

Abstract
LMO2 was discovered via chromosomal translocations in T-cell leukaemia and shown normally to be essential for haematopoiesis. LMO2 is made up of two LIM only domains (thus it is a LIM-only protein) and forms a bridge in a multi-protein complex. We have studied the mechanism of formation of this complex using a single domain antibody fragment that inhibits LMO2 by sequestering it in a non-functional form. The crystal structure of LMO2 with this antibody fragment has been solved revealing a conformational difference in the positioning and angle between the two LIM domains compared with its normal binding. This contortion occurs by bending at a central helical region of LMO2. This is a unique mechanism for inhibiting an intracellular protein function and the structural contusion implies a model in which newly synthesized, intrinsically disordered LMO2 binds to a partner protein nucleating further interactions and suggests approaches for therapeutic targeting of LMO2.
The LIM-only family of proteins has four members (LMO1-4) and are implicated in a spectrum of cancers1, including T cell leukaemia (LMO1, 2, 3)2,3,4,5,6,7, diffuse large B cell lymphoma (LMO2)8,9, prostate cancer (LMO2)10,11, neuroblastoma (LMO1 and LMO3)12,13,14 and breast carcinoma (LMO4)15. LMO2 was originally cloned from a chromosomal translocation breakpoint in T-cell acute leukaemia (T-ALL)3,5 that involved T-cell receptor loci (either TCRδ at 14q11 or TCRβ 7q35 in respectively t(11;14)(p13;q11) or t(7;11)(q35;p13)) and the LMO2 gene on chromosome 11 at band p13. Aberrant LMO2 expression is oncogenic in T cells16,17,18, while its normal role is in haematopoiesis, being required for both primitive and definitive haematopoiesis19,20, as well as a role in endothelial cell remodeling21. The LMO2 gene encodes a cysteine-rich protein, comprising two zinc-binding LIM domains22, each with two LIM fingers, that acts as a protein binding module interacting with a number of different proteins to form bipartite DNA-binding multi-protein complexes23.
In the normal development of T cells, LMO2 expression is down regulated during the initial stages of T-cell development24 and is not required for normal development of this lineage25. T-ALL is characterized by an accumulation of immature T-cells in the bone marrow. The effect of enforced LMO2 expression in T-cells was analysed with a transgenic mouse model in which Lmo2 was expressed in thymocytes using the CD2 promoter. Analysis of thymocytes from these mice showed an asymptomatic (pre-leukaemic) phase associated with an increase in the proportion of CD4-;CD8- double negative cells18,26 with a majority block at the double negative 3 (DN3; CD44-;CD25+) stage of development preceding the appearance of clonal T cell tumours27. Transplantation studies using the T cells from a CD2-Lmo2 transgenic mouse showed that the DN3 cells (but not DN1, DN2 or DN4) could be transplanted into irradiated recipients and had properties of self-renewal27, further supporting the concept that the initial function of LMO2 in T cell neoplasia is to cause a population cells (DN3) to expand as the site of secondary mutations to cause overt neoplasia. The Lmo2-transgenic mice develop clonal thymic tumours from around 6–9 months18,26 implying that LMO2 is necessary but not sufficient for T cell neoplasia. Conversely, it was shown that intracellular antibody fragments and peptide aptamers that interfere with LMO2 in tumour cells are effective in suppressing tumour growth28,29,30, indicating that sustained expression is required to maintain T cell tumourigenesis.
Since LMO2 functions as a transcription factor that assembles multi-protein complexes in normal settings (e.g. erythropoiesis) or abnormal settings (e.g. T cell cancer where chromosomal translocations cause aberrant LMO2 expression), approaches are needed to interrogate the mechanisms by which these protein complexes form. This will lead to an understanding of the biochemistry of this transcription factor complex assembly and the controls that are exerted in aberrant settings such as tumours and will potentially add to understanding of how protein complexes form in normal and abnormal cell metabolism. We previously compared the DNA binding properties of the LMO2-complex in erythroid cells with an LMO2-complex in T cell tumours from a strain of CD2-Lmo2 transgenic mice using the CASTing methodology31. We observed that in erythroid cells (a normal LMO2 expression setting) the LMO2 complex comprised LDB1, TAL1/SCL, E47 and GATA1 with the TAL1/SCL-E47 basic-helix-loop-helix (bHLH) heterodimer binding an E-box DNA motif and the GATA1 protein binding the GATA site23. Conversely, the T cell complex could comprise LMO2, LDB1 with two TAL1/SCL-E47 heterodimers, each binding to E-box motifs in T cells from Lmo2-transgenic mice32. Further binding studies with LMO2 showed it has the capability to bind to other bHLH proteins such as LYL1 and TAL233 both of which have been implicated in T cell tumours34 suggesting a possible inter-connectivity of LMO2 and bHLH proteins, dictated by aberrant expression of LMO2 in human cancer. LMO2 is down regulated by the DN3 stage in mice18,26,27 and evidently it is enforced expression of LMO2 that results in a block in T-cell development.
In addition to the intricacies of the protein-protein interactions (PPIs), a central question about LMO2 is the way in which it, and other specific proteins, find each other in the cellular milieu and locate the DNA binding sites. As a corollary, there are currently no clear mechanisms that could be exploited to develop therapies based on interfering with these LMO2-dependent PPI. The central role of LMO2 prompted us to develop an anti-LMO2 single VH domain intracellular antibody fragment (referred to as VH#576) to interfere with LMO2 protein function35. Antibodies and derivative fragments have the capacity to bind a huge variety of antigens with high specificity and affinity. Antibody binding sites comprise heavy chain variable domain (VH) and light chain variable domains (VL), each with three complementarity determining regions (CDRs), directly contacting antigen and involved in binding affinity and specificity. Single domain antibody fragments encompass a minimal antigen recognition and can also be used for intracellular applications. The single domain comprising specific V region framework scaffolds have excellent properties of solubility, stability, and high expression levels within the cell.
VH#576 moderates the tumourigenic function of LMO2 in vivo as it prevents Lmo2-dependent tumour growth in a transplantation assay. We have determined the crystal structure of human LMO2 in complex with VH#576 at 2.9 Å resolution (PDB ID 4KFZ) and found that LMO2 adopts a stable but contorted structure whose conformation is altered around central, short alpha helical region between the two LIM domains36. All three complementarity determining regions (CDRs) of the VH#576 are involved in direct interaction with LMO2. This suggests a mechanism for the normal role of LMO2 in protein complex formation and a mechanism for the perturbation of LMO2 structure-function by the VH. Our work locates specific interacting residues on the surface of both LMO2 and VH#576 that could inform the design of small molecules to disrupt leukaemia-associated LMO2 complexes.
Results
Interaction with the single antibody domain stabilises LMO2 structure
We previously described a single domain (VH) antibody fragment (VH#57635), identified by the Intracellular Antibody Capture procedure37,38, that displays inhibitory function when binding to LMO2 in T cell neoplasias or erythroid cells. As an approach to determine the mechanism of LMO2 inhibition by VH#576, we co-expressed LMO2 with VH#576 as a recombinant protein complex in E. coli for structural studies. Whereas previous attempts to make soluble LMO2 recombinant protein have been unsuccessful, co-expression with VH#576 in E. coli drastically increases the solubility, due in part to shielding of hydrophobic regions. Parenthetically, this property of single antibody domains, or scFv, to assist in deriving soluble recombinant proteins, should be a generally useful property for structural studies of otherwise intransigent proteins.
