

Abstract
Human mutations in the gonadotropin releasing hormone receptor (GNRHR) gene cause autosomal recessive, normosmic idiopathic hypogonadotropic hypogonadism (IHH). At least 19 different mutations have been identified in this G-protein coupled receptor, which consist mostly of missense mutations. The Gln106Arg and Arg262Gln mutations comprise nearly half of the identified alleles. Most mutations impair ligand binding and all compromise cell signaling events. Some of the mutations also adversely affect activation of gonadotropin subunit or Gnrhr gene promoters. Interestingly, a number of the mutant GnRHRs can be rescued in vitro from misfolding and degradation within the cell by the addition of a GnRHR antagonist IN3. Most affected patients have compound heterozygous GNRHR mutations that may cause either complete IHH (no evidence of puberty) or incomplete IHH (partial evidence of puberty), although some genotypes are associated with mild disease in some families and severe disease in others. GNRHR mutations also appear to cause constitutional delay of puberty, and one genotype (homozygosity for Gln106Arg) may be reversible in patients with IHH. Mutations in the human GNRHR have contributed greatly to the understanding of normosmic IHH, as well as the structure and function of the GnRHR.
INTRODUCTION
It is has been a decade since mutations in the human gonadotropin releasing hormone receptor gene (GNRHR) were first identified. The discovery that the receptor GNRHR, rather than the ligand GNRH1, was the first gene found to cause autosomal recessive idiopathic hypogonadotropic hypogonadism (IHH) [1, 2] was somewhat surprising. This was particularly true given that the hypogonadal mouse, a naturally occurring mouse with an IHH phenotype, demonstrated a Gnrh1 gene deletion[3]. In this mouse, gonadal function was restored by transplanting functional gonadotropin releasing hormone (GnRH) neurons or by gene therapy using the wild type Gnrh1 cDNA [4]. In this review, we will discuss the structure and function of the human GnRHR and describe each of the different identified GNRHR mutations in humans, along with their functional analyses. Lastly, we will examine the genotype/phenotype correlations and the prevalence of GNRHR gene mutations in patients with IHH/KS.
THE HUMAN GNRHR—STRUCTURE AND FUNCTION
The human GNRHR gene spans 18.7 kb of genomic sequence on chromosome 4q13.2 and consists of three exons [5-7]. The GnRHR protein is a G-protein coupled receptor (GPCR) that is expressed on the cell surface of pituitary gonadotropes [8]. The GNRHR gene is also expressed in many other tissues including the placenta, brain, ovary, testis, endometrium, myometrium, prostate, kidney, and liver [9]. The encoded 328 amino acid protein (NP 000397) contains the characteristic seven transmembrane domains (TMD), along with an N-terminal extracellular domain (ECD), three extracellular (ECL) and three intracellular (ICL) loops, but notably lacks a large intracellular cytoplasmic tail (Figure 1). The extracellular domains and/or transmembrane domains are involved in the formation of the ligand-binding pocket. Amino acid residues Asp98, Asn102, Lys121, Asp302, and Asn305 are thought to directly contact GnRH, [10] whereas the cytoplasmic regions interact with G proteins and other intracellular regulatory proteins. Pulsatile GnRH released from hypophyseal-portal capillaries is delivered to the pituitary where it interacts with the GNRHR to initiate a cascade of events culminating in the synthesis and secretion of gonadotropins follicle stimulating hormone (FSH) and luteinizing hormone (LH). Gonadotropins then stimulate the testes or ovaries to produce sex steroids and gametes.
Figure 1.
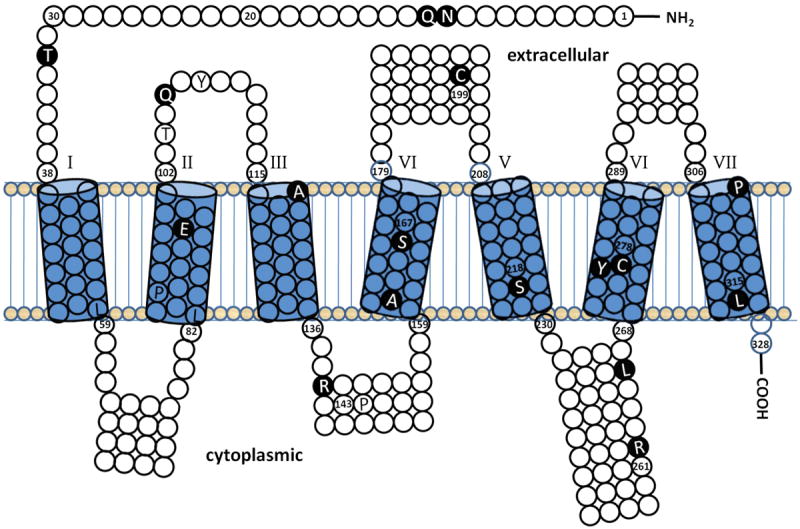
Shown is a topology diagram of the amino acid sequence of the human GnRHR protein that was generated using the HMMTOP transmembrane topology prediction server [53, 54]. The predicted seven transmembrane domains are indicated as cylinders, numbered 1-7. Also shown are the predicted extracellular loops above and the intracellular loops below the transmembrane domains. All missense mutations confirmed by in vitro analysis and one nonsense mutation (L314X) are depicted in white amino acid abbreviations on black circles. Missense mutations not studied in vitro are depicted in black letters on white or shaded circles. Residue numbers are given at the boundary of transmembrane domains and at other locations near mutated residues.
