

Abstract
It has been previously suggested that high amounts of oxalate in plasma could play a role in autism by binding to the bilobal iron transport protein transferrin (hTF) thereby interfering with iron metabolism by inhibiting iron delivery to cells. By examining the effect of the substitution of oxalate for the physiologically utilized synergistic carbonate anion in each lobe of hTF we sought to provide a molecular basis for or against such a role. Our work clearly shows both qualitatively (6 M urea gels) and quantitatively (kinetic analysis by stop flow spectrofluorimetry) that the presence of oxalate in place of carbonate in each binding site of hTF does indeed greatly interfere with iron removal from each lobe (both in the absence and presence of the specific hTF receptor). However, we also clearly demonstrate that once the iron is bound within each lobe of hTF, neither anion can displace the other. Additionally, as verified by urea gels and electrospray mass spectrometry, formation of completely homogeneous hTF-anion complexes requires that all iron must first be removed and hTF then reloaded with iron in the presence of either carbonate or oxalate. Of significance, experiments described herein show that carbonate is the preferred binding partner, i.e., even if an equal amount of each anion is available during the iron loading process the hTF-carbonate complex is formed.
Autism spectrum disorders (ASD) are complex neurological/developmental disorders which severely impact the social, behavioral and communication skills of affected children. Recent findings from the Centers for Disease Control suggest that as many as 1 in 88 children may be affected with some form of ASD (1). Despite the increasing frequency of diagnosis and numerous suggestions and/or theories as to the origin(s) of this apparent sudden epidemic of ASD, no single cause has been identified. In fact, it seems most likely that multiple genetic and environmental factors are involved in the pathology of ASD (2–5).
Interestingly, along with the typical behavioral and communication issues, gastrointestinal symptoms often exist in children with ASD (6, 7). This observation could indicate that a more complex and global metabolic disorder exists. Obviously, at least some of the same metabolic imbalances in children with ASD could be related to, or responsible for, the impairment in cognitive development and function observed.
More recently, largely anecdotal evidence suggests that the metabolism and homeostasis of oxalate may be a factor in ASD. Providing some scientific support for the involvement of oxalate in ASD is a study showing that children with ASD displayed a 3-fold increase in plasma oxalate levels compared to age and gender matched controls (7). Nevertheless, the precise molecular origin and potential effect of hyperoxalemia in children with ASD remains unclear. Given these findings, it is imperative to provide a more solid scientific basis, either for or against, a possible role of oxalate in ASD since information currently available on the Internet touts the benefits of a low oxalate diet to improve the cognitive function and behavior of children with ASD.
Oxalate (C2O4−2) is a relatively simple molecule that, when in complex with divalent metallic cations such as Ca2+, forms insoluble crystals that eventually can result in kidney stones. Oxalate is acquired by the body through dietary intake (various foods, especially plant materials including grains, seeds, nuts as well as vegetables such as chard, beet roots and spinach and various fruits such as rhubarb are high in oxalate). Additionally, oxalate is also produced within the body (8, 9) as the final degradation product of a number of amino acids (including the aromatic amino acids tyrosine, phenylalanine and tryptophan) and also ascorbate.
In addition to the single report on hyperoxalemia (7), a number of studies outside the United States have highlighted the increased prevalence of iron deficiency (ID) and/or iron deficiency anemia (IDA) in children with ASD in comparison to age-matched controls (10–13). However, this finding was not confirmed in a recent study of children with ASD in the United States (14). Of interest, a small subset of children with ASD and ID/IDA also appeared to be non-responsive to oral iron therapy (15), i.e., the ID/IDA was iron-refractory. It is well established that iron is critical to many essential biological processes, including oxygen and electron transport (16) and that it exerts both positive and negative effects on brain function (17–19). Therefore, ID/IDA could possibly contribute to the adverse effects on cognitive development and function observed in children with ASDs.
Intriguingly, it is possible that these seemingly unrelated metabolic imbalances (gastrointestinal dysfunction, hyperoxalemia and IDA) might be connected. With the task of binding iron (in the form of Fe3+) and safely delivering it to cells, the bilobal glycoprotein human serum transferrin (hTF) comprises the most dynamic iron pool within the human body, turning over nearly ten times per day (20). Following production of hTF in the liver and secretion into the blood, each lobe (referred to as the N- and C-lobe) binds Fe3+ very tightly (Kd ~1022 M−1), yet reversibly (21). Two tyrosine residues, one aspartic acid and one histidine residue (Tyr95, Tyr188, Asp63 and His249 in the N-lobe; Tyr426, Tyr517, Asp392 and His585 in the C-lobe) coordinate the Fe3+ in each lobe of hTF. The distorted octahedral coordination of the iron is completed by the final two ligands provided by a synergistic anion. With its relatively high concentration in serum, bicarbonate/carbonate (HCO3−2/CO3−2) was originally suggested, and has since been confirmed by a number of x-ray crystallographic structures to serve as the physiologically relevant synergistic anion (22, 23). The crystal structures further revealed that the synergistic carbonate is anchored by a conserved hTF residue (Arg124 in the N-lobe and Arg456 in the C-lobe). As its name implies, a suitable synergistic anion is an absolute requirement for high affinity Fe3+ binding by hTF within the binding cleft of each lobe (24). Consistent with the dynamic nature of many proteins in solution (25), equilibrium probably exists between the open and closed cleft in each lobe of hTF (26). It has also been suggested that the synergistic anion may bind first to pre-form the binding site (27, 28), because as indicated by the residue numbers above, the iron binding residues are distant in the primary sequence. Thus, high affinity binding of iron is accomplished when a suitable synergistic anion and ferric iron are present to drive the equilibrium into the fully iron-bound closed conformation.
At the neutral pH of ~7.4 of the blood, iron-bearing hTF binds with nanomolar affinity to the transferrin receptor (TFR), located on the cell surface of all iron-requiring, i.e., activity dividing cells. The hTF/TFR complex enters the cell via clathrin-dependent endocytosis (29). An ATP-dependent H+ pump lowers the pH within the endosome where along with salt and an unidentified chelator initiate TFR-mediated iron release from hTF (30). At endosomal pH, apohTF remains tightly bound to the TFR. Return of the apohTF/TFR complex to the cell surface initiates the release of apohTF into the blood (pH 7.4), where it can bind more Fe3+. Importantly, removal of Fe3+ from hTF in solution requires a decrease in the pH (from 7.4 to < 6.0) to protonate the synergistic anion as well as pH-sensitive ‘second-shell’ residues. These residues do not directly coordinate the Fe3+, but form hydrogen bonds with the primary iron-binding ligands and are involved in the mechanism of iron release from each lobe (31–33). In the N-lobe, Lys206 and Lys296 on opposite sides of the binding cleft share a hydrogen bond when Fe3+ is bound (34). In response to low pH, protonation causes repulsion of these lysine residues, triggering cleft opening and allowing iron release. A triad of amino acid residues occupies equivalent positions in the C-lobe (34). Lys534 (corresponding to Lys206 in the N-lobe) is located across the binding cleft from Arg632 (equivalent to Lys296 in the N-lobe) in the C1 subdomain. Similar to the dilysine trigger in the N-lobe, it has been suggested that Lys534 may share a hydrogen bond with Arg632 (34). The triad is completed by Asp634 which has been shown to stabilize the interaction of Lys534 and Arg632 (35). The role of these residues in the mechanism of iron release has been confirmed by mutagenesis studies (32, 33). In fact, substitution of Lys206 in the N-lobe by glutamate or of Lys534 or Arg 632 by alanine has allowed the creation of hTF constructs in which the iron is literally locked in the cleft (36).