Crystal trials were carried out with the purified heterodimer and diffracting crystals were obtained (Supplementary Fig. 1). This resulted in a 2.9 Å structure of human LMO2 in complex with VH#576 (Fig. 1 and data collection statistics in Supplementary Table 1). The architecture of VH#576 is that of the common immunoglobulin fold and was solved by molecular replacement. The LIM1 and LIM2 domains of LMO2 were positioned according to experimentally determined zinc positions. Structure refinement was completed using PHENIX-REFINE and AUTOBUSTER software. The LIM1 domain of LMO2 possesses the characteristic LIM domain structure of four β-strands and this is followed by a short α-helix, encompassing the hinge region residue Phe8836. Each zinc atom was positioned between a pair of anti-parallel β-strands. All four zinc ions are present in the structure and each is coordinated in a tetrahedral geometry by the side chains of three cysteine residues and one histidine or aspartic acid residue. Furthermore, zinc ions are stable in a cellular environment due to the lack of redox activity39.
Figure 1. Structure of the LMO2 in complex with the anti-LMO2 VH#576.
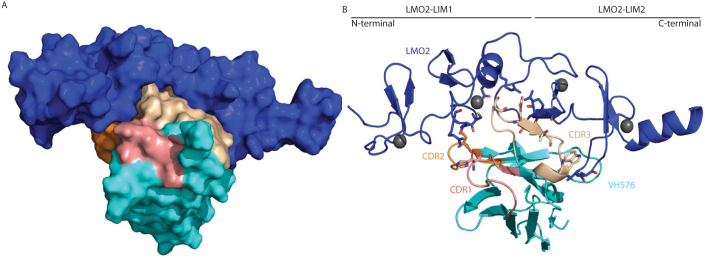
The crystal structure of the dimeric complex of LMO2 and anti-LMO2 VH is shown either in space filling (A) or ribbon form (B). In both, the LMO2 protein is shown in blue and the VH framework region in cyan with CDR regions one, two and three highlighted in salmon, orange and cream respectively. In panel B, the zinc atoms are shown as grey spheres and sticks are used to represent residues involved in inter-molecular hydrogen bonds with oxygen and nitrogen atoms coloured red and blue respectively. For VH#576, there is one residue of CDR one (His31) forming a hydrogen bond, one in CDR two (Ser57) and four in CDR3 (Ser103, Glu105, Thr107 and Trp110).
LMO2 has a long N-terminal tail (residue 8–26, see Supplementary Fig. 2B) with no defined secondary structure in the crystal. The first 20 residues of LMO2 are predicted to be disordered by regional order neural network software40. This region has a high content of charged/hydrophilic residues and few bulky hydrophobic residues characteristic of intrinsic disordered proteins41. The function of this N-terminal region may be in transcriptional control, as we have shown that it can act as a transcriptional trans-activator in two-hybrid reporter assays42.
Each of the VH#576 CDR loops is involved in the binding to LMO2 (Fig. 1) and these span amino-acid contacts in both LIM domains (contact residues are listed in Supplementary Table 2). In addition, there is an interacting loop from a framework region that binds to the LIM2 domain (Fig. 1B); this region of hydrophobic contacts lies at the former VH-VL interface of VH#576 spanning Val37 to Tyr50 (see Supplementary Fig. 2A). The close proximity of residues Leu45 and Tryp47 of VH#576 to Met108 and Met106 of Lmo2 indicate the hydrophobic effect to be a driving force in the LMO2-VH interaction. Both the methionine residues of LMO2 are located in the finger 3 region. Other hydrophobic amino acids interactions are in finger 4 and between the zinc-coordination residues of the second zinc atom of finger 3 (Supplementary Table 2).
The potency of interaction of the individual residues in the VH#576 CDRs was elucidated by their mutagenesis to Gly and/or Ala and the effect on interaction between the mutant VH#576 and LMO2 was determined by a mammalian two-hybrid luciferase reporter assay (Fig. 2A–C). To assess effects of the CDR mutations on expression levels, each mutant VH made as a fusion with VP16 at the C-terminal and expression assessed by Western blotting using anti-VP16 antibody (shown at the bottom of each panel of Fig. 2A–C and in Supplementary Fig. 3 with controls of VH#576 and/or VHY#6). These data show that residues in all three CDRs are critical for VH binding as specific mutations ablate interaction of the VH and LMO2 in the mammalian intracellular assay. The critical CDR residues are indicated in Supplementary Fig. 2A. Residues of CDR3 are particularly important (8 residues in this region are hot spots) when analysed in the two hybrid assay, as it has the highest proportion of residues involved in the interaction with LMO2. This concurs with the crystal structure (Fig. 2D, E). A hydrogen bonding network occurs between the LIM finger three of LIM2 and CDR3 of VH#576 residues Ser103, Glu105 and Thr107 (Fig. 2E, Supplementary Table 2). Arg109, a residue that is critical in LMO2-LDB1 LID interaction, forms a salt bridge with Glu105 of VH#576. In effect the VH#576 exploits naturally used loops (CDRs) as well as a region generally masked by VH-VL interaction to perform its role in inhibiting LMO2 function.
Figure 2. Probing the interaction surface of VH#576 with LMO2 using site directed mutagenesis.

The CDR residues of VH#576 were mutated to glycine or alanine and the mutant sequences cloned into pEF-VP16 vector (prey vector) for use in a mammalian two-hybrid luciferase reporter assay. These assays were performed by transient transfection into CHO cells by co-transfecting the prey vector with a vector expressing a Gal4-LMO2 fusion bait (pM1-δLMO2); luciferase signals in the histograms are the ratio of Firefly luciferase to Renilla luciferase where the latter was used as a transfection control. Controls were performed using VH#576 (positive control) and anti-RAS VH#6 (negative control). The expression levels of each mutant VH#576 protein were established by Western detection with an anti-VP16 antibody (shown at the bottom of panels A, B, C). Panel A shows data for mutations of CDR1 residues, panel B mutations of CDR2 residues and panel C mutations of CDR3 residues. Each bar represents an average of luciferase activity measured for two wells (replicates) and the bar extensions indicate the standard deviations. Panels D and E highlight the key residues in the VH#576 that emerged from the mutation analysis shown in A-C. The CDR1, CDR2 and CDR3 of VH#576 are shown in salmon, orange and cream respectively. In panel D, LMO2 is shown in yellow (space filling format) and the framework region of VH in cyan, with the indicated amino acids being from the VH. Red patches are areas of oxygen atoms, blue are areas of nitrogen atoms. In panel E the interactions of VH#576 CDR3 are highlighted. A majority of the interactions occur through the CDR3 region (cream) with a total of seven CDR3 residues identified as critical for the interaction. A series of interactions occur across the hinge region (Phe88) of LMO2 including a salt bridge between Glu105 and Arg86, and polar contacts, represented by dashed lines. Zinc atoms are shown as yellow spheres.