Once the decapeptide GnRH binds to the GnRH receptor, cell signaling involves the G proteins, typically Gαq, which activates phospholipase Cβ resulting in the production of second messengers inositol trisphosphate (IP3), diacyl glycerol, and calcium with subsequent secretion of both FSH and LH.[9] However, additional G-proteins—Gαs (pituitary) and Gαi (cancer cells)—may be involved in GnRH signaling depending upon the cell types [ 9]. Gαs stimulates and Gαi inhibits the adenylate cyclase/cyclic adenosine monophosphate (cAMP) pathway. There is also evidence that the pulse frequency of GnRH plays a large role in determining which of the two gonadotropins is released, with faster GnRH pulses preferentially stimulating LH release and slower pulses favoring FSH secretion [11]. Although a second GNRHR gene has been identified in many species, in the human ortholog on chromosome 1q21, the open reading frame is disrupted by a frameshift resulting in a truncated protein. However, it is interesting that this gene is transcriptionally active and may interfere with normal processing of the type I receptor in the nucleus, endoplasmic reticulum, or Golgi [12].
THE FIRST IDENTIFICATION OF GNRHR MUTATIONS
Mutations in the GNRHR gene were identified by two independent groups utilizing different strategies.[1, 2] De Roux et al[1] hypothesized that partial loss of function mutations would be expected in patients with incomplete pubertal development similar to those seen with luteinizing hormone receptor (LHR) and thyroid stimulating hormone receptor (TSHR) mutations. They studied a 22-year-old male with delayed puberty at 18, who had decreased libido, small (8 cc) testes (normal testes size 15-25cc), and a small penis. His testosterone was low at 80 ng/dL (normal 260-690 ng/dL) and his gonadotropins were low. LH pulse frequency was normal, but the amplitude was decreased. His semen analysis revealed a normal sperm concentration of 39 million/cc with low motility (5%). He was found to harbor compound heterozygous GNRHR mutations Gln106Arg/Arg262Gln (Figure 1), which were then studied in vitro upon transfection into COS-7 cells. The Gln106Arg mutation in ECL1 markedly impaired GnRH agonist binding to the receptor. Both the Gln106Arg and the Arg262Gln in ICL3 lowered IP3 production by 50% and the IP3 efficiency 50-fold in that 50 times the dose of GnRH agonist was needed to effect a 50% rise in IP3 production [1]. This male had a sister, who had thelarche at age 14 years and menarche at age 18 (with only one period). Similar to her brother, she exhibited compound heterozygosity for these two mutations [9]. The unaffected parents were heterozygous for either of the two mutations and an unaffected daughter was heterozygous, indicating autosomal recessive inheritance.
Just over a month later, Layman et al[2] identified a family with compound heterozygous GNRHR mutations. They reasoned that since GNRH1 mutations had not been identified and that some IHH patients were resistant to GnRH treatment, the GNRHR gene represented a promising candidate gene for mutations in IHH. Forty-six normosmic IHH patients were studied using polymerase chain reaction (PCR) and denaturing gradient gel electrophoresis (DGGE). Two variant fragments were identified in one family with four severely affected IHH patients [2]. Upon DNA sequencing, compound heterozygous GNRHR mutations Arg262Gln/Tyr284Cys (Figure 1) were identified in all four affected patients, but not in unaffected sibs (parents were deceased), indicating autosomal recessive inheritance.
The proband was a 30-year-old female without any breast development (Tanner 1). She had two sisters, ages 17 and 21, both with Tanner 1 breasts, and a brother who did not shave and had a serum testosterone of 75ng/dL (he did not permit a genital exam). Neither mutation had any effect upon receptor binding; however, similar to De Roux et al[1], the Arg262Gln impaired signal transduction following transfection using COS-1 cells. The Arg262Gln demonstrated a 25% reduction in receptor expression, a 40% reduction in IP3 production, and a 10-fold increase in the amount of GnRH agonist needed to effect a 50% maximal increase in IP3 production. The Tyr284Cys in TMD7 had an even more pronounced detrimental effect upon GnRH signaling with an 80% reduction in receptor expression, a 75% reduction in IP3 production, and a 20-fold increase in the amount of GnRH agonist to result in a 50% maximal IP3 response [2].
These two publications set the tone for those which followed, underscoring the phenotypic variation in mutations. In the first family, both affected members displayed a phenotype of incomplete IHH, whereby some evidence of sexual development and reproductive potential was present [1]. This family had the combination of a mutation in the ECL1 (Gln106Arg) that affected binding and signal transduction and a mutation in ICL3 that impaired signal transduction. However, in the second family, there was no evidence of any pubertal development in any of the four affected individuals, as they had complete IHH [2]. Both alleles impaired signal transduction that resulted in a more severe phenotype. Two of the three mutations identified, Gln106Arg and the Arg262Gln, have been determined to represent the most common alleles in all reported cases of human GNRHR mutations [13].
ADDITIONAL GNRHR MUTATIONS IN HUMANS
Following these two initial reports [1, 2], our review of the literature revealed a total of 19 different GNRHR alleles, all confirmed by functional analyses, that have been described in IHH patients (Table 1; Figure 1) [13-35]. Of these confirmed mutant alleles, 17 were missense mutations, one was a nonsense mutation, and one was a splice site mutation (Table 1). In addition, five missense mutations were identified which have not been studied in vitro, and are therefore, considered presumptive mutations (Table 1) [36-38]. Of the confirmed mutations, ten were found in exon 1, two were identified in exon 2, and six were seen in exon 3. The splice mutant was identified at the splice donor site in intron 1, which resulted in exon skipping [27]. Although concentrated in two exons, the mutations seem to be spread throughout most of the different domains of the protein—the ECD, ECL 1 and 2, TMD 2-7, and ICL 3. Nevertheless, two mutations appear to be particularly common, even among different ethnic groups: Gln106Arg represents 13 of 48 (27.1%) mutant alleles and Arg262Gln constitutes 8/46 (17.4%) mutant alleles (Table 1).[13] It is interesting that only one new causative mutation, Arg139Cys [34], has been identified in the GNRHR since the previous study of Bhagavath et al [13] in 2005.