As reported nearly 40 years ago, oxalate (C2O4−2) can substitute for carbonate to promote high affinity Fe3+ binding to hTF (37). Previous findings from our laboratory indicate that when oxalate serves as the synergistic anion within the isolated N-lobe of recombinant hTF (hTF/2N(OX)), iron release is much slower and requires lower pH (38). Additionally, full-length hTF with oxalate bound as the synergistic anion (hTF(OX)) completely prevents iron delivery to HeLa cells (38). The lower pKa of oxalate (4.2) compared to carbonate (6.4), allowed us to rationalize these results. Moreover, based on the 1.2 Å crystal structure of (hTF/2N(OX)), oxalate binds within the iron-binding cleft in a bidentate manner providing nearly perfect symmetry and further stabilizing its resistance to protonation in comparison to the more vulnerable carbonate anion (38).
The reports that: (1) Plasma oxalate levels in children with ASD tend to be high (7); (2) The prevalence of iron-refractory ID/IDA in children with ASD is high (10–13, 15); (3) Children with ASD have gastrointestinal problems that could affect both iron and oxalate uptake in the gut (6); and (4) hTF/2N(OX) and Fe2 hTF(OX) do not readily relinquish iron (38) collectively suggest a potential link between oxalate and iron deficiency anemia in autism. Thus, high plasma oxalate levels and iron-refractory ID/IDA in children with ASD might be mediated via hTF(OX). Therefore, in the current study, we set out to investigate the hypothesis that hTF provides a crucial link between high plasma oxalate levels and iron deficiency in children with ASD. First, we quantitatively examined the iron release properties of diferric hTF and the two monoferric hTF with oxalate bound as the synergistic anion using standardized methods and recombinant hTF that we have developed over the past several years (35). We then evaluated whether the two synergistic anions (carbonate and oxalate) were able to displace and/or compete with one another at physiologically relevant concentrations of each. This is critical to determining the possible relevance of high oxalate as a causative agent of IDA in children with ASDs (if it exists). And finally, we further characterized the oxalate containing hTF samples by mass spectrometry analysis on a high resolution instrument.
MATERIALS AND METHODS
Materials
Dulbecco’s modified Eagle’s medium-Ham F-12 nutrient mixture (DMEM-F12) and fetal bovine serum (FBS) were obtained from the GIBCO-BRL Life Technologies Division of Invitrogen. Antibiotic-antimycotic solution (100X) solution and trypsin were from Mediatech, Inc. Both Pro293A-CDM serum-free medium, and L-glutamine were purchased from Lonza. All tissue culture dishes, flasks, and Corning expanded surface roller bottles were obtained from local distributors. Ultracel 30 kDa molecular weight cutoff (MWCO) membrane microconcentrator devices were manufactured by Amicon. Ni- nitrilotriacetic acid (NTA) resin came from Qiagen. Hi-prep 26/60 Sephacryl S-200HR and S-300HR columns were acquired from GE Healthcare. Ethylenediaminetetraacetic acid (EDTA) was from Fisher. NTA, K2C2O4·H2O and ferrous ammonium sulfate were from Sigma (St. Louis, MO). Novex 6% Tris(hydroxymethyl)aminomethane–borate–EDTA (TBE) urea mini-gels, TBE running buffer (5X) and TBE-urea sample buffer (2X) were from Invitrogen.
Expression and Purification of hTFs and the soluble portion of the TFR (sTFR)
The expression and purification of baby hamster kidney (BHK) cell derived recombinant NHis-tagged hTF (diferric hTF, Fe2hTF; FeNhTF, monoferric N-lobe hTF in which mutation of iron binding ligands, Y426F/Y517F, prevents iron binding in the C-lobe; FeChTF monoferric C-lobe hTF in which mutation of iron binding ligands, Y95F/Y188F, prevents iron binding in the N-lobe) has been previously described (39). The K206E mutation (31, 32, 36), which prevents iron release from the N-lobe, as well as the Lys534 (32)and Arg632 mutations (32, 36), which prevent iron release from the C-lobe, have also been previously reported. To ensure iron saturation and stabilize the recombinant hTF, ferric iron (in the form of Fe3+-NTA) is added to collected tissue culture medium prior to purification. The Fe-NTA solution is prepared by mixing an appropriate amount of 25 mM ferrous ammonium sulfate (in 0.01 N HCl) with 100 mM NTA to obtain a Fe:NTA ratio of 1:2. Collected medium is reduced in volume using a tangential flow device with a 30 kDa molecular weight cutoff (MWCO) membrane and partially exchanged into 5 mM Tris-HCl buffer, pH 8.0, containing 0.02% sodium azide. Following cotton filtration and/or centrifugation to remove any cellular debris (6000 g for 15 min), 5X Qiagen start buffer is added to the supernatant to yield a final concentration of 1X Qiagen start buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 20 mM imidazole, 10% glycerol and 0.05% sodium azide). The hTF is then captured by passage over a Qiagen Ni-NTA column (8 mL of resin, 2 mL/min) and eluted by the addition of 250 mM imidazole to the 1X Qiagen start buffer. Pooled fractions are concentrated using 30 kDa MWCO Ultracel microconcentrators and final purification is accomplished by passage over a Sephacryl S-200 HR gel filtration column in 100 mM NH4HCO3. Pooled fractions of purified carbonate containing hTF are concentrated to 15 mg/mL using a 30 kDa MWCO Ultracel microconcentrator.
