

Abstract
Angiogenesis is essential for primary tumor growth and metastatic dissemination. E2F1, frequently upregulated in advanced cancers, was recently shown to drive malignant progression. In an attempt to decipher the molecular events underlying this behavior, we demonstrate that the tumor cell-associated vascular endothelial growth factor-C/receptor-3 (VEGF-C/VEGFR-3) axis is controlled by E2F1. Activation or forced expression of E2F1 in cancer cells leads to the upregulation of VEGFR-3 and its ligand VEGF-C, whereas E2F1 depletion prevents their expression. E2F1-dependent receptor induction is crucial for tumor cells to enhance formation of capillary tubes and neovascularization in mice. We further provide evidence for a positive feedback loop between E2F1 and VEGFR-3 signaling to stimulate pro-angiogenic platelet-derived growth factor B (PDGF-B). E2F1 or VEGFR-3 knockdown results in reduced PDGF-B levels, while the coexpression synergistically upregulates promoter activity and endogenous protein expression of PDGF-B. Our findings delineate an as yet unrecognized function of E2F1 as enhancer of angiogenesis via regulation of VEGF-C/VEGFR-3 signaling in tumors to cooperatively activate PDGF-B expression. Targeting this pathway might be reasonable to complement standard anti-angiogenic treatment of cancers with deregulated E2F1.
Keywords: E2F1, hypoxia, neovascularization, PDGF-B, VEGFR-3, VEGF-C, regulatory feedback loop
Introduction
E2F1 was the first identified member of the E2F family of transcription factors situated downstream of growth factor signaling cascades and required for S-phase progression (Chen et al., 2009b). E2F1 induces programmed cell death as part of an anti-tumorigenic fail-safe mechanism critical for protecting the organism from malignant transformation and suppressing tumor formation (Engelmann and Putzer, 2010; Putzer and Engelmann, 2013). Yet, recent findings indicate that E2F1 overexpression and the amplification of its gene locus, phenomena frequently observed during cancer progression, are associated with unfavorable patient survival, metastatic disease, and resistance to chemotherapy (Han et al., 2003; Imai et al., 2004). This gives rise to the conclusion that enhanced E2F1 levels may reflect gradual acquisition of biological capabilities fostering different cancer hallmarks. According to clinical data, high expression of this transcription factor and its related target genes predict the change from superficial to invasive bladder cancer (Lee et al., 2010). Binding of E2F1 to the gene promoter of the human androgen receptor (AR) that is essential for the initiation and progression of prostate cancer occurs in the absence of retinoblastoma (RB) function (Sharma et al., 2010). Thus, loss of RB during cancer progression correlates with increased E2F1 and AR levels in patients with castrate-resistant prostate cancer metastases. Similarly, epidermal growth factor receptor (EGFR) known as a metastatic marker of cutaneous melanoma was shown to be directly regulated by E2F1 (Alla et al., 2010). Others reported that the ability of E2F1 to promote an aggressive phenotype is controlled by transcriptional coregulators such as ATAD2 (Revenko et al., 2010). High levels of E2F1 and ATAD2 are strongly associated with triple-negative breast cancer, tumor metastasis, disease recurrence, and poor survival (Kalashnikova et al., 2010). Additional functional evidence that endogenously high E2F1 levels cause malignant progression was demonstrated in a human metastatic melanoma model where E2F1 depletion abrogates tumor invasion and pulmonary metastasis (Alla et al., 2010).
The family of vascular endothelial growth factor (VEGF) receptors modulates many endothelial cell (EC) functions during angiogenesis. One member, VEGFR-3 (also known as FLT-4), is present in all endothelia during embryonic development (Kaipainen et al., 1995). Cancers that overexpress VEGF-C induce lymphangiogenesis by activating VEGFR-3 signaling in lymphatic ECs, which enhances metastatic spread (Skobe et al., 2001). Upregulation of VEGFR-3 in the microvasculature of tumors and wounds is required for angiogenic sprouting and vascular network formation (Tammela et al., 2008). Intriguingly, solid tumors and certain blood cancers also express this receptor often together with its ligand VEGF-C (Su et al., 2006; Chien et al., 2009; Kurenova et al., 2009). Observations in cancer patients suggest that high levels of both VEGFR-3 and VEGF-C cause poorer prognosis (Su et al., 2006, 2007). Su and co-workers demonstrated that the autocrine VEGF-C/VEGFR-3 axis enhances cancer cell mobility, invasiveness, and metastasis via up-regulation of contactin-1 in a C/EBP-dependent manner (Su et al., 2006). However, we still lack knowledge about mechanisms activating the VEGF-C/VEGFR-3 system in tumor cells and further downstream functions.
In this study, we demonstrate that VEGF-C and its cognate receptor VEGFR-3 are highly upregulated in malignant cells with abundant E2F1 expression but downregulated in those cells depleted for E2F1. This occurs via transcriptional control of both genes by E2F1. Our results further define VEGFR-3 as a positive regulator of E2F1 in a feedback circuit, which directly coregulates angiogenic gene expression in an E2F1-dependent manner. The identified E2F1-VEGF-C/VEGFR-3 loop is a pivotal mechanism for tumor vascularization in vivo and therefore carries high therapeutic potential.
Results
E2F1 regulates expression of VEGFR-3 and VEGF-C
We explored the influence of E2F1 transcriptional activity on the expression of angiogenesis-regulating genes in cancer cells by using TaqMan arrays. We applied this approach on human melanoma cell lines whose metastatic potential ranges from low in SK-Mel-29 cells to high in SK-Mel-147 cells depending on endogenous E2F1 protein levels (Alla et al., 2010). Angiogenic gene expression was analyzed either in SK-Mel-147 cells transduced with E2F1-specific shRNA or in SK-Mel-29 cells stably expressing 4OHT-inducible ER-E2F1 fusion protein. Arrays revealed that E2F1 inhibition in SK-Mel-147 cells was clearly associated with the reduction of VEGFR-3 and VEGF-C transcript levels (Figure 1A, left). Vice versa, activation of E2F1 in SK-Mel-29.ER-E2F1 cells caused a significant upregulation of both VEGFR-3 and VEGF-C mRNAs comparable with their endogenous levels in SK-Mel-147 cells. Overall, both genes showed an E2F1-dependent expression pattern that resembled the pattern for known E2F target genes like follistatin used as positive control as verified by qPCR (Figure 1A, right). In contrast to VEGF-C, expression of another VEGFR-3 ligand, VEGF-D, was not affected by E2F1 regulation (Figure 1A).
Figure 1.
