

Abstract
Hyaluronic acid (HA) has been shown to promote angiogenesis. However, the mechanism behind this effect remains largely unknown. Therefore, in this study, the mechanism of HA-induced angiogenesis was examined. CD44 and PKCδ were shown to be necessary for induction of the receptor for HA-mediated cell motility (RHAMM), a HA-binding protein. RHAMM was necessary for HA-promoted cellular invasion and endothelial cell tube formation. Cytokine arrays showed that HA induced the expression of plasminogen activator-inhibitor-1 (PAI), a downstream target of TGFβ receptor signaling. The induction of PAI-1 was dependent on CD44 and PKCδ. HA also induced an interaction between RHAMM and TGFβ receptor I, and induction of PAI-1 was dependent on RHAMM and TGFβ receptor I. Histone deacetylase 3 (HDAC3), which is decreased by HA via rac1, reduced induction of plasminogen activator inhibitor-1 (PAI-1) by HA. ERK, which interacts with RHAMM, was necessary for induction of PAI-1 by HA. Snail, a downstream target of TGFβ signaling, was also necessary for induction of PAI-1. The down regulation of PAI-1 prevented HA from enhancing endothelial cell tube formation and from inducing expression of angiogenic factors, such as ICAM-1, VCAM-1 and MMP-2. HDAC3 also exerted reduced expression of MMP-2. In this study, we provide a novel mechanism of HA-promoted angiogenesis, which involved RHAMM-TGFβRI signaling necessary for induction of PAI-1.
Keywords: CD44, hyaluronic acid, PAI-1, RHAMM, TGFβ signaling
INTRODUCTION
Hyaluronic acid (HA) is involved in various cellular processes, such as cellular invasion (Bourguignon et al., 2007; Golshani et al., 2008; Kim et al., 2008), tissue regeneration (Contreras et al., 2009) and angiogenesis (Savani et al., 2001). HA suppresses NGF-induced cell differentiation through RHAMM (Washio et al., 2009). The CD44-EGFR interaction is necessary for HA-promoted cancer cell motility (Kim et al., 2008). Additionally, the HA interaction with CD44 activates Rac1 signaling and increases the production of reactive oxygen species (Bourguignon et al., 2007; Kim et al., 2008). HA also promotes angiogenesis through recruitment of stromal cells (Koyama et al., 2007). HA employs CD44 and RHAMM to induce angiogenesis (Golshani et al., 2008). RHAMM compensates for the loss of CD44 in inflamed CD44 knock out mice (Nedvetzki et al., 2004) and is necessary for basic FGF-induced neovascularization in mice (Savani et al., 2001).
RHAMM, which is a HA-binding protein, regulates ras signaling and is responsible for breast cancer progression (Wang et al., 1998). RHAMM also interacts with ERK1/2 to sustain basal motility (Hamilton et al., 2007) and RHAMM is necessary for CD44-mediated skin wound healing (Tolg et al., 2006). RHAMM is also a tumor antigen that shows expression in a cycle-specific manner and is suppressed by p53 (Sohr and Engeland, 2008). There have not been extensive studies aimed at understanding the role of RHAMM in HA-promoted angiogenesis in relation with CD44.
Histone deacetylase 3 (HDAC3) plays an important role in transcriptional repression of various genes (Evert et al., 2006; Jayne et al., 2006; Villagra et al., 2007). P300, in cooperation with HDAC3, represses c-myc expression (Sankar et al., 2007). HDAC3 also acts as a corepressor of NF-κB and negatively regulates HIF-1-alpha in breast carcinoma cells (Bendinelli et al., 2009). Down regulation of HDAC3 has been shown to lead to activation of STAT3 (Togi et al., 2009). However, the role of HDAC3 in angiogenesis has not been studied. Given the fact that HDAC3 acts as a negative regulator of HIF-1-alpha, it is reasonable that HDAC3 may act as a negative regulator of angiogenesis.
In this study, we hypothesized that multiple signaling pathways were involved in HA-promoted angiogenesis. We describe the role for RHAMM in HA-promoted angiogenesis and the mechanism of induction of RHAMM by HA. We also provide evidence of the involvement of TGFβ receptor in HA-promoted angiogenesis. Here, we identified a novel role of PAI-1, a downstream target of TGFβ receptor, in HA-promoted angio genesis and the mechanism of PAI-1 induction in relation with epigenetic factors, such as HDAC3. The mechanism of down regulation of HDAC3 by HA is provided. The combined results of this study show that multiple signaling pathways converge to mediate HA-promoted angiogenesis. We also identified novel targets of HA-prompted angiogenesis.
MATERIALS AND METHODS
Cell lines and culture
HUVECs were isolated from human umbilical cord veins by collagenase treatment and used in passages 3–6. The cells were grown in M199 medium supplemented with 20% fetal bovine serum, 100 U/ml penicillin G, 100 μg/ml streptomycin, 3 ng/ml bFGF (Upstate, USA), and 5 U/ml heparin at 37°C under 5% CO2/95% air.
To ensure that the hyaluronic acid used in this study was free of contaminants, cells were treated with hyaluronic acid in the presence or absence of hyaluronidase (30 U/ml, Sigma). Hyaluronidase was activated by preincubation at 37°C for 30 min and was inactivated by preincubation at 65°C for 30 min.
Hyaluronic acid
Each size of hyaluronic acid (HA) was purchased from CHABio & Diostech (Korea). In this study, we used HA of 6 kDa (Lot No.HSB-ULHA-10-001), 100 kDa (Lot No. HSB-LHA-09-001) and 1 mDa (Lot No. HSB-HHA-09-003). We also purchased HA of 1 mDa from Sigma Chemical Company.
Intravial microscope angiogenesis assays
Male BALB/c mice (6–8 week old) were obtained from Daehan Biolink (Korea). The mice were maintained at the specific pathogen-free housing facility at the School of Medicine, Kangwon National University. In vivo angiogenesis was assessed as follows. The mice were anesthetized with 2.5% avertin (v/v) via intraperitoneal injection (Surgivet, USA), and abdominal wall windows were implanted. Next, a titanium circular mount with eight holes on the edge was inserted between the skin and the abdominal wall. Growth factor-reduced Matrigel containing VEGF (100 ng/ml) or HA (1 mg/ml) was applied to the space between the windows, and a circular glass cover slip was placed on top and fixed with a snap ring. After four days, the animals were anesthetized and injected intravenously with 50 μl of 25 ng/ml fluorescein isothiocyanate-labeled dextran (molecular weight, Mr ∼2,000,000) via the tail vein. The mice were then placed on a Zeiss Axiovert 200 M microscope. The epi-illumination microscopy setup included a 100-W mercury lamp and filter set for blue light. Fluorescence images were recorded at random locations of each window using an electron-multiplying charge-coupled device camera (Photo Max 512, Princeton Instruments, USA) and digitalized for subsequent analysis using the Metamorph program (Universal Imaging, USA). The assay was scored from 0 (negative) to 5 (most positive) in a double-blinded manner.
Histological analyses
Eight-week-old Male BALB/c mice were anesthetized, and PBS, VEGF (100 ng/ml) or HA (200 μg/ml) was injected into their ears. After 3–5 days, ear samples were fixed in 10% (v/v) buffered formalin, embedded in paraffin, sectioned at 4 μm, and then stained with hematoxylin and eosin.
Endothelial cell tube formation assay
Growth factor-reduced Matrigel was pipetted into prechilled 24-well plates (200 μl matrigel per well) and polymerized for 30 min at 37°C. HUVECs were first incubated in M199 containing 1% FBS for 6 h and then treated with various concentrations of inhibitor for 60 min before seeding. The HUVECs were collected and placed onto the layer of Matrigel in 1 ml of M199 containing 1% FBS followed by the addition of 200 μg/ml of HA. After 6 to 8 h of incubation at 37°C in a 95%:5% (v/v) mixture of air and CO2, the endothelial cells were photographed using an inverted microscope (magnification, X100; Olympus). All experiments were conducted in triplicate. To determine the effect of RHAMM or MMP-2 on endothelial cell tube formation, HUVECs were transfected with control SiRNA (10 nM), RHAMM SiRNA (10 nM) or MMP-2 SiRNA (10 nM). At 48 h after transfection, cells were treated with or without HA (200 μg/ml). Endothelial cell tube formation assays were then performed. To determine the effect of CD44 on endothelial cell tube formation, HUVECs were pre-incubated with IgG (5 μg/ml) or human CD44-blocking antibody (A3D8, 5 μg/ml) for 3 h, prior to HA treatment.
Tube formation was observed using an inverted phase contrast microscope. Images were captured with a video graphic system. The degree of tube formation was quantified by measuring the length of tubes in five randomly chosen low-power fields (×100) from each well using the Image-Pro plus v4.5 (Media Cybernetics, USA).
Transwell migration assay
The chemotactic motility of HUVECs was determined using a Transwell migration assay (BD Biosciences) with 6.5-mm-diameter polycarbonate filters (8-μm pore size). Briefly, the filter of the Transwell plate was coated with 0.1% gelatin, after which, the bottom chambers were filled with 500 μl of M199 containing 1% FBS supplemented with 20 ng/ml VEGF. HUVECs (4 × 104) suspended in 100 μl of M199 containing 0.5% FBS plus HA (200 μg/ml) were seeded in the upper chambers. The cells were allowed to migrate for 20 to 24 h, after which, non-migrated cells were removed with cotton swabs, and the migrated cells were fixed with cold 4% paraformaldehyde and stained with 1% crystal violet. Images were taken using an inverted microscope (Olympus), and migrated cells were quantified by manual counting.
Flow cytometry
To detect RHAMM on cell surface, the trypsinized HUVECs (1 × 106) were harvested, washed with PBS containing 5% BSA and then fixed in PBS containing 3% formaldehyde. After removal of fixing solution, cells were incubated with anti-RHAMM antibody (1:100, Santa Cruz) for 1 h. The cells were washed with PBS and incubated with Alexa Fluor 488-conjugated secondary antibody (1:1000, Santa Cruz) for 30 min. After incubation, cells were washed and resuspended in PBS, and then analyzed by flow cytometry (FACS Scan, BD Biosciences).
Immunofluorescence staining
HUVECs were seeded on 10-mm cover slips at a density of 2 × 105 cells/35-mm plate. After treatment with or without HA for 1 h, cells were treated as indicated and then fixed as described previously (Urbich et al., 2009). Cover slips were incubated with a primary antibody to pERK (Santa Cruz: 1/500 dilution) or RHAMM (Santa Cruz: 1/500 dilution) at 4°C for 24 h. After washing with PBS, slides were incubated with Alexa Fluor 488 (for RHAMM) or Alexa Fluor 568 (for pERK)-conjugated secondary antibody for 1 h. After washing and mounting, fluorescence staining was visualized using confocal microscopy.
