

Abstract
Spermatogonial stem cells (SSCs) are the foundation of spermatogenesis throughout postnatal life in male and have the ability to transmit genetic information to the subsequent generation. In this study, we have optimized the transduction efficiency of SSCs using a lentiviral vector by considering different multiplicity of infection (MOI), duration of infection, presence or absence of feeder layer and polycationic agents. We tested MOI of 5, 10 or 20 and infection duration of 6, 9 or 12 h respectively. After infection, cells were cultured for 1 week and as a result, the number of transduced SSCs increased significantly for MOI of 5 and 10 with 6 h of infection. When the same condition (MOI of 5 with 6 hours) was applied in presence or absence of STO feeder layer and infected SSCs were cultured for 3 weeks on the STO feeder layer, a significant increase in the number of transduced cells was observed for without the feeder layer during infection. We subsequently studied the effects of polycationic agents, polybrene and dioctadecylamidoglycyl spermine (DOGS), on the transduction efficiency. Compared with the polybrene treatment, the recovery rate of the transduced SSCs was significantly higher for the DOGS treatment. Therefore, our optimization study could contribute to the enhancement of germ-line modification of SSCs using lentiviral vectors and in generation of transgenic animals.
Keywords: germline modification, Lentivirus, polycationic agent, spermatogonial stem cell, transgenesis
INTRODUCTION
Spermatogenesis, a series of complex processes that produce spermatozoa in the testes, occurs through consecutive cell divisions and differentiation of germ cells (Russell et al., 1990). A spermatogonial stem cell (SSC) in an adult rat testis is known to have high productive ability, yielding up to 4,096 spermatozoa (Russell et al., 1990). As the germ-line stem cells that are the foundation of spermatogenesis, SSCs are the vehicle for delivering the male genetic information to the offspring (Meistrich and van Beek, 1993). Similar to the characteristics found in other adult stem cells, SSCs can self-renew and differentiate (Meistrich and van Beek, 1993). Therefore, SSCs are the source of life-long postnatal fertility in males. The SSCs found in the male testes maintain numerical equilibrium through self-renewal after birth. Therefore, when genetic modification is performed at the SSC stage, transgenic offspring are produced, and germline modification allows the stable delivery of the transformed character to the descendants. Early studies regarding the SSCs depended solely on morphological observations due to the absence of a reliable marker until the development of SSC transplantation technique that allowed measurement of biological activity of SSCs (Brinster and Avarbock, 1994; Brinster and Zimmermann, 1994). Utilizing the SSC transplantation technique, the self-renewal activity and the productive capability of differentiated germ cells derived from transplanted SSC within the recipient seminiferous tubule can be directly analyzed. During the recent years, the SSC transplantation technique has been further improved with purification and efficient recipient animal production technique for SSC (Brinster et al., 2003; Kubota et al., 2003; Ryu et al., 2003; 2004; Shinohara et al., 2000). These techniques have allowed creation of transgenic animals via genetic modification and transplantation of SSCs that can pass on their traits to the progeny. The use of SSCs is more advantageous compared with pronuclear embryos or embryonic stem cells (ES cell) where the germ-line transmission is only 40–50% (Gordon and Ruddle, 1981). Therefore, the transgenic animal production using SSCs and the transplantation technique can be used to enhance the production efficiency of transgenic animals.
Several groups have produced transgenic animal using lentivirus-mediated SSC transduction (Hamra et al., 2002; Ryu et al., 2007). For most of these studies, the transgenic animals were produced by lentiviral infection of highly purified SSCs and transplanting them right into testes of the recipient animals. With the recent developments of the in vitro culture technique of rodent SSCs, the possibility of germ-line modification for cultured cells has been proposed (Kanatsu-Shinohara et al., 2003; Kubota et al., 2004). If genetic modification and culture techniques of SSC are used simultaneously with a drug selection method, improvements in purity and mass propagation of transduced cells are possible. However, specific studies for the transduction of SCCs under in vitro culture conditions are lacking. In this study, as an effort to refine in vitro transduction conditions, we evaluated transduction efficiency of SSCs for various durations of lentiviral treatment, various multiplicities of infection (MOI), and after the addition of polycationic agents or feeder layer.