Table 1.
All reported (n=19) different human GNRHR mutant alleles to date are shown, including mutation location in the gene and protein, the number of times the allele has been reported, and the effects upon binding and IP3 signaling.
| Missense Mutations | ||||||||
|---|---|---|---|---|---|---|---|---|
| No. | Author | Exon | Allele | n | Protein | Binding | IP3 Signaling | Other |
| 1 | [23] | 1 | Asn10Lys | 1 | ECD | ↓ | ↓ | |
| 2 | [33] | 1 | Asn10Lys+Gln11Lys | 1 | ECD | ↓ | ↓ | |
| 3 | [21, 29] | 1 | Thr32lle | 1 | ECD | ↓ | ↓ | |
| 4 | [20, 25] | 1 | Glu90Lys | 2 | TMD2 | ↓ | ↓ | |
| 5 | [1, 17, 21-24] | 1 | Gln106Arg | 13 | ECL1 | ↓ | ↓ | |
| 6 | [14, 26] | 1 | Ala129Asp | 2 | TMD3 | ↓ | ↓ | |
| 7 | [23, 31] | 1 | Arg139His | 4 | TMD3/ICL2 | Absent | ↓ | ↓ receptor # |
| 8 | [34] | 1 | Arg139Cys | 2 | TMD3/ICL2 | Normal | ↓ | |
| 9 | [16] | 1 | Ser168Arg | 2 | TMD4 | ↓ | ↓ | |
| 10 | [32] | 1 | Ala171Thr | 1 | TMD4 | Absent | ↓ | |
| 11 | [21, 29] | 2 | Cys200Tyr | 1 | ECL2 | ↓ | ↓ | |
| 12 | [15] | 2 | Gln106Arg/Ser217Arg | 1 | ECL1/TMD5 | ↓ | ↓ | |
| 13 | [1, 2, 14, 17, 21, 26, 35] | 3 | Arg262Gln | 8 | ICL3 | Normal | ↓ | |
| 14 | [21, 29] | 3 | Leu266Arg | 2 | ICL3 | ↓ | ↓ | |
| 15 | [21, 29] | 3 | Cys279Tyr | 2 | TMD6 | ↓ | ↓ | |
| 16 | [2] | 3 | Tyr284Cys | 1 | TMD7 | Normal | ↓ | |
| 17 | [33] | 3 | Pro320Leu | 1 | TMD7 | Absent | ↓ | |
| Other Mutations | ||||||||
| 18 | [27] | IVS1 | IVS1-1G→A | 2 | Exon2 skip | ND | ND | |
| 19 | [18] | 3 | Leu314X | 1 | TMD7 | ↓ | ↓ | |
| Other Reported Alleles Not Studied In Vitro | ||||||||
| 20 | [37] | 1 | Leu83Val | 1 | ND | ND | ||
| 21 | [37] | 1 | Pro96Ser | 1 | ND | ND | ||
| 22 | [36] | 1 | Thr104Ile | 1 | ND | ND | ||
| 23 | [36] | 1 | Tyr108Cys | 1 | ND | ND | ||
| 24 | [38] | 1 | Pro146Ser | 2 | ND | ND | ||
Missense mutations are ordered by codon number. Five additional alleles (#20-24) have also been reported, but not studied in vitro. Only newly identified alleles were included from Cerrato et al[37] since it was not possible to determine which patients with known causative alleles had been reported previously.
ND= not done;ECD = extracellular domain; ECL = extracellular loop, TMD = transmembrane domain; ICL = intracellular loop.
All of the reported GNRHR mutations were identified by PCR-based methods and DNA sequencing. However, these techniques do not detect heterozygous intragenic deletions. Deletions (and duplications) would be missed by PCR and DNA sequencing if a deletion occurred outside the boundary defined by the primers since the other (normal) allele would be amplified. A number of techniques have been reported to determine if heterozygous intragenic deletions occur, such as multiplex ligation-dependent amplification (MLPA) [39]. However, in a study of 100 IHH/KS using MLPA, no intragenic deletions were identified in the GNRHR, or any of the other autosomal genes studied (GNRH1, GPR54, NELF, or FGFR1), suggesting that deletions are not as common as in some other genetic disorders [39].
FUNCTIONAL ANALYSIS OF HUMAN GNRHR MUTATIONS
Most GNRHR mutations were studied in vitro utilizing transfection of the wild type and mutant separately into COS cells since they do not express the GNRHR [1, 2]. Ligand binding studies were performed upon membrane fractions by radioreceptor assays using labeled and unlabeled GnRH agonist [1, 2]. IP3 radioreceptor studies were also performed on cytosolic components using labeled and unlabeled IP3 to determine second messenger production and efficiency [1, 2].