LMO2 structure differs when bound to the VH or to LDB1 LID
We generated an in silico structure of the LMO2 protein using mouse Lmo4 and partial mouse Lmo2 structural data43,44 that identified a central helical region between the two LIM domains28. This included the hinge region residue Phe88 that was identified when the structure of LMO2 was solved in complex with LDB1-LID36. LDB1 is a widely expressed nuclear adaptor protein that binds LMO proteins through its C-terminal LID (LIM-interacting domain) and forms part of the LMO2 protein complex first identified in erythroid cells23 and later shown to be of similar composition in T cell neoplastic cells32. Superimposition of the respective shapes of LMO2 was carried out for our current structural analysis of LMO2-VH complex with the LMO2:LDB1-LID fusion protein36 and using the first LIM domain of LMO2 in both crystal structures for alignment (Fig. 3A). This shows that the LIM domains of both dimeric structures have close structural similarity but the relative orientation of the LIM1 and LIM2 domain varies markedly (Fig. 3A, B).
Figure 3. Comparison of the geometry of the LMO2:VH dimer with the LMO2:LDB1-LID protein.

Superimpositions are shown of LMO2 with the LDB1 LID domain or with anti-LMO2 VH#576 and were performed using the first LIM domain for alignment. Panel A depicts the structure of LMO2 in green (with the zinc atoms shown as green spheres) when bound to LID or in blue when bound to VH#576. Panel B shows the dimeric LMO2 structures with LDB1-LID shown in red bound to LMO2 (green) compared with the structure of the LMO2 (shown in dark blue) complexed with the VH (shown in cyan). The angle between LIM1 and LIM2 domains differs by 23° between the two LMO2 structures. LMO2 wraps around VH#576 and forms a strong hydrogen bonding network including the β-strand of VH#576 typical of protein-protein interactions. The β-strands at this interaction site are parallel and an analogous interaction occurs between β-strand 7 of LMO2 (see Supplementary fig. 2) and LDB1-LID, with LMO2 residues Thr107 and Arg109, being key residues in both interactions. Hydrophobic amino acids at the protein-protein interface are indicated in panel C, focused around LMO2 residues Met106, Met108, Leu117 and Phe120 with VH#576 residues Trp47 and Leu45 indicating the hyrdrophobic effect contributes to binding. Zinc atoms are shown as green or blue spheres respectively where LMO2 interacts with LDB1 or with VH#576.
The LIM2 is mis-oriented compared with LMO2:LDB1-LID allowing interactions between VH#576 and the conserved β-strand, b7 of LMO2 (see Supplementary Fig. 2 for β strand nomenclature). In figure 3C, hydrophobic amino acids are represented in ball and stick form at the former VH/VL interface of anti-LMO2 and residues Met106 and Leu117 indicating the hydrophobic contribution to binding at the protein-protein interface. Specifically, the angle between LIM1 and LIM2 varies by 23° between the two structures. The zinc positions within the electron density map were located using PHENIX AutoSol and are shown in Supplementary figure 4. Although an angle of motion between the LIM domains of LMO2 was observed between two independently determined crystal structures of LMO2:LDB1-LID fusion protein45,36 when comparing LMO2 in complex with LID or with VH#576 an overall root mean square deviation (RMSD) of 5.3 Å was calculated. The conformational change in LMO2 bound by the VH is more profound than that between LMO2:LDB1-LID structures where there is a maximum overall RMSD of 2.6 Å. In essence, VH#576 acts to contort LMO2 through a series of high affinity interactions, particularly with LIM2 of LMO2. Thus we conclude that the VH#576 interaction with LMO2 results in contortion of the LMO2 protein because LMO2 is highly dynamic and flexible such that its stable configuration is dependent upon its interaction partner. This may reflect a mechanism of allosteric control mediating associations with a broad range of natural protein partners.
The anti-LMO2 VH sequesters LMO2 from normal binding partners in vivo
The alternative conformations that the LMO2 flexibility allows suggests that protein-protein interactions can be influenced by this and that the mechanism by which the anti-LMO2 VH impairs function is by removing newly synthesized LMO2 into a separate complex. This hypothesis was tested using transfection studies in CHO cells that assessed individual interactions. It has been shown that LMO2 binds directly to a number of T-ALL associated bHLH proteins, including TAL1/SCL, TAL2 and LYL118,23. LMO2 does not bind directly to the E2A bHLH E12 or E4733 but forms a complex with E47 via LMO2-TAL1/SCL interaction in which the latter hetero-dimerises with E47. Luciferase assays were performed in CHO cells that were transfected with two LMO2 baits comprising the Gal4DBD fused to LMO2 or the LMO2-LDB1 LID fusion and a TAL1/SCL-VP16 fusion prey, with or without a vector expressing E47 bHLH (Fig. 4A, B). TAL1/SCL binds to LMO2 but does so relatively poorly compared with the LMO2-LID fusion (Fig. 4A). In addition, the binding has more efficacy when the TAL1/SCL partner E47 is present (Fig. 4A) although again the LMO2-LID bait is more efficacious. Little evidence of interaction was found with the LMO2-VH#576 when the LMO2 was expressed as the fusion with the LDB1 LID (Fig. 4B) since the luciferase activation was reduced to control levels (e.g. when LMO2-LID was co-expressed with an anti-RAS VH#Y6).
Figure 4. Intracellular binding of anti-LMO2 VH#576 to the LMO2 protein.

A mammalian luciferase two hybrid assay was used to assess interactions of LMO2 with partners (panel A) or VH (panel B). COS7 cells were transfected with GAL4-DBD bait plasmids expressing GAL4-LMO2 or GAL4-LMO2:LDB1-LID and VP16 activation domain prey plasmids expressing TAL1/SCL-VP16, VH#576-VP16 or VH#6-VP16. Where indicated, a plasmid expressing the E47 bHLH was co-transfected. Triplicate samples were analysed. p values of <0.005 were found for the difference between the interaction of LMO2 and TAL1/SCL with E47 or LMO2:LDB1-LID and TAL1/SCL with E47; <0.001 for the difference between the interaction of LMO2 and TAL1/SCL or LMO2:LDB1-LID and TAL1/SCL (both indicated by *) and 0.0001 for the difference between the interaction of TAL1/SCL and LMO2/LMO2:LDB1-LID with and without the presence of E47. The antibody fragment (VH#576) that binds LMO2 with high affinity reflected in the high luciferase signal in the two-hybrid reporter assay (panel B). However, the same VH has a low binding with the LMO2-LID fusion protein that comprises the LMO2 LIM1 and LIM2 domains fused to the LDB1 LIM-interacting domain (almost basal level compared to luciferase signal with a non-relevant control, anti-RAS VH, VH#6, panel B). (C) Western blotting of pull-down proteins. COS-7 cells were co-transfected with LMO2, LDB1, TAL1, E47 and GATA-1 and Flag-tagged VH#576 or Flag-tagged VH#6 (a non-relevant VH). Protein complexes were isolated with anti-Flag antibody beads and purified proteins separated on SDS-PAGE. The presence of LMO2, LDB1, GATA-1 or TAL1 was detected by Western blotting. (D). The effect on protein stability of binding VH#576 to LMO2 was measured using MEL585 cells stably expressing VH#576 or VH#6 (anti-RAS). Protein extracts from these or untransfected MEL585 (-) was separated by SDS-PAGE and transferred to membrane for Western analysis with anti-flag mouse monoclonal antibody (a); anti-tubulin as a protein loading control (b); or anti-LMO2 monoclonal antibody (AbD Serotec) (c) and anti-tubulin as a protein loading control (d). The Western blots have been cropped for clarity; please see Supplementary figures 5 and 6 for full length blots.