MATERIALS AND METHODS
Animals
B6.129S7-Gtrosa26 (designated ROSA; The Jackson Laboratory) and C57BL/6 mice (designated C57; The Jackson Laboratory) were used as SSC donors. W/Wv (designated W; The Jackson Laboratory) male adult mice were used as recipients. All procedures were performed according to the approved guidelines for the ethical treatment of animals as established in the regulations of Chung-Ang University and in compliance with standard international regulations.
SSC isolation and culture
The donor cells were cultured based on the method presented by Kubota et al. (2004) with minor modifications. Donor cells were extracted from ROSA and C57 mice 6–8 days after birth. After removal of the tunica albuginea, the exposed seminiferous tubules were digested with a 2:1 mixture of 0.25% trypsin-EDTA (Invitrogen) and DNase I (7 mg/ml in DPBS; Sigma). Fetal bovine serum (FBS, 10% of trypsin) was added to stop further enzymatic digestion, the resulting cell suspension was filtered with a strainer (pore size, 40 um; BD) and then centrifuged at 600 × g for 7 min at 4°C. SSCs that were isolated from the testicular cell suspension were enriched through magnetic-activated cell sorting (MACS) using an anti-Thy-1 antibody (Miltenyi Biotec). Using mouse serum-free medium (MSFM) (Kubota et al., 2004) and mitotically-inactivated STO cell feeders, enriched SSCs were cultivated in the presence of human glial cell-derived neurotrophic factor (GDNF) (R & D Systems), rat GDNF family receptor alpha 1-Fc fusion protein (GFRa1-Fc, R & D Systems), and human basic fibroblast growth factor (bFGF) (BD Biosciences) at final concentrations of 40 ng/ml, 300 ng/ml, and 1 ng/ml, respectively. The SSCs were then subcultured every 7-days with 0.25% trypsin-EDTA at a 1:2–4 dilution. All cultures were maintained with full medium change every 2–3 days at 37°C with 5% CO2.
Lentiviral transduction of mouse SSCs
For transduction, the lentiviral vector (pLV-TH) containing the green fluorescent protein (GFP) gene driven by the elongation factor 1 (EF1) promoter was used (Sandrin et al., 2002). With minor modifications, the lentivirus vector was produced in 293T cells according to the method presented by Watson and Wolfe et al. (2003). The basic conditions for the viral transduction procedure were as follows; 5 × 105 cells were plated per well in a 24-well plate and infected with MSFM containing 10% FBS. The infected SSCs were harvested by gentle pipetting and plated in newly prepared STO feeder cells and cultured 1–3 weeks. The cultured SSCs were dissociated into single cells with trypsin-EDTA and analyzed under the fluorescence microscope to evaluate transduction efficiency. The conditions for lentiviral transduction are as follows; SSCs were infected with lentivirus on mitomycin C-treated STO feeder cells with three different MOIs (5, 10 or 20) for different durations (6, 9, or 12 h). After determining the optimal condition for infection, we examined the effect of feeder layer on the proficiency of transduction. The SSCs were plated in a 24-well plate and infected with in the presence or absence of feeder layer. Third, we studied the effects of different polycationic agents on the transduction efficiency. The SSCs were infected with lentivirus with an MOI of 5 for 6 h in the absence of feeder cells with different polycationic agents. The polycationic agents studied were polybrene (Sigma) at concentration of 4 or 8 ug/ml and dioctadecylamidoglycyl spermine (DOGS; Promega) at concentration of 2 or 4 ug/ml. After viral infection, the SSCs were harvested by pipetting and then washed with MSFM 3 times and cultured for 1 or 3 weeks on STO feeder cells with MSFM. A higher nucleus/cytoplasm ratio and smaller cell size of SSCs were distinguished from STO feeder cells. The transduction efficiency was evaluated based on the number of GFP expressing SSCs that were scored under the fluorescence microscope (Fig. 1).
Fig. 1.