Whole mount staining
Mouse ears were fixed in 4% (vol/vol) paraformaldehyde and blocked with TNB buffer (NEN Life Science Products) containing 0.3% Triton X-100. Primary antibodies, rat anti-Receptor for Hyaluronan-Mediated Motility ([RHAMM]; Santa Cruz biotechnology), rabbit anti-CD31 or anti-mouse smooth muscle actin (Sigma) were diluted in TNB buffer, and ear samples were incubated with primary antibodies overnight at 4°C. The samples were then washed three times and subsequently incubated with the secondary antibodies anti-rat IgG Alex 546, anti-rabbit IgG Alex 488, or anti-mouse IgG Alex 488 (Molecular Probes). To determine effect of HDAC3 on angiogenesis, ears of BALB/c mice were given injection of control siRNA (100 nM) or HDAC3 siRNA (100 nM) twice in a total of eight days. Mouse ears were fixed in 4% (v/v) paraformaldehyde and blocked with TNB buffer. Ear samples were incubated with rabbit anti-CD31 (PECAM-1) overnight at 4°C. The samples were then washed three times and subsequently incubated with the secondary antibody, anti-rabbit IgG Alex 488 (Molecular probes).
Western blot analysis
For PAGE and Western blot, cell lysates were prepared using lysis buffer [62.5 mM Tris-HCl, pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 50 mM dithiothreitol, 0.01% (w/v) bromphenol blue, 10 mM NaF, 1% (v/v) protease inhibitor mixture, 1 mM sodium orthovanadate]. The samples were then boiled for 5 min, after which equal amounts of protein (20 μg/well) were analyzed by 10% SDS-PAGE. Following electrophoresis, proteins were transferred onto a nitrocellulose membrane and subjected to immunoblotting. The antibodies and dilutions included anti-RHAMM (1:1,000) and anti-GAPDH (1:2,000). After extensive washing, the blots were further incubated with an anti-mouse or anti-rabbit IgG-horseradish peroxidase-conjugated antibody at a 1:3,000 dilution for 1 h and then developed using an enhanced chemiluminescence kit (Amersham Biosciences).
Rac1 activity assay
Cells were grown in 100-mm dishes and treated with HA (200 ng/ml) for the indicated times. The cells were then quickly rinsed in phosphate-buffered saline and lysed using 500 μl of lysis buffer (50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mM NaCl, 10 mM MgCl2) plus protease inhibitors. Next, the cells were scraped and the cell lysates were centrifuged for 5 min at 14,000 rpm. To determine the levels of Rac1-GTP, 400 μl of each cell lysate was incubated with PAK (CRIB)-GST beads for 1 h with rocking at 4°C. The samples were then centrifuged for 2 min at 2000 rpm, after which the supernatant was discarded. The beads were then washed three times with wash buffer (50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 150 mM NaCl, 10 mM MgCl2). Finally, 50 μl of 2× Laemmli buffer was added to the beads, and the mixture was boiled for 10 min. The levels of active Rac1, Rac1-GTP, were detected by Western blotting using specific monoclonal anti-Rac1 antibody (BD Transduction Laboratories). Approximately 10 μg of total lysate were analyzed to examine the levels of total Rac1 or GAPDH (Sigma).
Reactive oxygen species (ROS) measurement
Serum-starved HUVECs on cover slips were treated with or without various concentrations of HA (200 μg/ml) for 20 min. DCFH-DA (5 μM) was then added for 10 min to measure the production of hydrogen peroxide. Cells were then immediately observed by the use of laser scanning instead of DCFH-DA. ROS measurements were performed as described.
Cellular fractionation
Nuclear and cytosolic extracts were prepared with a nuclear/cytosol fractionation kit (Biovision, USA). Cells were collected by centrifugation at 600 × g for 5 min at 4°C. Cell pellets were washed twice with ice-cold PBS, followed by the addition of 0.2 ml of Cytosol Extraction Buffer A and vigorous mixing for 5 s. Ice-cold Cytosol Extraction Buffer B (11 μl) was then added to the solution. After mixing, the nuclear and cytosolic fractions were separated by centrifugation at 16,000 × g for 5 min (supernatants were the cytosolic fraction). Nuclear extraction buffer was then added to the nuclei. After vortexing for a total of 40 min, the nuclei were centrifuged at 16,000 × g for 10 min. The supernatants obtained were the nuclear fraction. The protein concentration of each fraction was determined using a DC Protein Assay Kit (Bio-Rad). Equal amounts of nuclear/cytosolic extracts were loaded for SDS-PAGE, after which Western blot analysis was performed. The purity of the cytosolic and nuclear fraction was confirmed by GAPDH and histone H1, respectively. For fractionation of the cytosol and membrane, cells were resuspended in a buffer containing 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 10 μg/ml leupeptin and then lysed by sonication. The lysates were then centrifuged at 100,000 × g for 1 h at 4°C. The supernatants constitute the cytosolic fraction. The pellet was resuspended in the above buffer, which also contained 0.1% Triton X-100, and the mixture was lysed by sonication and centrifuged again at 100,000 × g for 1 h at 4°C to obtain the membrane fraction (supernatant). The purity of the cytosol and membrane fraction was confirmed by GAPDH and EGFR, respectively.
ChIP assays
Assays were performed according to the manufacturer’s instruction (Upstate). Briefly, 25% of the chromatin solution was reserved for to Total input. The remaining solution was pre-cleared with protein A-agarose, subsequently incubated with respective antibody (2 μg/ml each) for 12 h at 4°C with shaking, and then further incubated with protein A-sepharose for 2 h. The immunoprecipitates were reverse cross-linked. PCR was performed on the phenol-chloroform-extracted DNA. For detection of c-fos binding to RHAMM promoter sequences, specific primers of RHAMM promoter [5′-TGCGCAAATGCTAGGT TACA-3′ (sense) and 5′-ATTAGCTGGCCTGCGAGTTT-3′ (antisense)] were used. For detection of Snail binding to PAI-1 promoter sequences, specific primers of PAI-1 promoter [5′-CCTTCACCAGCCCTCTTTCCATTGC-3′ (sense) and 5′-ACC GAGGCCACCCCATAGGGT-3′ (antisense)] were used. For detection of HDAC3 binding to PAI-1 promoter sequences, specific primers of PAI-1 promoter [5′-CGGGCTTAAGGCAG AGAACT-3′ (sense) and 5′-TTTTTGGGAGAGCAGGGTTT-3′ (antisense)] were used.
For detection of HDAC3, c-jun, histone H3-AcK9/14 or H4-AcK16 binding to MMP-2 promoter sequences, specific primers of MMPI-2 promoter [5′-TGTGACAACCGTCTCTGA-3′ (sense) and 5′-ATCTCTGGGCCATTGTCA-3′ (antisense)] were used.
Preparation of SiRNA duplexes and transfection
The SiRNA duplexes were constructed with the following target sequences HDAC3, sense (5′-AAGGCTTCACCAAGAGTCT TACCTGTCTC-3′); antisense (5′-AATAAGACTCTTGGTGAA GCCCCTGTCTC-3′); RHAMM, sense (5′-AATGACCCTTCT GGTTGTGCACCTGTCTC-3′); antisense (5′-AATGCACAAC CAGAAGGGTCACCTGTCTC-3′); c-fos, sense (5′-AAC AGACTT CTC ATC TTC TAG CCT GTC TC-3′); antisense (5′-AAC TAG AAG ATG AGA AGT CTG CCT GTC TC-3′); DNMT1, sense (5′-AATTTTCCCTTGCCCTTCCCTCCTGTCTC-3′); antisense (5′-AAAGGGAAGGGCAAGGGAAAACCTGTCTC-3′); PAI-1, sense (5′-CCTTCACCAGCCCTCTTTCCATTGC-3′); antisense (5′-ACCGAGGCCACCCCATAGGGT-3′); MMP-2, sense (AAATAGTCCTGAATGCCCCCTGTCTC); antisense (AAGG GCATTC AGGAGCTCTATCCTGTCTC); control, sense (5′-AATTCTCCGAACGTGTCACGTCCTGTCTC-3′); antisense (5′-AAACGTGACACGTTCGGAGAACCTGTCTC-3′). The construction of SiRNAs was performed according to the instruction manual provided by the manufacturer (Ambion, USA). The transfection of the SiRNA construct was performed by using lipofectamine 2000 (Invitrogen). Transfection of the plasmids was conducted using Lipofectamine Plus reagent and Lipofectamine reagent (Invitrogen).
Immunoprecipitation
Cells (1 × 107) were lysed in immunoprecipitation buffer [50 mmol/ml HEPES (pH 7.6), 150 mmol/ml NaCl, 5 mmol/ml EDTA, 0.1% NP40]. After centrifugation (10 min at 15,000 × g) to remove the particulate material, the supernatant was incubated with each antibody (2 μg/ml) with constant agitation at 4°C. The immunocomplexes were then precipitated with protein A/G Sepharose (Sigma) and analyzed by Western blot analysis.
Gelatin zymography
Gelatin zymogrpahy was performed according to the standard procedures (2).
Human angiogenesis antibody array
Expression levels of angiogenic factors were determined by using a Proteom Profiler™ Human Angiogenesis Antibody Array Kit (R&D Systems, USA) according to the manufacturer’s instructions. Briefly, HUVECs were treated with or without HA (200 μg/ml) for 1 h, after which the cells were then solubilized (1 × 107 cells/ml) in lysis buffer. Each membrane was incubated with blocking buffer, followed by treatment with cell lysates (50 μg/ml) and antibody cocktail for 12–14 h at 4°C on a rocking platform. The membrane was then washed and incubated with horseradish peroxidase-conjugated streptavidin, and the protein spots were detected using the ECL Western blotting detection reagents.
In vivo matrigel plug assay
Matrigel plug assay was performed as previously described (Min et al., 2007). Briefly, 7-week-old BALB/C mice (DBL Co., Ltd, Korea) were injected subcutaneously with 0.5ml of Matrigel containing PBS, HA (200 μg), VEGF (50 ng), TSA (50 nM, 250 nM) or SB (2 mM, 10 mM) and 10 units of heparin (Sigma). The injected Matrigel rapidly formed a single, solid gel plug. After 8 days, the skin of the mouse was easily pulled back to expose the Matrigel plug, which remained intact.