Pull-down experiments were carried out to further analyse the mutually exclusive in vivo protein binding. COS-7 cells were co-transfected with LMO2, LDB1, TAL1, E47 and GATA-1 and either Flag-tagged VH#576 or Flag-tagged VH#Y6 (a non-relevant VH). Protein complexes associated with the VH proteins were analysed by capturing protein with anti-Flag antibody beads, fractionating on SDS-PAGE and Western blotting with the antibodies shown in Fig. 4C. The pull-down data mirror those obtained in the two-hybrid reporter systems since the Western shows that expressed VH#576 associates with predominant amounts of LMO2 (Fig. 4C), only a very small, proportional amount of LDB1 but we could not detect associated TAL1/SCL or GATA1 in this assay.
Finally, it does not appear that the binding of LMO2 by VH#576 simply results in removal of LMO2 by proteolysis as MEL cells transfected with VH#576 expression vectors display approximately similar levels of LMO2 protein as do untransfected cells (Fig. 4D). This means that LMO2-VH complex is located in the cells as a stable dimer that is not per se subject to increased instability. These data support the concept that binding of LMO2 by the VH#576 limits its natural interactions by sequestration and that LDB1 presents the LIM domains in such a way to enable further native protein complex assembly.
Discussion
The LMO2 gene is a paradigm of a chromosomal translocation master gene46 as it encodes a developmental regulator whose function is subverted by the contextual alteration of expression following translocation to T cell receptor loci. This property, manifest in transgenic mouse models by a blockade of differentiation of thymocytes18,26, and is exerted by LMO2 mediating the formation multi-protein DNA-binding complexes23. Our data show that the anti-LMO2 intracellular VH antibody fragment substantially interferes with the formation of this protein complex by a mechanism that does not simply involve enhanced degradation of the LMO2 protein itself47. Rather, structural determination of LMO2 in complex with the VH suggests a mechanism of functional inhibition whereby the conformation of LMO2, when in complex with VH, is distinct from that normally found and that this impairs binding of LMO2 to its natural partners. While the first LIM domain seems to be structurally unaltered by interaction with the VH compared to interaction with LDB1 (Figs. 1, 3), the second LIM domain lies at an angle compared to LMO2-LDB1. The degree of distortion of the second LIM domain is reminiscent of the inhibitory effect a peptide aptamer that we discovered that also interacts with the finger 4 of LMO228. In this case, the peptide seems to bind competitively to the fourth zinc atom. These observations suggest that targeting this region of the LMO2 protein would be an effective drug strategy.
In normal circumstances, interaction of LMO2 with LDB1 confers a structure on LMO2 that enables the assembly of an active transcription factor complex. The conserved hinge region between the two LIM domains of LMO2 has been identified which enables interaction with other proteins, such as bHLH that may occur in contact with DNA. The anti-LMO2 VH exploits this flexible region to allow contortion of LMO2 into a conformation that impairs the binding of natural partners such as TAL1/E47 and GATA-1, thereby inhibiting formation of a functional transcription factor complex.
Mammalian two-hybrid data also show that VH#576 binds poorly to LMO2:LDB1-LID (Fig. 4) and cannot compete with LDB1 for LMO2 binding whereas LDB1 can compete VH#576 binding to LMO235. This is consistent with the higher affinity of LDB1 than VH#576 for LMO2 (the former being kd = 2.0×10−8 M and latter kd = 9.4×10−8 M35. Therefore, inhibition of LMO2 by VH#576 presumably occurs by sequestering newly synthesized protein. Further, the small size of LMO proteins suggests they should be capable of freely moving between the cytoplasm and the nucleus)30,48 however they are found predominantly in the nucleus, possibly through nuclear retention by LDB1. The interaction between VH#576 and LMO2 may occur in the cytoplasm following protein synthesis, preventing the interaction between LMO2 and LDB1.
These studies into anti-LMO2 VH suggest a model (Fig. 5) in which alternative LMO2 structures dictate functional outcome. Our studies suggest that initially synthesized LMO2 protein is intrinsically disordered and that binding to a partner protein confers a configuration that nucleates subsequent protein complex formation. The structure of LMO2:LDB1-LID36,49, demonstrates a relatively flat, extended rod structure, able to “present” the LIM fingers to allow for other protein-protein interactions such as with GATA150. By contrast, interaction with VH confers a distinct structure that renders LMO2 inactive due to inefficient binding of natural partners. Recent data reveal the N-terminal zinc finger of GATA1 mediates the interaction between GATA1 and LMO251. These data, and our in silico model of the pentameric complex generated from individual structural information (Fig. 5), indicate that a single molecule of LMO2 could bridge the DNA binding proteins GATA1 and TAL1/SCL-E47, in keeping with our original model of the LMO2 complex23. These data suggest a model in which LMO2 binds LDB1 in the cytoplasm and is then transported to the nucleus. The LDB1 interaction “tethers” the LIM domains in such a confirmation to allow TAL1/SCL-E47 to interact with the LMO2 LIM1 domain and for GATA-1 to interact with the LIM 2 domain and also other protein-protein interactions.
Figure 5. The sequestration model for building of LMO2 protein complexes.
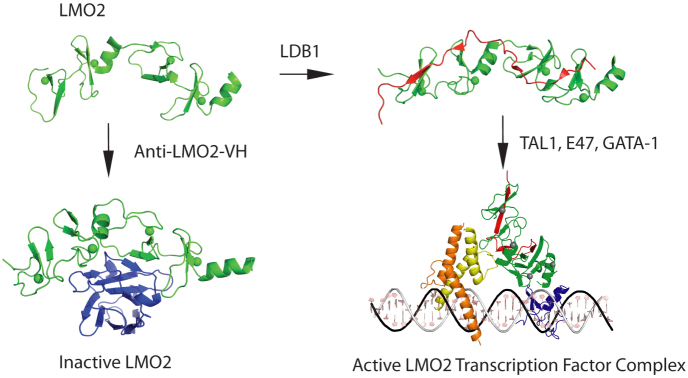
In the structural data of this paper and previous publications, it seems that the newly synthesized LMO2 protein (shown in green) is intrinsically unstable, with few structured regions. One structured region is the central short α-helix between LIM1 and LIM2 domains, giving the LMO2 protein options for interacting with partner proteins. When it binds to a natural partner such as LDB1 (shown in red), a structural constraint is imposed on LMO2 and the heterodimer can nucleate subsequent protein complex formation, depicted here the complex found in erythroid cells including GATA1. The inhibitory effect of the VH#576 (shown in blue) exploits the LMO2 hinge region and the unstable properties by sequestering LMO2 (shown in green) into a complex that has poor interacting properties with natural partners. TAL1/SCL is shown in yellow, E47 in orange and GATA1 in blue. The diagrammatic model of the pentameric complex of LMO2 (green), LDB1 (red), E47 (orange), TAL1 (yellow) and GATA-1 (blue) was generated by in silico modeling. The structure of a guide DNA including the binding motifs for E47-TAL1 (CAGGTG) and GATA-1 (GATA) transcription factors was generated using Nucleic Acid Builder of AMBER tools69. Subsequently, the structures of GATA-1, E47 and TAL1 bound DNA were derived by homology modeling70 using GATA-1 (PDB codes 1gat71 and 1gnf72) and heterodimer E47/NeuroD1 transcription factor (PDB code 2ql273) structures as templates for GATA-1 and E47-TAL1 respectively. The orientation of LMO2-LDB1 was manually adjusted by maximizing the overlap of interactions regions described in the literature: LMO2-E47-TAL136 and LMO2-GATA-174.