The production and purification of the His-tagged sTFR consisting of residues 121–760 is also as previously described (40).
hTF/sTFR Complex Formation and Purification
The hTF/sTFR complexes are prepared by adding a small molar excess (~20%) of hTF to 1.5 mg of each mutant sTFR. Following equilibration at room temperature for ~5 min, hTF/mutant sTFR complexes are purified by passage over a Sephacryl S300HR gel filtration column in 100 mM NH4HCO3 to remove excess hTF. Fractions containing the complex are concentrated to 15 mg/mL with respect to hTF.
hTF(OX) Formation
Oxalate containing samples have been prepared by two different methods as follows. In the first method, carbonate containing hTF (Fe2hTF, FeNhTF and FeChTF) or hTF/sTFR complexes were exchanged into 200 mM K2C2O4·H2O using microconcentrators (30 kDa MWCO). After incubation (3–4 days at 4 °C) in 200 mM K2C2O4·H2O, additional Fe-NTA was added to the hTF samples to ensure complete iron saturation. Samples were then exchanged into 200 mM K2C2O4·H2O to remove excess Fe-NTA. In the second method apohTF was produced by incubating the hTF samples (Fe2hTF, FeNhTF and FeChTF) in 500 mM sodium acetate, pH 4.9 containing 1 mM NTA and 1 mM EDTA overnight at 4°C. Following an exchange into 100 mM KCl, the apo samples were then exchanged into 200 mM K2C2O4·H2O before an excess amount of iron (in the form of ferrous sulfate) was added.
Analysis by ESI MS
Samples prepared as described above were exhaustively exchanged into 50 mM ammonium acetate pH 6.7 on microconcentrators (30 kDa MWCO) and adjusted to a final protein concentration of 5 μM. Mass spectra were recorded on a solariX (Bruker Daltonics, Billerica, MA) 7T FT-ICR instrument with collisional energy parameters in the source kept at minimal values in order to preserve ligand-protein complexes in the gas state. Ligand binding was disrupted by adding a solution of formic acid to lower the sample pH to 3.7 and incubation for 15 min at 25°C. All spectra consist of 500 averaged scans collected over a total acquisition time of 6 min. External calibration from 2,000 to 5,000 m/z was performed using sodium perfluoroheptonate clusters.
Urea gel analysis
The iron status of hTF and hTF/sTFR complexes was evaluated by urea gel electrophoresis using Novex 6% TBE-urea mini-gels in 90 mM Tris–borate, pH 8.4, containing 16 mM EDTA as previously described (36, 41). Iron-containing complexes were mixed 1:1 with 2X TBE-urea gel sample buffer (final concentration 0.5 μg/μL). To determine the extent of iron removal, an aliquot of each sample was added to iron removal buffer (100 mM MES buffer, pH 5.6, containing 300 mM KCl and 4 mM EDTA) and incubated at room temperature for 5 min. The iron removal process was halted by addition of 2X TBE-urea gel sample buffer. Samples (2.5 μg) were loaded and the gel was electrophoresed for 2.25 h at 125 V. Protein bands were visualized by staining with Coomassie blue (42).
Kinetic Analysis of Iron Release from hTF ± sTFR at pH 5.6
Iron release from hTF mutants, hTF(OX), hTF mutant and hTF(OX) complexes was monitored at 25° C as previously described using an Applied Photophysics SX.20MV stopped-flow spectrofluorimeter (36, 43). The content of one syringe (hTF sample or hTF/sTFR complex (375 nM) in 300 mM KCl) was rapidly mixed with the iron removal buffer in the second syringe, MES buffer (200 mM, pH 5.6), KCl (300 mM) and EDTA (8 mM). Rate constants were determined by fitting the change in fluorescence intensity versus time using Origin software (version 7.5) to standard models as described in detail previously (36). All data were corrected to zero fluorescence intensity at time zero before fitting.
Displacement of Carbonate by Oxalate
Carbonate containing Fe2hTF in 25 mM NH4HCO3 was incubated with increasing concentrations of K2C2O4·H2O (0–50 μM) for 2 h at 37 °C. Samples (2.5 μg) were removed and exposed to iron removal buffer (100 mM MES buffer, pH 5.6, containing 300 mM KCl and 4 mM EDTA) and incubated at room temperature for 15 min. The iron removal process was halted by addition of 2X TBE-urea gel sample buffer and samples (2.5 μg) were run and visualized as described above.
Displacement of Oxalate by Carbonate
Oxalate containing Fe2hTF(OX) in increasing concentrations of K2C2O4·H2O (0–50 μM) was incubated with 25 mM NH4HCO3 for 2 h at 37 °C. Samples (2.5 μg) were removed and exposed to iron removal buffer (100 mM MES buffer, pH 5.6, containing 300 mM KCl and 4 mM EDTA) and incubated at room temperature for 15 min. The iron removal process was halted by addition of 2X TBE-urea gel sample buffer and samples (2.5 μg) were run and visualized as described above.
Competitive Synergistic Anion Binding to ApohTF
ApohTF was produced by incubating Fe2hTF in 500 mM sodium acetate, pH 4.9 containing 1 mM NTA and 1 mM EDTA overnight at 4°C followed by extensive exchange into 100 mM KCl. ApohTF in 100 mM KCl (5 mM final concentration) was incubated with 25 mM NH4HCO3 containing increasing concentrations of K2C2O4·H2O (0–50 μM) and excess Fe-NTA for 2 h at 37 °C. Samples (2.5 μg) were removed and exposed to iron removal buffer (100 mM MES buffer, pH 5.6, containing 300 mM KCl and 4 mM EDTA) and incubated at room temperature for 15 min. The iron removal process was halted by addition of 2X TBE-urea gel sample buffer and samples (2.5 μg) were run and visualized as described above.
RESULTS AND DISCUSSION
Effect of Oxalate on Iron Release from hTF
The results of a qualitative evaluation (by 6% TBE urea gels) of iron release from various hTF and hTF(OX) constructs are presented in Fig. 1. In comparison to the respective carbonate control, the presence of oxalate in the three hTF constructs significantly decreases the amount of iron that is released within the tested time frame. Although in this format a small amount of iron remains bound in each lobe of the diferric control (i.e., both monoferric hTF species are present), after a 15 min treatment with our standard iron removal buffer (100 mM MES, pH 5.6, containing 300 mM KCl and 4 mM EDTA) very little Fe2hTF remains (Fig. 1A). Under identical conditions, it appears that a small amount of Fe3+ is removed from the N-lobe (as indicated by the presence of FeChTF(OX)), however, most of the Fe2hTF(OX) sample remains in the diferric state. Treatment of FeNhTF(OX) and FeChTF(OX) with our iron removal regimen also results in far less iron removal in comparison to the carbonate control samples (Fig. 1B and 1C). To provide a basis for comparison, we have included the results of the same experiment with our LockN/LockChTF constructs in which both lobes are compromised by substitution of key residues that help to trigger iron release (Lys206 in the N-lobe and Lys534 and Arg632 in the C-lobe) (32, 33, 36). As clearly shown in Fig S1 these constructs are extremely resistant to iron removal under the conditions used. In fact, only the K206E/K534E Fe2hTF mutant releases any iron, apparently from the C-lobe, since the migration of the band corresponds to FeNhTF.
Figure 1.