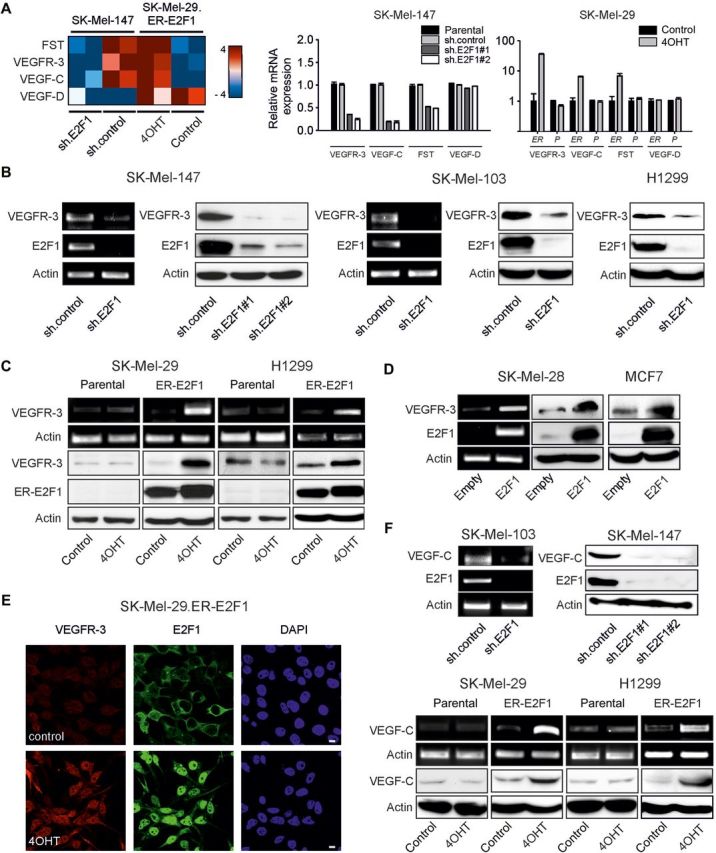
E2F1 regulates expression of tumor cell-associated VEGF-C/VEGFR-3. (A) Heat map illustration of VEGFR-3 and VEGF-C transcript abundance determined by TaqMan qRT–PCR arrays in SK-Mel-147 cells expressing control or E2F1-specific shRNA, and in stable SK-Mel-29.ER-E2F1 cells treated with 4OHT or solvent (control; left panel). Normalized mRNA expression is reported as a log2 ratio. Blue shading represents low and red shading indicates high levels of mRNA. VEGFR-3 and VEGF-C levels were verified by qPCR in SK-Mel-147 cells (using two sh.E2F1 constructs compared with sh.control as well as parental cells) and SK-Mel-29 cells (ER, stable SK-Mel-29.ER-E2F1 cells; P, parental SK-Mel-29 cells; right panels). VEGF-D was used as negative control and follistatin (FST) as positive control. (B–D) RT–PCR and immunoblotting analyses of VEGFR-3 and E2F1 expression after sh.E2F1 treatment (B), E2F1 activation by 4OHT (C), or E2F1 overexpression (D) in indicated cancer cell lines. Parental and sh.control cells served as controls. (E) Subcellular localization of VEGFR-3 (red) and E2F1 (green) in SK-Mel-29.ER-E2F1 cells treated for 24 h with 4OHT. Nuclei were counter-stained with DAPI (blue) and fluorescence was visualized by confocal laser scanning microscopy (CLSM). Scale bar, 10 µm. (F) Detection of VEGF-C mRNA and protein expression upon E2F1 depletion (top) or activation (bottom). Actin served as loading control.
To ascertain that VEGF-C/VEGFR-3 regulation is dependent on E2F1, we inhibited, activated, or overexpressed E2F1 in different cancer cell lines. Consistent with the TaqMan array data, depletion of E2F1 severely impaired VEGFR-3 expression in tumor cells (Figure 1B). No difference on protein levels was observed between parental and sh.control-expressing cells (Supplementary Figure S1). In contrast, VEGFR-3 mRNA and protein levels were substantially upregulated in response to E2F1 in SK-Mel-29, H1299 (Figure 1C), SK-Mel-28, and MCF7 cells (Figure 1D). The link between E2F1 and receptor expression was further substantiated by confocal microscopy on SK-Mel-29.ER-E2F1 cells after 4OHT treatment where enhanced VEGFR-3 staining was visible (Figure 1E). Likewise, transcript and protein levels of VEGF-C increased in melanoma and lung cancer cells upon E2F1 activation, but reduced in metastatic melanoma cell lines expressing sh.E2F1 (Figure 1F). These results imply that E2F1 is essential for tumor cells to promote expression of the VEGF-C/VEGFR-3 system.
E2F1 binds and activates human VEGFR-3 and VEGF-C gene promoters
To gain further insight into the mechanism of E2F1-dependent VEGF-C/VEGFR-3 induction, SK-Mel-29 cells were cotransfected with individual promoter reporter constructs and E2F1 expression plasmid or empty vector. As shown in Figure 2A (left), the VEGFR-3−849/+55 promoter was stimulated through E2F1 in a dose-dependent manner, which was in clear contrast to the DNA-binding-deficient mutant. In coherence with enhanced luciferase levels, sequence analysis revealed six putative E2F-binding motifs, which clustered in the region from −849 to −118 of the VEGFR-3 promoter (Figure 2B, left). The 5′-deletion mutants were used to identify the core element required for direct VEGFR-3 regulation. Truncation from −849 to −331 did not significantly affect promoter activation through cotransfected E2F1. Deletion of the region between −331 and −118 that covered three putative binding sites completely abolished the transactivating effect of E2F1 (Figure 2B, left). Promoter specificity was confirmed by an empty expression vector and a promoter construct lacking the transcription start site (Δ309). Chromatin immunoprecipitation (ChIP) assays in SK-Mel-29.ER-E2F1 cells demonstrated that E2F1 directly bound within the 153 bp element (−311 to −158) bearing the proximal E2F motifs (Figure 2C, left).
Figure 2.

E2F1 directly interacts with human VEGFR-3 and VEGF-C promoters. (A) Relative luciferase activities measured 30 h after cotransfection of VEGFR-3−849/+55 or VEGF-C−49/+419 promoter construct with increasing amounts (0.5, 1, 2, and 3 µg) of E2F1 expression plasmid or DNA-binding-deficient E132 mutant (3 µg). Western blots confirm E2F1 and E132 protein expression after transfection (right). Actin was used for equal loading. (B) Schemes of VEGFR-3 and VEGF-C promoter deletion constructs with relative locations of predicted transcription factor binding sites. Luciferase activity was measured in response to E2F1 overexpression. (C) ChIP in SK-Mel-29.ER-E2F1 cells cultured 24 h in the presence or absence of 4OHT using anti-E2F1 or IgG. Input represents 10% of sheared chromatin prior to immunoprecipitation (left). E2F1-dependent VEGFR-3 and VEGF-C promoter reporter activities were detected in the absence or presence of 4OHT (right). (D) Cotransfection of VEGFR-3−849/+55 or VEGF-C−49/+419 promoter reporters and different E2F family members. E2F1, E2F2, and E2F3a expression was verified by western blotting. Promoter activity is normalized to per mg protein and relative to empty vector.
Based on the prior findings (Figure 1F), we also tested whether the VEGF-C gene directly responds to E2F1. To this end, increasing amounts of E2F1 were coexpressed with a reporter plasmid in which the VEGF-C−49/+419 promoter region harboring a putative E2F1-binding element consisting of two directly adjacent motifs near the transcriptional start site (+19 to +35) regulates luciferase transcription. Our results indicated that dose-dependent activation of this promoter by E2F1 occurred in a similar manner as that to the VEGFR-3 promoter (Figure 2A, middle), whereas deletion of the E2F motifs completely abolished the E2F1 response (Figure 2B, right). The relevance of the 169 bp stretch +24 to +193 within the first exon of the VEGF-C gene for E2F1-binding was confirmed by ChIP (Figure 2C, left). In accordance with the recruitment of E2F1 to the promoters of VEGF-C and VEGFR-3, we observed increased luciferase levels in SK-Mel-29.ER-E2F1 cells after transfection of promoter reporter constructs and treatment with 4OHT (Figure 2C, right).
To determine whether other E2F family members are able to activate VEGFR-3−849/+55 or VEGF-C−49/+419, luciferase assays were performed after cotransfection of E2F2 and E2F3a expression vectors. We found that the VEGFR-3 promoter was, although to a less extent, activated by both proteins. Whereas E2F3a increased the activity of the VEGF-C promoter at a level comparable with that of the VEGFR-3 promoter, E2F2 had no effect. However, compared with E2F2 and E2F3a, E2F1 seems to be the primary activator of VEGF-C/VEGFR-3 (Figure 2D).