RESULTS
Hyaluronic acid induces angiogenic response independent of VEGF
We wanted to investigate effect of HA on angiogenesis and the mechanisms behind this effect. Intravital microscopy showed that HA induced angiogenesis (Fig. 1A). The 1 mDa HA, but not 6 kDa or 100 kDa HA, induced angiogenesis (Fig. 1A). Each HA at equimolar concentration was used. HA (1 mDa) induced angiogenesis in a dose-dependent manner, based on intravtital microscopy (data not shown). Hyaluronidase prevented HA (1 mDa) from inducing endothelial cell tube formation in HUVECs (Fig. 1B, left panel). The 6 kDa HA and 100 kDa HA, when used at higher concentrations, also enhanced endothelial cell tube formation (data not shown). In this study, we found that HA (1 mDa) purchased from Sigma Chemical Company also enhanced endothelial cell tube formation, which was comparable to the effects by our 1 mDa HA (data not shown). This further confirmed that high molecular weight form of HA was as good as low molecular weight form of HA in terms of inducing angiogenesis. Quantitation of the tube length showed the specific effect of HA on endothelial cell tube formation (Fig. 1B, right panel). We examined whether HA-induced angiogenesis required VEGF. To accomplish this, AVASTIN, a VEGF-neutralizing antibody, was added along with HA to matrigel bloc and blood vessel formation was evaluated by intravital microscopy. HA promoted angiogenesis regardless of AVASTIN (Fig. 1C), suggesting that HA promotes angiogenesis in a different manner than VEGF. Injection of HA into the ears of BALB/c mouse induced blood vessel formation based on H&E staining (Fig. 1D, left panel). Whole mount staining of ear tissue of BALB/c mouse injected with HA showed increased expression of RHAMM (Fig. 1D, right panel). However, VEGF did not induce expression of RHAMM, a HA-binding protein (Fig. 1D, right panel). Both HA and VEGF induced expression of CD31, an angiogenic marker protein (Fig. 1D, middle panel). Flow cytometry analysis showed increased cell surface expression of RHAMM by HA in HUVECs (Fig. 1E). These results showed the VEGF-independent angiogenic potential of HA and the possible involvement of RHAMM in HA-promoted angiogenesis.
Fig. 1.

HA promotes angiogenesis independent of VEGF. (A) Intravital microscopy analysis was conducted as described. An equimolar concentration of each size of HA was mixed with matrigel. Four days later, FITC-dextran was injected via tail vein to visualize blood vessel formation. Each figure shows a representative example of three independent experiments. (B) Hyaluronidase (30 U/ml) exerts a negative effect on endothelial cell tube formation induced by HA (1 mDa). Hyaluronidase (30 U/ml) was activated by preincubation at 37°C for 30 min and was inactivated by pre-incubation at 65°C for 30 min. **P < 0.005; ***P < 0.0005. Each value represents the average of three independent experiments. NS denotes not significant. Endothelial cell tube formation assays were performed 12 h after addition. (C) Intravital microscopy shows that HA promotes angiogenesis independent of VEGF. AVASTIN (4 μg/ml), a VEGF neutralizing antibody, was added with or without HA (1 mg/ml). Each figure shows a representative example of three independent experiments. (D) H&E staining of ear tissue of BALB/c mouse that received HA (200 μg/ml) or VEGF (100 ng/ml) was performed (left panel). Arrows denote blood vessels. Whole mount staining of ear tissue of BALB/c mouse that received HA (200 μg/ml) or VEGF (100 g/ml) was performed by using anti-CD31 antibody (middle panel) or anti-RHAMM antibody (right panel). Each figure shows a representative example of three independent experiments. (E) HUVECs were treated with or without HA (1 MDa, 200 μg/ml) for 1 h. Flow cytometry analysis using anti-RHAMM antibody was performed as described.
CD44-PKCδ is responsible for induction of RHAMM by HA
HA promotes tumor cell motility by PKC signaling (Washio et al., 2009). Therefore we examined the role of PKC signaling in HA-promoted angiogenesis. HA increased phosphorylation of PKCδ, but not PKCα (Fig. 2A). HA also induced an interaction between CD44 and PKCδ (Fig. 2A). Inactivation of PKCδ by the dominant negative construct prevented HA from increasing expression of RHAMM, a HA-binding protein (Fig. 2B). PKCδ was necessary for activation of rac1 and the increased expression of c-fos (Fig. 2B). This suggests that AP-1 may be involved n the induction of RHAMM by HA. This also suggests that rac1 is involved in HA-promoted angiogenesis. The down regulation of RHAMM did not prevent HA from increasing phosphorylation of PKCδ (Fig. 2C), which suggests that PKCδ acts upstream of RHAMM to promote angiogenesis.
Fig. 2.

HA induces expression of RHAMM, a HA-binding protein, by PKCδ. (A) HUVECs were treated with or without HA (1 mDa, 200 μg/ml) for various time intervals. Cell lysates were immunoprecipitated with anti-CD44 antibody (2 μg/ml), followed by Western blot analysis. (B) HUVECs were transiently transfected with control vector (1 μg) or dominant negative PKCδ (1 μg). At 48 h after transfection, cell lysates were prepared and subjected to Western blot analysis. (C) HUVECs were transfected with control SiRNA (10 nM) or RHAMM SiRNA (10 nM). At 48 h after transfection, cell lysates were prepared and subjected to Western blot analysis. (D) HUVECs were treated with or without various sizes of HA at an equi-molar concentration for various time intervals (upper panel). HUVECs were treated with or without each size of HA for 1 h at various concentrations (lower panel). Cell lysates were then subjected to Western blot analysis.
We examined whether 6 kDa or 100 kDa HA would induce expression of RHAMM. When used at equimolar concentration, 1 mDa HA, but not 6 kDa or 100 kDa induced expression of RHAMM (Fig. 2D, upper panel). However, 6 kDa and 100 kDa HA induced expression of RHAMM and activation of PKCδ, when used at higher concentrations (Fig. 2D, lower panel). These results suggest that CD44-PKCδ interaction by HA induces expression of RHAMM to promote angiogenesis.
RHAMM, induced by AP-1, mediates HA-promoted angiogenesis
Because HA induced the expression of RHAMM (Fig. 2B), we examined the mechanism of induction of RHAMM. HA induced the expression of c-fos and c-jun, but not SP-1, and increased phosphorylation of JNK (Fig. 3A). A ChIP assay showed the binding of c-fos and c-jun to RHAMM promoter sequences (Fig. 3B), suggesting that AP-1 acts as a regulator of RHAMM expression. SP-1 did not show binding to RHAMM promoter sequences, as expected (data not shown). The down regulation of c-fos prevented HA from inducing the expression of RHAMM (Fig. 3C). The down regulation of RHAMM exerted negative effects on HA-promoted endothelial cell tube formation (Fig. 3D) and invasion potential of HUVEC (Fig. 3E). The blocking of CD44 prevented HA from inducing endothelial cell tube formation (Fig. 3F, upper panel) and prevented HA from enhancing the invasion potential of HUVECs (Fig. 3F, lower panel). Our results indicate that both CD44 and RHAMM are required for HA to enhance invasion potential and endothelial cell tube formation.
Fig. 3.

RHAMM, induced by HA via AP-1, and CD44 mediate HA-promoted angiogenesis. (A) HUVECs were treated with or without HA (1 mDa, 200 μg/ml) for various time intervals. Western blot analysis was performed. (B) HUVECs were treated with or without HA (1 mDa, 200 μg/ml) for 1 h and ChIP assays using anti-IgG (2 μg/ml), anti-c-jun (2 μg/ml) or anti-c-fos antibody (2 μg/ml) was performed. (C) HUVECs were transfected with control SiRNA (10 nM) or c-fos SiRNA (10 nM). At 48 h after transfection, cell lysates were prepared and subjected to Western blot analysis. (D) HUVECs were transfected with control SiRNA (10 nM) or RHAMM SiRNA (10 nM). At 48 h after transfection, HUVECs were then treated with or without HA (1 mDa, 200 μg/ml). Endothelial cell tube formation assays were performed as described. Means ± SEM of three independent experiments are depicted. ***p < 0.0005. (E) is the same as (D) except that cellular invasion assays were performed as described. The trypsinized HUVECs (2 × 104 cells) were subjected to transwell migration assays as described. Means ± SEM of three independent experiments are depicted. ***p < 0.0005. (F) HUVECs were pre-incubated with IgG antibody (5 μg/ml) or CD44-blocking antibody (A3D8, 5 μg/ml) for 3 h, followed by treatment with or without HA (200 μg/ml). Endothelial cell tube formation assays were performed 12 h after addition of HA (left panel). Effect of blocking of CD44 on the invasion potential of HUVECs was also determined as described (right panel). Means ± SEM of three independent experiments are depicted. **p < 0.005.