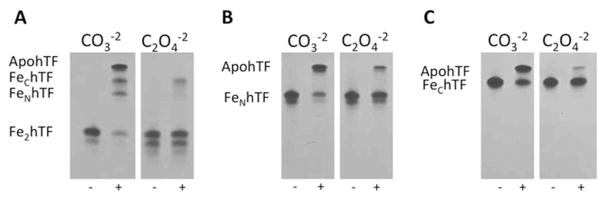
Urea gel analysis of carbonate (CO3−2) and oxalate (C2O4−2) containing Fe2hTF (A), FeNhTF (B) and FeChTF (C). Samples were electrophoresed before and after (+) incubation with iron removal buffer (100 mM MES, pH 5.6, containing 300 mM KCl and 4 mM EDTA) for 15 min.
We are able to provide a far more quantitative estimate of the dramatic decrease in the rate of iron release from the hTF(OX) samples from kinetic rate constants obtained by fitting the increase in the intrinsic tryptophan fluorescence of hTF as Fe3+ is released. Although as clearly shown in the urea gel analysis, very little iron is released from the hTF(OX) constructs a small increase in the intrinsic tryptophan fluorescence was observed under our standard iron removal conditions in the stopped-flow format. In contrast to the respective controls, which yield either two or three rate constants assigned to iron release and conformational events (35, 36), all of the oxalate samples fit to a simple A→B model (Table 1) and yielded a single very slow kinetic rate constant which we assign to an unknown pH-induced conformational event. As an additional control, an increase in the intrinsic tryptophan fluorescence is observed in the LockN/LockChTF constructs, which also do not release iron upon exposure to low pH. As shown in Table S1, three out of the four LockN/LockChTF constructs fit to a more complex A→B→C model than the hTF(OX) constructs. Noting the unusually large 95% confidence intervals obtained with the locked constructs, the first rate (k1) is similar to the rate constant obtained from the hTF(OX) constructs (Table S1 vs. Table 1). Interestingly, the second rate (k2) of these LockN/LockChTF constructs is very slow and additionally is the only rate obtained for the (K206E/R632A Fe2hTF) LockN/LockChTF construct (Table S1). Given the drastically slowed rate constants and the fact that little iron is removed from any of the LockN/LockChTF constructs or hTF(OX) samples, these very slow rates are assigned to small pH induced conformational events. Collectively, it is very clear from the kinetic evaluation (both by urea gel and intrinsic tryptophan fluorescence) that little or no iron is removed from either the oxalate containing hTF samples or the LockN/LockChTF constructs.
Table 1.
Kinetic Rate Constants for Iron Release from hTF(OX) samples.
| Construct | K1 (min−1) | K2 (min−1) | |
|---|---|---|---|
| Fe2hTFa | 17.7 ± 2.2 (kN) | 0.65 ± 0.06 (kC) | |
| Fe2hTF(OX) | - | 0.29 ± 0.01 | |
| Construct | K1 (min−1) | K2 (min−1) | K3 (min−1) |
| FeNhTFa | 24.8 ± 3.2 (kN) | 5.8 ± 1.2 | 1.1 ± 0.1 |
| FeNhTF(OX) | - | - | 0.37 ± 0.01 |
| Construct | K1 (min−1) | K2 (min−1) | |
| FeChTFa | 0.79 ± 0.11 (kC) | 1.9 ± 0.5 | |
| FeChTF(OX) | - | 0.30 ± 0.01 |
From (36). See supporting information Figure 4 for iron dependant changes as a function of time and the fit of this kinetic data to yield the rate constants in this table.
Effect of Oxalate on Iron Release from hTF/sTFR Complexes
To determine whether the substitution of oxalate as the synergistic anion affected iron removal from hTF/sTFR complexes, the hTF(OX)/sTFR complexes were electrophoresed on a urea gel after a 5 min incubation in our standard iron removal buffer (pH 5.6). As reported previously, the sTFR significantly impacts the kinetics and mechanism of iron release from each lobe of hTF (36, 41, 44–46). Specifically, when bound to the soluble portion of the TFR, the rate of iron release from the C-lobe is increased and the rate of iron release from the N-lobe is decreased, making the two rates more equivalent (35, 36). As clearly shown in Fig. 2, iron release from hTF is almost entirely inhibited when oxalate serves as the synergistic anion in the hTF/sTFR complexes. As expected these results are replicated in three of the four LockN/LockChTF/sTFR complexes (Fig. S2), which release very little, if any iron even after a15 min incubation in our standard iron removal buffer. Interestingly, the K534E mutation does not appear to completely lock Fe3+ in the C-lobe in the presence of the sTFR, as indicated by observation of a band corresponding to FeNhTF in the K206E/K534E Fe2hTF/sTFR complex (Fig. S2).
Figure 2.
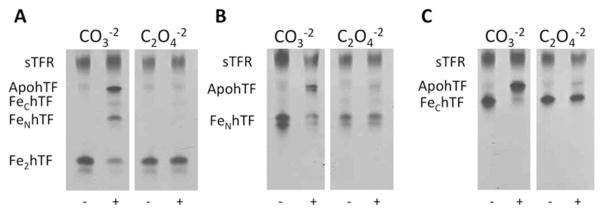
Urea gel analysis of carbonate (CO3−2) and oxalate (C2O4−2) containing Fe2hTF/sTFR (A), FeNhTF/sTFR (B) and FeChTF/sTFR (C) complexes. Samples were electrophoresed before and after (+) incubation with iron removal buffer (100 mM MES, pH 5.6, containing 300 mM KCl and 4 mM EDTA) for 5 min.
The dramatic decrease in the rate of iron release from the oxalate samples is further validated by the kinetic rate constants obtained from fitting the increase in intrinsic tryptophan fluorescence after exposing the hTF(OX)/sTFR complexes to our “endosomal” conditions (Table 2). While the conformational change preceding iron release from the FeChTF/sTFR complex (k1) is maintained in the FeChTF(OX)/sTFR complex, a similar conformational change is lost in the FeNhTF(OX)/sTFR complex in comparison to the FeNhTF/sTFR control. Additionally, a similar rate constant is obtained from the fitting of one of the LockN/LockChTF complexes (K206E/R632E Fe2hTF/sTFR, Table S2). All the hTF(OX)/sTFR complexes give rise to a similar rate constant (k ≈ 0.6 min−1, Table 2) that is slower than any of the rates obtained for the four LockN/LockChTF constructs (Table S2). Intriguingly, the Fe2hTF(OX)/sTFR complex fits to an A → B → C model where k1=k2 (k = 0.6 min−1, Table 2). Since very little if any Fe3+ is removed from the Fe2hTF(OX)/sTFR complex, the actual events to which these two kinetic rate constants correspond remain unclear, but could potentially be the combination of rates observed for the FeNhTF(OX)/sTFR and FeChTF(OX)/sTFR complexes (Table 2). We conclude that the sTFR is unable to overcome either the presence of oxalate as the synergistic anion or the effect of the mutations in the LockN/LockChTF constructs as very little iron can be released from these hTF/sTFR complexes.