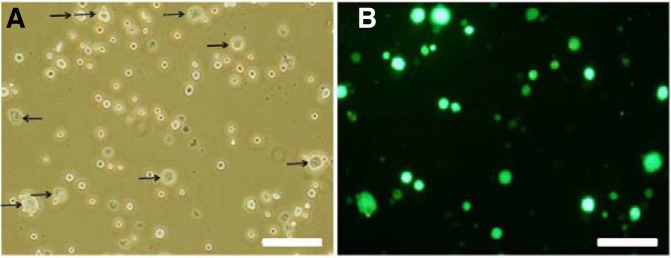
Microscopic images of lentivirus-infected cells after 1 week in culture (MOI 5, 6 h). (A, B): Light and corresponding fluorescence photomicrograph (arrows: STO feeder cells, Scale bar = 100 um)
Transplantation
Donor lentivirus-infected SSCs were introduced into the W recipient testis via the efferent duct. Two months after transplantation, the recipient mouse testes were collected and examined using a fluorescent microscope to find the GFP-positive colonies of donor SSCs-derived spermatogenesis (see Fig. 6). We used W mutant mice as recipients for transplantation to examine whether transduced SSCs could restore the spermatogenesis of the recipient mouse that was congenitally infertile and lack endogenous spermatogenesis due to mutations in the c-Kit receptor tyrosine kinase gene. W mice were also chosen as it has been previously shown that efficient colonization by transplanted SSCs was enhanced in the W testis environment (Chabot et al., 1988; Geissler et al., 1988; Shinohara et al., 2001). Therefore, the presence of spermatogenesis inside the testes of W mice could only be derived from the transplanted donor SSCs.
Fig. 6.

Histology of W sterile recipient testes injected with spermatogonial stem cells expres-sing lentiviral vector EF1-GFP (2 ug/ml of DOGS, MOI 5, 6 h infection) viewed under a microscope. Whole-mount images of GFP colonies from donor-derived spermatogenesis at 2 months after transplantation (A, B) and immunohistochemical evaluation of transplanted seminiferous tubules (C–H). (A, B) Light and correspond-ing fluorescence photomicrographs, left testis, each GFP expressing area indicates donor-derived spermatogenesis, right testis, control testis without transplantation did not show fluorescence; (C–H) Fluorescent image showing multiple layers of GFP-expressing donor-derived germ cells and sperm. Cross-section through a colony of donor spermatogenesis stained by GFP antibody and the incidence of sperm detected by PNA lectin and tubulin antibodies. (C–D) and (E–H) images represent a right next section from the same tubule. (C, E) DAPI, (D) Alexa flour 488 labeled GFP, (F) Alexa flour 488 labeled PNA lectin, (G) Alexa flour 647 labeled tubulin, (H) Overlay of E–G fluorescent signals. Inset displays a higher magnification of sperm heads in the dashed white box (Arrow, PNA stained acrosome of sperm; Arrow head, tubulin stained sperm tail, Scale bars = A, B: 5 mm, C–H: 50 um).
Immunohistochemistry
For immunofluorescence staining, testes were fixed in Bouin’s solution (sigma) at room temperature overnight, washed in 70% ethanol, embedded in paraffin wax and sliced into 4 μm sections in turn. Sections were dewaxed with zylene for 5 min and rehydrated in 100, 90, 80, and 70% ethanol. The sections were subjected to antigen retrieval in boiling sodium citrate buffer (0.01 M, pH 6.0) for 10 min under microwave and incubated with 0.1% trypsin for 10 min at 37°C. Sections were incubated with PBS containing 0.1% Tween-20 for 5 min at room temperature. Nonspecific binding was blocked by incubating sections in PBS containing 5% BSA and 0.1% Tween-20 for 30 min. After additional washes with PBS containing 0.1% Tween-20, section were incubated for overnight at 4°C with monoclonal mouse anti-tubulin antibody (Abcam), polyclonal rabbit anti-GFP (Abcam) and Alexa flour 488 conjugated peanut agglutinin (PNA) lectin (Invitrogen) with PBS containing 5% BSA and 0.1% Tween-20. The secondary antibodies used were Alexa flour 647 goat anti-mouse (invitrogen) for tubulin and Alexa flour 488 donkey anti-rabbit (invitrogen) for GFP. To preserve fluorescent signals and counter staining, we used Vectashield mounting solution containing DAPI (Vector Laboratories).
Statistical analysis
The analysis of the differences in transduction efficiency between the feeder cell plate and the feeder cell-free plate was conducted using a t-test in SPSS (ver. 12.0, Standard Software Package Inc., USA). The other data were analyzed using ANOVA. If the p-value was < 0.05, then Tukey’s HSD was performed. Data were expressed as the mean ± SEM.