VEGFR-3 expression is required for E2F1-induced angiogenic tubule formation
VEGFR-3 is involved in the regulation of tumor angiogenesis (Laakkonen et al., 2007). Considering our and others' previous findings suggesting that some E2F1 target genes participate in the process of angiogenesis (Stanelle et al., 2002; Pillai et al., 2010), we hypothesized that the E2F1-VEGF-C/VEGFR-3 axis facilitates the angiogenic potential of solid tumors. We first examined whether overexpression or depletion of E2F1 in cancer cells alters its ability to stimulate endothelial tubule formation. HUVECs were plated on matrigel-coated wells and cultured with conditioned media from SK-Mel-28 cells transfected with E2F1 or SK-Mel-147 cells expressing shRNA against E2F1. Enforced E2F1 expression significantly increased tubule formation (Figure 3A, left). In contrast, conditioned media from sh.E2F1-expressing SK-Mel-147 cells reduced the capacity of ECs to form tubule-like structures (Figure 3A, right), underscoring the pro-angiogenic activity of E2F1 in these cells. Furthermore, HUVECs were cultured with supernatant from SK-Mel-147 cells depleted for VEGFR-3 or expressing a dominant-negative (DN) receptor mutant that abrogates wild-type autophosphorylation (Karkkainen et al., 2000). Interference with VEGFR-3 activity substantially reduced the angiogenic potential of cancer cells leading to a decrease in HUVEC tubule formation, mimicking the effect of E2F1 depletion (Figure 3B). Increased tubule formation was detectable in the culture with the supernatant from SK-Mel-28 cells expressing wild-type VEGFR-3 (Figure 3C). Next, we tested the relative contribution of tumor-associated VEGFR-3 expression on E2F1 to the angiogenic potential. Strikingly, E2F1-induced formation of endothelial tubule-like structures could be completely abolished by conditioned media from SK-Mel-29.ER-E2F1 and H1299.ER-E2F1 cells in the presence of sh.VEGFR-3 (Figure 3D). Conditioned media from parental cells exposed to 4OHT did not have any influence on HUVECs. Finally, an add-back experiment was conducted where VEGFR-3 and VEGF-C were restored in SK-Mel-147 E2F1 knockdown cells and showed a partial recovery of HUVEC tubule formation in the absence of E2F1, demonstrating that E2F1 is important for VEGF-C/VEGFR-3 induced angiogenesis (Figure 3E). Overall, our data clearly indicate that induction of VEGFR-3 by E2F1 in cancer cells is required for ECs to form capillary tubes.
Figure 3.

Expression of E2F1 and VEGFR-3 in tumor cells is crucial to stimulate tubule formation of HUVECs. (A–D) HUVECs seeded on matrigel were cultured with conditioned media from tumor cells overexpressing E2F1 or treated with sh.E2F1 (A), sh.VEGFR-3, DN-VEGFR-3 (B), wild-type VEGFR-3 (C), or 4OHT in the presence of sh.VEGFR-3 (D). (E) Add-back experiment where VEGFR-3 and VEGF-C were restored to parental levels in E2F1 knockdown cells. Capillary tube formation was monitored by phase contrast microscopy and quantified by ImageJ (scale bar, 100 µm). Each panel shows representative images. Quantitative data are shown as bar graphs. Asterisks indicate statistical significance (P < 0.05) calculated by two-sided t-test.
E2F1 and its transcriptional target VEGFR-3 promote neovascularization in vivo
To assess whether the E2F1-VEGFR-3 axis in cancer cells affects vessel formation in vivo, the dorsal skinfold chamber was used to model tumor-induced angiogenesis. Endogenous E2F1 in SK-Mel-147 cells was silenced by retrovirally delivered sh.E2F1 RNAs (#1, #2) and cells were transplanted below the skinfolds of athymic nude mice. Epifluorescence video microscopy revealed first signs of neovascularization on Day 3 and Day 5 in all groups, characterized by formation of capillary buds and sprouts that originated from venular segments of host striated muscle capillaries and postcapillary venules. The intensity of the angiogenic response markedly differed in different groups. In mice that had received sh.control-expressing SK-Mel-147 cells, the newly formed capillary network was denser and displayed enhanced vessel ingrowth and maturation compared with that in mice receiving sh.E2F1-expressing cells (Figure 4A, right, IVM images). Quantitative analysis of tumor vascularization over 2 weeks showed an increased relative functional capillary density (FCD) of 291 ± 77 cm/cm2 in the control group on Day 14 after transplantation versus 189 ± 38 cm/cm2 (#1) and 196 ± 58 cm/cm2 (#2) in the E2F1-depleted groups (Figure 4A, left). Differences in blood vessel formation were also evident in histological sections from tumor tissues of the dorsal skinfolds, demonstrating a lower micro-vessel density in the E2F1-depleted groups than in the control group (Figure 4A, right, H&E and CD31).
Figure 4.
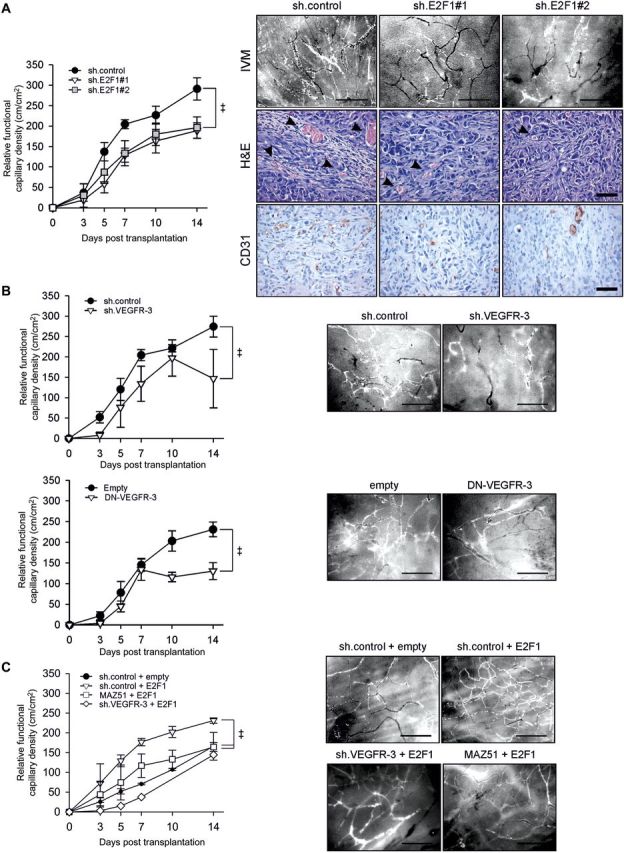
E2F1 and VEGFR-3 depleted tumors exhibit reduced neovascularization in vivo. (A) Quantitative analysis of rFCD after transplantation of SK-Mel-147 cells expressing sh.E2F1 (left; sh.control n = 8, sh.E2F1#1 n = 4, sh.E2F1#2 n = 5). Results are given in mean ± SEM. Two-way ANOVA and appropriate post hoc tests were performed. Statistical significance is P < 0.05 (‡). Representative images of intratumoral microvasculature visualized by epi-illumination fluorescence video microscopy (right, IVM). Scale bar, 200 µm. Histology of SK-Mel-147 xenografts expressing sh.control or sh.E2F1 on Day 14 after transplantation on striated muscle tissue of the dorsal skinfold in nude mice (right). Tissue sections were stained with hematoxylin-eosin (H&E) and CD31 (ab28364). Arrows indicate intratumoral vessels with erythrocytes evident in the lumen. Scale bar, 50 µm. (B) Quantitative analysis of rFCD after transplantation of SK-Mel-147 cells expressing sh.VEGFR-3 (top; n = 3/group) or DN-VEGFR-3 (bottom; n = 5/group). Representative IVM image of each group is shown. (C) The rFCD after transplantation of SK-Mel-29 cells overexpressing E2F1 (n = 2), co-treated with MAZ51 (n = 3) or co-expressing E2F1 with sh.VEGFR-3 (n = 1) compared with controls (n = 3). Representative IVM image of each group is shown.