HA induces expression of plasminogen activator inhibitor-1 (PAI-1) to promote angiogenesis
In an effort to understand the mechanism of HA-promoted angiogenesis, we attempted to identify angiogenic factor(s) regulated by HA. Cytokine array analysis showed that HA increased expression of plasminogen activator inhibitor-1 (PAI-1), a TGFβ-responsive gene, among various angiogenic factors (Fig. 4A). However, HA did not induce expression of TGFβ (Fig. 4A). This suggests that induction of PAI-1 may occur via transactivation of TGFβ receptor. HA did not affect the expression of VEGF or MMP-9 (Fig. 4A), which confirmed that HA did not require VEGF to promote angiogenesis. PAI-1 protects endothelial cells from Fas-mediated apoptosis (Bajou et al., 2008), and facilitates retinal angiogenesis (Basu et al., 2009). These reports indicate the potential role of PAI-1 in angiogenesis. Blocking of CD44 using a blocking antibody (A3D8) prevented HA from increasing expression of PAI-1 and RHAMM (Fig. 4B, left panel). A3D8 prevented the interaction between CD44 and PKCδ, and prevented HA from increasing phosphorylation of PKCδ (Fig. 4B, right panel). These results indicate that the induction of PAI-1 by HA occurs via the CD44/PKCδ interaction. Western blot analysis showed that PAI-1 was induced by HA (Fig. 4C). The decreased expression of HDAC3 and induction of Snail and HDAC2 occurred earlier than the induction of PAI-1 by HA (Fig. 4C). This suggests that expression of PAI-1 may be under the control of HDAC2, HDAC3 and Snail. Snail is required for TGFβ-induced epithelial-mesenchymal transition (Kokudo et al., 2008), and functional blockade of Snail decreases the expression of PAI-1 in breast cancer cells and exerts a negative effect on cancer cell migration (Fabre-Guillevin et al., 2008). PAI-1 is induced by TGFβ (Liu et al., 2010) and TGF-β1 utilizes RHAMM to promote cell motility (Samuel et al., 1993). Histone deacetylase (HDAC), such as HDAC5, acts as a negative regulator of angiogenesis (Urbich et al., 2009). Phopshorylation of HDAC5 mediates VEGF-promoted angiogenesis (Ha et al., 2008). In our study, HA did not increase or induce phosphorylation of HDAC5 (data not shown). HDAC3 is known to interact with HDAC5 (Fischle et al., 2002). HDAC3 has been shonw to mediate TGF-β-induced expression in C3H10T1/2 fibroblasts (Barter et al., 2010). Because decreased expression of HDAC3 occurred earlier than induction of PAI-1, we hypothesized that HDAC3 might exert a negative regulation on PAI-1 expression. The down regulation of HDAC3 induced expression of PAI-1 (Fig. 4D), suggesting that HDAC3 may act as a negative regulator of PAI-1. Over expression of HDAC3 prevented HA from inducing expression of PAI-1 (Fig. 4E), indicating that HDAC3 plays a negative regulatory role in the induction of PAI-1 by HA. Over expression of HDAC3 did not prevent HA from increasing the phosphorylation of PKCδ (Fig. 4E), confirming that PKCδ acts upstream of HDAC3. The ChIP assay showed the binding of Snail and HDAC3 to PAI-1 promoter sequences (Fig. 4F), suggesting that HDAC3 and Snail regulate the expression of PAI-1. This also indicates that HA relieves transcriptional repression of PAI-1 exerted by HDAC3. The down regulation of PAI-1 prevented HA from enhancing endothelial cell tube formation (Fig. 4G). The down regulation of PAI-1 also prevented HA from inducing expression of angiogenic factors, such as ICAM-1, VCAM-1and MMP-2 (Fig. 4H). We examined whether HDAC3 could act as a negative regulator of angiogenesis. The down regulation HDAC3 enhanced endothelial cell tube formation (Supplementary Fig. 1A). The down regulation HDAC3 also enhanced expression of PAI-1 (Supplementary Fig. 1A). Over expression of HDAC3 prevented HA from enhancing expression of PAI-1 and exerted a negative effect on HA-promoted endothelial cell tube formation (Supplementary Fig. 1B). Over expression of HDAC3 did not affect expression of RHAMM (Supplementary Fig. 1B), suggesting that HDAC3 acts downstream of RHAMM. The down regulation of HDAC3 enhanced blood vessel formation based on whole mount staining (Supplementary Fig. 2A). VEGF enhanced blood vessel formation (Supplementary Fig. 2A). HDAC inhibitors, such as trichostatin A (TSA) and sodium butyrate (SB), did not enhance blood vessel formation (Supplementary Fig. 2A). Western blot of BALB/c mouse ear tissue lysates showed that TSA or SB did not affect expression of PAI-1 (Supplementary Fig. 2B). The down regulation of HDAC3 enhanced expression of PAI-1 (Supplementary Fig. 2B). The matrigel plug assay showed that HA and VEF displayed angiogenic potential while TSA or SB did not display angiogenic potential (Supplementary Fig. 2C). Taken together, these results suggest that HDAC3, which was decreased by HA, may act as a negative regulator of angiogenesis by regulating expression of PAI-1.
Fig. 4.

Induction of PAI-1, an angiogenic factor, by HA results from down regulation of HDAC3. (A) HUVECs were treated with or without HA (1 mDa, 200 μg/ml) for 1 h, after which the cell lysates were subjected to cytokine array analysis as described. (B) HUVECs were pre-incubated with IgG antibody (5 μg/ml) or CD44-blocking antibody (A3D8, 5 μg/ml) for 3 h, followed by treatment with or without HA (1 mDa, 200 μg/ml) for 1 h. Cell lysates were prepared and subjected to Western blot analysis (left panel). Cell lysates were also immunoprecipitated with anti-actin antibody (2 μg/ml) or anti-CD44 antibody (2 μg/ml), followed by Western blot analysis and cell lysates were also subjected to Western blot analysis (right panel). (C) HUVECs were treated with or without HA (1 mDa, 200 μg/ml) for various time intervals. Cell lysates prepared at each time point were subjected to Western blot analysis. (D) HUVECs were transfected with control SiRNA (10 nM) or HDAC3 SiRNA (10 nM). At 48 h after transfection, cell lysates were prepared and subjected to Western blot analysis. (E) HUVECs were transfected with control vector (1 μg) or HDAC3-Flag (1 μg). At 48 h after transfection, cells were treated with or without HA (1 mDa, 200 μg/ml) for 1 h, followed by Western blot analysis. (F) HUVECs were treated with or without HA (1 mDa, 200 μg/ml) for 1 h. ChIP assays were performed as described. (G) HUVECs were transfected with control SiRNA (10 nM) or PAI-1 SiRNA (10 nM). At 48 h after transfection, cells were treated with or without HA (1 mDa, 200 μg/ml). Endothelial cell tube formation assays were performed as described. (H) HUVECs were transfected with control SiRNA (10 nM) or various concentrations of PAI-1 SiRNA. The 48 h after transfection, cells were treated with or without HA (1 mDa, 200 μg/ml) for 1 h. Cell lysates were prepared and subjected to Western blot analysis. Numbers in parentheses represent concentrations of SiRNAs.
Rac1 is necessary for decreased expression of HDAC3 by HA
HA has been shown to promote cancer cell motility, which is accompanied by activation of PKC signaling involving rac1 (2). HA was shown to increase rac1 activity by PKCδ (Fig. 2B). Therefore, we examined whether rac1 is involved in HA-promoted angiogenesis. HA increased reactive oxygen species levels (Supplementary Fig. 3A). Cellular fractionation showed translocation of cytosolic rac1 into the membrane by HA (Supplementary Fig. 1B, upper panel). HA induced an interaction between RHAMM and rac1 in the membrane (Supplementary Fig. 3B, lower panel). Inactivation of rac1 by the dominant negative construct prevented HA from increasing reactive oxygen species levels (Supplementary Fig. 3C). We also investigated the mechanism behind the decreased expression of HDAC3 by HA. We examined whether the decreased expression of HDAC3 was related with posttranslational modification of HDAC3. HA decreased expression of HDAC3, and this decreased expression of HDAC3 was related with increased tyrosine nitration of HDAC3 (Supplementary Fig. 3D). Inactivation of rac1 by the dominant negative construct prevented HA from decreasing expression of HDAC3 and also prevented HA from increasing tyrosine nitration of HDAC3 (Supplementary Fig. 3E).
This result suggests that tyrosine nitration of HDAC3 may signal for degradation of HDAC3 by HA.
Inactivation of rac1 restored the interaction between HDAC3 and its co-repressor N-CoR (Supplementary Fig. 3E). Taken together, these results indicate that rac1 mediates HA-promoted angiogenesis by decreasing expression of HDAC3, which in turn increases expression of PAI-1 and promote angiogenesis.
MMP-2, induced by HA, mediates HA-promoted angiogenesis
CD44-EGFR interaction induced by HA increases expression of MMP-2 and was shown to promote cancer cell motility (Kim et al., 2008). The down regulation of PAI-1 prevents HA from increasing expression of MMP-2 (Fig. 4G). Therefore, we examined whether MMP-2 could mediate HA-promoted angiogenesis. PKCδ was found to be responsible for the induction of MMP-2 by HA (Supplementary Fig. 4A). Because HDAC3 acts as a transcriptional repressor of various genes, we hypothesized that HDAC3 could repress expression of MMP-2. The down regulation of HDAC3 induced the expression of MMP-2, as well as histone H3-AcK9/14 and histone H4-AcK16 (Supplementary Fig. 4B, upper panel). The down regulation of HDAC3 also increased secretion of MMP-2 (Supplementary Fig. 4B, lower panel). We examined the mechanism of induction of MMP-2 by HA. As expected, the ChIP assays revealed the binding of c-jun to MMP-2 promoter sequences (Supplementary Fig. 4C). The ChIP assays also showed the binding of histone H3-AcK9/14 and histone H4-AcK16 to MMP-2 promoter sequences (Supplementary Fig. 4C). Furthermore, MMP2 was necessary for HA-promoted endothelial cell tube formation (Supplementary Fig. 4D) and enhanced invasion potential of HUVECs (Supplementary Fig. 4E). The down regulation of MMP-2 decreased expression of PAI-1 (Supplementary Fig. 4F), suggesting that PAI-1 and MMP-2 are interdependent. The down regulation of MMP-2 did not influence phosphorylation of PKCδ (Supplementary Fig. 4F), confirming that PKCδ functions upstream of MMP-2.
HA employs TGFβRI-RHAMM interaction to induce PAI-1
Because HA induced both RHAMM (Fig. 4B) and PAI-1 (Fig. 4C), we examined the relationship between RHAMM and TGFβRI/II. HA increased phosohorylation of TGFβRI (Fig. 5A, upper panel) and also induced an interaction between RHAMM and TGFβRI (Fig. 5A, upper panel). This suggests that the RHAMM-TGFβRI interaction is necessary for induction of PAI-1 by HA. The soluble form of TGFβRII (TGFβRII-Fc chimera) prevented TGFβI from increasing phosphorylation of TGFβRI and Smad2 (Fig. 5A, lower panel). TGFβRII was also necessary for activation of TGFβRI by HA (Fig. 5A, lower panel). Activation of TGFβRI by TGFβ1 involved the interaction between TGFβRI and Smad2 (Fig. 5A, lower panel). However, activation of TGFβRI by HA did not involve the interaction between TGFβRI and Smad2 or induction of phospho-Smad2 (Fig. 5A, lower panel). HA did not affect expression of phospho-Smad 2 (Fig. 5A, lower panel), suggesting that activation of TGFβRI and thereby induction of PAI-1 by HA may occur in a Smad-independent manner.
Fig. 5.

PKCδ mediates the effect of HA on activation of TGFβ receptor signaling, and the induction of PAI-1 by HA occurs independently of Smad2. (A) HUVECs were treated with or without HA (1 mDa, 200 μg/ml) for various time intervals. Cell lysates were immunoprecipitated with anti-TGFβRI antibody (2 μg/ml), followed by Western blot analysis (upper panel). HUVECs were treated with HA (1 mDa, 200 μg/ml) or human recombinant TGFβ1 (Sigma, 50 ng/ml) for 1 h. To determine effect of TGFβRII on activation of TGFβRI, HUVECs were pre-incubated with soluble form of TGFβRII (TGFβRII-Fc chimera, 50 ng/ml) for 3 h, prior to incubation with HA (1 mDa, 200 μg/ml) or TGFβ1 (50 ng/ml). Cell lysates were prepared and immunoprecipitated with anti-actin antibody (2 μg/ml) or anti-TGFβRI antibody (2 μg/ml), followed by Western blot analysis (lower panel). Cell lysates prepared were also subjected to western blot analysis (lower panel). The rTGFβ denotes human recombinant TGFβ and sTGFβRII denotes the soluble form of TGFβRII. (B) HUVECs were pretreated with or without various concentrations of SB431542, an inhibitor of TGFβRI, for 30 min, followed by treatment with or without HA (1 mDa, 200 μg/ml) for 1 h. Cell lysates were then subjected to Western blot analysis (upper panel). HUVECs were transfected with control SiRNA (10 nM) or RHAMM SiRNA (10 nM). At 48 h after transfection, cells were pretreated with or without SB431542 (1 μM) for 30 min, followed by treatment with or without HA (1 mDa, 200 μg/ml) for 1 h. Cell lysates were prepared and immunoprecipitated with anti-TGFβRI antibody, followed by Western blot analysis (lower panel). C denotes control SiRNA and R denotes RHAMM SiRNA. (C) HUVECs were transiently transfecetd with control vector (1 μg) or dominant negative PKCδ (1 μg). At 48 h after transfection, cell lysates were immunoprecipitated with anti-actin antibody (2 μg/ml) or anti-TGFβRI antibody (2 μg/ml), followed by Western blot analysis using the indicated antibodies (upper panel). Cell lysates were also subjected to Western blot analysis (lower panel).