RESULTS
Transduction efficiency for different MOIs and infection times
We first examined the effects of MOI for lentivirus and the duration of infection on transduction efficiency and total number of transduced cells for SSCs. Lentiviral transduction efficiency (the number of GFP positive SSCs/the total number of SSCs counted × 100) and the number of transduced cells (transduction efficiency (%) × the total number of SSCs recovered/100) were determined 7 days after transduction by scoring GFP positive cells under the fluorescent microscope. The 5, 10, or 20 MOI of lentivirus was transduced into SSCs for 6 h, and the cells were examined after one week of cultivation. The transduction efficiencies among different MOIs were not significant: MOI 5; 35.3 ± 5.1% (mean ± SEM, n = 3), MOI 10; 41.0 ± 2.2% (n = 3), and MOI 20; 43.4 ± 3.4% (n = 3) (Fig. 2A). For the number of transduced cells, significant difference was not observed between cells infected with MOI 5 (4.5 ± 0.2 × 104, n = 3) and MOI 10 (4.3 ± 0.6 × 104, n = 3). However, the number of transduced cells was significantly lower for MOI 20 (2.3 ± 0.2 × 104, n = 3) (Fig. 2B), which could be the result of higher viral toxicity. Based on these results, we chose MOI 5 for the determination of the optimal infection time (6, 9, or 12 h). Again, no significant difference in transduction efficiency was observed for duration of infection of 6, 9, and 12 h groups, displaying transduction efficiency of 35.3 ± 5.1% (n = 3), 46.0 ± 4.6% (n = 3) and 42.8 ± 0.5% (n = 3), respectively (Fig. 2C). The number of transduced cells was decreased as infection time increased with no significant differences between the infection groups (Fig. 2D); in the 6, 9, and 12 h groups, the resultant transduced cells were 4.5 ± 0.2 × 104 (n = 3), 4.3 ± 0.9 × 104 (n = 3) and 3.0 ± 0.6 × 104 (n = 3), respectively. Based on the findings, the effect of feeder layer on transduction efficiency was performed with SSCs that were infected at MOI 5 for 6 h.
Fig. 2.

Transduction efficiency and number of transduced cells for different MOIs and infection times (Bar: mean ± SEM; n = 3). Different lowercase letters were significantly different by Tukey’s HSD (P < 0.05).
The effect of feeder layer on SSC transduction
To examine the effect of feeder layer on lentiviral infection, SSC transduction was carried out in cultures containing monolayer of feeder cells (feeder group) and was compared with no feeder cells (feeder-free group). After transduction in the presence or absence of feeder cells, both groups of cells were transferred to the dishes containing STO feeder cells. After 3 weeks of culture, the transduction efficiencies of SSCs in the feeder-free group and feeder group were 44.3 ± 2.3% (n = 3) and 44.0 ± 1.5% (n = 3), respectively, showing no significant difference between two (Fig. 3A). However, the feeder-free group significantly showed a 1.6-fold increase (3.9/2.5) in the number of transduced cells compared with the feeder group (Fig. 3B). The number of transduced cells in the feeder-free group was 3.9 ± 0.7 × 105 (n = 3) whereas that of the feeder group was 2.5 ± 0.2 × 105 (n = 3). Based on these findings, the effect of polycationic agents on transduction was tested in the absence of feeder layer.
Fig. 3.

The effects of feeder layer on transduction efficiency and in vitro culture of transduced cells (Bar: mean ± SEM; n = 3). Asterisk indicates significant difference between two groups by t-test (P < 0.05).
The effect of polycationic agents on SSC transduction
To determine the effect of the polycationic agents, polybrene (at 4 and 8 ug/ml) or DOGS (at 2 and 4 ug/ml) was added to the culture medium for the lentiviral infection of cells and the transduced cells were cultured for 3 weeks on STO feeder cells with MSFM. Transduction efficiency of the control group treated with no polycationic agent (virus only) was 32.1 ± 0.6% (n = 3), while those of treated with 4 and 8 ug/ml polybrene groups were 47 ± 1.2% (n = 3) and 44 ± 0.1% (n = 3), respectively (Fig. 4A). Furthermore, transduction efficiencies of the 2 or 4 ug/ml DOGS groups were significantly higher than those of the polybrene groups, showing 53.7 ± 0.9% (n = 3) and 60.5 ± 1.4% (n = 3), respectively (Fig. 4A).
