these regions experience the greatest change in %D when both ligand and coactivator are bound, as is reflected in Fig. 7.
Based on these results, we examined if these four structural features were present in other RXR structures contained in the Protein Data Bank. Twenty-six structures of hRXRα-LBD were found (data not shown). For the 15 structures of hRXRα-LBD homodimers containing a bound agonist and a coactivator peptide (GRIP-1 or SCR-1), we found each of the four structural features listed above (12, 30, 48–55). Two structures were of hRXRα-LBD homodimers or tetramers with corepressor peptides bound (56), and the four interactions identified for coactivator binding with agonist were missing. In a similar manner, the crystal structures of apo-hRXRα-LBD homodimer (7) or apo-hRXRα-LBD tetramer (57) did not contain these interactions. These structures support the importance of each of these interactions in establishing a linkage between the agonist in the LBP and the coactivator binding site on the surface on the receptor.
The crystallographic structures of the coactivator peptide complex with holo-hRXRα-LBD (containing rexinoid or pan-agonist) are very similar. In contrast to these subtle structural changes, we made use of comparative HDX MS to reveal large changes in dynamics between these two structures. Three helices (H3, H11, and H12) in holo-hRXRα-LBD homodimers were stabilized when GRIP-1 bound to hRXRα-LBD containing either 9cUAB30 or Targretin. The dynamics of peptides corresponding to essentially all of the H11 residues (spanning Leu419–Phe438) were significantly reduced (up to 39%) with GRIP-1 binding (Fig. 6 and supplemental Table S1). H11 tilts by 10°, and Phe437 at its carboxyl end makes a major conformational change and swings toward the interior of the LBD. As shown in Fig. 4B, Phe437 is central to a molecular bridge that connects H12 residues (Leu455 and Glu456) and residue His687 on the coactivator peptide GRIP-1 in one direction to the rexinoid ring in the other direction.
In light of this extensive change in H11 dynamics, we compared the contacts between the retinoid rings with H11 residues more closely. 9cRA interacts with H11 residues through its gem-dimethyl group on C1 (C16 and C17). We had reported that these interactions become more enhanced when coactivator peptide is present because the 9cRA ring rotates toward H11 on the GRIP-1-bound structure (18). (We previously reported that the trimethylcyclohexenyl ring inverts conformation, but we were in error.) Leu436, Phe439, His435, and Cys432 make favorable van der Waals contacts with the gem-dimethyl groups of 9cRA (C16 and C17) (Fig. 5A). For 9cUAB30, the tetralone ring does not contain methyl substituents, but the six-member cyclohexenyl ring (C5, C6, C7, C2′, C1′, and C18 with an exocyclic double bond) interacts well with many H11 residues. In particular, the methylene groups of this ring (C2′, C1′, and C18) make substantial contact with the four H11 residues in the LBP (Fig. 5C; C2′ forms van der Waals contact with Leu436, C1′ makes contact with Cys432, and C18 interacts with Phe439). For Targretin, the three methyl groups (C23, C24, and C25) contact H11 residues, but there is little contact from other carbon atoms in this ring. Targretin is more twisted in the LBP than the other two retinoids, and contact to H11 occurs only through these methyl groups (Fig. 5, B–D).
In addition to differences in H11 dynamics, we observed changes in dynamics for peptides containing Tyr249–Leu279 residues. These residues are contained in the loop connecting H1 to H3 and the amino end of H3 (Fig. 2). (H2 is resolved in x-ray structures of apo-hRXRα-LBD, but it is unresolved in all holostructures.) This region of the LBD includes Asp273 and Phe277 (both on H3), which change conformation and establish another molecular bridge connecting the H12 residues and the retinoids. Unlike the bridge for H11 residues, Phe277 interacts strongly with Phe450 on H12, which is a key residue for forming the hydrophobic pocket of Ile689 and Leu693 in the ILXXLL motif of GRIP-1 (Fig. 4B). Asp273 on H3 forms a strong salt bridge with Arg302 on H4. Whereas H3 residues substantially reduce dynamics upon GRIP-1 binding, the dynamics of H4 residues, including Arg302, are already low, and they do not significantly change with GRIP-1 binding (Fig. 6A). Ile268, Cys269, Ala271, and Ala272 on H3 are involved in forming the LBP for the rexinoids. Cys269 interacts very strongly with the C27 methyl group of Targretin, which occupies space that the other agonists do not (see Fig. 5, C and D). A conformational change occurs at the surface of the LBD receptor for Lys686 and Ile689 on GRIP-1 (Fig. 3, B and C), which may be communicated by this network of interactions.