To determine the impact of VEGFR-3 activity in cells with high E2F1 expression, sh.VEGFR-3 or DN-VEGFR-3 was stably integrated into SK-Mel-147 cells. The modified cells were transplanted into skinfolds and neovascularization was monitored as described above. After 2 weeks, tumors originated from cells depleted for VEGFR-3 (Figure 4B, top) or overexpressing the DN mutant (Figure 4B, bottom) showed ∼45% reduction in FCD compared with each control group. Importantly, high vessel density induced by E2F1 overexpression in SK-Mel-29 cells was significantly decreased on Day 14 upon MAZ51 treatment or knockdown of VEGFR-3 by shRNA to parental levels (Figure 4C). These results show that E2F1 expression favors tumor vascularization via the VEGFR-3.
VEGF-C/VEGFR-3 signaling promotes a positive feedback loop affecting E2F1-dependent induction of platelet-derived growth factor B
Adaption to hypoxia is a critical step for neovascularization, since oxidative stress promotes the expression of pro-angiogenic cytokines in tumors. Previously, it was shown that low levels of oxygen activate the VEGF-C/VEGFR-3 system in MCF7 cells, while other tumoral loops between VEGF family members and cognate receptors remained unaffected (Simiantonaki et al., 2008). We cultured MCF7 and SK-Mel-28 cells with low endogenous E2F1 and VEGFR-3 levels under hypoxic conditions and observed a strong increase in mRNA (Figure 5A) and protein (Figure 5B) levels of both receptor and ligand. In addition, E2F1 accumulated upon hypoxia, suggesting a causative link to VEGF-C/VEGFR-3 upregulation (Figure 5B). Increased expression of the VEGFC/VEGFR-3 system and E2F1 in these cells was associated with enhanced levels of the hypoxia-inducible cytokine platelet-derived growth factor B (PDGF-B), which was stimulated by the VEGFR-3 pathway (Onimaru et al., 2009) and was also found upregulated in our TaqMan arrays from tumor cells with activated E2F1. Hypoxic cells showed significantly lower VEGF-C/VEGFR-3 expression when E2F1 was depleted and this was accompanied by downregulation of PDGF-B (Figure 5C, left). Importantly, inhibition of VEGFR-3 by its chemical inhibitor MAZ51 prevented E2F1 accumulation in response to oxidative stress and abrogated PDGF-B induction (Figure 5C, right). This argues for a positive feedback loop between E2F1 and VEGF-C/VEGFR-3 that regulates PDGF-B expression.
Figure 5.

E2F1 and VEGFR-3 establish a positive feedback loop in tumor cells that promotes PDGF-B expression. (A and B) Detection of VEGF-C, VEGFR-3, E2F1, and PDGF-B mRNA (A) and protein (B) levels in MCF7 and SK-Mel-28 cells cultured under normoxic or hypoxic conditions. (C) Mutual regulation of E2F1 and VEGF-C/VEGFR-3 and the effect on PDGF-B expression in MCF7 cells are shown by E2F1 knockdown using sh.E2F1 (left) and selective inhibition of VEGFR-3 signaling using 10 µM MAZ51 (right). (D and E) Impact of VEGFR-3 knockdown (D, left), VEGFR-3 overexpression (D, right), and VEGF-C(Cys156Ser) treatment at 50 ng/ml (E) on endogenous E2F1 and PDGF-B levels. (F) Analysis of PDGF-B protein level in E2F1-depleted melanoma cells exposed to Cys156Ser. Actin was used for equal loading.
In line with this, we observed a decrease in E2F1 and PDGF-B expression after knockdown of VEGFR-3 in metastatic SK-Mel-147 cells that exhibit an E2F1-dependent pro-angiogenic phenotype in vivo (Figure 5D, left), whereas VEGFR-3 overexpression in SK-Mel-28 cells led to the upregulation of both E2F1 and PDGF-B protein levels (Figure 5D, right). Receptor stimulation with a recombinant VEGF-C(Cys156Ser) mutant that selectively binds and activates VEGFR-3 resulted in a considerable induction of E2F1, PDGF-B, as well as VEGFR-3 expression (Figure 5E). VEGF-C(Cys156Ser)-dependent upregulation of PDGF-B, however, was not accomplished in E2F1-depleted cells (Figure 5F), indicating that PDGF-B regulation by VEGF receptor signaling occurs via E2F1.
Next, we examined whether PDGF-B is directly targeted by E2F1. The experiment shown in Figure 6A revealed that this cytokine was secreted into the supernatant of tumor cells overexpressing E2F1 (Figure 6A). PDGF-B was downregulated after efficient knockdown of E2F1 (Figure 6B, left) and became upregulated upon E2F1 activation (Figure 6B, right). For luciferase assays, the proximal promoter region of the human PDGF-B gene ranging from −583 to +80 was cloned into the pGL3-basic reporter plasmid and cotransfected with wild-type or mutant E2F1 expression plasmid in SK-Mel-29 and MCF-7 cells. As shown in Figure 6C, only E2F1 that bound to the PDGF-B−583/+80 promoter induced a strong activation, while the E132 mutant did not. Consistent with these results, we found that E2F1 directly associated with a 360 bp DNA element (−223 to −583) in the proximal PDGF-B promoter region spanning four putative E2F motifs (Figure 6D). According to recent studies demonstrating that activated VEGF receptors are translocated to the nucleus (Santos et al., 2007), and in case of EGFR interacting with DNA-binding transcription factors such as E2F1 to activate target gene expression (Hanada et al., 2006), we anticipated that VEGFR-3 might cooperate with E2F1 to regulate PDGF-B transactivation. Fluorescence microscopy and Western blots of SK-Mel-147 cells showed that E2F1 colocalizes with phosphorylated VEGFR-3 in the nucleus of SK-Mel-147 cells (Figure 6E). ChIP assays using VEGFR-3 antibody led to an enrichment of the proximal PDGF-B promoter region bound by E2F1 (Figure 6F, top). The association of VEGFR-3 with the PDGF-B E2F-site was abrogated in sh.VEGFR-3-expressing cells, verifying the specificity of the ChIP results. Moreover, binding of VEGFR-3 to the PDGF-B E2F-site was solely detectable in SK-Mel-28 cells when exposed to VEGF-C(Cys156Ser) (Figure 6F, bottom). We performed reporter assays to examine whether VEGFR-3 facilitates E2F1-mediated gene expression. As demonstrated in Figure 6G, overexpression of VEGFR-3 in SK-Mel-29 cells stimulated transcription of the PDGF-B promoter at a level comparable with that induced by E2F1 (∼15-fold). Furthermore, we showed that E2F1 and VEGFR-3 acted synergistically on the PDGF-B promoter, resulting in a strong (∼70-fold) increase in luciferase activity, which was also evident from Western blots of endogenous PDGF-B. As VEGFR-3, like other receptor tyrosine kinases (RTKs), lacks a DNA-binding domain, our data clearly suggest that VEGFR-3 functions as a transcriptional coactivator in a complex with E2F1 targeting the PDGF-B promoter.
Figure 6.