The data described in this paper have implications for both the mechanism of LMO2 transcription factor complex formation and how the structure of LMO2 may be exploited in a therapeutic approach. All our previous attempts to produce recombinant LMO2 protein have proved unsuccessful due to the intransigent insolubility of the protein in induction systems (unpublished). However, we found that co-expressing LMO2 with the anti-LMO2 single domain resulted in efficient production of soluble protein that was readily purified and crystallized in the dimeric state35. This has two main implications. First, the use of single domain VH (or indeed scFv) to facilitate co-expression of otherwise intransigent proteins is likely to be a useful general strategy. Molecules like LMO2, that show significant intrinsic flexibility52,49, could be stabilized by binding with antibody fragments aided by the masking of hydrophobicity in the interaction with the VH interface.
Keeping in mind that the VH#576 antibody single domain was isolated from a diverse library of VH segments merely by its binding property with an LMO2 bait, the mechanism of functioning is significant. The manifest stabilization of the LMO2 structure by interaction with the VH#576 and the sequestration of the bound LMO2 into a state that reduces the interaction of LMO2 with its natural partners such as LDB1 and TAL1/SCL has implications for the natural formation of the LMO2 multi-protein complex. The noted hinge region between the two LIM domains provides the motional flexibility that locks LMO2-LDB1 interactions and places LMO2 in a configuration that facilitates the surrounding protein complex formation involving variously TAL1/SCL, E47 and GATA1. This final complex may require contact with DNA binding sites. In similar vein, when the anti-LMO2 VH#576 binds to the unstructured LMO2, the dimer complex adopts a configuration that distorts the LMO2, particularly the LIM2 domain is bent around Phe88 and the fourth zinc atom is misaligned from the normal context. This structurally serves to sequester LMO2 protein away from the natural complex. The LMO2-VH dimer is not wholly degraded but rather is isolated.
The nature of the VH#576 single domain binding to LMO2 is unusual as there is binding from framework residues (listed in Supplementary Table 2), some of which come from former VH-VL interface residues. The region of VH#576 binds to the LIM2 domain of LMO2 assisting in the stable configuration of LMO2. Residues from all three VH CDRs are involved in binding LMO2 such that there are contacts on both LIM1 and LIM2 domains across the Phe88 hinge. The imposition of this ‘four-handed' binding by VH#576 is sufficient to impose functional ablation by sequestration.
VH#576 is a macromolecule that could be used as a drug per se if methods for efficient delivery into cells in vivo can be achieved53. There are several options whereby this might be achievable for the use of macromolecules as drugs (macrodrugs)54. The use of chemical emulators of macrodrug binding is an enticing, but not yet achieved goal, of this field of work. It has been thought that obtaining small molecule drugs that prevent protein-protein interaction would not be possible but more examples of this are becoming known55. One issue will be the relatively low affinity that can be achieved with a small molecule to bind at a PPI interface and prevent interaction of two larger proteins. Our findings with the LMO2-VH#576 complex offers an possible strategy that would be based on obtaining two compounds to emulate, respectively, the binding sites of VH#576 on LIM1 and LIM2. If these can be isolated and chemically joined56, the affinity of the final compound would be the product of the two affinities, potentially giving a high affinity compound that can emulate the LMO2 bending and effective inactivation.
Methods
Protein expression and purification
For co-expression of recombinant LMO2 and the anti-LMO2 VH576 single domain, an initial bicistronic expression vector (pRK-HIS-TEV-VH576-LMO2) was constructed by sub-cloning the anti-LMO2 VH#576 in-frame35. Truncated LMO2 (LMO2Δ8NΔ11C residues 9–147) cDNA was used to replace LMO2 in the pRK-HIS-TEV-VH576-LMO2 construct using restriction enzyme sites NheI and EcoRI. pRK-HIS-TEV-VH576-LMO2 was subsequently engineered to create expression vector pRK-HIS-TEV-VH576-LMO2 (LMO2Δ8N residues 9-158). The construct was transformed into E. coli C41(DE3) and cultured in 1 L of LB plus ampicillin (100 μg/ml) at 37°C, aerated at 225 rpm until the absorbance at 600 nm reached 0.6. ZnSO4 was added prior to induction to a final concentration of 0.1 mM. Protein expression was induced by adding Isopropyl β-D-1-thiogalactopyranoside to a final concentration of 0.5 mM, and incubated at 16°C for 14 hours. The VH#576-LMO2 protein complex was extracted after cell disruption (Constant cell disruption system) in 20 mM Tris-HCl pH 8, 250 mM NaCl, 20 mM imidazole, 0.1 mM ZnSO4, 10 mM β-mercaptoethanol and EDTA-free protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). LMO2 and VH#576 were co-purified using a Ni2+ charged HiTrap chelating HP column (5 ml, GE Healthcare) using a gradient elution from 20 to 300 mM imidazole. To remove His-tag from VH#576, the purified proteins were digested with his-tagged Tobacco Etch virus protease at 4°C overnight during dialysis against the buffer without imidazole. The proteins were passed through a nickel-nitrilotriacetic acid agarose column (Qiagen), the bound material eluted and concentrated using an Amicon Ultra-15 centrifugal filter device, 10 KDa cut-off (Millipore, MA, USA). For further purification of LMO2-VH#576 complex, the proteins were size-excluded by gel filtration chromatography with a Hi-Load Superdex 75 column (GE Healthcare, Uppsala, Sweden) in 20 mM Tris-HCl pH 8, 150 mM NaCl, 1 mM DTT. Fractions containing VH#576/LMO2 complex were pooled and concentrated to 8 mg/ml and used for crystallisation (Supplementary Fig. 1A).
Crystallization and structure determination of LMO2 in complex with VH#576
VH#576-LMO2Δ8NΔ11C was crystallised using the sitting-drop vapour-diffusion method at 294 K. Precipitant solutions (100 μl per well) were dispensed into reservoirs of a Greiner 96 well plate. Using a robot (Cartesian) 100 nl from each reservoir was dispensed onto each platform along with 100 nl of protein to form a single droplet. Each plate was sealed manually, using self adhesive transparent film (Viewseal, Greiner). Crystals appeared after a day and further optimization was performed in 24-well Cryschem plates (Hampton Research, CA, USA), mixing 2 ml protein solution with 1 ml reservoir solution and equilibrating the drop against 500 ml reservoir solution. The best diffracting crystals grew within one week of setup in 100 mM MES monohydrate pH 6.0, 0.8 M ammonium sulfate, and additive 1, 6 hexanediol. Crystals typically measured 100 μm by 40 μm (Supplementary Fig. 1B).