Table 2.
Kinetic Rate Constants for Iron Release from hTF(OX)/sTFR Complexes.
| hTF/sTFR complex | K1 (min−1) | K2 (min−1) |
|---|---|---|
| Fe2hTF/sTFRa | 5.5 ± 0.9 (kC) | 1.4 ± 0.2 (kN) |
| Fe2hTF(OX)/sTFR | 0.6 ± 0.1 | 0.6 ± 0.1 |
| hTF/sTFR complex | K1 (min−1) | K2 (min−1) |
| FeNhTF/sTFRa | 22.0 ± 0.7 | 1.7 ± 0.6 (kN) |
| FeNhTF(OX)/sTFR | - | 0.6 ± 0.01 |
| hTF/sTFR complex | K1 (min−1) | K2 (min−1) |
| FeChTF/sTFRa | 20.6 ± 1.2 | 7.2 ± 0.4 (kC) |
| FeChTF(OX)/sTFR | 22.8 ± 10.9 | 0.6 ± 0.1 |
From (36). See supporting information Figure 5 for iron dependant changes as a function of time and the fit of this kinetic data to yield the rate constants in this table.
Synergistic Anion Displacement from Fe2hTF
The ability of one synergistic anion (carbonate or oxalate) to displace the other from Fe2hTF under physiologic conditions was evaluated using urea gel analysis since (as clearly shown above) CO3−2 and C2O4−2 containing hTF are easily distinguished by this method. Even at relatively high (not physiologically relevant) concentrations of oxalate (50 μM), CO3−2 was not displaced from Fe2hTF (Fig. 3). Likewise, carbonate was unable to displace oxalate as the synergistic anion of Fe2hTF even in the absence of added oxalate (0 μM C2O4−2) (Fig. 4). These results suggest that once bound to hTF, one synergistic anion (carbonate or oxalate) cannot displace the other (at least in the absence of other factors that are perhaps present in the blood). This statement appears to hold true not only at physiologically relevant normal oxalate concentrations, but also at significantly elevated concentrations of oxalate, much higher than those observed in children with ASD.
Figure 3.
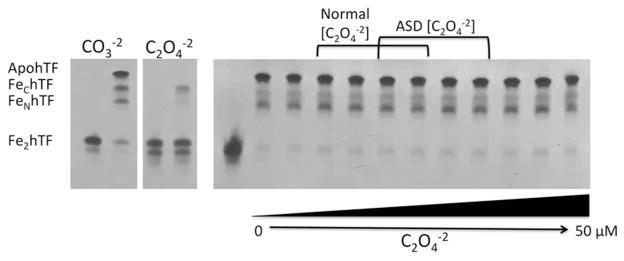
Displacement of carbonate (CO3−2) from Fe2hTF by oxalate (C2O4−2). Carbonate-bound Fe2hTF was incubated in normal [CO3−2] (25 mM NH3HCO3−2) (47) with increasing concentrations of C2O4−2 ranging from normal C2O4−2 concentrations (1.8–4.7 μM) (7), ASD C2O4−2 concentrations (3.5–7.5 μM)(7) and above (lanes 2–12 are 0, 1.0, 1.8, 2.5, 3.5, 4.7, 5.6, 7.5, 10, 25 and 50 μM C2O4−2 respectively). Samples were removed and electrophoresed following incubation with iron removal buffer (100 mM MES, pH 5.6, containing 300 mM KCl and 4 mM EDTA) for 15 min. The control carbonate (CO3−2) and oxalate (C2O4−2) containing Fe2hTF samples from Figure 1A are shown on the left as a reference.
Figure 4.
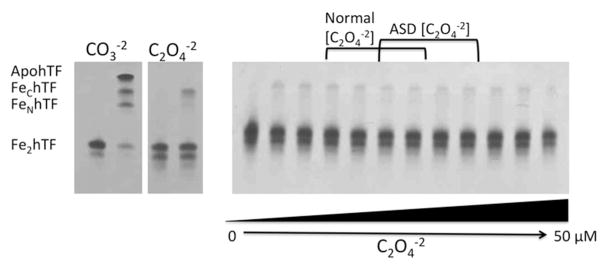
Displacement of oxalate (C2O4−2) from Fe2hTF by carbonate (CO3−2). Oxalate-bound Fe2hTF was incubated in normal serum CO3−2 concentration (25 mM NH3HCO3−2) (47) with increasing concentrations of C2O4−2 ranging from normal C2O4−2 concentrations (1.8–4.7 μM) (7), ASD C2O4−2 concentrations (3.5–7.5 μM) (7) and above (lanes 2–12 are 0, 1.0, 1.8, 2.5, 3.5, 4.7, 5.6, 7.5, 10, 25 and 50 μM C2O4−2 respectively). Samples were removed and electrophoresed following incubation with iron removal buffer (100 mM MES, pH 5.6, containing 300 mM KCl and 4 mM EDTA) for 15 min. The control carbonate (CO3−2) and oxalate (C2O4−2) containing Fe2hTF samples from Figure 1A are shown on the left as a reference.
ApohTF Synergistic Anion Competition
The ability of one synergistic anion (carbonate or oxalate) to compete with the other for concomitant binding with Fe3+ to apohTF was also assessed by urea gel. As shown in Fig. 5, when apohTF is exposed to a mixture of carbonate and oxalate at varying concentrations in the presence of an available iron source, Fe-NTA, carbonate appears to be the preferred synergistic anion for both lobes of hTF. These results suggest that, even at very high concentrations (50 μM) of oxalate, the much higher concentrations of carbonate present in the blood (20–30 mM) (47), would probably preclude the formation of hTF(OX).
Figure 5.
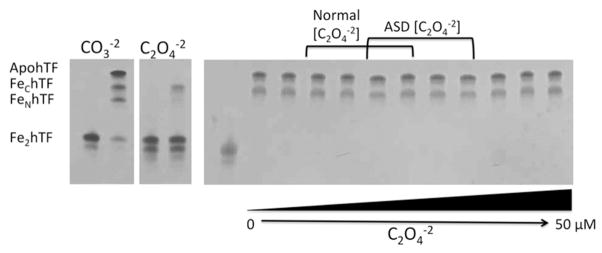
Synergistic anion competition for binding to apohTF. ApohTF was incubated in 25 mM (CO3−2) with increasing concentrations of C2O4−2 ranging from normal C2O4−2 concentrations (1.8–4.7 μM) (7), ASD C2O4−2 concentrations (3.5–7.5 μM) (7) and above (lanes 2–12 are 0, 1.0, 1.8, 2.5, 3.5, 4.7, 5.6, 7.5, 10, 25 and 50 μM C2O4−2 respectively) in the presence of excess Fe3+. Samples were removed and electrophoresed following incubation with iron removal buffer (100 mM MES, pH 5.6, containing 300 mM KCl and 4 mM EDTA) for 15 min. The control carbonate (CO3−2) and oxalate (C2O4−2) containing Fe2hTF samples from Figure 1A are shown on the left as a reference.