H12 is necessary for recruitment of coactivator proteins and activation of transcription (58). Several studies of RXR activation have shown that H12 is not locked in a single active conformation, suggesting that H12 of RXR is only weakly influenced by the presence of the agonist (59, 60). For example, the structure of RXRβ bound by RXR-selective agonist LG268 showed that H12 did not adopt the active conformation when crystallized without coactivator (61). This is supported in our HDX MS studies of hRXRα-LBD dynamics. H12 stabilized only when GRIP-1 and rexinoids (Targretin or 9cUAB30) were present (Fig. 6 and supplemental Table S2). In contrast to the two rexinoids, the HDX MS analysis of the pan-agonist 9cRA binding clearly demonstrated that only GRIP-1 binding stabilizes H12 (12) and that 9cRA does not play a direct role (Fig. 6). Thus, our report provides the first evidence that retinoid agonists influence the H12 dynamics of hRXRα-LBD differently to recruit GRIP-1 coactivator peptide.
RXR tetramers form at physiologically relevant concentrations, and they are a transcriptionally silent form of the receptor (62). Tetramers dissociate into dimers upon ligand binding (63, 64). An mRXRΔH12 mutant does not have transcriptional activity, but it displays WT oligomeric activity (tetramer-dimer association), and cancer-relevant genes are up-regulated (65). Each of the regions of RXR LBD identified by their differential HDX MS profiles here (H3, H11, and H12) are important for RXR tetramer formation. The crystal structure of RXR tetramer demonstrated that the RXR tetrameric structure is stabilized by H11-H11 and H3-H3 interactions as well as interactions between the H12 AF-2 domain of one dimer and the coactivator binding groove of the other dimer (57). A recent tetramer structure that includes the silencing mediator for retinoid and thyroid hormone receptors (SMRT) and an antagonist, rhein, identified H3 and H11 in the autorepression of RXR (56). In fact, two key residues (Asp273 and Phe437), which we propose connect the LBP and the coactivator, were also shown to stabilize the antagonist and corepressor in the tetramer. This would lead us to hypothesize that the structurally different rexinoid agonists may interact with H3 and H11 residues uniquely and destabilize the hRXRα tetramer at different rates.
Reduction in H3 and H11 dynamics in this NR is a hallmark of coactivator peptide recruitment by an agonist-bound LBD. HDX MS is often used to screen potential drug libraries and predict potency for NR agonist activity. The capacity for an agonist to stabilize H3 and H11 should be a reflection of its potency, which makes HDX a valuable drug screen. Because the reduced dynamics are dependent on both the presence of agonist and coactivator peptide, the results presented here indicate that drug libraries are best screened when coactivator peptides are included rather than the more common approach, which compares NR dynamics in the presence and absence of only agonist. Importantly, our HDX MS results clearly demonstrate that changes in dynamics upon ligand binding alone are not necessarily indicative of the changes observed when the coactivator is present.
This study integrates structural with dynamical analyses in order to understand the manner by which two RXR-selective agonists, Targretin and 9cUAB30, recruit the coactivator peptide GRIP-1 to the surface of the LBD. Although these two rexinoids have significantly different chemical structures, each fills the LBP of hRXRα-LBD and allows the LBD to fold to nearly identical tertiary structures in the presence of the GRIP-1 coactivator peptide in the x-ray crystal structures. A review of all x-ray structures with and without coactivator peptides bound to this NR LBD allows us to define four structural changes that form two molecular bridges that connect agonist and coactivator binding sites. These structural changes are hallmarks that define how agonist and coactivator binding stabilize H3, H11, and H12 and initiate transcription.
Acknowledgment
We thank Bruce Pascal for assistance with data analysis software.
This work was supported, in whole or in part, by National Institutes of Health Grants 2 P50 CA089019 (to D. D. M.), 5 P50 CA089019 (to M. B. R.), RR17261 (to M. B. R.). This work was also supported by a grant from the Alabama Drug Discovery Alliance (to M. B. R.) and Komen Foundation Grant BCTR 20000690 (to D. D. M.).