More recently, additional studies have been performed on some GNRHR mutants to determine more clearly the mechanisms involved in their dysfunction. Cellular localization has been analyzed using immunofluorescence in GH3 cells transfected with the wild type or mutant receptor [29]. The effect of the mutant receptor has also been studied with regard to the ability to activate promoters of the gonadotropin genes in GH3 cells, including the Cga (chorionic gonadotropin-alpha), follicle stimulating hormone-beta (Fshb), and luteinizing hormone-beta (Lhb) promoters using the corresponding promoter sequence cloned upstream to luciferace (Luc) reporter sequence [29]. Cyclic AMP dependent activity has been studied by cloning the cAMP response element (CRE) upstream to Luc, and the MAPK pathway has been studied by determining extracellular signal-regulated kinase (ERK) phosphorylation by western blot analysis [30]. Finally, the effect of the mutant GNRHR upon Gnrhr/Luc promoter activity was studied to determine the effects of the mutants upon its own expression [29].
Fourteen of 19 (73.6%) of the reported GNRHR mutations interfere with ligand binding, while all interfere with affect IP3 signal transduction (Table 1). If binding is reduced, signal transduction would also be predicted to be impaired. One of the 17 missense mutations was found to markedly reduce cell surface expression, but not binding. For this Arg139Cys mutation, expression was increased by the administration of IN3 (a GnRHR antagonist), which has been reported to rescue degradation when mutant, misfolded GnRHRs have been misrouted to the endoplasmic reticulum rather than the cell surface. Once there was a sufficient increase in mutant receptors, ligand binding assays were found to be normal [34]. Even though 14 of the 19 mutations interfered with binding to GnRH agonist, it is interesting that none of them appear to be residues known to bind to GnRH [9].
In addition to the phospholipase C pathway via Gαq, GnRH signaling may occur by the Gαs pathway via protein kinase A (PKA)/cAMP pathway and extracellular signal-regulated kinase (ERK) activation [9]. Although both the Gln106Arg and Arg262Gln mutations had similar detrimental effects upon stimulation of the Fshb (14 and 15-fold, respectively) and the Lhb (8.8 and 8.1-fold, respectively) promoters, Arg262Gln had a more significant effect upon Cga and Cre promoter activity, and phosphorylation of ERK (Table 2) [30]. These findings indicate that both mutations may have differential effects upon these pathways, further providing a better mechanistic understanding of the pathogenesis of these mutations. In addition, both Leu266Arg and Cys279 were unable to stimulate gonadotropin subunit or GNRHR promoter activity in vitro (Table 2), while Cys200Tyr and Thr32Ile were able to stimulate gonadotropin subunit and GNRHR promoter activity, demonstrating the complexity of these mutants upon different signaling pathways [29].
Table 2.
Other in vitro analyses of the mutant GNRHRs reported in patients with normosmic IHH.
| No. | Allele | Protein | Fshb P | Lhb P | Cga P | Gnrhr P | Cre P | ERK Phos | IN3 Rescue |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Asn10Lys | ECD | Yes[42] | ||||||
| 2 | Asn10Lys+Gln11Lys | ECD | |||||||
| 3[29] | Thr32lle | ECD | ↓4 | ↓2.2 | ↓3 | ↓3.3 | ↓1.5 | ↓11.8 | Yes[41] |
| 4 | Glu90Lys | TMD2 | Yes[41] | ||||||
| 5[30] | Gln106Arg | ECL1 | ↓14 | ↓8.8 | ↓3.8 | ↓2.2 | ↓2.7 | ↓2.7 | Yes[42] |
| 6 | Ala129Asp | TMD3 | Partial[42] | ||||||
| 7 | Arg139His | TMD3/ICL2 | Partial[42] | ||||||
| 8 | Arg139Cys | TMD3/ICL2 | |||||||
| 9 | Ser168Arg | TMD4 | No[42] | ||||||
| 10 | Ala171Thr | TMD4 | |||||||
| 11[29] | Cys200Tyr | ECL2 | None | ↓3.4 | ↓4.5 | None | ↓3.9 | None | Partial[41, 42] |
| 12 | Ser217Arg | ECL1/TMD5 | No[42] | ||||||
| 13[30] | Arg262Gln | ICL3 | ↓15 | ↓8.1 | ↓10.6 | ↓3.7 | ↓6.7 | ↓5.1 | Yes[41] |
| 14[29] | Leu266Arg | ICL3 | None | None | None | None | None | None | Partial[41, 42] |
| 15[29] | Cys279Tyr | TMD6 | None | None | None | None | None | None | Partial[41, 42] |
| 16 | Tyr284Cys | TMD7 | Yes[42] | ||||||
| 17 | Pro320Leu | TMD7 |
The fold reduction in sensitivity (i.e., the fold increase in the ED50) to the promoters (Fshb, Lhb, Cga, Cre) are show, as well as the effects of the mutant receptor upon ERK phosphorylation with GnRH agonist administration and whether or not rescue occurred with the administration of the GNRHR antagonist IN3. Numbering of the mutations is the same as in Table 1. Only those studied are shown. References are included in the mutation No. column.
Interestingly, studies utilizing site-directed mutagenesis and in vitro analysis have shown that some GNRHR mutations appear to act as dominant negative mutations [40]. Since rescue of the Glu90Lys mutation was accomplished by either deleting Lys191 or adding a carboxy-terminal sequence [25], 13 of the human GNRHR mutations were studied further [41, 42]. Most (11 of 13) of the mutations shown in Table 1 can be rescued using the GnRHR antagonist IN3, which the authors suggested corrects improper trafficking of the misfolded, mutant receptors from degradation and allows them to assume their normal localization in the cell membrane (Table 2). These findings do suggest that perhaps several different mechanisms could be involved in the pathophysiology of GNRHR mutations. Of note, a Gnrhr mutant mouse (created by ENU mutagenesis) has a phenotype consistent with IHH, but the mutant was not able to be rescued by IN3 [43].