The chemical inhibition of TGFβ receptor by SB431542 prevented HA from inducing expression of RHAMM and PAI-1 (Fig. 5B, upper panel), indicating that HA activates the TGFβ receptor and induces PAI-1. SB431542 prevented HA from activating TGFβRI (Fig. 5B, upper panel). The down regulation of RHAMM by SiRNA prevented HA from increasing phosphorylation of TGFβRI (Fig. 5B, lower panel). Inactivation of PKCδ by the dominant negative construct prevented HA from increasing phosphorylation of TGFβ receptor (Fig. 5C, upper panel). Inactivation of PKCδ also prevented HA from inducing the interaction between TGFβRI and RHAMM as well as the interactions between TGFβRI and rac1 and between TGFβRI and ERK (Fig. 5C, upper panel). Inactivation of PKCδ also prevented HA from inducing expression of PAI-1, as expected (Fig. 5C, lower panel). These results suggest that PKCδ and ERK transactivate TGFβRI, which in turn leads to the interaction between TGFβRI and RHAMM and induction of PAI-1.
RHAMM-ERK interaction induced by HA is necessary for activation of TGFβRI and induction of PAI-1
The interaction between TGFβRI and ERK (Fig. 5C) suggests that RHAMM and ERK may interact. ERK is known to mediate the angiogenic effect of neuromedin B (Park et al., 2009). We hypothesized that ERK might be necessary for activation of TGFβRI by HA.
Immunofluorescence staining showed co-localization of RHAMM and ERK (Fig. 6A, upper panel). Induction of RHAMM by HA was correlated with the activation of ERK and Elk-1, a substrate of ERK (Fig. 6A, lower left panel). Immunoprecipitation showed an interaction between RHAMM and ERK (Fig. 6A, lower right panel). Because HA was shown to promote cancer cell motility through the activation of FAK (Kim et al., 2008), we examined the possible interaction between RHAMM and FAK. However, HA did not induce an interaction between RHAMM and FAK (Fig. 6A, lower right panel). Inactivation of ERK by the dominant negative construct exerted a negative effect on activation of TGFβRI by HA (Fig. 6B, upper panel). ERK acted downstream of PKCδ and inactivation of ERK prevented HA from inducing expression of PAI-1 (Fig. 6B, lower panel). Taken together, these results suggest that HA employs RHAMM-TGFβ receptor-ERK signaling to induce expression of PAI-1 and promote angiogenesis.
Fig. 6.

RHAMM-ERK interaction, induced by HA, is necessary for activation of TGFβRI and the induction of PAI-1. (A) HUVECs were treated with or without HA (1 mDa, 200 μg/ml) for 1 h. Immunofluorescence staining using anti-RHAMM (2 μg/ml) or anti-phospho ERK antibody (2 μg/ml) was performed (upper panel). HUVECs were treated with HA (1 mDa, 200 μg/ml) for various time intervals. Cell lysates prepared at each time point were subjected to Western blot analysis (lower left panel). HUVECs were treated with or without HA (1 mDa, 200 μg/ml) for 1 h. Cell lysates were immunoprecipitated with anti-RHAMM antibody (2 μg/ml), followed by Western blot analysis (lower right panel). (B) HUVECs were transiently transfected with control vector (1 μg) or dominant negative ERK construct (1 μg). At 48 h after transfection, cell lysates were immunoprecipitated with anti-actin antibody (2 μg/ml) or anti-TGFβRI antibody (2 μg/ml), followed by Western blot analysis using the indicated antibody (upper panel). Cell lysates were also subjected to Western blot analysis (lower panel).
DISCUSSION
Hyaluronic acid synthase-1 (HAS-1) promotes angiogenesis through CD44 (Golshani et al., 2008). There are three HAS isoforms, HAS1, HAS2, and HAS3, which synthesize HA at different catalytic rates, resulting in different size polymers (Itano et al., 1999). For example, HAS3 is catalytically more active and synthesizes smaller HA polymers (1 × 105 to 1 × 106 Da) than HAS1 and HAS2 (2 × 105 to 2 × 106 Da). HA can be cleaved by hyaluronidases into small oligosaccharides which have been shown to exert distinct biological activity relative to their high-molecular-weight counterparts (West and Kumar, 1989; West et al., 1985). High molecular weight (HMW) HA (900–1200 kDa), but not low molecular weight form of HA, induces the Snail2 protein via NF-κB to enhance the invasion potential of mouse embryo fibroblasts (Craig et al., 2009).
This functional difference between HAs of varying sizes is a matter of controversy since many studies have reported opposing results in regard to which type of HA can bring about cellular changes (McKee et al., 1997). These discrepancies may be due to differences in experimental settings, purity of HA (McDonald and Camenisch, 2003), and the possibility of diverse responses to HA depending on the cell type. In this study, 6 kDa HA and 100 kDa HA, when used at equimolar concentration, did not induce angiogenesis based on intravital microscopy (Fig. 1A). However, these HAs, when used at higher concentration, en-hanced endothelial cell tube formation (personal observation). Therefore, there was no difference among HA of different sizes in terms of inducing angiogenesis.
Both CD44 and RHAMM bind to HA. Blocking of CD44, but not RHAMM, using blocking antibody prevents adhesion of endothelial cells to HA (Savani et al., 2001). However, blocking of RHAMM does not affect adhesion (Savani et al., 2001). RHAMM, but not CD44, is necessary for endothelial cell migration through matrigel (Savani et al., 2001). These results suggest that there are differential requirements for CD44 and RHAMM in HA-promoted angiogenesis. Down regulation of RHAMM exerts a negative effect on HA-induced endothelial cell tube formation (Matou-Nasri et al., 2009). RHAMM mediates the effect of HA on endothelial cell tube formation (Fig. 3D) and is also necessary for the enhanced invasion potential of endothelial cells by HA (Fig. 3E). Our results in this study indicate that both RHAMM and CD44 contribute to endothelial cell tube formation and invasion of HUVEC (Figs. 3D–3F). Rac1 is required for activation of hypoxia inducible factor 1 (Hirota et al., 2001). Rac1 is also necessary for the induction of VEGFR-2 in vascular endothelial cells (Meissner et al., 2009) and is necessary for endothelial cell migration (Lee et al., 2007). VEGF activates Rac1 via vav-2 (Garrett et a l., 2007) and activation of Rac1 is involved in the hypoxia-induced production of angiogenesis-promoting factors (Xue et al., 2006). Thrombin-induced angiogenesis requires Rac1 and NADPH oxidase 2 (Diebold et al., 2009). Inhibition of Rac1 reduces VEGF-induced tyrosine phosphorylation of VEGFR-2 (Ushio-Fukai et al., 2002). Immunoprecipitation shows an interaction between RHAMM and Rac1 by HA (Supplementary Fig. 1B). HA also induces activation of rac1 (supplemental Fig. 1B).
Epigenetic factor(s) has been known to be involved in angiogenesis. Overexpression of HDAC1 promotes angiogenesis by down regulation of tumor suppressor genes, such as p53 and VHL genes (Dai et al., 2009). HDAC inhibitors target HIF1 alpha to exert anti-angiogenic effect (Dai et al., 2009). These reports suggest that HDAC(s) play a role in angiogenesis.
Cigarette smoke-induced reduction of HDAC2 is related with phosphorylation and ubiquitination in macrophage and epithelial cells (Adenuga et al., 2009). Reduction of HDAC2 expression is closely related to the nitration of distinct tyrosine residues (Osoata et al., 2009). Because inactivation of Rac1 by the dominant negative construct prevents HA from decreasing the expression of HDAC3 (Supplementary Fig. 3E) and inducing tyrosine nitration (supplemental Fig. 3E), it is reasonable that HA induces modification, such as tyrosine nitration on HDAC3 via rac1, which may lead to proteasomal degradation of HDAC3. Cigarette smoke increases reactive oxygen species production, resulting in posttranslational modifications of HDAC3 (Yang et al., 2006).
Down regulation of HDAC3 enhances the endothelial cell tube formation, increases reactive oxygen species levels and Rac1 activity (personal observations). Over expression of HDAC3 prevents HA-promoted endothelial cell tube formation (personal observation). These results indicate that HDAC3 may act as a negative regulator of angiogenesis. Liver specific knockout of HDAC3 results in an enlarged organ, hepatocyte hypertrophy and disturbed fat metabolism (Knutson et al., 2008). HDAC2-/-mice die within 24 h after birth and show severe cardiac defects (Montgomery et al., 2007). HDAC3 is critical in endothelial cell survival and atherosclerosis development in response to disturbed flow (Zampetaki et al., 2007). There have been no reports concerning role of HDAC2 or HDAC3 in angiogenesis.
Placenta growth factor (PIGF) induces expression of PAI-1 in Sickle cell disease (Patel et al., 2010). In this process, PIGF induces binding of AP-1 to PAI-1 promoter sequences. PIGF also induces expression of PAI-1 through JNK, NADPH oxidase and HIF-1 (Lin et al., 208). Based α on these reports, we hypothesized that HA might induce expression of PAI-1 to promote angiogenesis. In fact, HA did induce expression of PAI-1 based on cytokine array analysis (Fig. 4A). Thus, it is probable that AP-1, induced by HA (Fig. 3A), may mediate the effect of HA on induction of PAI-1. Recently, it was reported that expression of PAI-1 is at least partially related to the methylation status of promoter sequences (Gao et al., 2010). We found down regulation of DNA methyltransferase I (DNMT1) by HA (personal observation). This suggests that DNMT1 may act as a negative regulator of PAI-1. However, the methylation status of PAI-1 still needs to be investigated.