Fig. 4.

The effects of polycationic agents on the transduction efficiency and in vitro culture of transduced cells (PB: polybrene, Bar: mean ± SEM; n = 3). Different lowercase letters were significantly different by Tukey’s HSD (P < 0.05).
When the proliferation pattern of transduced SSCs was analyzed by measuring the number of transduced cells under the fluorescence microscope during the 3 week in vitro culture after lentiviral infection, a numerical increase of transduced SSCs was observed in all treatments as the culture period increased. Although the number of transduced cells in the control [4.8 ± 0.2 × 105 (n = 3)] and polybrene-treated groups [5.5 ± 0.3 × 105 for 4 ug/ml and 4.7 ± 0.5 × 105 for 8 ug/ml (n = 3)] did not show any significant difference, those in DOGS-treated groups [9.0 ± 0.7 × 105 for 2 ug/ml and 7.0 ± 0.5 × 105 for 4 ug/ml (n = 3)] were significantly enhanced. The 2 ug/ml DOGS-treated group was the highest in the number of transduced cells among all treatment groups. Taken together, the optimal condition for lentiviral infection was proposed to be MOI 5 for 6 h with 2 ug/ml of DOGS.
Stable expression of transduced GFP was also determined after 3 weeks of culture (Fig. 5). When compared with 1 week cultures, the number of transduced cells had significantly increased by approximately 6-fold (9.0 ± 0.6/1.5 ± 0.8 × 105) during the additional 2 weeks of culture period (Fig. 4B).
Fig. 5.
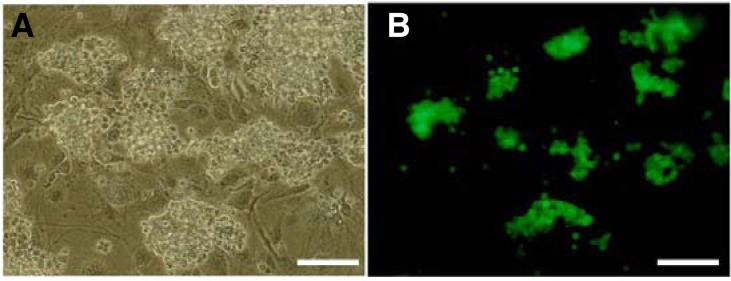
Microscopic images of lentivirus-infected cells after three weeks of culture using 2 ug/ml of DOGS (MOI 5, 6 h). (A, B) Light and corresponding fluorescence photomicrographs (Scale bar = 100 um)
Transplantation of transduced SSCs
To test whether the GFP-expressing SSCs could transplant into the testis and generate the mature sperm, we performed functional transplantation assay. SSCs were infected with lentivirus in the optimal infection condition and the GFP-expressing SSCs were isolated using Fluorescence-Activated Cell Sorting technique (FACS, Aria II, BD Biosciences, Center for Research Facilities, Chung-Ang University) at 3 weeks after transduction. Highly purified the GFP-expressing SSCs were transplanted into the seminiferous tubule of the W mouse, and the recipient testes were collected after 2 months for analysis.
We could find normal GFP-expressing germ cell colonies in W recipient testis (Figs. 6A and 6B). Because the donor transduced SSCs and multiple layered spermatogenic germ cells express GFP, we could unequivocally identify the donor-derived spermatogenesis (Fig. 6D). Furthermore, the donor-derived germ cells and spermatozoa showing affinity for PNA lectin, that labels post-meiotic germ cells and acrosomal membrane, were observed in the seminiferous tubule (Figs. 6F and 6H). Also, sperm tails in the lumen of the seminiferous tubule were detected by tubulin immunostaining (Figs. 6G and 6H). Therefore, our transduction protocol of SSCs is effective to generate normal transgenic spermatogenesis in recipient testis with no functional defects.