For data collection, the crystals were flash-cooled to 100 K in a cryo-protectant solution consisting of mother liquor and 30% glycerol. A three wavelengths MAD data set was collected on the Zn K-edge using an ADSC Q315 detector on beamline I02 at the Diamond Light Source (Didcot, UK). Analysis of the diffraction data using HKL200057 showed that the crystals belonged to space group P6 (unit cell dimensions a, b = 124.3, c = 81.4 Å and α, β = 90.0°, γ = 120.0°) with two molecules in the asymmetric unit (Matthews coefficient, VM = 2.87 Å3/Da−1 with a solvent content of 57%58. The structure of VH#576/LMO2 was solved by a combination of molecular replacement and SAD phasing. A molecular replacement solution for the VH#576 was determined with the program PHASER59 using the anti-RAS VH#Y6 heavy chain as a search model (PDB 2UZI). PHENIX AUTOSOL60 was used to locate the LMO2 intrinsic zinc positions using a three-wavelength anomalous dispersion experiment at the peak, inflection and remote wavelengths of the Zn X-ray absorption edge. The molecular replacement program MOLREP61 was used to search within this map for two LIM1 domains (using the isolated LIM1 from LMO2:LDB1-LID as a search model (PDB 2XJY)36 and successfully positioned the two molecules. The LIM2 domain (from LMO2:LDB1-LID) was manually positioned in the electron density map using the determined PHENIX zinc positions as anchor points. Manual model building of the structure was done using the COOT (Crystallographic Object-Oriented Toolkit) software62 and restrained refinement was performed with PHENIX-REFINE and then AUTOBUSTER63,64 taking care of keeping the same Rfree test set in both programs. Non-crystallographic symmetry (NCS) restrains were imposed on the 2 VH#576/LMO2 heterodimers. Towards the end of refinement, TLS (Translation/Libration/Screw) vibrational motion refinement was used. At a later date diffraction data was collected from crystals made up of VH#576-LMO2Δ8N, formed using the same conditions. Data was processed and refined to give a final Rwork/Rfree of 23.9/25.8%. Refinement statistics are listed in Supplementary Table I.
Mutation of the anti-LMO2 VH and mammalian two-hybrid analysis
Transient mammalian two-hybrid assays were carried out as described elsewhere65. Selected residues in the CDR regions of VH#576 were mutated, from the base vector template pEF-VH#576-VP16 to either glycine or alanine by point mutation followed by assembly PCR. The final PCR product was cloned back into pEF-VP16 using SfiI and NotI as a fusion with the VP16 activation domain. Truncated LMO2 bait (amino acids 28-150, see Supplementary Fig. 2) was cloned into the pM3 vector66 as a fusion with the GAL4 DNA-binding domain. CHO cells were co-transfected using Lipofectamine 2000 (Invitrogen) as previously described67 with appropriate plasmids pM-LMO2 bait, pEFVP16-prey, pG5-Fluc, and pRL-CML. Forty-eight hours after transfection, the cells were lysed and assayed with the Dual-Luciferase reporter assay system (Promega) according to the manufacturer's instructions. All luminescence values by firefly luciferase were normalized using Renilla luciferase levels. Expression of VH#576 was analysed by Western blotting using anti-VP16 antibody, (14–5 Santa Cruz biotechnology).
An LMO2-LDB1-LID construct was made as a fusion encoding LMO2 (residues 26 to 156) fused to LDB1-LID domain residues 336 to 368; the LMO2 was coupled C-terminally to LID via a ten residue linker (GSGGSGGSGG). The insert was amplified by PCR from pGBT9-LMO2:LDB1-LID (a kind gift from Dr Jacqueline Matthews) and cloned into SalI/NotI sites of pM3. To compare the interaction between LMO2 or LMO2:LDB1-LID with TAL1 or VH#576, pM3-bait (pM3-LMO2 or pM3-LDB1-LID:LMO2), pEF-VP16-prey (pEF-TAL1-VP16, pEF-VH#576-VP16 or pEF-VH#6-VP16), pG5-luc, pEF-BOS-E4723 and pRL-CML plasmids were transiently co-transfected into COS-7 cells using Lipofectamine 2000 (Invitrogen). Forty-eight hours post-transfection, Firefly and Renilla luciferase enzyme activities were assayed with the Dual-Luciferase reporter assay system.
Transfection of MEL cells with VH#576
pFUW-3XFlag-VH576-GFP and pFUW-3XFlag-VHY6-GFP vectors were prepared through a series of sub-cloning steps. Assembly PCR was used to generate BamHI-P2A-AgeI-GFP-NheI-STOP-EcoRI. The final PCR product was digested with BamHI-EcoRI and cloned into the same sites of the parent vector pFUW68 3XFlag-VH#576 or 3XFlag-VH#Y6 were amplified and the PCR products were cloned into restriction enzyme sites SalI and XbaI. The vectors expressing the Flag-tagged anti-LMO2 VH#576 or anti-RAS VH#Y6 into murine erythroleukaemia MEL(585) cells were transiently transfected using an Amaxa Nucleofector™ apparatus (Amaxa), according to the manufacturer's instructions. 1×107 cells were re-suspended in the specified Amaxa electroporation buffer (buffer R) plus 5 μg of the plasmid. GFP-positive cells were separated by sorting using Cytomation MOFLOW cytometer 24 hours after transfection. Equal number of cells were lysed by 30minute incubation on ice with RIPA buffer (150 mM sodium chloride 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) and equivalent amounts of protein extracts were separated by SDS-PAGE prior to transfer to membranes. Western analysis used anti-FLAG (clone M2, Sigma catalog number F3165), anti-LMO2 (clone 1A9-1, AbD SeroTec) or anti-tubulin (clone AA13, Sigma).
Pull-down with anti-Flag beads
COS-7 cells were seeded in a 12 well plate and co-transfected, using Lipofectamine 2000 (Invitrogen) with pGL3-ElbLUC-(Ebox-GATA)2, pEF-LDB1, pEF-LMO2, pEF-TAL1, pEF-E47, and either pEF-3XFLAG-VH576-NLS-neo or pEF-3XFLAG-VHY6-NLS-neo. Each transfection was done in triplicate. Forty-eight hours post-transfection, cells were removed from the wells using trypsin and triplicate wells pooled. Cells were pelleted by centrifugation at 1000Xg for 5 minutes and the supernatant removed. The cells were re-suspended in 200 μl lysis buffer (50 mM Tris HCL, pH7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton 100, protease inhibitor) and rotated at 4°C for 30minutes. Samples were spun down at 12,000Xg for 10 minutes. The supernatant removed and saved for the next step. Anti-Flag antibody beads (Sigma, A2220) were washed and re-suspended in PBS at a ratio of 1:2. 30 μl of re-suspended resin was added to each sample and rotated for 2 hours at 4°C. Samples were spun down at 5000Xg for 5 minutes and the supernatant removed for analysis. Anti-Flag beads were washed three times in 1 ml of PBS followed each time by spinning down at 5000Xg for 5 minutes and removing the supernatant. The beads were finally suspended in 90 μl of PBS and analysed by Western blotting.
The crystal structure of LMO2 with the anti-LMO2 VH has been deposited in the protein database with PDB ID code 4KFZ.
Author Contributions
H.S., T.T. and T.H.R. designed experiments. H.S., T.T., J.C. carried out the work. H.S., T.T., K.E.O., E.M., A.C. solved the crystal structure. N.F.F. carried out computational biology, T.H.R. designed, supervised the project and interpreted data. All authors have reviewed the manuscript and H.S., T.T. and T.H.R. wrote the paper.