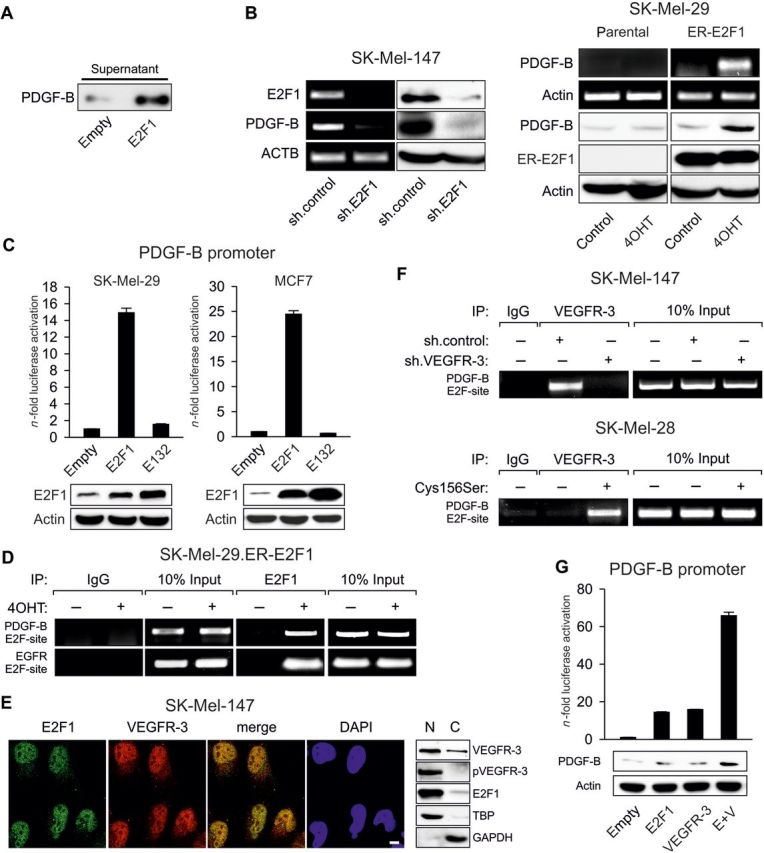
VEGFR-3 and E2F1 cooperatively regulate PDGF-B gene expression. (A) Detection of PDGF-B cytokine in the supernatant of MCF7 cells overexpressing E2F1. (B) Endogenous PDGF-B expression after E2F1 knockdown (left) or in response to E2F1 activation by 4OHT (right). Parental SK-Mel-29 cells were used as control. (C) Luciferase activity of the human PDGF-B−583/+80 promoter at 30 h after cotransfection of wild-type E2F1 or mutant E132. (D) In vivo binding of E2F1 to the PDGF-B promoter was detected by ChIP using E2F1 antibody or control IgG. In the precipitates by anti-E2F1 antibody, the EGFR promoter serves as positive control. Input lane, 10% of total chromatin. (E) Nuclear colocalization of E2F1 and VEGFR-3 visualized by CLSM. Scale bar, 10 μm. Nuclear and cytosolic fractions of SK-Mel-147 cells were analyzed for total or phosphorylated VEGFR-3 and E2F1. TBP and GAPDH are used as positive controls for protein separation. (F) Interaction of nuclear VEGFR-3 with the E2F-site of the PDGF-B promoter in cells expressing control or VEGFR-3 shRNA (top) or after Cys156Ser treatment at 50 ng/ml (bottom). ChIP assays were performed using anti-VEGFR-3 antibody and primers flanking the E2F1-binding region of the PDGF-B promoter. Input was used as loading control; IgG served as negative control. (G) SK-Mel-29 cells were cotransfected with PDGF-B−583/+80 reporter construct, E2F1 and/or VEGFR-3 expression plasmid. Western blots show endogenous PDGF-B expression of transfected SK-Mel-29 cells.
Discussion
The RB-E2F pathway is primarily known for the regulation of the cell cycle. Surprisingly, in vivo studies using gene-targeting knockout strategies showed that E2Fs are indispensable for proliferation (Chen et al., 2009a), suggesting that these transcription factors drive tumor development also by other cellular processes (Chen et al., 2009b). Regulation of tumor angiogenesis seems to be an unanticipated E2F function. Initially, E2F1 was proposed to modulate neovascularization based on the pattern of target genes identified by microarray profiling (Stanelle et al., 2002). However, E2f1−/− mice displayed enhanced angiogenesis of tumor xenografts and increased EC proliferation as a result of VEGF-A overproduction (Qin et al., 2006). This unexpected observation is due to the cooperation between E2F1 and p53 at the promoter level to repress VEGF-A transcription in vascular cells, although other studies report a p53-independent mechanism (Merdzhanova et al., 2010). These results were in context with unique tumor suppressor function of E2F1 within the RB-E2F pathway. On the contrary, VEGF-A stimulation of ECs leads to the inhibition of Rb followed by E2F1 activation that is crucial for proper EC function (Pillai et al., 2010), and corroborates previous research indicating that E2F1 promotes EC growth and suppresses apoptosis (Goukassian et al., 2003). It is therefore apparent that the role of E2F1 in vascular biology is more complex than expected and might be exploited in cancers.
E2F1 is frequently overexpressed in aggressive tumors and associated with increased cancer morbidity. We and others recently demonstrated its pro-metastatic activity (Alla et al., 2010; Johnson et al., 2012) and the relevance to chemoresistance (Millour et al., 2011; Alla et al., 2012), and suggested a functional switch of E2F1 from a tumor suppressor in untransformed tissues to an oncogene capable to promote cancer progression independent of its proliferative activity in malignant cells (Chen et al., 2009b; Alla et al., 2010; Engelmann and Putzer, 2012). Based on our prior data and attributed to its potential role as a positive regulator of pro-angiogenic target genes, we proposed that deregulated E2F1 can induce angiogenesis, invasion, and metastasis from original tumor (Stanelle et al., 2002). In this regard, RTKs expressed in cancer cells play an important role in tumor progression via autocrine interaction with corresponding ligands. Since many tumors coexpress VEGFR-3 and VEGF-C, tumor cell-autonomous VEGFR-3 signaling receives growing attention (Su et al., 2006; Kurenova et al., 2009). Our study unravels a hitherto unknown E2F1-dependent mechanism of activation of the VEGF-C/VEGFR-3 axis in cancer cells. We demonstrate that upregulation of receptor and ligand depends on E2F1 by direct interaction with their gene promoters. Coregulation of VEGF-C/VEGFR-3 by E2F1 stimulates cultured ECs to form tubule-like structures and promotes formation of new blood vessels in vivo. In melanoma xenografts, specific inhibition of either E2F1 or VEGFR-3 considerably impaired tumor neo-angiogenesis. Thus, our results extend previous findings that VEGF-C promotes angiogenesis in leukemic cells via the VEGFR-3/JNK/AP-1 pathway (Chien et al., 2009) and point towards the relevance of the E2F1-VEGF-C/VEGFR-3 axis in cancer biology by acting directly on tumor cells. In addition, the data provide first evidence for the contribution of this axis to solid tumor angiogenesis.