HDAC2 interacts with Snail to regulate expression of various genes (Peinado et al., 2004; von Burstin et al., 2009). We found that HDAC2 and snail were induced by HA (Fig. 4C). Therefore HDAC2-Snail complex may bind to promoter sequences of PAI-1 to induce expression of PAI-1. Snail binds to PAI-1 promoter sequences (Fig. 4F). The ChIP assay showed that HDAC3 complex binds to the promoter sequences of PAI-1 in the absence of HA (Fig. 4F), suggesting that HA leads to the displacement of HDAC3 from the promoter sequences of PAI-1. The down regulation of PAI-1 prevents HA from inducing expression of MMP-2, ICAM-1 and VCAM-1 (Fig. 4H). ICAM-1/CD18 and VCAM-1/integrin α4 are necessary for recruitment of endothelial precursor cells (EPC) to the ischemic myocardium and also are necessary for angiogenesis (Wu et al., 2006). MMP-2 alters VEGF expression via PI3kianse signaling in lung cancer cells (Chetty et al., 2010). This suggests that MMP2 is involved in angiogenesis. TGFβ receptor signaling is necessary for up regulation of MMP-2 (Marquez-Aguirre et al., 2009). Chemical inhibition of MMP-2 suppresses TGFβ1-induced PAI-1 expression in rat kidney fibroblast cells (Cho et al., 2012). Because induction of PAI-1 expression is dependent on TGFβ receptor signaling (Fig. 5B), it is reasonable that PAI-1 and MMP-2 are interdependent. There have been no reports concerning the effect of PAI-1 on expression of MMP-2 or vice versa. It would be necessary to identify transcription factors that are affected by down regulation of PAI-1 or MMP-2.
The CD44-EGFR interaction by HA activates PKC signaling, which involves MMP-2, in mouse melanoma cells (Kim et al., 2008). HDAC3 exerts a negative effect on MMP-2 expression (Liu et al., 2003). MMP-2 is necessary for bFGF-induced angiogenesis (Boosani et al., 2010). In vivo tube formation is associated with increased MMP-2 activity in caveolae fraction in endothelial cells (Cavallo-Medved et al., 2009). Therefore, we examined the role of MMP-2 in HA-promoted angiogenesis in relation with HDAC3. The deletion of HDAC3 was shown to lead to increased acetylation of histone H3 at Lys9/14 and histone H4 at Lys5/12(Bhaskara et al., 2010). In this study, the down regulation of HDAC3 increased acetylation of histone H3 at Lys9/14 and histone H4 at Lys16 (Supplementary Fig. 2B). AP-1 is necessary for the induction of MMP-2 (Hasegawa et al., 2009). Because HA induces expression of MMP-2 (Fig. 4H), it is reasonable that the down regulation of HDAC3 may induce AP-1 and increase expression of MMP-2. In fact, the down regulation of HDAC3 induces binding of c-jun to MMP-2 promoter sequences (Supplementary Fig. 4C).
Ski co-repressor complex exerts a negative regulation on TGFβ-target gene, SMAD7, via HDAC3 and protein arginine methyl transferase 5 (PRMT5) (Tabata et al., 2009), suggesting that PAI-1 expression maybe under epigenetic regulation involving histone acetylation/deacetylation and methylation.
EGF, just like HA, induces expression of PAI-1 by PKC delta in glioblastoma cells (Paugh et al., 2008). TNF-α regulates TGFβ-promoted epithelial mesenchymal transition (EMT) by inducing HA-CD44-moesin interaction (Takahashi et al., 2010), suggesting transactivation of TGFβRI by TNF-α. We hypothesized that HA would activate TGFβ receptor signaling to induce expression of PAI-1. HA was shown to activate TGFβRI without inducing phosphorylation of Smad2 (Fig. 5A) and induced the interaction between RHAMM and TGFβRI via PKCδ (Fig. 5C). This suggests that CD44-PKCδ transactivates TGFβRI. RHAMM regulates ERK kinase activity (Ha et al., 2008; Zhang et al., 1998). In this study, we examined whether different sizes of HAs have an effect on TGFβRI.
The 1 mDa HA, but not 6 kDa or 100 kDa HA, induced activation of TGFβRI and expression of PAI-1 (Supplementary Fig. 5). This suggests that HAs of different sizes may promote angiogenesis in a different way. The role of ERK1/2 in osteopontin-induced angiogenesis in endothelial cells has been reported (Kim et al., 2001). The role of ERK, in relation with RHAMM, on HA-promoted angiogenesis has not been studied. HA induces co-localization of RHAMM with ERK (Fig. 6A, upper panel). This indicates that HA induces TGFβRI-RHAMM-ERK complex via PKCδ to promote angiogenesis. Because HA induces activation of TGFβRI without inducing phosphorylation of Smad2 (Fig. 5A, lower panel), we hypothesized that ERK would be also necessary for transactivation of TGFβR1. In fact, ERK was necessary for activation of TGFβRI, induction of RHAMM and PAI-1 (Fig. 6B).
In conclusion, HA-promoted angiogenesis is mediated by interplay between CD44/PKCδ, acting upstream, and RHAMM-ERK-TGFβRI, and involves induction of PAI-1 which results from the down regulation of HDAC3 by rac1 (Fig. 7). The down regulation of HDAC3 induces expression of MMP-2, which mediates HA-promoted angiogenesis (Fig. 7). Our study provides a novel mechanism of HA-promoted angiogenesis and potential targets for the development of therapeutics for angiogenesis-related diseases.
Fig. 7.
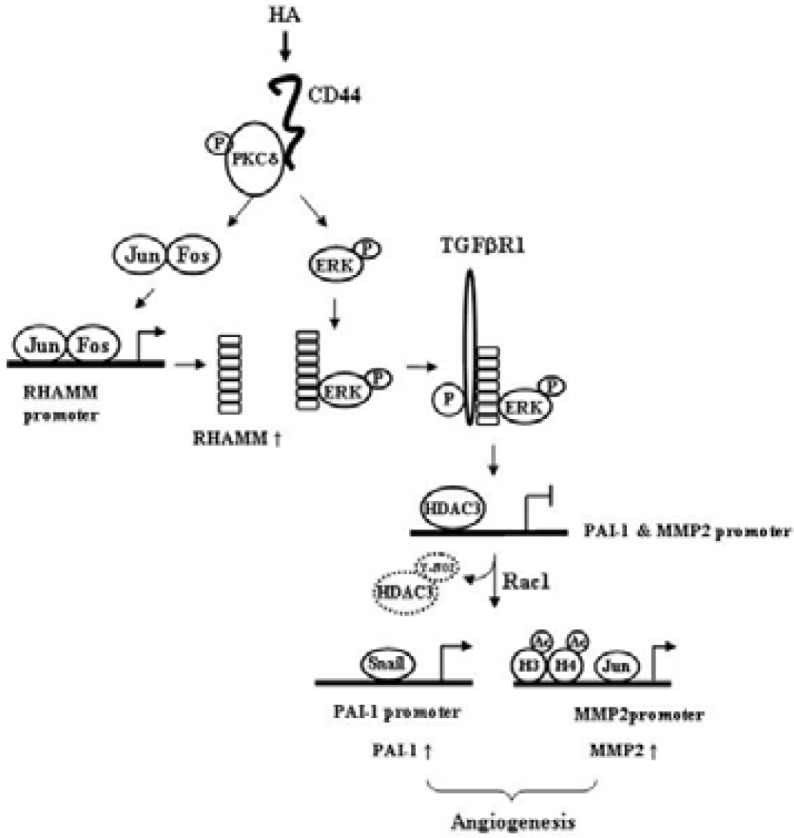
Proposed model of HA-promoted angiogenesis. HA induces an interaction between CD44 and PKCδ. Activation of PKCδ leads to the induction of RHAMM via AP-1. PKCδ is also necessary for the activation of ERK. HA induces an interaction between RHAMM and ERK. HA transactivates TGFβRI via PKCδ and ERK. The activation of TGFβRI by HA is closely associated with degradation of HDAC3, which results from tyrosine nitration induced by rac1. The decreased expression of HDAC3 is responsible for the induction of PAI-1 and MMP-2.
Acknowledgments
This work was supported by the Korea Research Foundation (C1001478-01-01, C1006272-01-01, C1007565-01-01 and C1006625-01-02); Korea Research Foundation Grant funded by the Korean Government [Ministry of Education, Science and Technology (MEST)]” (The Regional Research Universities Program/Medical & Bio-Materials Research Center); and the Regional Innovation Center Program (MEST).
Note:
Supplementary information is available on the Molecules and Cells Website (www.molcells.org).