This article contains supplemental Tables S1 and S2.
The atomic coordinates and structure factors (codes 4K4J and 4K6I) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NR
- nuclear receptor
- RXR
- retinoid X receptor
- hRXRα
- human retinoid X receptor α
- LBD
- ligand-binding domain
- 9cUAB30
- 9-cis-UAB30
- 9cRA
- 9-cis-retinoic acid
- GRIP-1
- glucocorticoid receptor-interacting protein-1
- HDX
- hydrogen/deuterium exchange
- LBP
- ligand binding pocket
- Hn
- helix n
- ITC
- isothermal titration calorimetry
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- %D
- percentage of deuterium incorporation
- RAR
- retinoic acid receptor
- RK3E cells
- rat kidney epithelial cells
- KLF4
- Krüppel-like factor 4
- Bis-Tris
- 2-Bis(2-hydroxyethyl)amino-2-(hydroxymethyl)-1,3-propanediol.
REFERENCES
- 1. Kroenke K., Arrington M. E., Mangelsdorff A. D. (1990) The prevalence of symptoms in medical outpatients and the adequacy of therapy. Arch. Intern. Med. 150, 1685–1689 [DOI] [PubMed] [Google Scholar]
- 2. Heyman R. A., Mangelsdorf D. J., Dyck J. A., Stein R. B., Eichele G., Evans R. M., Thaller C. (1992) 9-cis-retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 68, 397–406 [DOI] [PubMed] [Google Scholar]
- 3. Levin A. A., Sturzenbecker L. J., Kazmer S., Bosakowski T., Huselton C., Allenby G., Speck J., Kratzeisen C., Rosenberger M., Lovey A. (1992) 9-cis-Retinoic acid stereoisomer binds and activates the nuclear receptor RXRα. Nature 355, 359–361 [DOI] [PubMed] [Google Scholar]
- 4. Allegretto E. A., McClurg M. R., Lazarchik S. B., Clemm D. L., Kerner S. A., Elgort M. G., Boehm M. F., White S. K., Pike J. W., Heyman R. A. (1993) Transactivation properties of retinoic acid and retinoid X receptors in mammalian cells and yeast. Correlation with hormone binding and effects of metabolism. J. Biol. Chem. 268, 26625–26633 [PubMed] [Google Scholar]
- 5. Allenby G., Bocquel M. T., Saunders M., Kazmer S., Speck J., Rosenberger M., Lovey A., Kastner P., Grippo J. F., Chambon P. (1993) Retinoic acid receptors and retinoid X receptors. Interactions with endogenous retinoic acids. Proc. Natl. Acad. Sci. U.S.A. 90, 30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Egea P. F., Mitschler A., Rochel N., Ruff M., Chambon P., Moras D. (2000) Crystal structure of the human RXRα ligand-binding domain bound to its natural ligand: 9-cis-retinoic acid. EMBO J. 19, 2592–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bourguet W., Ruff M., Chambon P., Gronemeyer H., Moras D. (1995) Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-α. Nature 375, 377–382 [DOI] [PubMed] [Google Scholar]
- 8. Chalmers M. J., Busby S. A., Pascal B. D., He Y., Hendrickson C. L., Marshall A. G., Griffin P. R. (2006) Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 78, 1005–1014 [DOI] [PubMed] [Google Scholar]
- 9. Lu J., Cistola D. P., Li E. (2006) Analysis of ligand binding and protein dynamics of human retinoid X receptor α ligand-binding domain by nuclear magnetic resonance. Biochemistry 45, 1629–1639 [DOI] [PubMed] [Google Scholar]
- 10. Yan X., Broderick D., Leid M. E., Schimerlik M. I., Deinzer M. L. (2004) Dynamics and ligand-induced solvent accessibility changes in human retinoid X receptor homodimer determined by hydrogen deuterium exchange and mass spectrometry. Biochemistry 43, 909–917 [DOI] [PubMed] [Google Scholar]