Further insight was provided into the mechanism by which E2F1 regulates tumor-related angiogenic VEGFR signaling downstream of the receptor. Our study revealed that E2F1 is activated by VEGFR-3 in a positive feedback loop and they cooperate in the nucleus to transactivate PDGF-B directly implicated in EC activation (Battegay et al., 1994; Guo et al., 2003). That VEGF-C/VEGFR-3 pro-angiogenic signaling strongly relies on the cooperation with E2F1 within a feedback loop is also visible from the add-back experiment in E2F1 knockdown cells, where full recovery of HUVEC tubule formation by VEGF-C/VEGFR-3 was not achieved in the absence of E2F1. We detected high levels of E2F1 together with phosphorylated VEGFR-3 in the nuclear fraction of metastatic SK-Mel-147 cells. Overexpression of E2F1 in MCF-7 cells at levels comparable with endogenous E2F1 in aggressive melanoma cells mediates strong PDGF-B secretion. Moreover, this cytokine is significantly reduced in SK-Mel-147 cells following E2F1 depletion. This implies striking similarities of the E2F1-VEGF-C/VEGFR-3-PDGF-B axis with nuclear EGFR signaling. It has been reported that activated EGFR translocates to the nucleus and drives expression of genes associated with cancer progression (Lo and Hung, 2006). As EGFR lacks a DNA-binding domain, nuclear EGFR cooperates with promoter-bound E2F1 to transactivate B-Myb target gene expression (Hanada et al., 2006). Together with the observed nuclear colocalization of E2F1 and VEGFR-3, our ChIP data provide confirmatory evidence that the VEGFR-3, like EGFR, functions as transcription cofactor in PDGF-B promoter activation. Although we have not proven how E2F1-induced VEGF-C/VEGFR-3 signaling upregulates E2F1 expression in this feedback circuit, it might be accomplished through the cooperative activity of E2F1/VEGFR-3 on the E2F1 promoter as well. On the background that E2F1 is a driving force of melanoma metastasis, which occurs via direct upregulation of EGFR (Alla et al., 2010), we suggest a model in which a single transcription factor initiates different aspects of cancer progression by exploiting several RTKs. These findings substantiate observations that poor survival of cancer patients is strongly associated with co-expression of PDGF-B and VEGFR-3 (Donnem et al., 2010).
Moreover, the contribution of E2F1 to the angiogenic potential of tumor cells was previously attributed to its interaction with mutant p53 that positively controls ID4 expression as shown in p53-deficient H1299 lung cancer cells overexpressing mutant p53 (Fontemaggi et al., 2009). In these cells, we found that E2F1 upregulates VEGF-C/VEGFR-3 in the absence of either wild-type or mutated p53, underscoring the significance of the E2F1-VEGF-C/VEGFR-3-PDGF-B cascade as an independent mechanism by which solid tumors recruit their vascularization.
Normal anti-angiogenic therapy causes hypoxic responses of tumors that trigger a series of adaptive changes leading to a resistant phenotype (Abdollahi and Folkman, 2010). Hypoxia is an essential stimulus for the upregulation of autocrine signaling in tumor cells (Simiantonaki et al., 2008). Under hypoxia, VEGF-C/VEGFR-3 is upregulated via E2F1 in several cancer cell lines resulting in the induction of PDGF-B. In line with clinical observations (Bergers and Hanahan, 2008), our results explain how current anti-angiogenic treatment rather promotes tumor progression than ameliorates patient survival. Thus, specific interruption of the angiogenic E2F1-VEGF-C/VEGFR-3 pathway should be the focus of future drug development.
Materials and methods
Cell culture and reagents
SK-Mel-28, -29, -103, and -147 cells were maintained as previously described (Alla et al., 2010). HEK293T, H1299, and MCF7 cells were obtained from ATCC and maintained in DMEM with 10% serum. HUVECs purchased from Invitrogen were cultured in EBM-2 supplemented with growth factors (EGM-2 bullet kit, Lonza). Stable SK-Mel-29.ER-E2F1 and H1299.ER-E2F1 cells were described previously (Alla et al., 2010; John et al., 2011) and were treated with 1 µM 4OHT. For VEGF-C(Cys156Ser) stimulation, serum-starved cells were stimulated at 50 ng/ml for 24 h. Oxygen deprivation was carried out in an incubator with 1% O2, 5% CO2, and 94% N2 for 24 h. Transfections were performed using TurboFect (Thermo Scientific). E2F1 and E132 plasmids were described before (Alla et al., 2010). The pCMV-VEGF-C expression plasmid was purchased from OriGene (SC116730). VEGFR-3 and VEGF-C promoter reporter plasmids, cDNAs for wild-type and DN VEGFR-3(G857R) were described before (Suzuki et al., 2005; Sapoznik et al., 2009; Flister et al., 2010). The VEGF-C promoter deletion mutant was PCR-amplified from wildtype promoter plasmid and cloned into p-Luc using HindIII and XhoI restriction sites. The mutant clone was verified via sequencing (for oligos see Supplementary Table S1).
Viral vectors
Lentiviral plasmids (pLKO.1-puro) encoding sh.E2F1 (TRCN250, TRCN253) and sh.control (SHC002) were purchased from Sigma-Aldrich. VEGFR-3-specific shRNA was described before (Kurenova et al., 2009). The cDNA of DN-VEGFR3(G857R) was cut from pcDNA3 by BamHI/XbaI restriction and ligated into pWPXL (Addgene). VSV-G enveloped pseudotyped lentiviral vectors were generated by cotransfection of HEK293T cells with plasmids of pMD2.G and psPAX2 (Addgene) using calcium phosphate.
qRT–PCR
Quantitative RT–PCR was performed as previously described (Engelmann et al., 2010). Briefly, RNA was reverse-transcribed using First Strand cDNA Synthesis Kit (Thermo Scientific) and Oligo-dT primer. cDNA samples were mixed with iTaq Universal SYBR Green Supermix (Bio-Rad) and analyzed on iQ™5 Multicolor Real-Time PCR Detection System. For primer sequences see Supplementary Table S1.
Tube formation assay
Cancer cells were cultured in EBM for 24 h and supernatant was collected. Matrigel (BD) was added to µ-Slides (ibidi) followed by incubation for 60 min at 37°C. HUVECs (7000 cells/10 µl gel) grew in conditioned media for 8 h. Capillary tube formation was monitored by phase contrast microscopy and quantified using ImageJ (NIH).
Immunoblotting and immunofluorescence
Equal amounts of proteins were separated by SDS–PAGE, transferred to nitrocellulose membranes (Amersham), and probed with primary antibodies against E2F1 (KH95, BD), E2F2 (C-20), E2F3 (C-18), PDGF-B (H-55), VEGFR-3 (C-20), VEGF-C (H-190) from Santa Cruz, phospho-VEGFR-3 (pc460, Calbiochem), and actin (Sigma). For immunofluorescence, cells grown on glass slides were fixed with paraformaldehyde, permeabilized with Triton X-100, blocked with BSA, and incubated with primary antibodies overnight. After washing with PBS, cells were incubated with fluorescence-labeled (Alexa-488, Cy5) secondary antibody (Molecular Probes) for 45 min. Images were obtained using an inverted confocal laser scanning microscope (Zeiss, ELYRA PS.1).
RNA isolation, RT–PCR, and TaqMan human angiogenesis array
Total RNA was extracted with NucleoSpin® (Machery-Nagel) and reverse-transcribed using cDNA Synthesis Kit (Applied Biosystems). PCR amplification was performed by using peqGOLD Master-Mix (Peqlab). Primer sequences are available in Supplementary material. TaqMan Human Angiogenesis Arrays (384-well microfluidic card) were purchased from Applied Biosystems. Real-time PCR was performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems).
PDGF-B promoter cloning, luciferase reporter assay and chromatin immunoprecipitation
A DNA fragment containing 583 bp of the 5′ upstream region of PDGF-B and 80 bp of exon 1 was amplified by PCR from human genomic DNA and subcloned into pcDNA3.1, digested with KpnI/XhoI and ligated into pGL3-basic. Luciferase activity was measured 30 h after transfection using the Luciferase Reporter Assay System (Promega) and normalized to total protein concentration in cell extracts. ChIP assays were performed essentially as described (Engelmann et al., 2010). Protein-DNA complexes were immunoprecipitated using antibodies described above or control IgG. For primer sequences see Supplementary Table S1.