Evaluation of the hTF complexes by ESI MS
Oxalate binding to hTF was also examined by ESI MS under near native conditions allowing the bound iron and synergistic anion to be observed directly (48). Based on mass differences a distinction can be made between the oxalate and carbonate bound forms of hTF. Additionally, acidifying the sample typically allows the simultaneous displacement of both the iron and the synergistic anion from each lobe of hTF. Of interest, we found that simple buffer exchange into the pH 5.6 oxalate-buffer was not effective at replacing all carbonate (particularly in the C-lobe). Residual carbonate was clearly observed in the FeChTF(OX) and Fe2hTF(OX) samples, but not the FeNhTF(OX) sample suggesting that carbonate in the C-lobe was not as readily displaced (Fig S3). This result appears to be consistent with the higher affinity of the C-lobe of hTF for iron (21, 36). In contrast, only the oxalate bound form was observed when iron was removed prior to the buffer exchange to replace carbonate (Fig 6). The strong binding affinity of iron/oxalate in the C-lobe was also evident upon acidification of the sample to a pH of 3.7. Despite using a pH well below that reported for the endosome to facilitate formation of the apo species, a significant fraction of Fe2hTF retained iron and oxalate.
Figure 6.

ESI MS analysis of oxalate bound forms of hTF. Spectra for hTF and each monoferric variant (FeC, and FeN) were recorded at pH 6.7 and then at pH 3.7. Oxalate binding is demonstrated in the zoomed view of each 20+ charge state (insets); theoretically calculated positions for the various ligand bound states are labeled.
Iron uptake and circulation within the body: loading of hTF with iron
A small amount of iron (~2–3 mg) enters the body daily through uptake by the enterocytes lining the intestine. The remainder of the iron that enters the circulation comes from reticuloendothelial macrophages which recycle iron from senescent erythrocytes. Thus ~30 mg of iron is recovered and recycled each day in order to produce hemoglobin in reticulocytes (20). The redox pair, Fe2+ and Fe3+, undergoes a series of oxidations and reductions to allow transport through cells, across cell membranes and around the circulatory system (as Fe3+ within each cleft of hTF). A number of proteins are engaged in the inter-conversion, including ceruloplasmin and hephastin for oxidation and the cytochrome b ferrireductase, Dcytb for reduction (20). Although a good deal is known, some crucial details have yet to be elucidated including the precise location of the loading, i.e. within a cell or at the cell surface or both? Thus, if the local levels of oxalate at the sites of iron loading are high or the pH is low the possibility of oxalate entering the binding cleft of hTF certainly exists.
In the current work we have endeavored to investigate the potential link between high plasma oxalate levels and iron deficiency in children with ASD. Using techniques which have been developed and validated in our laboratory, we have been able to confirm previous findings that hTF with oxalate bound as the synergistic anion (hTF(OX)), releases far less Fe3+ at pH 5.6 than hTF in which carbonate serves as the synergistic anion. This finding holds true both in the absence and presence of the sTFR and is significant because our previous work shows that the sTFR overwhelms and in fact trumps other contributions to the mechanism of iron release including both variations in pH and salt (35, 36). Moreover, based on our perhaps overly simple in vitro system, oxalate (even at pathologically relevant concentrations) appears to be unable to displace or compete with carbonate for synergistic anion binding to hTF. Therefore, we must conclude that it is unlikely that high oxalate concentrations promoting the formation of hTF(OX) are responsible for iron-refractory ID/IDA in children with ASD. Obviously, further studies are required to validate these findings in vivo. Finally, as previously suggested (49), the very slow rate of iron release from hTF(OX)/sTFR complexes could increase dwell time of these complexes within endocytic compartments of cells, potentially making hTF(OX) an effective carrier for targeted drug delivery.
Supplementary Material
Acknowledgments
This work was supported by USPHS (grant R01 DK 21739) to A.B.M, and by R01GM061666 to I.A.K. A.N.L was funded by an AHA Predoctoral Fellowship (10PRE4200010).). Acquisition of an FT ICR mass spectrometer was supported by an NSF grant CHE-0923329 (through the Major Research Instrumentation program).
Abbreviations
- ASD
Autism spectrum disorders
- hTF
human serum transferrin
- TFR
transferrin receptor
- apohTF
iron-free hTF
- Fe2hTF
recombinant N-terminal hexa-His tagged non-glycosylated diferric hTF
- FeNhTF
recombinant N-terminal hexa-His tagged non-glycosylated monoferric hTF that binds iron only in the N-lobe (Y426F/Y517F mutations prevent iron binding in C-lobe)
- FeChTF
recombinant N-terminal hexa-His tagged non-glycosylated monoferric hTF that binds iron only in the C-lobe (Y95F/Y188F mutations prevent iron binding in N-lobe)
- LockN/LockChTF
constructs in which both lobes are unable to release iron due to substitution of K206E in the N-lobe and either K534E or K534A or R632E or R632A in the C-lobe
- hTF(OX)
human serum transferrin with oxalate in place of carbonate as the synergistic anion
- hTF/2N(OX)
the N-lobe of hTF with oxalate as the synergistic anion
- sTFR
glycosylated N-terminal hexa-His tagged soluble recombinant transferrin receptor (residues 121–760)
- BHK cells
baby hamster kidney cells
- NTA
nitrilotriacetic acid
- EDTA
ethylenediaminetetraacetic acid
- TBE
Tris(hydroxymethyl)aminomethane–borate–EDTA
- ESI MS
electrospray mass spectrometry
Footnotes
Tables 1 and 2, Kinetic constants for locked Fe2hTF constructs in the absence and presence of the sTFR, respectively. Figures 1 and 2, Urea gels for double locked Fe2hTF constructs in the absence and presence of the sTFR, respectively. Figure 3, ESI MS analysis of Fe2hTF and the two monoferric hTF constructs showing incomplete exchange of the synergistic anions oxalate and carbonate. Figures 4 and 5, Kinetic curves and fits for the oxalate complexes of Fe2hTF and the two monoferric hTF constructs in the absence and presence of the sTFR, respectively. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Prevention CfDCa. Prevalence of Autism Spectrum Disorders-Autism and Developmental Disabilities Monitoring Network, 14 Sites, United States, 2008. Morbidity and Mortality Weekly Report. 2012;61:1–19. [PubMed] [Google Scholar]