- 11. Chalmers M. J., Pascal B. D., Willis S., Zhang J., Iturria S. J., Dodge J. A., Griffin P. R. (2011) Methods for the analysis of high precision differential hydrogen deuterium exchange data. Int. J. Mass Spectrom. 302, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xia G., Boerma L. J., Cox B. D., Qiu C., Kang S., Smith C. D., Renfrow M. B., Muccio D. D. (2011) Structure, energetics, and dynamics of binding coactivator peptide to the human retinoid X receptor α ligand binding domain complex with 9-cis-retinoic acid. Biochemistry 50, 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lehmann J. M., Jong L., Fanjul A., Cameron J. F., Lu X. P., Haefner P., Dawson M. I., Pfahl M. (1992) Retinoids selective for retinoid X receptor response pathways. Science 258, 1944–1946 [DOI] [PubMed] [Google Scholar]
- 14. Boehm M. F., Zhang L., Badea B. A., White S. K., Mais D. E., Berger E., Suto C. M., Goldman M. E., Heyman R. A. (1994) Synthesis and structure-activity relationships of novel retinoid X receptor-selective retinoids. J. Med. Chem. 37, 2930–2941 [DOI] [PubMed] [Google Scholar]
- 15. Bischoff E. D., Gottardis M. M., Moon T. E., Heyman R. A., Lamph W. W. (1998) Beyond tamoxifen. The retinoid X receptor-selective ligand LGD1069 (TARGRETIN) causes complete regression of mammary carcinoma. Cancer Res. 58, 479–484 [PubMed] [Google Scholar]
- 16. Bischoff E. D., Heyman R. A., Lamph W. W. (1999) Effect of the retinoid X receptor-selective ligand LGD1069 on mammary carcinoma after tamoxifen failure. J. Natl. Cancer Inst. 91, 2118. [DOI] [PubMed] [Google Scholar]
- 17. Gottardis M. M., Bischoff E. D., Shirley M. A., Wagoner M. A., Lamph W. W., Heyman R. A. (1996) Chemoprevention of mammary carcinoma by LGD1069 (Targretin). An RXR-selective ligand. Cancer Res. 56, 5566–5570 [PubMed] [Google Scholar]
- 18. Lubet R. A., Christov K., Nunez N. P., Hursting S. D., Steele V. E., Juliana M. M., Eto I., Grubbs C. J. (2005) Efficacy of Targretin on methylnitrosourea-induced mammary cancers. Prevention and therapy dose-response curves and effects on proliferation and apoptosis. Carcinogenesis 26, 441–448 [DOI] [PubMed] [Google Scholar]
- 19. Duvic M., Martin A. G., Kim Y., Olsen E., Wood G. S., Crowley C. A., Yocum R. C. (2001) Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphoma. Arch. Dermatol. 137, 581–593 [PubMed] [Google Scholar]
- 20. Miller V. A., Benedetti F. M., Rigas J. R., Verret A. L., Pfister D. G., Straus D., Kris M. G., Crisp M., Heyman R., Loewen G. R., Truglia J. A., Warrell R. P., Jr. (1997) Initial clinical trial of a selective retinoid X receptor ligand, LGD1069. J. Clin. Oncol. 15, 790–795 [DOI] [PubMed] [Google Scholar]
- 21. Muccio D. D., Brouillette W. J., Breitman T. R., Taimi M., Emanuel P. D., Zhang X., Chen G., Sani B. P., Venepally P., Reddy L., Alam M., Simpson-Herren L., Hill D. L. (1998) Conformationally defined retinoic acid analogues. 4. Potential new agents for acute promyelocytic and juvenile myelomonocytic leukemias. J. Med. Chem. 41, 1679–1687 [DOI] [PubMed] [Google Scholar]
- 22. Dawson M. I., Jong L., Hobbs P. D., Cameron J. F., Chao W. R., Pfahl M., Lee M. O., Shroot B., Pfahl M. (1995) Conformational effects on retinoid receptor selectivity. 2. Effects of retinoid bridging group on retinoid X receptor activity and selectivity. J. Med. Chem. 38, 3368–3383 [DOI] [PubMed] [Google Scholar]
- 23. Grubbs C. J., Lubet R. A., Atigadda V. R., Christov K., Deshpande A. M., Tirmal V., Xia G., Bland K. I., Eto I., Brouillette W. J., Muccio D. D. (2006) Efficacy of new retinoids in the prevention of mammary cancers and correlations with short-term biomarkers. Carcinogenesis 27, 1232–1239 [DOI] [PubMed] [Google Scholar]
- 24. Atigadda V. R., Vines K. K., Grubbs C. J., Hill D. L., Beenken S. L., Bland K. I., Brouillette W. J., Muccio D. D. (2003) Conformationally defined retinoic acid analogues. 5. Large-scale synthesis and mammary cancer chemopreventive activity for (2E,4E,6Z,8E)-8-(3′,4′-dihydro-1′(2′H)-naphthalen-1′-ylidene)-3,7-dimethyl-2,4,6-octatrienoic acid (9cUAB30). J. Med. Chem. 46, 3766–3769 [DOI] [PubMed] [Google Scholar]