Since most human GNRHR mutations are missense, we used SIFT (sorting intolerant from tolerant) [44], which is used to compare all of the amino acid residues from different species to assign a probability of whether an amino acid change is likely to be tolerated or not (Table 3). If the P<0.05 for a particular amino acid substitution, then it is predicted to not be tolerated. It is interesting that SIFT analysis predicted that only 9/17 of the reported missense mutations would not be tolerated, and therefore would be predicted to impair protein function. This analysis was based upon the comparisons of 14 available species for the which the GNRHR has been cloned. If more distant species are excluded, 12/17 (70.6%) of the demonstrated functional GNRHR mutations would have been predicted to be detrimental. Only one missense mutation (Ala184Pro) was found in SNP database, and as expected, it was predicted to be tolerated. SIFT has been successfully used by a number of investigators to identify potential mutations to further study by functional assays [44-46], but its predictive value is somewhat limited in the human GNRHR gene.
Table 3.
SIFT analysis for the GNRHR missense mutations using the following 14 species: human, mouse, rat, cow, dog. boar, platypus, monkey, chimpanzee, chicken, rabbit, trout, sheep, and C. elegans.
| No. | Exon | Mutation | Letter Code Mutation | SIFT 1 | SIFT 2&3 | SIFT 4 | SIFT 5 |
|---|---|---|---|---|---|---|---|
| 1 | 1 | Asn10Lys | N10K | I | |||
| 2 | 1 | Gln11Lys | Q11K | T | |||
| 3 | 1 | Thr32lle | T32I | I | |||
| 4 | 1 | Glu90Lys | E90K | T | T | I | I |
| 5 | 1 | Gln106Arg | Q106R | T | |||
| 6 | 1 | Ala129Asp | A129D | I | |||
| 7 | 1 | Arg139His | R139H | I | |||
| 8 | 1 | Arg139Cys | R139C | I | |||
| 9 | 1 | Ser168Arg | S168R | T | |||
| 10 | 1 | Ala171Thr | A171T | I | |||
| 11 | 2 | Cys200Tyr | C200Y | T | I | I | I |
| 12 | 2 | Ser217Arg | S217R | T | T | T | I |
| 13 | 3 | Arg262Gln | R262Q | I | |||
| 14 | 3 | Leu266Arg | L266R | I | |||
| 15 | 3 | Cys279Tyr | C279Y | T | |||
| 16 | 3 | Tyr284Cys | Y284C | T | |||
| 17 | 3 | Pro320Leu | P320L | I | |||
| Mutations Not Studied In Vitro | |||||||
| 18 | 1 | Leu83Val | L83V | I | |||
| 19 | 1 | Pro96Ser | P96S | I | |||
| 20 | 1 | Thr104Ile | T104I | I | |||
| 21 | 1 | Tyr108Cys | Y108C | T | |||
| 22 | 1 | Pro146Ser | P146S | T | I | I | I |
SIFT 1 includes all 14 orthologs; SIFT 2 is without C. elegans; SIFT 3 is without C. elegans and trout; SIFT 4 is without C. elegans, trout, and chicken; SIFT 5 is without C. elegans, trout, chicken, & platypus.
GENOTYPE/PHENOTYPE CORRELATION IN GNRHR MUTATIONS
In our analysis of reported GNRHR mutations, we identified 17 different reported genotypes, some of which were reported in two or three different families (Table 4). These genotypes were grouped based upon severity into complete IHH (no evidence of puberty), incomplete IHH (partial evidence of puberty), constitutional delay of puberty, or not reported. As shown in Table 4, 9/17 (52.9%) different genotypes (seen in 11 families) resulted in a phenotype of complete IHH, and 5/17 (29.4%) from 8 different families had incomplete IHH. Two genotypes (Gln106Arg/Arg262Gln and Gln106Arg/Leu266Arg) identified in four unrelated families could cause either complete or incomplete IHH. It is interesting that the one family with compound heterozygous Gln106Arg/Arg262Gln mutations and complete IHH also had a heterozygous FGFR1 mutation (Table 4) [47].
Table 4.
Genotype/phenotype correlations are shown for patients with GNRHR mutations that have been supported by functional analysis.