DISCUSSION
Lentivirus, a subclass of retrovirus, can transmit genetic information to quiescent, non-dividing cells. Therefore lentiviral vector could be useful vector for transduction of cells that have a slow cell cycle, such as SSCs (Kim et al., 2010; Kubota et al., 2004; Nagano et al., 2002). In previous studies, transgenic animals were also produced by lentiviral transduction and transplantation of SSCs (Hamra et al., 2002; Ryu et al., 2007). However, they used different MOI and duration of infection, making it difficult to directly compare each other. Regardless, as observed in the results of the present study, the transduction efficiency tends to increase as the MOI and infection time increases. However, the number of transduced cells quantified after 1 week in culture tended to decrease as increase the MOI and duration of infection (Fig. 2). Therefore, it is important to determine an optimal MOI and duration of infection for lentiviral vectors. Ryu et al. (2007) has transplanted SSCs directly into the recipient testis without additional in vitro culture where no significant difference was observed for neither the survival rate nor the transduction efficiency for MOI of 20 (44.8%) and MOI of 80 (35.8%) with 15 h of infection. In this study, the transduction efficiencies at different levels of MOI infection were not significantly different: the transduction efficiency was in the range of 35.3–43.4%. However, the number of transduced cells was significantly reduced for MOI 20 after 1 week culture among the infected groups (Fig. 2B). This finding is implicated by the cytotoxicity caused by increased number of lentivirus, which lowers the survival rate of SSCs. Also, differences in these two studies on the survival rate of SSC after viral infection is thought to be due to environmental differences between in vivo transplantation and in vitro culture. The in vivo environment may have had a better buffer effect than the in vitro environment because of the cytotoxic actions resulting from outside factors such as viral infection.
For the in vitro culture of stem cells including SSCs, feeder layer with supplements that promote cell growth, self-renewal and signal transduction are important elements. In the case of SSCs, commonly used feeder cells are STO and mouse embryonic fibroblast (MEF) (Kanatsu-Shinohara et al., 2003; Kubota et al., 2004; Ryu et al., 2005). However, the effect of STO and MEF on the transduction and survival rate of stem cells including SSCs has not been studied. This study has addressed this question by demonstrating that feeder layer did not affect transduction efficiency during lentiviral infection, but absence of it significantly increased proliferation of infected cells during 3 weeks of culture. While, further studies are needed to determine the effect of feeder layer on SSCs during the duration of infection, these results suggest feeder layer is not required for lentiviral infection of SSCs.
Polycationic agents such as polybrene and DEAE dextran are often used to increase the transduction efficiency in mammalian cells (Hodgson et al., 1996; Singh and Rigby, 1996). In the absence of polycationic agents, virus and cells tend to repel each other as they both have negatively charged surfaces. Therefore, polycationic agents mask the negatively charges on the virus to facilitate the passage of virus through the negatively charged cell membrane. Both the surface of the virus and the membrane of animal cell have a negative charge. The general transduction method uses polybrene as a polycationic agent for the transduction of SSCs using lentivirus (Nagano et al., 2002; Ryu et al., 2007). Another method of gene delivery into the target cell involves injection of a plasmid DNA using a cationic liposome. The cationic liposome can stably transmit genes into the cell through fusion with the cell membrane due to the shielding of genes that have a negative charge (Gao and Huang, 1995; Smith et al., 1993). It was reported that the liposome DOGS increases the transduction efficiency by having the membrane fusion-promoting lipid in its basic structure (Felgner et al., 1987). Since the composition of a liposome is similar to the composition of the cell membrane, the transduction efficiency is higher and the cytotoxicity is less compared with polycationic agents (Fraley and Papahadjopoulos, 1982; Nicolau et al., 1987). Indeed, when polybrene 4 ug/ml and DOGS 2 ug/ml were micro-injected into rat testes, and the testes were histologically analyzed after 2–5 months, DOGS-treated group displayed spermatogenesis in the seminiferous tubules of more than 80% of the recipients while in the polybrene-treated groups showed spermatogenesis in only 20% of the recipients (unpublished data). Themis and Forbes (1998) performed internal transduction by injecting retrovirus directly into the body, and showed efficient transduction into tissue cells can be achieved by the addition of DOGS. Similarly our results for this study showed lower cytotoxicity for the DOGS-treated group compared with the polybrene-treated group (Fig. 4), thus highlighting the usefulness of DOGS as a polycationic agent in the transduction of SSCs.