- 25. Lindeblad M., Kapetanovic I. M., Kabirov K. K., Dinger N., Mankovskaya I., Morrisey R., Martín-Jiménez T., Lyubimov A. (2011) Assessment of oral toxicity and safety of 9-cis-UAB30, a potential chemopreventive agent, in rat and dog studies. Drug Chem. Toxicol. 34, 300–310 [DOI] [PubMed] [Google Scholar]
- 26. Vedell P. T., Lu Y., Grubbs C. J., Yin Y., Jiang H., Bland K. I., Muccio D. D., Cvetkovic D., You M., Lubet R. (2013) Effects on gene expression in rat liver after administration of RXR agonists. UAB30, 4-methyl-UAB30, and Targretin (Bexarotene). Mol. Pharmacol. 83, 698–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kolesar J. M., Hoel R., Pomplun M., Havighurst T., Stublaski J., Wollmer B., Krontiras H., Brouillette W., Muccio D., Kim K., Grubbs C. J., Bailey H. E. (2010) A pilot, first-in-human, pharmacokinetic study of 9cUAB30 in healthy volunteers. Cancer Prev. Res. (Phila.) 3, 1565–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang W., Deng W., Bailey S. K., Nail C. D., Frost A. R., Brouillette W. J., Muccio D. D., Grubbs C. J., Ruppert J. M., Lobo-Ruppert S. M. (2009) Prevention of KLF4-mediated tumor initiation and malignant transformation by UAB30 rexinoid. Cancer Biol. Ther. 8, 289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Egea P. F., Moras D. (2001) Purification and crystallization of the human RXRα ligand-binding domain-9-cisRA complex. Acta Crystallogr. D Biol. Crystallogr. 57, 434–437 [DOI] [PubMed] [Google Scholar]
- 30. Egea P. F., Mitschler A., Moras D. (2002) Molecular recognition of agonist ligands by RXRs. Mol. Endocrinol. 16, 987–997 [DOI] [PubMed] [Google Scholar]
- 31. Sobolev V., Sorokine A., Prilusky J., Abola E. E., Edelman M. (1999) Automated analysis of interatomic contacts in proteins. Bioinformatics 15, 327–332 [DOI] [PubMed] [Google Scholar]
- 32. Kleywegt G. J., Jones T. A. (1994) Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D Biol. Crystallogr. 50, 178–185 [DOI] [PubMed] [Google Scholar]
- 33. Kleywegt G. J., Jones T. A. (1996) xdlMAPMAN and xdlDATAMAN. Programs for reformatting, analysis and manipulation of biomacromolecular electron-density maps and reflection data sets. Acta Crystallogr. D Biol. Crystallogr. 52, 826–828 [DOI] [PubMed] [Google Scholar]
- 34. Pascal B. D., Chalmers M. J., Busby S. A., Griffin P. R. (2009) HD desktop. An integrated platform for the analysis and visualization of H/D exchange data. J. Am. Soc. Mass Spectrom. 20, 601–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kang S., Poliakov A., Sexton J., Renfrow M. B., Prevelige P. E., Jr. (2008) Probing conserved helical modules of portal complexes by mass spectrometry-based hydrogen/deuterium exchange. J. Mol. Biol. 381, 772–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ascano J. M., Beverly L. J., Capobianco A. J. (2003) The C-terminal PDZ-ligand of JAGGED1 is essential for cellular transformation. J. Biol. Chem. 278, 8771–8779 [DOI] [PubMed] [Google Scholar]
- 37. Capobianco A. J., Zagouras P., Blaumueller C. M., Artavanis-Tsakonas S., Bishop J. M. (1997) Neoplastic transformation by truncated alleles of human NOTCH1/TAN1 and NOTCH2. Mol. Cell. Biol. 17, 6265–6273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Foster K. W., Ren S., Louro I. D., Lobo-Ruppert S. M., McKie-Bell P., Grizzle W., Hayes M. R., Broker T. R., Chow L. T., Ruppert J. M. (1999) Oncogene expression cloning by retroviral transduction of adenovirus E1A-immortalized rat kidney RK3E cells. Transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF. Cell Growth Differ. 10, 423–434 [PubMed] [Google Scholar]