Dorsal skinfold chamber assay, intravital microscopy, and histology
Dorsal skinfold chambers were surgically implanted in male NMRI nude mice as reported previously (Abshagen et al., 2009). Animals were allowed 3 days to recover from surgery. Subsequently, SK-Mel-147 cells (4 × 105) were transplanted into the center of the chamber. IVM was performed on Day 3, 5, 7, 10 and 14 after transplantation. Animals were anesthetized by an intraperitoneal injection of ketamine/xylazine prior to the procedure. Then, 0.1 ml of fluoresceinisothiocyanate-conjugated dextran (FITC-dextran, MW 150 kDa; 2%) was administered via retro-bulbar injection shortly before microscopy to attain contrast enhancement of individual microvessels by blue light epi-illumination. Images were captured by a CCD video camera (Pieper) and transferred to a digital versatile disc system (Pioneer). IVM was followed by offline computer assisted assessment of the dynamic tumor neo-angiogenesis (CapImage, Zeintl) including measurement of tumor area (TA) and vascularized tumor area (VTA) as well as FCD, i.e. the length of perfused capillaries per area of vascularized tumor. Relative VTA (rVTA) and relative FCD (rFCD) were calculated from VTA, TA, and FCD according to the formulae rVTA = VTA/TA and rFCD = FCD × rVTA. Animals were sacrificed on Day 14 after transplantation and tumor-bearing skinfolds were dissected en bloc, fixed in formalin, and embedded in paraffin. Paraffin-embedded tumor-bearing skinfolds were cut for 4 µm sections, mounted on slides, and stained with hematoxylin-eosin and anti-CD31 antibody (ab28364, Abcam) according to standard procedures. All animal experiments were approved by the local animal care committee.
Supplementary material
Supplementary material is available at Journal of Molecular Cell Biology online.
Funding
The work was supported by Deutsche Krebshilfe, Dr. Mildred Scheel Stiftung (109801 to B.M.P and D.E.) and the Rostock University Medical Center for the project Systems Medicine of Cancer Invasion and Metastasis.
Conflict of interest: none declared.
Supplementary Material
Acknowledgments
The authors thank Dr M. Neeman (Weizmann Institute of Science, Rehovot, Israel) for the kind gift of the VEGF-C promoter construct, Dr H. Suzuki (University of Tsukuba, Japan) for the VEGFR-3 cDNAs, Drs W.C. Lin (BCM, Houston, TX, USA) and R. Bernards (NKI, Amsterdam, Netherlands) for providing the E2F2 and E2F3a expression plasmids, and Dr E.V. Kurenova (RPCI, Buffalo, NY, USA) for sh.VEGFR-3 construct. We greatly appreciate the help of Dr B. Kowtharapu (IEGT, Rostock, Germany) with cloning and production of lentiviral delivery systems.
References
- Abdollahi A., Folkman J. Evading tumor evasion: current concepts and perspectives of anti-angiogenic cancer therapy. Drug Resist. Updat. 2010;13:16–28. doi: 10.1016/j.drup.2009.12.001. doi:10.1016/j.drup.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Abshagen K., Schrodi I., Gerber T., et al. In vivo analysis of biocompatibility and vascularization of the synthetic bone grafting substitute NanoBone. J. Biomed. Mater. Res. A. 2009;91:557–566. doi: 10.1002/jbm.a.32237. [DOI] [PubMed] [Google Scholar]
- Alla V., Engelmann D., Niemetz A., et al. E2F1 in melanoma progression and metastasis. J. Natl Cancer Inst. 2010;102:127–133. doi: 10.1093/jnci/djp458. doi:10.1093/jnci/djp458. [DOI] [PubMed] [Google Scholar]
- Alla V., Kowtharapu B.S., Engelmann D., et al. E2F1 confers anticancer drug resistance by targeting ABC transporter family members and Bcl-2 via the p73/DNp73-miR-205 circuitry. Cell Cycle. 2012;11:3067–3078. doi: 10.4161/cc.21476. doi:10.4161/cc.21476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battegay E.J., Rupp J., Iruela-Arispe L., et al. PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF beta-receptors. J. Cell Biol. 1994;125:917–928. doi: 10.1083/jcb.125.4.917. doi:10.1083/jcb.125.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G., Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. doi:10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D., Pacal M., Wenzel P., et al. Division and apoptosis of E2f-deficient retinal progenitors. Nature. 2009a;462:925–929. doi: 10.1038/nature08544. doi:10.1038/nature08544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.Z., Tsai S.Y., Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat. Rev. Cancer. 2009b;9:785–797. doi: 10.1038/nrc2696. doi:10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien M.H., Ku C.C., Johansson G., et al. Vascular endothelial growth factor-C (VEGF-C) promotes angiogenesis by induction of COX-2 in leukemic cells via the VEGF-R3/JNK/AP-1 pathway. Carcinogenesis. 2009;30:2005–2013. doi: 10.1093/carcin/bgp244. doi:10.1093/carcin/bgp244. [DOI] [PubMed] [Google Scholar]
- Donnem T., Al-Saad S., Al-Shibli K., et al. Co-expression of PDGF-B and VEGFR-3 strongly correlates with lymph node metastasis and poor survival in non-small-cell lung cancer. Ann. Oncol. 2010;21:223–231. doi: 10.1093/annonc/mdp296. doi:10.1093/annonc/mdp296. [DOI] [PubMed] [Google Scholar]
- Engelmann D., Putzer B.M. Translating DNA damage into cancer cell death-A roadmap for E2F1 apoptotic signalling and opportunities for new drug combinations to overcome chemoresistance. Drug Resist. Updat. 2010;13:119–131. doi: 10.1016/j.drup.2010.06.001. doi:10.1016/j.drup.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Engelmann D., Putzer B.M. The dark side of E2F1: in transit beyond apoptosis. Cancer Res. 2012;72:571–575. doi: 10.1158/0008-5472.CAN-11-2575. doi:10.1158/0008-5472.CAN-11-2575. [DOI] [PubMed] [Google Scholar]
- Engelmann D., Knoll S., Ewerth D., et al. Functional interplay between E2F1 and chemotherapeutic drugs defines immediate E2F1 target genes crucial for cancer cell death. Cell. Mol. Life Sci. 2010;67:931–948. doi: 10.1007/s00018-009-0222-0. doi:10.1007/s00018-009-0222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flister M.J., Wilber A., Hall K.L., et al. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1. Blood. 2010;115:418–429. doi: 10.1182/blood-2008-12-196840. doi:10.1182/blood-2008-12-196840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontemaggi G., Dell'Orso S., Trisciuoglio D., et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat. Struct. Mol. Biol. 2009;16:1086–1093. doi: 10.1038/nsmb.1669. doi:10.1038/nsmb.1669. [DOI] [PubMed] [Google Scholar]
- Goukassian D.A., Kishore R., Krasinski K., et al. Engineering the response to vascular injury: divergent effects of deregulated E2F1 expression on vascular smooth muscle cells and endothelial cells result in endothelial recovery and inhibition of neointimal growth. Circ. Res. 2003;93:162–169. doi: 10.1161/01.RES.0000082980.94211.3A. doi:10.1161/01.RES.0000082980.94211.3A. [DOI] [PubMed] [Google Scholar]
- Guo P., Hu B., Gu W., et al. Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am. J. Pathol. 2003;162:1083–1093. doi: 10.1016/S0002-9440(10)63905-3. doi:10.1016/S0002-9440(10)63905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S., Park K., Bae B.N., et al. E2F1 expression is related with the poor survival of lymph node-positive breast cancer patients treated with fluorouracil, doxorubicin and cyclophosphamide. Breast Cancer Res. Treat. 2003;82:11–16. doi: 10.1023/B:BREA.0000003843.53726.63. doi:10.1023/B:BREA.0000003843.53726.63. [DOI] [PubMed] [Google Scholar]