As we have verified the optimum conditions, when we transduced SSCs and transplanted them into recipient testis, the transduced SSCs retained the GFP-expressing multiple germ cell colonies 2 months after transplantation and generated viable sperms from complete spermatogenesis (Fig. 6). Thus, the optimized protocol of lentiviral transduction for SSCs in the present study may be applicable in other stem cells and species, and useful to promote aggressive implementation of transgenic animal production using SSCs.
Acknowledgments
We thank Y.-J. Park for assistance in developing the figures. This study was supported by the Next-Generation BioGreen 21 Program (No. PJ0080482011), the Rural Development Administration, a National Research Foundation of Korea (NRF) grant funded by the Ministry of Education, Science and Technology (MEST) (2010-0023848), the MEST/Stem Cell Research Center of the 21st Century Frontier Research Program (SC-5140).
REFERENCES
- Brinster R.L., Avarbock M.R. Germline transmission of donor haplotype following spermatogonial transplantation. Proc. Natl. Acad. Sci. USA. 1994;91:11303–11307. doi: 10.1073/pnas.91.24.11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster R.L., Zimmermann J.W. Spermatogenesis following male germ-cell transplantation. Proc. Natl. Acad. Sci. USA. 1994;91:11298–11302. doi: 10.1073/pnas.91.24.11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster C.J., Ryu B.Y., Avarbock M.R., Karagenc L., Brinster R.L., Orwig K.E. Restoration of fertility by germ cell transplantation requires effective recipient preparation. Biol. Reprod. 2003;69:412–420. doi: 10.1095/biolreprod.103.016519. [DOI] [PubMed] [Google Scholar]
- Chabot B., Stephenson D.A., Chapman V.M., Besmer P., Bernstein A. The proto-oncogene c-kit encoding a trans-membrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335:88–89. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- Felgner P.L., Gadek T.R., Holm M., Roman R., Chan H.W., Wenz M., Northrop J.P., Ringold G.M., Danielsen M. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA. 1987;84:7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley R., Papahadjopoulos D. Liposomes: the development of a new carrier system for introducing nucleic acid into plant and animal cells. Curr. Top. Microbiol. Immunol. 1982;96:171–191. doi: 10.1007/978-3-642-68315-2_11. [DOI] [PubMed] [Google Scholar]
- Gao X., Huang L. Cationic liposome-mediated gene transfer. Gene Ther. 1995;2:710–722. [PubMed] [Google Scholar]
- Geissler E.N., Ryan M.A., Housman D.E. The dominant white spotting (W) locus of the mouse encodes the c-kit protooncogene. Cell. 1988;55:185–192. doi: 10.1016/0092-8674(88)90020-7. [DOI] [PubMed] [Google Scholar]
- Gordon J.W., Ruddle F.H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science. 1981;214:1244–1246. doi: 10.1126/science.6272397. [DOI] [PubMed] [Google Scholar]
- Hamra F.K., Gatlin J., Chapman K.M., Grellhesl D.M., Garcia J.V., Hammer R.E., Garbers D.L. Production of transgenic rats by lentiviral transduction of male germ-line stem cells. Proc. Natl. Acad. Sci. USA. 2002;99:14931–14936. doi: 10.1073/pnas.222561399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson C.P., Solaiman F. Virosomes: cationic liposomes enhance retroviral transduction. Nat. Biotechnol. 1996;14:339–343. doi: 10.1038/nbt0396-339. [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M., Ogonuki N., Inoue K., Miki H., Ogura A., Toyokuni S., Shinohara T. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol. Reprod. 2003;69:612–616. doi: 10.1095/biolreprod.103.017012. [DOI] [PubMed] [Google Scholar]
- Kim B.G., Cho C.M., Lee Y.A., Kim B.J., Kim K.J., Kim Y.H., Min K.S., Kim C.G., Ryu B.Y. Enrichment of testicular gonocytes and genetic modification using lentiviral transduction in pigs. Biol. Reprod. 2010;82:1162–1169. doi: 10.1095/biolreprod.109.079558. [DOI] [PubMed] [Google Scholar]