- 39. Kolligs F. T., Hu G., Dang C. V., Fearon E. R. (1999) Neoplastic transformation of RK3E by mutant β-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol. Cell. Biol. 19, 5696–5706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jung D., Inagaki T., Gerard R. D., Dawson P. A., Kliewer S. A., Mangelsdorf D. J., Moschetta A. (2007) FXR agonists and FGF15 reduce fecal bile acid excretion in a mouse model of bile acid malabsorption. J. Lipid Res. 48, 2693–2700 [DOI] [PubMed] [Google Scholar]
- 41. Rahman L., Voeller D., Rahman M., Lipkowitz S., Allegra C., Barrett J. C., Kaye F. J., Zajac-Kaye M. (2004) Thymidylate synthase as an oncogene. A novel role for an essential DNA synthesis enzyme. Cancer Cell 5, 341–351 [DOI] [PubMed] [Google Scholar]
- 42. Tonon G., Modi S., Wu L., Kubo A., Coxon A. B., Komiya T., O'Neil K., Stover K., El-Naggar A., Griffin J. D., Kirsch I. R., Kaye F. J. (2003) t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat. Genet. 33, 208–213 [DOI] [PubMed] [Google Scholar]
- 43. Foster K. W., Liu Z., Nail C. D., Li X., Fitzgerald T. J., Bailey S. K., Frost A. R., Louro I. D., Townes T. M., Paterson A. J., Kudlow J. E., Lobo-Ruppert S. M., Ruppert J. M. (2005) Induction of KLF4 in basal keratinocytes blocks the proliferation-differentiation switch and initiates squamous epithelial dysplasia. Oncogene 24, 1491–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rocha G. M., Michea L. F., Peters E. M., Kirby M., Xu Y., Ferguson D. R., Burg M. B. (2001) Direct toxicity of nonsteroidal antiinflammatory drugs for renal medullary cells. Proc. Natl. Acad. Sci. U.S.A. 98, 5317–5322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghosh J. C., Yang X., Zhang A., Lambert M. H., Li H., Xu H. E., Chen J. D. (2002) Interactions that determine the assembly of a retinoid X receptor/corepressor complex. Proc. Natl. Acad. Sci. U.S.A. 99, 5842–5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dai S. Y., Chalmers M. J., Bruning J., Bramlett K. S., Osborne H. E., Montrose-Rafizadeh C., Barr R. J., Wang Y., Wang M., Burris T. P., Dodge J. A., Griffin P. R. (2008) Prediction of the tissue-specificity of selective estrogen receptor modulators by using a single biochemical method. Proc. Natl. Acad. Sci. U.S.A. 105, 7171–7176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang J., Chalmers M. J., Stayrook K. R., Burris L. L., Wang Y., Busby S. A., Pascal B. D., Garcia-Ordonez R. D., Bruning J. B., Istrate M. A., Kojetin D. J., Dodge J. A., Burris T. P., Griffin P. R. (2011) DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat. Struct. Mol. Biol. 18, 556–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. le Maire A., Bourguet W., Balaguer P. (2010) A structural view of nuclear hormone receptor. Endocrine disruptor interactions. Cell. Mol. Life Sci. 67, 1219–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. le Maire A., Grimaldi M., Roecklin D., Dagnino S., Vivat-Hannah V., Balaguer P., Bourguet W. (2009) Activation of RXR-PPAR heterodimers by organotin environmental endocrine disruptors. EMBO Rep. 10, 367–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lippert W. P., Burschka C., Götz K., Kaupp M., Ivanova D., Gaudon C., Sato Y., Antony P., Rochel N., Moras D., Gronemeyer H., Tacke R. (2009) Silicon analogues of the RXR-selective retinoid agonist SR11237 (BMS649). Chemistry and biology. ChemMedChem 4, 1143–1152 [DOI] [PubMed] [Google Scholar]