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported by grants from the Leukaemia and Lymphoma Research and the Medical Research Council. E.J.M. is a Royal Society University Research Fellow. We would like to thank Dr. Jacqueline Matthews for the expression clone pGBT9-LMO2:LDB1-LID.
References
- Rabbitts T. H. Chromosomal translocation master genes, mouse models and experimental therapeutics. Oncogene 20, 5763–77 (2001). [DOI] [PubMed] [Google Scholar]
- Boehm T. et al. The mechanism of chromosomal translocation t(11;14) involving the T-cell receptor Cd locus on human chromosome 14q11 and a transcribed region of chromosome 11p15. EMBO J. 7, 385–394 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm T., Foroni L., Kaneko Y., Perutz M. P. & Rabbitts T. H. The rhombotin family of cysteine-rich LIM-domain oncogenes: Distinct members are involved in T-cell translocations to human chromosomes 11p15 and 11p13. Proc. Natl. Acad. Sci. USA 88, 4367–4371 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire E. A. et al. The t(11;14)(p15;q11) in a T-cell acute lymphoblastic leukemia cell line activates multiple transcripts, including Ttg-1, a gene encoding a potential zinc finger protein. Mol. Cel. Biol. 9, 2124–2132 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer-Pokora B., Loos U. & Ludwig W.-D. TTG-2, a new gene encoding a cysteine-rich protein with the LIM motif, is overexpressed in acute T-cell leukaemia with the t(11;14)(p13;q11). Oncogene 6, 1887–1893 (1991). [PubMed] [Google Scholar]
- Uebelhart B. & Rizzoli R. [Osteoporosis and anti-androgenic therapy in case of prostate cancer]. Revue Med Suisse 1, 2261–2, 2264–5 (2005). [PubMed] [Google Scholar]
- Simonis M. et al. High-resolution identification of balanced and complex chromosomal rearrangements by 4C technology. Nature Meth. 6, 837–42 (2009). [DOI] [PubMed] [Google Scholar]
- Natkunam Y. et al. LMO2 protein expression predicts survival in patients with diffuse large B-cell lymphoma treated with anthracycline-based chemotherapy with and without rituximab. J Clin Oncol 26, 447–54 (2008). [DOI] [PubMed] [Google Scholar]
- Natkunam Y. et al. The oncoprotein LMO2 is expressed in normal germinal-center B cells and in human B-cell lymphomas. Blood 109, 1636–42 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo S., Attard G. & Hudson D. L. Prostate epithelial stem cells. Cell Prolif. 38, 363–74 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S. et al. The significance of LMO2 expression in the progression of prostate cancer. J. Pathology 211, 278–85 (2007). [DOI] [PubMed] [Google Scholar]
- Wang K. et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature 469, 216–20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai E. et al. Oncogenic LMO3 collaborates with HEN2 to enhance neuroblastoma cell growth through transactivation of Mash1. PLoS One 6, e19297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama M. et al. LMO3 interacts with neuronal transcription factor, HEN2, and acts as an oncogene in neuroblastoma. Cancer Res. 65, 4587–97 (2005). [DOI] [PubMed] [Google Scholar]
- Visvader J. E. et al. The LIM domain gene LMO4 inhibits differentiation of mammary epithelial cells in vitro and is overexpressed in breast cancer. Proc. Natl. Acad. Sci USA 98, 14452–7 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisch P. et al. T-cell acute lymphoblastic lymphoma induced in transgenic mice by the RBTN1 and RBTN2 LIM-domain genes. Oncogene 7, 2389–2397 (1992). [PubMed] [Google Scholar]
- Larson R. et al. T cell tumours with disparate phenotypes develop with long latency in mice transgenic for rbtn2. Oncogene 9, 3675–3681 (1994). [PubMed] [Google Scholar]
- Larson R. C. et al. Protein dimerisation between Lmo2 (Rbtn2) and Tal1 alters thymocyte development and potentiates T cell tumorigenesis in transgenic mice. EMBO J. 15, 1021–1027 (1996). [PMC free article] [PubMed] [Google Scholar]
- Warren A. J. et al. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell 78, 45–58 (1994). [DOI] [PubMed] [Google Scholar]
- Yamada Y. et al. The T cell leukaemia LIM protein Lmo2 is necessary for adult mouse haematopoiesis. Proc. Natl. Acad. Sci. USA 95, 3890–3895 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y., Pannell R. & Rabbitts T. H. The oncogenic LIM-only transcription factor Lmo2 regulates angiogenesis but not vasculogenesis. Proc. Natl. Acad. Sci. USA 97, 320–324 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer V. E. et al. Cysteine-rich LIM domains of LIM-homeodomain and LIM-only proteins contain zinc but not iron. Proc. Natl. Acad. Sci. USA a 91, 316–20 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadman I. A. et al. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J. 16, 3145–3157 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herblot S., Steff A. M., Hugo P., Aplan P. D. & Hoang T. SCL and LMO1 alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T alpha chain expression. Nature Imm. 1, 138–44 (2000). [DOI] [PubMed] [Google Scholar]
- McCormack M. P., Forster A., Drynan L. F., Pannell R. & Rabbitts T. H. The LMO2 T-cell oncogene is activated via chromosomal translocations or retroviral insertion during gene therapy but has no mandatory role in normal T-cell development. Mol Cell Biol 23, 9003–9013 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson R. C., Osada H., Larson T. A., Lavenir I. & Rabbitts T. H. The oncogenic LIM protein Rbtn2 causes thymic developmental aberrations that precede malignancy in transgenic mice. Oncogene 11, 853–862 (1995). [PubMed] [Google Scholar]
- McCormack M. P. et al. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science 327, 879–83 (2010). [DOI] [PubMed] [Google Scholar]
- Appert A. et al. Targeting LMO2 with a peptide aptamer establishes a necessary function in overt T-cell neoplasia. Cancer Res 69, 4784–90 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam C. H. et al. An antibody inhibitor of the LMO2-protein complex blocks its normal and tumorigenic functions. Oncogene 27, 4962–8 (2008). [DOI] [PubMed] [Google Scholar]
- Kadrmas J. L. & Beckerle M. C. The LIM domain: from the cytoskeleton to the nucleus. Nature Rev. Mol Cell Biol. 5, 920–31 (2004). [DOI] [PubMed] [Google Scholar]
- Wright W. E. & Funk W. D. CASTing for multicomponent DNA-binding complexes. TIBS 18, 77–80 (1993). [DOI] [PubMed] [Google Scholar]
- Grutz G. et al. The oncogenic T cell LIM-protein Lmo2 forms part of a DNA-binding complex specifically in immature T cells. EMBO J. 17, 4594–4605 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadman I. et al. Specific in vivo association between the bHLH and LIM proteins implicated in human T cell leukemia. EMBO J 13, 4831–9 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y. S., Khoury H., Dick J. E. & Minden M. D. Oncogenic potential of the transcription factor LYL1 in acute myeloblastic leukemia. Leukemia 19, 1941–7 (2005). [DOI] [PubMed] [Google Scholar]