- Kubota H., Avarbock M.R., Brinster R.L. Spermatogonial stem cells share some, but not all, phenotypic and functional characteristics with other stem cells. Proc. Natl. Acad. Sci. USA. 2003;100:6487–6492. doi: 10.1073/pnas.0631767100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota H., Avarbock M.R., Brinster R.L. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. USA. 2004;101:16489–16494. doi: 10.1073/pnas.0407063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meistrich M., van Beek E.A.B. Cell and molecular biology of the testis. New York, USA: Oxford Univ. Press; 1993. pp. 266–295. [Google Scholar]
- Nagano M., Watson D.J., Ryu B.Y., Wolf J.H., Brinster R.L. Lentiviral vector transduction of male germ line stem cells in mice. FEBS Lett. 2002;524:111–115. doi: 10.1016/s0014-5793(02)03010-7. [DOI] [PubMed] [Google Scholar]
- Nicolau C., Legrand A., Grosse E. Liposomes as carriers for in vivo gene transfer and expression. J. Biol. Chem. 1987;261:16722–16726. doi: 10.1016/0076-6879(87)49054-x. [DOI] [PubMed] [Google Scholar]
- Russell L.D., Ettlin R.A., Hikim A.P., Clegg E.D. Histological and Histopathological Evaluation of the Testis. Clearwater FL, USA: Cache River Press; 1990. pp. 1–40. [Google Scholar]
- Ryu B.Y., Orwig K.E., Avabock M.R., Brinster R.L. Stem cell and niche development in the postnatal rat testis. Dev. Biol. 2003;263:253–263. doi: 10.1016/j.ydbio.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Ryu B.Y., Orwig K.E., Kubota H., Avarbock M.R., Brinster R.L. Phenotypic and functional characteristics of spermatogonial stem cells in rats. Dev. Biol. 2004;274:158–170. doi: 10.1016/j.ydbio.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Ryu B.Y., Kubota H., Avarbock M.R., Brinster R.L. Conservation of spermatogonial stem cell self-renewal signaling between mouse and rat. Proc. Natl. Acad. Sci. USA. 2005;102:14302–14307. doi: 10.1073/pnas.0506970102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu B.Y., Orwig K.E., Oatley J.M., Lin C.C., Chang L.J., Avarbock M.R., Brinster R.L. Efficient generation of transgenic rats through the male germline using lentiviral transduction and transplantation of spermatogonial stem cells. J. Androl. 2007;28:353–360. doi: 10.2164/jandrol.106.001511. [DOI] [PubMed] [Google Scholar]
- Sandrin V., Boson B., Salmon P., Gay W., Negre D., Le Grand R., Trono D., Cosset F.L. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood. 2002;100:823–832. doi: 10.1182/blood-2001-11-0042. [DOI] [PubMed] [Google Scholar]
- Shinohara T., Orwig K.E., Avarbock M.R., Brinster R.L. Spermatogonial stem cell enrichment by multiparameter selection of mouse testis cells. Proc. Natl. Acad. Sci. USA. 2000;97:8346–8351. doi: 10.1073/pnas.97.15.8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara T., Orwig K.E., Avarbock M.R., Brinster R.L. Remodeling of the postnatal mouse testis is accompanied by dramatic changes in stem cell number and niche accessibility. Proc. Natl. Acad. Sci. USA. 2001;98:6186–6191. doi: 10.1073/pnas.111158198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D., Rigby P.W. The use of histone as a facilitator to improve the efficiency of retroviral gene transfer. Nucleic Acids Res. 1996;24:3113–3114. doi: 10.1093/nar/24.15.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.G., Walzem R.L., German J.B. Liposomes as agents of DNA transfer. Biochem. Biophys. Acta. 1993;1154:327–340. doi: 10.1016/0304-4157(93)90004-8. [DOI] [PubMed] [Google Scholar]
- Themis M., Forbes S.J., Chan L., Cooper R.G., Etheridge C.J., Miller A.D., Hodgson H.J.F., Coutelle C. Enhanced in vitro and in vivo gene delivery using cationic agent complexed retrovirus vectors. Gene Ther. 1998;5:1180–1186. doi: 10.1038/sj.gt.3300715. [DOI] [PubMed] [Google Scholar]
- Watson D.J., Wolfe J.H. Viral vectors for gene therapy: methods and protocols. Totowa, NJ, USA: Humana Press; 2003. pp. 383–404. [Google Scholar]