- 2.Liu J, Nyholt DR, Magnussen P, Parano E, Pavone P, Geschwind D, Lord C, Iversen P, Hoh J, Ott J, Gilliam TC. A genomewide screen for autism susceptibility loci. Am J Human Genetics. 2001;69:327–340. doi: 10.1086/321980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Volkmar FR, Pauls D. Autism. Lancet. 2003;362:1133–1141. doi: 10.1016/S0140-6736(03)14471-6. [DOI] [PubMed] [Google Scholar]
- 4.Kim SJ, Brune CW, Kistner EO, Christian SL, Courchesne EH, Cox NJ, Cook EH. Transmission disequilibrium testing of the chromosome 15q11-q13 region in autism. Am J Med Genetics Part B, Neuropsychiatric. 2008;147B:1116–1125. doi: 10.1002/ajmg.b.30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duchan E, Patel DR. Epidemiology of autism spectrum disorders. Ped Clinics of North America. 2012;59:27–43. ix–x. doi: 10.1016/j.pcl.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Jyonouchi H. Food allergy and autism spectrum disorders: is there a link? Current Allergy and Asthma Reports. 2009;9:194–201. doi: 10.1007/s11882-009-0029-y. [DOI] [PubMed] [Google Scholar]
- 7.Konstantynowicz J, Porowski T, Zoch-Zwierz W, Wasilewska J, Kadziela-Olech H, Kulak W, Owens SC, Piotrowska-Jastrzebska J, Kaczmarski M. A potential pathogenic role of oxalate in autism. Eur J Paediatr Neurol. 2012;16:485–491. doi: 10.1016/j.ejpn.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Holmes RP, Assimos DG. Glyoxylate synthesis, and its modulation and influence on oxalate synthesis. J Urol. 1998;160:1617–1624. [PubMed] [Google Scholar]
- 9.Linster CL, Van Schaftingen E. Vitamin C. Biosynthesis, recycling and degradation in mammals. The FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- 10.Latif A, Heinz P, Cook R. Iron deficiency in autism and Asperger syndrome. Autism: Intnat J Res and Practice. 2002;6:103–114. doi: 10.1177/1362361302006001008. [DOI] [PubMed] [Google Scholar]
- 11.Dosman CF, Drmic IE, Brian JA, Senthilselvan A, Harford M, Smith R, Roberts SW. Ferritin as an indicator of suspected iron deficiency in children with autism spectrum disorder: prevalence of low serum ferritin concentration. Develop Med and Child Neurol. 2006;48:1008–1009. doi: 10.1017/S0012162206232225. [DOI] [PubMed] [Google Scholar]
- 12.Bilgic A, Gurkan K, Turkoglu S, Akca OF, Kilic BG, Uslu R. Iron Deficiency in Preschool Children with Autistic Spectrum Disorders. Research Autism Spectrum Disorders. 2010;4:639–644. [Google Scholar]
- 13.Herguner S, Kelesoglu FM, Tanidir C, Copur M. Ferritin and iron levels in children with autistic disorder. Eur J Pediatr. 2012;171:143–146. doi: 10.1007/s00431-011-1506-6. [DOI] [PubMed] [Google Scholar]
- 14.Reynolds A, Krebs NF, Stewart PA, Austin H, Johnson SL, Withrow N, Molloy C, James SJ, Johnson C, Clemons T, Schmidt B, Hyman SL. Iron Status in Children With Autism Spectrum Disorder. Pediatrics. 2012;130:S154–S159. doi: 10.1542/peds.2012-0900M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dosman CF, Drmic IE, Brian JA, Harford M, Sharieff W, Smith R, Moldofsky H, Zlotkin S, Roberts W. Response to Iron Supplementation in Children with Autism Spectrum Disorders. Canadian Paediatric Society’s 81st Annual Meeting; Montreal, Quebec. 2004. [Google Scholar]
- 16.Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem and Cell Biol. 2001;33:940–959. doi: 10.1016/s1357-2725(01)00063-2. [DOI] [PubMed] [Google Scholar]
- 17.Lozoff B, Georgieff MK. Iron Deficiency and Brain Development. Seminars in Pediatric Neurology. 2006;13:158–165. doi: 10.1016/j.spen.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Lukowski AF, Koss M, Burden MJ, Jonides J, Nelson CA, Kaciroti N, Jimenez E, Lozoff B. Iron deficiency in infancy and neurocognitive functioning at 19 years: evidence of long-term deficits in executive function and recognition memory. Nutritional Neuroscience. 2010;13:54–70. doi: 10.1179/147683010X12611460763689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acikyol B, Graham RM, Trinder D, House MJ, Olynyk JK, Scott RJ, Milward EA, Johnstone DM. Brain transcriptome perturbations in the transferrin receptor 2 mutant mouse support the case for brain changes in iron loading disorders, including effects relating to long-term depression and long-term potentiation. Neuroscience. 2013;235:119–128. doi: 10.1016/j.neuroscience.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 20.Sheftel AD, Mason AB, Ponka P. The long history of iron in the Universe and in health and disease. Biochim Biophys Acta. 2011;1820:161–187. doi: 10.1016/j.bbagen.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aisen P, Leibman A, Zweier J. Stoichiometric and site characteristics of the binding of iron to human transferrin. J Biol Chem. 1978;253:1930–1937. [PubMed] [Google Scholar]
- 22.Schade AL, Reinhart RW, Levy H. Carbon dioxide and oxygen in complex formation with iron and siderophilin, the iron-binding component of human plasma. Arch Biochem. 1949;20:170–172. [PubMed] [Google Scholar]
- 23.MacGillivray RT, Moore SA, Chen J, Anderson BF, Baker H, Luo Y, Bewley M, Smith CA, Murphy ME, Wang Y, Mason AB, Woodworth RC, Brayer GD, Baker EN. Two high-resolution crystal structures of the recombinant N-lobe of human transferrin reveal a structural change implicated in iron release. Biochemistry. 1998;37:7919–7928. doi: 10.1021/bi980355j. [DOI] [PubMed] [Google Scholar]
- 24.Price EM, Gibson JF. A re-interpretation of bicarbonate-free ferric transferrin E.P.R. spectra. Biochem Biophys Res Commun. 1972;46:646–651. doi: 10.1016/s0006-291x(72)80189-x. [DOI] [PubMed] [Google Scholar]
- 25.Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, Wriggers W. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 26.Gerstein M, Anderson BF, Norris GE, Baker EN, Lesk AM, Chothia C. Domain closure in lactoferrin. Two hinges produce a see- saw motion between alternative close-packed interfaces. J Mol Biol. 1993;234:357–372. doi: 10.1006/jmbi.1993.1592. [DOI] [PubMed] [Google Scholar]