REFERENCES
- Adenuga D., Yao H., March T.H., Seagrave J., Rahman I. Histone deacetylase 2 is phosphorylated, ubiquitinated, and degraded by cigarette smoke. Am. J. Respir. Cell Mol. Biol. 2009;40:464–473. doi: 10.1165/rcmb.2008-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajou K., Peng H., Laug W.E., Maillard C., Noel A., Foidart J.M., Martial J.A., DeClerck Y.A. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell. 2008;14:324–334. doi: 10.1016/j.ccr.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter M.J., Pybus L., Litherland G.J., Rowan A.D., Clark I.M., Edwards D.R., Cawston T.E., Young D.A. HDAC-mediated control of ERK- and PI3K-dependent TGF-β-induced extracellular matrix-regulating genes. Matrix Biol. 2010;29:602–612. doi: 10.1016/j.matbio.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Basu A., Menicucci G., Maestas J., Das A., McGuire P. Plasminogen activator inhibitor-1 (PAI-1) facilitates retinal angiogenesis in a model of oxygen-induced retinopathy. Invest. Ophthalmol. Vis. Sci. 2009;50:4974–4981. doi: 10.1167/iovs.09-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendinelli P., Matteucci E., Maroni P., Desiderio M.A. NF-kappaB activation, dependent on acetylation/deacetylation, contributes to HIF-1 activity and migration of bone metastatic breast carcinoma cells. Mol. Cancer Res. 2009;8:1328–1341. doi: 10.1158/1541-7786.MCR-08-0548. [DOI] [PubMed] [Google Scholar]
- Bhaskara S., Chyla B.J., Amann J.M., Knutson S.K., Cortez D., Sun Z.W., Hiebert S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara S., Knutson S.K., Jiang G., Chandrasekharan M.B., Wilson A.J., Zheng S., Yenamandra A., Locke K., Yuan J.L., Bonine-Summers A.R., et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436–447. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boosani C.S., Nalabothula N., Sheibani N., Sudhakar A. Inhibitory effects of arresten on bFGF-induced proliferation, migration, and matrix metalloproteinase-2 activation in mouse retinal endothelial cells. Curr. Eye Res. 2010;35:45–55. doi: 10.3109/02713680903374208. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bourguignon L.Y., Gilad E., Peyrollier K., Brightman A., Swanson R.A. Hyaluronan-CD44 interaction stimulates Rac1 signaling and PKN gamma kinase activation leading to cytoskeleton function and cell migration in astrocytes. J. Neurochem. 2007;101:1002–1017. doi: 10.1111/j.1471-4159.2007.04485.x. [DOI] [PubMed] [Google Scholar]
- Cavallo-Medved D., Rudy D., Blum G., Bogyo M., Caglic D., Sloane B.F. Live-cell imaging demonstrates extracellular matrix degradation in association with active cathepsin B in caveolae of endothelial cells during tube formation. Exp. Cell Res. 2009;315:1234–1246. doi: 10.1016/j.yexcr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetty C., Lakka S.S., Bhoopathi P., Rao J.S. MMP-2 alters VEGF expression via alphaVbeta3 integrin-mediated PI3K/AKT signaling in A549 lung cancer cells. Int. J. Cancer. 2010;127:1081–1095. doi: 10.1002/ijc.25134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H.J., Kang J.H., Jeong J.H., Jeong Y.J., Park K.K., Park Y.Y., Moon Y.S., Kim H.T., Chung I.K., Kim C.H., et al. Ascochlorin suppresses TGF-β1-induced PAI-1 expression through the inhibition of phospho-EGFR in rat kidney fibroblast cells. Mol. Biol. Rep. 2012;39:4597–4603. doi: 10.1007/s11033-011-1251-y. [DOI] [PubMed] [Google Scholar]
- Contreras E.G., Gaete M., Sánchez N., Carrasco H., Larraín J. Early requirement of Hyaluronan for tail regeneration in Xenopus tadpoles. Development. 2009;136:2987–2996. doi: 10.1242/dev.035501. [DOI] [PubMed] [Google Scholar]
- Craig E.A., Parker P., Camenisch T.D. Size-dependent regulation of Snail2 by hyaluronan: its role in cellular invasion. Glycobiology. 2009;19:890–898. doi: 10.1093/glycob/cwp064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J., Peng L., Fan K., Wang H., Wei R., Ji G., Cai J., Lu B., Li B., Zhang D., et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene. 2009;28:3412–3422. doi: 10.1038/onc.2009.189. [DOI] [PubMed] [Google Scholar]
- Diebold I., Djordjevic T., Petry A., Hatzelmann A., Tenor H., Hess J., Görlach A. Phosphodiesterase 2 mediates redox-sensitive endothelial cell proliferation and angiogenesis by thrombin via Rac1 and NADPH oxidase 2. Circ. Res. 2009;104:1169–1177. doi: 10.1161/CIRCRESAHA.109.196592. [DOI] [PubMed] [Google Scholar]
- Evert B.O., Araujo J., Vieira-Saecker A.M., de Vos R.A., Harendza S., Klockgether T., Wüllner U. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J. Neurosci. 2006;26:11474–11486. doi: 10.1523/JNEUROSCI.2053-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre-Guillevin E., Malo M., Cartier-Michaud A., Peinado H., Moreno-Bueno G., Vallée B., Lawrence D.A., Palacios J., Cano A., Barlovatz-Meimon G., et al. PAI-1 and functional blockade of SNAI1 in breast cancer cell migration. Breast Cancer Res. 2008;10:R100. doi: 10.1186/bcr2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W., Dequiedt F., Hendzel M.J., Guenther M.G., Lazar M.A., Voelter W., Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- Gao S., Nielsen B.S., Krogdahl A., Sørensen J.A., Tagesen J., Dabelsteen S., Dabelsteen E., Andreasen P.A. Epigenetic alterations of the SERPINE1 gene in oral squamous cell carcinomas and normal oral mucosa. Genes Chromosomes Cancer. 2010;49:526–538. doi: 10.1002/gcc.20762. [DOI] [PubMed] [Google Scholar]
- Garrett T.A., Van Buul J.D., Burridge K. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp. Cell Res. 2007;313:3285–3297. doi: 10.1016/j.yexcr.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golshani R., Lopez L., Estrella V., Kramer M., Iida N., Lokeshwar V.B. Hyaluronic acid synthase-1 expression regulates bladder cancer growth, invasion, and angiogenesis through CD44. Cancer Res. 2008;68:483–491. doi: 10.1158/0008-5472.CAN-07-2140. [DOI] [PubMed] [Google Scholar]
- Ha C.H., Wang W., Jhun B.S., Wong C., Hausser A., Pfizenmaier K., McKinsey T.A., Olson E.N., Jin Z.G. Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J. Biol. Chem. 2008;283:14590–14599. doi: 10.1074/jbc.M800264200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha C.H., Jhun B.S., Kao H.Y., Jin Z.G. VEGF stimulates HDAC7 phosphorylation and cytoplasmic accumu-lation modulating matrix metalloproteinase expression and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2008;28:1782–1788. doi: 10.1161/ATVBAHA.108.172528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton S.R., Fard S.F., Paiwand F.F., Tolg C., Veiseh M., Wang C., McCarthy J.B., Bissell M.J., Koropatnick J., Turley E.A. The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J. Biol. Chem. 2007;282:16667–16680. doi: 10.1074/jbc.M702078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa H., Senga T., Ito S., Iwamoto T., Hamaguchi M. A role for AP-1 in matrix metalloproteinase production and invadopodia formation of v-Crk-transformed cells. Exp. Cell Res. 2009;315:1384–1392. doi: 10.1016/j.yexcr.2009.02.019. [DOI] [PubMed] [Google Scholar]
- Hirota K., Semenza G.L. Rac1 activity is required for the activation of hypoxia-inducible factor 1. J. Biol. Chem. 2001;276:21166–21172. doi: 10.1074/jbc.M100677200. [DOI] [PubMed] [Google Scholar]
- Itano N., Sawai T., Yoshida M., Lenas P., Yamada Y., Imagawa M., Shinomura T., Hamaguchi M., Yoshida Y., Ohnuki Y., et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J. Biol. Chem. 1999;274:25085–25092. doi: 10.1074/jbc.274.35.25085. [DOI] [PubMed] [Google Scholar]
- Jayne S., Zwartjes C.G., van Schaik F.M., Timmers H.T. Involvement of the SMRT/NCoR-HDAC3 complex in transcriptional repression by the CNOT2 subunit of the human Ccr4-Not complex. Biochem. J. 2006;398:461–467. doi: 10.1042/BJ20060406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.S., Kwon H.J., Lee Y.M., Baek J.H., Jang J.E., Lee S.W., Moon E.J., Kim H.S., Lee S.K., Chung H.Y., et al. Histone deacetylases induce angiogenesis by negative regula-tion of tumor suppressor genes. Nat. Med. 2001;7:437–443. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- Kim Y., Lee Y.S., Hahn J.H., Choe J., Kwon H.J., Ro J.Y., Jeoung D. Hyaluronic acid targets CD44 and inhibits FcepsilonRI signaling involving PKCdelta, Rac1, ROS, and MAPK to exert anti-allergic effect. Mol. Immunol. 2008;45:2537–2547. doi: 10.1016/j.molimm.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Kim Y., Lee Y.S., Choe J., Lee H., Kim Y.M., Jeoung D. CD44-epidermal growth factor receptor interaction mediates hyaluronic acid-promoted cell motility by activating protein kinase C signaling involving Akt, Rac1, Phox, reactive oxygen species, focal adhesion kinase, and MMP-2. J. Biol. Chem. 2008;283:22513–22528. doi: 10.1074/jbc.M708319200. [DOI] [PubMed] [Google Scholar]
- Knutson S.K., Chyla B.J., Amann J.M., Bhaskara S., Huppert S.S., Hiebert S.W. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008;27:1017–1028. doi: 10.1038/emboj.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokudo T., Suzuki Y., Yoshimatsu Y., Yamazaki T., Watabe T., Miyazono K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008;121:3317–3324. doi: 10.1242/jcs.028282. [DOI] [PubMed] [Google Scholar]
- Koyama H., Hibi T., Isogai Z., Yoneda M., Fujimori M., Amano J., Kawakubo M., Kannagi R., Kimata K., Taniguchi S., et al. Hyperproduction of hyaluronan in neu-induced mammary tumor accelerates angiogenesis through stromal cell recruitment: possible involvement of versican/PG-M. Am. J. Pathol. 2007;170:1086–1099. doi: 10.2353/ajpath.2007.060793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Kunz J., Lin S.H., Yu-Lee L.Y. 16-kDa prolactin inhibits endothelial cell migration by down-regulating the Ras-Tiam1-Rac1-Pak1 signaling pathway. Cancer Res. 2007;67:11045–11053. doi: 10.1158/0008-5472.CAN-07-0986. [DOI] [PubMed] [Google Scholar]
- Lin M.T., Kuo I.H., Chang C.C., Chu C.Y., Chen H.Y., Lin B.R., Sureshbabu M., Shih H.J., Kuo M.L. Involvement of hypoxia-inducing factor-1alpha-dependent plasminogen activator inhibitor-1 up-regulation in Cyr61/CCN1-induced gastric cancer cell invasion. J. Biol. Chem. 2008;283:15807–15815. doi: 10.1074/jbc.M708933200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Liu L.T., Chang H.C., Chiang L.C., Hung W.C. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Res. 2003;63:3069–3072. [PubMed] [Google Scholar]