| No | Author | Genotype | Phenotype | n | Sex | Ethnicity |
|---|---|---|---|---|---|---|
|
| ||||||
|
Severe Phenotype—Complete IHH
| ||||||
| 1 | [14, 26] | Ala129Asp/Arg262Gln | Complete | 2 | M,F | NA |
| Complete | M | Caucasian | ||||
|
| ||||||
| 2 | [23, 31] | Arg139His (HMZ) | Complete | 2 | F | Brazilian |
| Complete | M | NA | ||||
|
| ||||||
| 3 | [18] | Leu314X/Gln106Arg | Complete | 1 | F | NA |
|
| ||||||
| 4 | [27] | IVS1, G-A, -1 | Complete | 1 | F | Indian |
|
| ||||||
| 5 | [2] | Arg262Gln/ Tyr284Cys | Complete | 1 | M,F | Caucasian |
|
| ||||||
| 6 | [16] | Ser168Arg (HMZ) | Complete | 1 | M | NA |
|
| ||||||
| 7 | [21] | Thr32Ile/Cys200Tyr | Complete | 1 | M | NA |
|
| ||||||
| 8 | [32] | Ala171Thr/Gln106Arg | Complete | 1 | M | Caucasian |
|
| ||||||
| 9 | [20, 25] | Glu90Lys (HMZ) | Complete | 1 | M | Mexican-mestizo |
|
| ||||||
|
Intermediate Phenotype within Families
| ||||||
| 10 | [1, 17, 21] | Gln106Arg/Arg262Gln | Incomplete | 2 | M,F | NA |
| Complete* | F,F | NA | ||||
|
| ||||||
| 11 | [21] | Gln106Arg/Leu266Arg | Incomplete | 2 | F | NA |
| Complete | F | Caucasian | ||||
|
| ||||||
|
Milder Phenotype—Incomplete IHH
| ||||||
| 12 | [21, 22, 24] | Gln106Arg (HMZ) | Incomplete# | 3 | M | NA |
| Incomplete | F | NA | ||||
| Incomplete$ | M | NA | ||||
|
| ||||||
| 13 | [15] | Arg262Gln/Gln106+Ser217Arg | Incomplete | 1 | M,F | NA |
|
| ||||||
| 14 | [23] | Asn10Lys/Gln106Arg | Incomplete | 1 | M,F | Brazilian |
|
| ||||||
| 15 | [33] | Asn10Lys+Gln11Lys/Pro320Leu | Incomplete | 1 | F | Caucasian |
|
| ||||||
| 16 | [35] | Arg262Gln (HMZ) | Incomplete or CDP | 1 | M, M | Asian Indian |
|
| ||||||
|
Unknown Severity
| ||||||
| 17 | [21] | Cys279Tyr (HMZ) | Unknown | 1 | M | NA |
They are categorized on the basis of disease severity (Complete IHH; Complete or Incomplete IHH; Incomplete IHH); the number of times the specific genotype has been reported in unrelated families, the sex of the proband (M=male; F=female) within the family, and ethnicity.
NA=not available. CDP=constitutional delay of puberty
Patient also had a heterozygous FGFR1 mutation. [47]
Patient had a spontaneous pregnancy (and therefore reversal).[24]
Given the small number of affected individuals with these different genotypes, it is difficult to make precise genotype/phenotype correlations. However, some genotype/phenotype correlations become apparent when examining Table 4. Perhaps the most transparent is the Gln106Arg genotype reflecting mild disease. Of the 11 families with complete IHH, only two (2/11) have a Gln106Arg allele, while five of the seven families with incomplete IHH have this allele. In the three unrelated probands who had homozygosity for the Gln106Arg allele, some degree of sexual development was present in all, suggesting that this is a mild allele.
Two unrelated probands demonstrating compound heterozygosity for the Ala129Asp/Arg262Gln had complete IHH, lacking any identifiable signs of steroid production. In the family described by Caron et al. [14], two affected males had micropenis and bilateral cryptorchidism, and the female completely lacked breast development. Layman et al [26] identified this same genotype in another unrelated male with complete IHH, but this affected male had prepubertal-sized, bilaterally descended testes with some spermatogenesis response to gonadotropin stimulation. Both unrelated probands with homozygosity for Arg139His also presented with complete IHH (Table 1). The other genotypes have only been reported once, making genotype/phenotype comparisons difficult.
As is common in most genetic diseases, clinical heterogeneity in phenotype was observed within families with GNRHR mutations. Some affected patients within the same family may show some signs of pubertal development, while others exhibit a total lack of pubertal development (complete IHH) [13]. Although all four sibs with Arg262Gln/Tyr284Cys compound heterozygous mutations had a complete absence of pubertal development, the response to exogenous GnRH varied among them [26]. This occurred despite the demonstration that both mutations markedly impaired IP3 signal transduction (20-fold decrease for Tyr284Cys and 10-fold decrease for Arg262Gln) [2]. When a second dose of GnRH was given to two of these patients, all FSH and LH levels were higher than the responses after the initial dose, suggesting that some priming and function of the receptor occurs with these mutant GNRHRs [26]. Interestingly, the affected male also had a ring chromosome 21 and there was also another sister with Down syndrome, whose pubertal status was not ascertained [2, 19]. This variation in phenotype in different families or even within the same family is likely influenced by other modifier genes, epigenetic events, and environmental factors.
Other investigators have reported successful priming of the GnRHR and even ovulation and pregnancy with exogenous GnRH [17, 24] or spontaneously [24] has been reported in a small number of patients. Although term deliveries have been reported, the risk of spontaneous abortion is also high. One female with compound heterozygosity for Gln106Arg/Arg262Gln had three children with gonadotropin stimulation, but her sister had three spontaneous abortions [17]. Another patient with homozygosity for the Gln106Arg mutation had several miscarriages and no term deliveries [24]. Nevertheless, term pregnancies have been reported in patients with GNRHR mutations, suggesting that receptor function is not mandatory for human pregnancy.
Now that the most severe phenotypes have been examined, it is reasonable to consider patients with a less severe phenotype. Lin al [35] described a homozygous Arg262Gln GNRHR mutation in a patient with constitutional delay of puberty (CDP) and his brother, who could either have CDP or IHH (this could be determined when he becomes age 18). It is less clear if GNRHR mutations cause adult-onset IHH, but this does appear to be reasonable. At least one IHH patient with homozygosity for Arg106Gln GNRHR mutation has been reported to have a spontaneous reversal of his IHH when treatment was discontinued [48]. This phenomenon was also reported in a another patient with Gln106Arg homozygosity, who had reversal with a spontaneous pregnancy [24]. These very interesting findings suggest that perhaps IHH patients, particularly with mild mutations, could have their steroid treatment discontinued periodically to determine if pulsatile gonadotropin responses ensue without treatment.