- 27.Gaber BP, Miskowski V, Spiro TG. Resonance Raman scattering from iron(3)- and copper(II)-transferrin and an iron(3) model compound. A spectroscopic interpretation of the transferrin binding site. J Am Chem Soc. 1974;96:6868–6873. doi: 10.1021/ja00829a010. [DOI] [PubMed] [Google Scholar]
- 28.Lin LN, Mason AB, Woodworth RC, Brandts JF. Calorimetric studies of the binding of ferric ions to human serum transferrin. Biochemistry. 1993;32:9398–9406. doi: 10.1021/bi00087a019. [DOI] [PubMed] [Google Scholar]
- 29.Morgan EH, Appleton TC. Autoradiographic localization of 125-I-labelled transferrin in rabbit reticulocytes. Nature. 1969;223:1371–1372. doi: 10.1038/2231371a0. [DOI] [PubMed] [Google Scholar]
- 30.Morgan EH. Inhibition of reticulocyte iron uptake by NH4Cl and CH3NH2. Biochim Biophys Acta. 1981;642:119–134. doi: 10.1016/0005-2736(81)90143-7. [DOI] [PubMed] [Google Scholar]
- 31.He QY, Mason AB, Tam BM, MacGillivray RT, Woodworth RC. Dual role of Lys206–Lys296 interaction in human transferrin N-lobe: iron-release trigger and anion-binding site. Biochemistry. 1999;38:9704–9711. doi: 10.1021/bi990134t. [DOI] [PubMed] [Google Scholar]
- 32.Halbrooks PJ, He QY, Briggs SK, Everse SJ, Smith VC, MacGillivray RT, Mason AB. Investigation of the mechanism of iron release from the C-lobe of human serum transferrin: mutational analysis of the role of a pH sensitive triad. Biochemistry. 2003;42:3701–3707. doi: 10.1021/bi027071q. [DOI] [PubMed] [Google Scholar]
- 33.Halbrooks PJ, Giannetti AM, Klein JS, Bjorkman PJ, Larouche JR, Smith VC, MacGillivray RT, Everse SJ, Mason AB. Composition of pH-sensitive triad in C-lobe of human serum transferrin. Comparison to sequences of ovotransferrin and lactoferrin provides insight into functional differences in iron release. Biochemistry. 2005;44:15451–15460. doi: 10.1021/bi0518693. [DOI] [PubMed] [Google Scholar]
- 34.Dewan JC, Mikami B, Hirose M, Sacchettini JC. Structural evidence for a pH-sensitive dilysine trigger in the hen ovotransferrin N-lobe: implications for transferrin iron release. Biochemistry. 1993;32:11963–11968. doi: 10.1021/bi00096a004. [DOI] [PubMed] [Google Scholar]
- 35.Steere AN, Byrne SL, Chasteen ND, Mason AB. Kinetics of iron release from transferrin bound to the transferrin receptor at endosomal pH. Biochim Biophys Acta. 2012;1820:326–333. doi: 10.1016/j.bbagen.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byrne SL, Chasteen ND, Steere AN, Mason AB. The unique kinetics of iron-release from transferrin: The role of receptor, lobe-lobe interactions and salt at endosomal pH. J Mol Biol. 2010;396:130–140. doi: 10.1016/j.jmb.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlabach MR, Bates GW. The synergistic binding of anions and Fe3+ by transferrin. Implications for the interlocking sites hypothesis. J Biol Chem. 1975;250:2182–2188. [PubMed] [Google Scholar]
- 38.Halbrooks PJ, Mason AB, Adams TE, Briggs SK, Everse SJ. The oxalate effect on release of iron from human serum transferrin explained. J Mol Biol. 2004;339:217–226. doi: 10.1016/j.jmb.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 39.Mason AB, He QY, Halbrooks PJ, Everse SJ, Gumerov DR, Kaltashov IA, Smith VC, Hewitt J, MacGillivray RT. Differential effect of a his tag at the N- and C-termini: functional studies with recombinant human serum transferrin. Biochemistry. 2002;41:9448–9454. doi: 10.1021/bi025927l. [DOI] [PubMed] [Google Scholar]
- 40.Byrne SL, Leverence R, Klein JS, Giannetti AM, Smith VC, MacGillivray RT, Kaltashov IA, Mason AB. Effect of glycosylation on the function of a soluble, recombinant form of the transferrin receptor. Biochemistry. 2006;45:6663–6673. doi: 10.1021/bi0600695. [DOI] [PubMed] [Google Scholar]
- 41.Steere AN, Byrne SL, Chasteen ND, Smith VC, MacGillivray RT, Mason AB. Evidence that His349 acts as a pH-inducible switch to accelerate receptor-mediated iron release from the C-lobe of human transferrin. J Biol Inorg Chem. 2010;15:1341–1352. doi: 10.1007/s00775-010-0694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Byrne SL, Mason AB. Human serum transferrin: a tale of two lobes. Urea gel and steady state fluorescence analysis of recombinant transferrins as a function of pH, time, and the soluble portion of the transferrin receptor. J Biol Inorg Chem. 2009;14:771–781. doi: 10.1007/s00775-009-0491-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Byrne SL, Chasteen ND, Steere AN, Mason AB. The unique kinetics of iron release from transferrin: The role of receptor, lobe-lobe interactions, and salt at endosomal pH. J Mol Biol. 2010;396:130–140. doi: 10.1016/j.jmb.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byrne SL, Steere AN, Chasteen ND, Mason AB. Identification of a kinetically significant anion binding (KISAB) site in the N-lobe of human serum transferrin. Biochemistry. 2010;49:4200–4207. doi: 10.1021/bi1003519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eckenroth BE, Steere AN, Chasteen ND, Everse SJ, Mason AB. How the binding of human transferrin primes the transferrin receptor potentiating iron release at endosomal pH. Proc Natl Acad Sci U S A. 2011;108:13089–13094. doi: 10.1073/pnas.1105786108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steere AN, Chasteen ND, Miller BF, Smith VC, Macgillivray RT, Mason AB. Structure-Based Mutagenesis Reveals Critical Residues in the Transferrin Receptor Participating in the Mechanism of pH-Induced Release of Iron from Human Serum Transferrin. Biochemistry. 2012;51:2113–2121. doi: 10.1021/bi3001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raphael KL, Wei G, Baird BC, Greene T, Beddhu S. Higher serum bicarbonate levels within the normal range are associated with better survival and renal outcomes in African Americans. Kidney International. 2011;79:356–362. doi: 10.1038/ki.2010.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaltashov IA, Bobst CE, Zhang M, Leverence R, Gumerov DR. Transferrin as a model system for method development to study structure, dynamics and interactions of metalloproteins using mass spectrometry. Biochim Biophys Acta. 2012;1820:417–426. doi: 10.1016/j.bbagen.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoon DJ, Chu DS, Ng CW, Pham EA, Mason AB, Hudson DM, Smith VC, MacGillivray RT, Kamei DT. Genetically engineering transferrin to improve its in vitro ability to deliver cytotoxins. J Control Release. 2009;133:178–184. doi: 10.1016/j.jconrel.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.