- 51. Nahoum V., Pérez E., Germain P., Rodríguez-Barrios F., Manzo F., Kammerer S., Lemaire G., Hirsch O., Royer C. A., Gronemeyer H., de Lera A. R., Bourguet W. (2007) Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. Proc. Natl. Acad. Sci. U.S.A. 104, 17323–17328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pérez Santín E., Germain P., Quillard F., Khanwalkar H., Rodríguez-Barrios F., Gronemeyer H., de Lera A. R., Bourguet W. (2009) Modulating retinoid X receptor with a series of (E)-3-[4-hydroxy-3-(3-alkoxy-5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)phenyl]acrylic acids and their 4-alkoxy isomers. J. Med. Chem. 52, 3150–3158 [DOI] [PubMed] [Google Scholar]
- 53. Zhang H., Li L., Chen L., Hu L., Jiang H., Shen X. (2011) Structure basis of bigelovin as a selective RXR agonist with a distinct binding mode. J. Mol. Biol. 407, 13–20 [DOI] [PubMed] [Google Scholar]
- 54. Zhang H., Xu X., Chen L., Chen J., Hu L., Jiang H., Shen X. (2011) Molecular determinants of magnolol targeting both RXRα and PPARγ. PLoS One 6, e28253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang Y., Zhang H., Yao X. G., Shen H., Chen J., Li C., Chen L., Zheng M., Ye J., Hu L., Shen X., Jiang H. (2012) (+)-Rutamarin as a dual inducer of both GLUT4 translocation and expression efficiently ameliorates glucose homeostasis in insulin-resistant mice. PLoS One 7, e31811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang H., Chen L., Chen J., Jiang H., Shen X. (2011) Structural basis for retinoic X receptor repression on the tetramer. J. Biol. Chem. 286, 24593–24598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gampe R. T., Jr., Montana V. G., Lambert M. H., Wisely G. B., Milburn M. V., Xu H. E. (2000) Structural basis for autorepression of retinoid X receptor by tetramer formation and the AF-2 helix. Genes Dev. 14, 2229–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barroso I., Gurnell M., Crowley V. E., Agostini M., Schwabe J. W., Soos M. A., Maslen G. L., Williams T. D., Lewis H., Schafer A. J., Chatterjee V. K., O'Rahilly S. (1999) Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402, 880–883 [DOI] [PubMed] [Google Scholar]
- 59. Renaud J. P., Rochel N., Ruff M., Vivat V., Chambon P., Gronemeyer H., Moras D. (1995) Crystal structure of the RAR-γ ligand-binding domain bound to all-trans-retinoic acid. Nature 378, 681–689 [DOI] [PubMed] [Google Scholar]
- 60. Lin B. C., Hong S. H., Krig S., Yoh S. M., Privalsky M. L. (1997) A conformational switch in nuclear hormone receptors is involved in coupling hormone binding to corepressor release. Mol. Cell. Biol. 17, 6131–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Love J. D., Gooch J. T., Benko S., Li C., Nagy L., Chatterjee V. K., Evans R. M., Schwabe J. W. (2002) The structural basis for the specificity of retinoid-X receptor-selective agonists. New insights into the role of helix H12. J. Biol. Chem. 277, 11385–11391 [DOI] [PubMed] [Google Scholar]
- 62. Kersten S., Kelleher D., Chambon P., Gronemeyer H., Noy N. (1995) Retinoid X receptor α forms tetramers in solution. Proc. Natl. Acad. Sci. U.S.A. 92, 8645–8649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kersten S., Pan L., Chambon P., Gronemeyer H., Noy N. (1995) Role of ligand in retinoid signaling. 9-cis-retinoic acid modulates the oligomeric state of the retinoid X receptor. Biochemistry 34, 13717–13721 [DOI] [PubMed] [Google Scholar]
- 64. Kersten S., Pan L., Noy N. (1995) On the role of ligand in retinoid signaling. Positive cooperativity in the interactions of 9-cis-retinoic acid with tetramers of the retinoid X receptor. Biochemistry 34, 14263–14269 [DOI] [PubMed] [Google Scholar]
- 65. Yasmin R., Kannan-Thulasiraman P., Kagechika H., Dawson M. I., Noy N. (2010) Inhibition of mammary carcinoma cell growth by RXR is mediated by the receptor's oligomeric switch. J. Mol. Biol. 397, 1121–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]