- Tanaka T., Sewell H., Waters S., Philips S. & Rabbitts T. H. Single domain intracellular antibodies from diverse libraries:emphasizing dual functions of LMO2 protein interactions using a VH single domian. J Biol Chem 286, 3707–3716 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Omari K. et al. Structure of the leukemia oncogene LMO2: implications for the assembly of a hematopoietic transcription factor complex. Blood 117, 2146–56 (2011). [DOI] [PubMed] [Google Scholar]
- Tse E. et al. Intracellular antibody capture technology: application to selection of single chain Fv recognising the BCR-ABL oncogenic protein. J. Mol. Biol. 317, 85–94 (2002). [DOI] [PubMed] [Google Scholar]
- Tanaka T. & Rabbitts T. H. Protocol for the selection of single-domain antibody fragments by third generation intracellular antibody capture. Nature Protocols 5, 67–92 (2010). [DOI] [PubMed] [Google Scholar]
- McCall K. A., Huang C. & Fierke C. A. Function and mechanism of zinc metalloenzymes. J. Nutr. 130, 1437S–46S (2000). [DOI] [PubMed] [Google Scholar]
- Yang Z. R., Thomson R., McNeil P. & Esnouf R. M. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 21, 3369–76 (2005). [DOI] [PubMed] [Google Scholar]
- Sickmeier M. et al. DisProt: the Database of Disordered Proteins. Nucleic Acids Res 35, D786–93 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Garcia I., Axelson H. & Rabbitts T. H. Functional diversity of LIM proteins: amino-terminal activation domains in the oncogenic proteins RBTN1 and RBTN2. Oncogene 10, 1301–1306 (1995). [PubMed] [Google Scholar]
- Deane J. E. et al. Tandem LIM domains provide synergistic binding in the LMO4:Ldb1 complex. EMBO J. 23, 3589–98 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews J. M., Visvader J. E. & Mackay J. P. 1H, 15N and 13C assignments of FLIN2, an intramolecular LMO2:ldb1 complex. J. Biomol. NMR 21, 385–6 (2001). [DOI] [PubMed] [Google Scholar]
- Ryan D. P. et al. Identification of the key LMO2-binding determinants on Ldb1. J Mol Biol 359, 66–75 (2006). [DOI] [PubMed] [Google Scholar]
- Rabbitts T. H. Translocations, Master genes, and Differences between the Origins of Acute and Chronic Leukemias. Cell 67, 641–644 (1991). [DOI] [PubMed] [Google Scholar]
- Xu Z. et al. Single-stranded DNA-binding proteins regulate the abundance of LIM domain and LIM domain-binding proteins. Genes & Devel. 21, 942–55 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol 8, 195–208 (2007). [DOI] [PubMed] [Google Scholar]
- Dastmalchi S. et al. Solution structure of a tethered Lmo2(LIM2)/Ldb1(LID) complex. Protein Sci 21, 1768–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada H., Grutz G., Axelson H., Forster A. & Rabbitts T. H. Association of erythroid transcription factors: Complexes involving the LIM protein RBTN2 and the zinc-finger protein GATA1. Proc. Natl. Acad. Sci. USA 92, 9585–9589 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson-White L. et al. Structural basis of simultaneous recruitment of the transcriptional regulators LMO2 and FOG1/ZFPM1 by the transcription factor GATA1. Proc Natl Acad Sci U S A 108, 14443–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores S. C., Lu L. J., Yang J., Carriero N. & Gerstein M. B. Hinge Atlas: relating protein sequence to sites of structural flexibility. BMC Bioinformatics 8, 167 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Martinez D., Tanaka T. & Rabbitts T. H. Intracellular antibodies and cancer: new technologies offer therapeutic opportunities. BioEssays 32, 589–98 (2010). [DOI] [PubMed] [Google Scholar]
- Forster A. et al. Chromosomal translocation engineering to recapitulate primary events of human cancer. Cold Spring Harb Symp Quant Biol 70, 275–82 (2005). [DOI] [PubMed] [Google Scholar]
- Wells J. A. & McClendon C. L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450, 1001–9 (2007). [DOI] [PubMed] [Google Scholar]
- Congreve M., Chessari G., Tisi D. & Woodhead A. J. Recent developments in fragment-based drug discovery. J. Med. Chem 51, 3661–80 (2008). [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. & Minor W. Porcessing of X-fay diffraction data collected in oscillation mode. Methods Ezymol 276 (1997). [DOI] [PubMed] [Google Scholar]
- Matthews B. W. Solvent content of protein crystals. J Mol Biol 33, 491–7 (1968). [DOI] [PubMed] [Google Scholar]
- McCoy A. J. et al. Phaser crystallographic software. J. Appl Cryst 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D. et al. The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. & Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66, 22–5 (2010). [DOI] [PubMed] [Google Scholar]
- Emsley P. & Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst 60, 2126–32 (2004). [DOI] [PubMed] [Google Scholar]
- Blanc E. et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Cryst 60, 2210–21 (2004). [DOI] [PubMed] [Google Scholar]
- Murshudov G. N., Vagin A. A. & Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst 53, 240–55 (1997). [DOI] [PubMed] [Google Scholar]
- Tanaka T., Lobato M. N. & Rabbitts T. H. Single domain intracellular antibodies: a minimal fragment for direct in vivo selection of antigen-specific intrabodies. J. Mol. Biol. 331, 1109–1120 (2003). [DOI] [PubMed] [Google Scholar]
- Sadowski I., Bell B., Broad P. & Hollis M. GAL4 fusion vectors for expression in yeast or mammalian cells. Gene 118, 137–141 (1992). [DOI] [PubMed] [Google Scholar]
- Tanaka T. & Rabbitts T. H. Intrabodies based on intracellular capture frameworks that bind the RAS protein with high affinity and impair oncogenic transformation. EMBO J 22, 1025–1035 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C., Hong E. J., Pease S., Brown E. J. & Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–72 (2002). [DOI] [PubMed] [Google Scholar]
- Cornell W. D. et al. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc 117, 5179–5197 (1995). [Google Scholar]
- Fernandez-Fuentes N., Rai B. K., Madrid-Aliste C. J., Fajardo J. E. & Fiser A. Comparative protein structure modeling by combining multiple templates and optimizing sequence-to-structure alignments. Bioinformatics 23, 2558–65 (2007). [DOI] [PubMed] [Google Scholar]
- Omichinski J. G. et al. NMR structure of a specific DNA complex of Zn-containing DNA binding domain of GATA-1. Science 261, 438–46 (1993). [DOI] [PubMed] [Google Scholar]
- Kowalski K., Czolij R., King G. F., Crossley M. & Mackay J. P. The solution structure of the N-terminal zinc finger of GATA-1 reveals a specific binding face for the transcriptional co-factor FOG. J Biomol NMR 13, 249–62 (1999). [DOI] [PubMed] [Google Scholar]
- Longo A., Guanga G. P. & Rose R. B. Crystal structure of E47-NeuroD1/beta2 bHLH domain-DNA complex: heterodimer selectivity and DNA recognition. Biochemistry 47, 218–29 (2008). [DOI] [PubMed] [Google Scholar]
- Wilkinson-White L. et al. Structural basis of simultaneous recruitment of the transcriptional regulators LMO2 and FOG1/ZFPM1 by the transcription factor GATA1. Proc. Natl Acad Sci USA 108, 14443–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information