PREVALENCE OF HUMAN GNRHR MUTATIONS
GNRHR mutations cause autosomal recessive, normosmic IHH. Compound heterozygous GNRHR mutations were first found in a single proband (and affected siblings) from a total of 46 normosmic IHH patients for a prevalence of 2.2%. When only females were considered, then 1/14 (7.1%) were affected [2]. When this study was updated to include a sample of 185 IHH/KS patients by Bhagavath et al [13], similar findings were seen. Findings from this largest study to date indicate that GNRHR mutations were found in 6-11% of IHH families demonstrating clear autosomal recessive inheritance [13]. Another potential indicator of an autosomal recessive disease would be to determine the prevalence of GNRHR mutations in all unrelated females studied. Of the 34 female IHH patients, two (5.9%) were affected, making the prevalence slightly higher in female patients [13]. If only normosmic females were considered, 2/18 (11.1%) had GNRHR mutations. No GNRHR mutations were found in any of the presumed autosomal dominant or X-linked recessive families. In another study by Beranova et al [21], 5/108 (4.6%) IHH patients had GNRHR mutations, including 5/48 (10.4%) with normosmic IHH, and none of 60 anosmic IHH patients [21].
In Table 5, findings from available studies suggest that the prevalence of GNRHR mutations in all IHH/KS patients ranges between 2-5%. If all patients (normosmic, anosmic/hyposmic, and unknown) are included, 1.5% of patients had GNRHR mutations. To date, there have been no reports of GNRHR mutations in anosmic or hyposmic IHH patients (n=125) [13, 21], indicating that GNRHR mutations are extremely rare in patients with disorders of olfaction. Overall, the prevalence of GNRHR mutations in normosmic IHH patients is 3.9%, ranging from 0-10.4% (Table 5). Although GNRHR mutations are inherited in an autosomal recessive fashion, this was not apparent from their pedigrees since no additional family members were affected. Since untreated IHH affects fertility, families typically do not contain a large number of affected individuals.
Table 5.
Studies of prevalence of GNRHR mutations.
| Author | Sample size of IHH/KS | Mutations in IHH/KS | Prevalence in Normosmic IHH | Method of Analysis |
|---|---|---|---|---|
| Bhagavath et al[13] | 185 | 3 | 3/85 (3.5%) | DGGE/ sequencing |
| Beranova et al[21] | 108 | 5 | 5/48 (10.4%) | TGGE/sequencing |
| Lanfranco et al[55] | 45 | 0 | 0/45 (0%) | SSCP/sequencing |
| Vagenakis et al[38] | 26 | 0 | 0/26 0 (0%) | DNA sequencing |
| Trarbach et al[56] | 17 | 0 | 0/5(0%) | DNA sequencing |
| Topaloglu et al | 22 | 1 | 1/22 (4.5%) | DNA sequencing |
| Total | 403 | 9/403 (1.5%) | 9/231 (3.9%) | |
| Determination of Heterozygous Deletions | ||||
| Pedersen-White et al[39] | 100 | No deletions | 0 | MLPA |
Although GNRHR mutations were initially thought to be the most common cause of normosmic IHH [1, 2, 13], it appears that the mutation frequency of two autosomal dominantly inherited genes FGFR1 (10%) [49, 50] and CHD7 (6%) appear more common and interestingly, cause similar frequencies of both normosmic IHH and Kallmann syndrome [45]. Autosomal recessive gene mutations have been reported in at least seven genes—GNRHR, LEP, LEPR, PCSK1, GPR54, and PROKR2, and PROK2, but only GNRHR mutations appear to be very common. Mutations in these other six genes have only been described in a few affected individuals [51, 52]. Mutations in the GNRHR gene constituted the first known autosomal recessive molecular cause of IHH [1, 2] and still play an important role in normosmic IHH.
Acknowledgments
Support by NIH grants HD33004 and HD040287 (L.C.L). We appreciate the help of Steven Walker, graduate student at MCG, for his preparation of Figure 1.
References
- 1.de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, Milgrom E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337:1597–602. doi: 10.1056/NEJM199711273372205. [DOI] [PubMed] [Google Scholar]
- 2.Layman LC, Cohen DP, Jin M, Xie J, Li Z, Reindollar RH, Bolbolan S, Bick DP, Sherins RJ, Duck LW, Musgrove LC, Sellers JC, Neill JD. Mutations in the gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism. Nat Genet. 1998;18:14–5. doi: 10.1038/ng0198-14. [DOI] [PubMed] [Google Scholar]
- 3.Mason AJ, Hayflick JS, Zoeller T, Young WS, Phillips HS, Nikolics K, Seeburg PH. A deletion truncating the gonadotropin-releasing hormone gene is responsible for hypogonadism in the hpg mouse. Science. 1986;234:1366–71. doi: 10.1126/science.3024317. [DOI] [PubMed] [Google Scholar]
- 4.Mason AJ, Pitts SL, Nikolics K, Szonyi E, Wilcox JN, Seeburg PH, Stewart TA. The hypogonadal mouse: reproductive functions restored by gene therapy. Science. 1986;234:1372–8. doi: 10.1126/science.3097822. [DOI] [PubMed] [Google Scholar]