- Liu R.M., Choi J., Wu J.H., Gaston Pravia K.A., Lewis K.M., Brand J.D., Mochel N.S., Krzywanski D.M., Lambeth J.D., Hagood J.S., et al. Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor beta1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J. Biol. Chem. 2010;285:16239–16247. doi: 10.1074/jbc.M110.111732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez-Aguirre A., Sandoval-Rodriguez A., Gonzalez-Cuevas J., Bueno-Topete M., Navarro-Partida J., Arellano-Olivera I., Lucano-Landeros S., Armendariz-Borunda J. Adenoviral delivery of dominant-negative transforming growth factor beta type II receptor up-regulates transcriptional repressor SKI-like oncogene, decreases matrix metalloproteinase 2 in hepatic stellate cell and prevents liver fibrosis in rats. J. Gene Med. 2009;1:207–219. doi: 10.1002/jgm.1303. [DOI] [PubMed] [Google Scholar]
- Matou-Nasri S., Gaffney J., Kumar S., Slevin M. Oligosaccharides of hyaluronan induce angiogenesis through distinct CD44 and RHAMM-mediated signaling pathways involving Cdc2 and gamma-adducin. Int. J. Oncol. 2009;35:761–773. doi: 10.3892/ijo_00000389. [DOI] [PubMed] [Google Scholar]
- McDonald J.A., Camenisch T.D. Hyaluronan: Genetic insights into the complex biology of a simple polysaccharide. Glyconjugate J. 2003;19:331–339. doi: 10.1023/A:1025369004783. [DOI] [PubMed] [Google Scholar]
- McKee C.M., Lowenstein C.J., Horton M.R., Wu J., Bao C., Chin B.Y., Choi A.M., Noble P.W. Hyaluronan fragments induce nitric-oxide synthase in murine macrophages through a nuclear factor kappaB-dependent mechanism. J. Biol. Chem. 1997;272:8013–8018. doi: 10.1074/jbc.272.12.8013. [DOI] [PubMed] [Google Scholar]
- Meissner M., Michailidou D., Stein M., Hrgovic I., Kaufmann R., Gille J. Inhibition of Rac1 GTPase downregulates vascular endothelial growth factor receptor-2 expression by suppressing Sp1-dependent DNA binding in human endothelial cells. Exp. Dermatol. 2009;18:863–869. doi: 10.1111/j.1600-0625.2009.00867.x. [DOI] [PubMed] [Google Scholar]
- Min J.K., Cho Y.L., Choi J.H., Kim Y., Kim J.H., Yu Y.S., Rho J., Mochizuki N., Kim Y.M., Oh G.T., et al. Receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) increases vascular permeability: impaired permeability and angiogenesis in eNOS-deficient mice. Blood. 2007;109:1495–1502. doi: 10.1182/blood-2006-06-029298. [DOI] [PubMed] [Google Scholar]
- Montgomery R.L., Davis C.A., Potthoff M.J., Haberland M., Fielitz J., Qi X., Hill J.A., Richardson J.A., Olson E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottet D., Bellahcène A., Pirotte S., Waltregny D., Deroanne C., Lamour V., Lidereau R., Castronovo V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res. 2007;101:1237–1246. doi: 10.1161/CIRCRESAHA.107.149377. [DOI] [PubMed] [Google Scholar]
- Nedvetzki S., Gonen E., Assayag N., Reich R., Williams R.O., Thurmond R.L., Huang J.F., Neudecker B.A., Wang F.S., Turley E.A., et al. RHAMM, a receptor for hyaluronan-mediated motility, compensates for CD44 in inflamed CD44-knockout mice: a different interpretation of redundancy. Proc. Natl. Acad. Sci. USA. 2004;101:18081–18086. doi: 10.1073/pnas.0407378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osoata G.O., Yamamura S., Ito M., Vuppusetty C., Adcock I.M., Barnes P.J., Ito K. Nitration of distinct tyrosine residues causes inactivation of histone deacetylase 2. Biochem. Biophys. Res. Commun. 2009;384:366–371. doi: 10.1016/j.bbrc.2009.04.128. [DOI] [PubMed] [Google Scholar]
- Park H.J., Kim S.R., Bae S.K., Choi Y.K., Bae Y.H., Kim E.C., Kim W.J., Jang H.O., Yun I., Kim Y.M., et al. Neuromedin B induces angiogenesis via activation of ERK and Akt in endothelial cells. Exp. Cell Res. 2009;315:3359–3369. doi: 10.1016/j.yexcr.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Patel N., Sundaram N., Yang M., Madigan C., Kalra V.K., Malik P. Placenta growth factor (PlGF), a novel inducer of plasminogen activator inhibitor-1 (PAI-1) in sickle cell disease (SCD) J. Biol. Chem. 2010;285:16713–16722. doi: 10.1074/jbc.M110.101691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh B.S., Paugh S.W., Bryan L., Kapitonov D., Wilczynska K.M., Gopalan S.M., Rokita H., Milstien S., Spiegel S., Kordula T. EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCdelta, and sphingosine kinase 1 in glioblastoma cells. FASEB J. 2008;22:455–465. doi: 10.1096/fj.07-8276com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H., Ballestar E., Esteller M., Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell. Biol. 2004;24:306–319. doi: 10.1128/MCB.24.1.306-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rössig L., Li H., Fisslthaler B., Urbich C., Fleming I., Förstermann U., Zeiher A.M., Dimmeler S. Inhibitors of histone deacetylation downregulate the expression of endo-thelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ. Res. 2002;91:837–844. doi: 10.1161/01.res.0000037983.07158.b1. [DOI] [PubMed] [Google Scholar]
- Samuel S.K., Hurta R.A., Spearman M.A., Wright J.A., Turley E.A., Greenberg A.H. TGF-beta 1 stimulation of cell locomotion utilizes the hyaluronan receptor RHAMM and hyaluronan. J. Cell Biol. 1993;123:749–758. doi: 10.1083/jcb.123.3.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankar N., Baluchamy S., Kadeppagari R.K., Singhal G., Weitzman S., Thimmapaya B. p300 provides a corepressor function by cooperating with YY1 and HDAC3 to repress c-Myc. Oncogene. 2008;27:5717–5728. doi: 10.1038/onc.2008.181. [DOI] [PubMed] [Google Scholar]
- Savani R.C., Cao G., Pooler P.M., Zaman A., Zhou Z., DeLisser H.M. Differential involvement of the hyaluro-nan (HA) receptors CD44 and receptor for HA-mediated motility in endothelial cell function and angiogenesis. J. Biol. Chem. 2001;276:36770–36778. doi: 10.1074/jbc.M102273200. [DOI] [PubMed] [Google Scholar]
- Sohr S., Engeland K. RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle. 2008;7:3448–3460. doi: 10.4161/cc.7.21.7014. [DOI] [PubMed] [Google Scholar]
- Tabata T., Kokura K., Ten Dijke P., Ishii S. Ski corepressor complexes maintain the basal repressed state of the TGF-beta target gene, SMAD7, via HDAC3 and PRMT5. Genes Cells. 2009;14:17–28. doi: 10.1111/j.1365-2443.2008.01246.x. [DOI] [PubMed] [Google Scholar]
- Takahashi E., Nagano O., Ishimoto T., Yae T., Suzuki Y., Shinoda T., Nakamura S., Niwa S., Ikeda S., Koga H., et al. Tumor necrosis factor-alpha regulates transforming growth factor-beta-dependent epithelial-mesenchymal transition by promoting hyaluronan-CD44-moesin interaction. J. Biol. Chem. 2010;285:4060–4073. doi: 10.1074/jbc.M109.056523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togi S., Kamitani S., Kawakami S., Ikeda O., Muromoto R., Nanbo A., Matsuda T. HDAC3 influences phosphorylation of STAT3 at serine 727 by interacting with PP2A. Biochem. Biophys. Res. Commun. 2009;379:616–620. doi: 10.1016/j.bbrc.2008.12.132. [DOI] [PubMed] [Google Scholar]
- Tolg C., Hamilton S.R., Nakrieko K.A., Kooshesh F., Walton P., McCarthy J.B., Bissell M.J., Turley E.A. Rhamm-/-fibroblasts are defective in CD44-mediated ERK1,2 mitogenic signaling, leading to defective skin wound repair. J. Cell Biol. 2006;175:1017–1028. doi: 10.1083/jcb.200511027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbich C., Rössig L., Kaluza D., Potente M., Boeckel J.N., Knau A., Diehl F., Geng J.G., Hoffmann W.K., Zeiher A.M., et al. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood. 2009;113:5669–5679. doi: 10.1182/blood-2009-01-196485. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M., Tang Y., Fukai T., Dikalov S.I., Ma Y., Fujimoto M., Quinn M.T., Pagano P.J., Johnson C., Alexander R.W. Novel role of gp91 (phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ. Res. 2002;91:1160–1167. doi: 10.1161/01.res.0000046227.65158.f8. [DOI] [PubMed] [Google Scholar]
- Villagra A., Ulloa N., Zhang X., Yuan Z., Sotomayor E., Seto E. Histone deacetylase 3 down-regulates cholesterol synthesis through repression of lanosterol synthase gene expression. J. Biol. Chem. 2007;282:35457–35470. doi: 10.1074/jbc.M701719200. [DOI] [PubMed] [Google Scholar]
- von Burstin J., Eser S., Paul M.C., Seidler B., Brandl M., Messer M., von Werder A., Schmidt A., Mages J., Pagel P., et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology. 2009;137:361–371. doi: 10.1053/j.gastro.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Wang C., Thor A.D., Moore D.H., 2nd, Zhao Y., Kerschmann R., Stern R., Watson P.H., Turley E.A. The overexpression of RHAMM, a hyaluronan-binding protein that regulates ras signaling, correlates with overexpression of mitogen-activated protein kinase and is a significant parameter in breast cancer progression. Clin. Cancer Res. 1998;4:567–576. [PubMed] [Google Scholar]
- Washio A., Kitamura C., Jimi E., Terashita M., Nishihara T. Mechanisms involved in suppression of NGF-induced neuronal differentiation of PC12 cells by hyaluronic acid. Exp. Cell Res. 2009;315:3036–3043. doi: 10.1016/j.yexcr.2009.07.006. [DOI] [PubMed] [Google Scholar]
- West D.C., Hampson I.N., Arnold F., Kumar S. Angiogenesis induced by degradation products of hyaluronic acid. Science. 1985;228:1324–1326. doi: 10.1126/science.2408340. [DOI] [PubMed] [Google Scholar]
- West D.C., Kumar S. The effect of hyaluronate and its oligosaccharides on endothelial cell proliferation and monolayer integrity. Exp. Cell Res. 1989;183:179–196. doi: 10.1016/0014-4827(89)90428-x. [DOI] [PubMed] [Google Scholar]
- Wu Y., Ip J.E., Huang J., Zhang L., Matsushita K., Liew C.C., Pratt R.E., Dzau V.J. Essential role of ICAM-1/CD18 in mediating EPC recruitment, angiogenesis, and repair to the infarcted myocardium. Circ. Res. 2006;99:315–322. doi: 10.1161/01.RES.0000235986.35957.a3. [DOI] [PubMed] [Google Scholar]
- Xue Y., Bi F., Zhang X., Zhang S., Pan Y., Liu N., Shi Y., Yao X., Zheng Y., Fan D. Role of Rac1 and Cdc42 in hypoxia induced p53 and von Hippel-Lindau suppression and HIF1alpha activation. Int. J. Cancer. 2006;118:2965–2972. doi: 10.1002/ijc.21763. [DOI] [PubMed] [Google Scholar]
- Yang S.R., Chida A.S., Bauter M.R., Shafiq N., Seweryniak K., Maggirwar S.B., Kilty I., Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;291:46–57. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- Zampetaki A., Zeng L., Margariti A., Xiao Q., Li H., Zhang Z., Pepe A.E., Wang G., Habi O., deFalco E., et al. Histone deacetylase 3 Is critical in endothelial survival and Atherosclerosis development in response to disturbed flow. Circulation. 2010;121:132–142. doi: 10.1161/CIRCULATIONAHA.109.890491. [DOI] [PubMed] [Google Scholar]
- Zhang S., Chang M.C., Zylka D., Turley S., Harrison R., Turley E.A. The hyaluronan receptor RHAMM regulates extracellular-regulated kinase. J. Biol. Chem. 1998;273:11342–11348. doi: 10.1074/jbc.273.18.11342. [DOI] [PubMed] [Google Scholar]