- Hanada N., Lo H.W., Day C.P., et al. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol. Carcinog. 2006;45:10–17. doi: 10.1002/mc.20147. doi:10.1002/mc.20147. [DOI] [PubMed] [Google Scholar]
- Imai M.A., Oda Y., Oda M., et al. Overexpression of E2F1 associated with LOH at RB locus and hyperphosphorylation of RB in non-small cell lung carcinoma. J. Cancer Res. Clin. Oncol. 2004;130:320–326. doi: 10.1007/s00432-003-0538-3. doi:10.1007/s00432-003-0538-3. [DOI] [PubMed] [Google Scholar]
- John K., Alla V., Meier C., et al. GRAMD4 mimics p53 and mediates the apoptotic function of p73 at mitochondria. Cell Death Differ. 2011;18:874–886. doi: 10.1038/cdd.2010.153. doi:10.1038/cdd.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.L., Pillai S., Pernazza D., et al. Regulation of matrix metalloproteinase genes by E2F transcription factors: Rb-Raf-1 interaction as a novel target for metastatic disease. Cancer Res. 2012;72:516–526. doi: 10.1158/0008-5472.CAN-11-2647. doi:10.1158/0008-5472.CAN-11-2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaipainen A., Korhonen J., Mustonen T., et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc. Natl Acad. Sci. USA. 1995;92:3566–3570. doi: 10.1073/pnas.92.8.3566. doi:10.1073/pnas.92.8.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalashnikova E.V., Revenko A.S., Gemo A.T., et al. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 2010;70:9402–9412. doi: 10.1158/0008-5472.CAN-10-1199. doi:10.1158/0008-5472.CAN-10-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkkainen M.J., Ferrell R.E., Lawrence E.C., et al. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat. Genet. 2000;25:153–159. doi: 10.1038/75997. doi:10.1038/75997. [DOI] [PubMed] [Google Scholar]
- Kurenova E.V., Hunt D.L., He D., et al. Vascular endothelial growth factor receptor-3 promotes breast cancer cell proliferation, motility and survival in vitro and tumor formation in vivo. Cell Cycle. 2009;8:2266–2280. doi: 10.4161/cc.8.14.9101. doi:10.4161/cc.8.14.9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakkonen P., Waltari M., Holopainen T., et al. Vascular endothelial growth factor receptor 3 is involved in tumor angiogenesis and growth. Cancer Res. 2007;67:593–599. doi: 10.1158/0008-5472.CAN-06-3567. doi:10.1158/0008-5472.CAN-06-3567. [DOI] [PubMed] [Google Scholar]
- Lee J.S., Leem S.H., Lee S.Y., et al. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J. Clin. Oncol. 2010;28:2660–2667. doi: 10.1200/JCO.2009.25.0977. doi:10.1200/JCO.2009.25.0977. [DOI] [PubMed] [Google Scholar]
- Lo H.W., Hung M.C. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br. J. Cancer. 2006;94:184–188. doi: 10.1038/sj.bjc.6602941. doi:10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merdzhanova G., Gout S., Keramidas M., et al. The transcription factor E2F1 and the SR protein SC35 control the ratio of pro-angiogenic versus antiangiogenic isoforms of vascular endothelial growth factor-A to inhibit neovascularization in vivo. Oncogene. 2010;29:5392–5403. doi: 10.1038/onc.2010.281. doi:10.1038/onc.2010.281. [DOI] [PubMed] [Google Scholar]
- Millour J., de Olano N., Horimoto Y., et al. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol. Cancer Ther. 2011;10:1046–1058. doi: 10.1158/1535-7163.MCT-11-0024. doi:10.1158/1535-7163.MCT-11-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onimaru M., Yonemitsu Y., Fujii T., et al. VEGF-C regulates lymphangiogenesis and capillary stability by regulation of PDGF-B. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H1685–H1696. doi: 10.1152/ajpheart.00015.2009. doi:10.1152/ajpheart.00015.2009. [DOI] [PubMed] [Google Scholar]
- Pillai S., Kovacs M., Chellappan S. Regulation of vascular endothelial growth factor receptors by Rb and E2F1: role of acetylation. Cancer Res. 2010;70:4931–4940. doi: 10.1158/0008-5472.CAN-10-0501. doi:10.1158/0008-5472.CAN-10-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzer B.M., Engelmann D. E2F1 apoptosis counterattacked: evil strikes back. Trends Mol. Med. 2013;19:89–98. doi: 10.1016/j.molmed.2012.10.009. doi:10.1016/j.molmed.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Qin G., Kishore R., Dolan C.M., et al. Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc. Natl Acad. Sci. USA. 2006;103:11015–11020. doi: 10.1073/pnas.0509533103. doi:10.1073/pnas.0509533103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revenko A.S., Kalashnikova E.V., Gemo A.T., et al. Chromatin loading of E2F-MLL complex by cancer-associated coregulator ANCCA via reading a specific histone mark. Mol. Cell. Biol. 2010;30:5260–5272. doi: 10.1128/MCB.00484-10. doi:10.1128/MCB.00484-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos S.C., Miguel C., Domingues I., et al. VEGF and VEGFR-2 (KDR) internalization is required for endothelial recovery during wound healing. Exp. Cell Res. 2007;313:1561–1574. doi: 10.1016/j.yexcr.2007.02.020. doi:10.1016/j.yexcr.2007.02.020. [DOI] [PubMed] [Google Scholar]
- Sapoznik S., Cohen B., Tzuman Y., et al. Gonadotropin-regulated lymphangiogenesis in ovarian cancer is mediated by LEDGF-induced expression of VEGF-C. Cancer Res. 2009;69:9306–9314. doi: 10.1158/0008-5472.CAN-09-1213. doi:10.1158/0008-5472.CAN-09-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A., Yeow W.S., Ertel A., et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Invest. 2010;120:4478–4492. doi: 10.1172/JCI44239. doi:10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simiantonaki N., Jayasinghe C., Michel-Schmidt R., et al. Hypoxia-induced epithelial VEGF-C/VEGFR-3 upregulation in carcinoma cell lines. Int. J. Oncol. 2008;32:585–592. [PubMed] [Google Scholar]
- Skobe M., Hawighorst T., Jackson D.G., et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 2001;7:192–198. doi: 10.1038/84643. doi:10.1038/84643. [DOI] [PubMed] [Google Scholar]
- Stanelle J., Stiewe T., Theseling C.C., et al. Gene expression changes in response to E2F1 activation. Nucleic Acids Res. 2002;30:1859–1867. doi: 10.1093/nar/30.8.1859. doi:10.1093/nar/30.8.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J.L., Yang P.C., Shih J.Y., et al. The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer Cell. 2006;9:209–223. doi: 10.1016/j.ccr.2006.02.018. doi:10.1016/j.ccr.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Su J.L., Yen C.J., Chen P.S., et al. The role of the VEGF-C/VEGFR-3 axis in cancer progression. Br. J. Cancer. 2007;96:541–545. doi: 10.1038/sj.bjc.6603487. doi:10.1038/sj.bjc.6603487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., Watabe T., Kato M., et al. Roles of vascular endothelial growth factor receptor 3 signaling in differentiation of mouse embryonic stem cell-derived vascular progenitor cells into endothelial cells. Blood. 2005;105:2372–2379. doi: 10.1182/blood-2004-07-2547. doi:10.1182/blood-2004-07-2547. [DOI] [PubMed] [Google Scholar]
- Tammela T., Zarkada G., Wallgard E., et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454:656–660. doi: 10.1038/nature07083. doi:10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.