- 5.Kakar SS, Musgrove LC, Devor DC, Sellers JC, Neill JD. Cloning, sequencing, and expression of human gonadotropin releasing hormone (GnRH) receptor. Biophys Biochem Res Commun. 1992;189:289–95. doi: 10.1016/0006-291x(92)91556-6. [DOI] [PubMed] [Google Scholar]
- 6.Fan NC, Jeung E-B, Peng C, Olofsson JI, Krisinger J, Leung PCK. The human gonadotropin-releasing hormone (GnRH) receptor gene: cloning, genomic organization and chromosomal assignment. Mol Cell Endocrinol. 1994;103:R1–R6. doi: 10.1016/0303-7207(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 7.Kaiser UB, Dushkin H, Altherr MR, Beier DR, Chin WW. Chromosomal location of the gonadotropin-releasing hormone receptor gene to human chromosome 4q13.1-q21.1 and mouse chromosome 5. Genomics. 1994;20:506–8. doi: 10.1006/geno.1994.1211. [DOI] [PubMed] [Google Scholar]
- 8.Fan NC, Peng C, Krisinger J, Leung PC. The human gonadotropin-releasing hormone receptor gene: complete structure including multiple promoters, transcription initiation sites, and polyadenylation signals. Mol Cell Endocrinol. 1995;107:R1–8. doi: 10.1016/0303-7207(94)03460-b. [DOI] [PubMed] [Google Scholar]
- 9.Bedecarrats GY, Kaiser UB. Mutations in the human gonadotropin-releasing hormone receptor: insights into receptor biology and function. Semin Reprod Med. 2007;25:368–78. doi: 10.1055/s-2007-984743. [DOI] [PubMed] [Google Scholar]
- 10.Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR. Gonadotropin-releasing hormone receptors. Endocr Rev. 2004;25:235–75. doi: 10.1210/er.2003-0002. [DOI] [PubMed] [Google Scholar]
- 11.Kaiser UB, Jakubowiak A, Steinberger A, Chin WW. Differential effects of gonadotropin-releasing hormone (GnRH) pulse frequency on gonadotropin subunit and GnRH receptor messenger ribonucleic acid levels in vitro. Endocrinology. 1997;138:1224–31. doi: 10.1210/endo.138.3.4968. [DOI] [PubMed] [Google Scholar]
- 12.Pawson AJ, Maudsley S, Morgan K, Davidson L, Naor Z, Millar RP. Inhibition of human type i gonadotropin-releasing hormone receptor (GnRHR) function by expression of a human type II GnRHR gene fragment. Endocrinology. 2005;146:2639–49. doi: 10.1210/en.2005-0133. [DOI] [PubMed] [Google Scholar]
- 13.Bhagavath B, Ozata M, Ozdemir IC, Bolu E, Bick DP, Sherins RJ, Layman LC. The prevalence of gonadotropin-releasing hormone receptor mutations in a large cohort of patients with hypogonadotropic hypogonadism. Fertil Steril. 2005;84:951–7. doi: 10.1016/j.fertnstert.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 14.Caron P, Chauvin S, Christin-Maitre S, Bennet A, Lahlou N, Counis R, Bouchard P, Kottler M-L. Resistance of hypogonadotropic patients with mutated GnRH receptor genes to pulsatile GnRH administration. J Clin Endocrinol Metab. 1999;84:990–6. doi: 10.1210/jcem.84.3.5518. [DOI] [PubMed] [Google Scholar]
- 15.de Roux N, Young J, Brailly-Tabard S, Misrahi M, Milgrom E, Chaison G. The same molecular defects of the gonadotropin-releasing hormone determine a variable degree of hypogonadism in affected kindred. J Clin Endocrinol Metab. 1999;84:567–72. doi: 10.1210/jcem.84.2.5449. [DOI] [PubMed] [Google Scholar]
- 16.Pralong FP, Gomez F, Castillo E, Cotecchia S, Abuin L, Aubert ML, Portmann L, Gaillard RC. Complete hypogonadotropic hypogonadism associated with a novel inactivating mutation of the gonadotropin-releasing hormone receptor. J Clin Endocrinol Metab. 1999;84:3811–6. doi: 10.1210/jcem.84.10.6042. [DOI] [PubMed] [Google Scholar]
- 17.Seminara SB, Beranova M, Oliveira LM, Martin KA, Crowley WF, Jr, Hall JE. Successful use of pulsatile gonadotropin-releasing hormone (GnRH) for ovulation induction and pregnancy in a patient with GnRH receptor mutations. J Clin Endocrinol Metab. 2000;85:556–62. doi: 10.1210/jcem.85.2.6357. [DOI] [PubMed] [Google Scholar]
- 18.Kottler ML, Chauvin S, Lahlou N, Harris CE, Johnston CJ, Lagarde JP, Bouchard P, Farid NR, Counis R. A new compound heterozygous mutation of the gonadotropin-releasing hormone receptor (L314X, Q106R) in a woman with complete hypogonadotropic hypogonadism: chronic estrogen administration amplifies the gonadotropin defect. J Clin Endocrinol Metab. 2000;85:3002–8. doi: 10.1210/jcem.85.9.6783. [DOI] [PubMed] [Google Scholar]
- 19.Layman LC, McDonough PG, Cohen DP, Maddox M, Tho SP, Reindollar RH. Familial gonadotropin-releasing hormone resistance and hypogonadotropic hypogonadism in a family with multiple affected individuals. Fertil Steril. 2001;75:1148–55. doi: 10.1016/s0015-0282(01)01782-4. [DOI] [PubMed] [Google Scholar]
- 20.Soderlund D, Canto P, de la Chesnaye E, Ulloa-Aguirre A, Mendez JP. A novel homozygous mutation in the second transmembrane domain of the gonadotrophin releasing hormone receptor gene. Clin Endocrinol (Oxf) 2001;54:493–8. doi: 10.1046/j.1365-2265.2001.01211.x. [DOI] [PubMed] [Google Scholar]
- 21.Beranova M, Oliveira LM, Bedecarrats GY, Schipani E, Vallejo M, Ammini AC, Quintos JB, Hall JE, Martin KA, Hayes FJ, Pitteloud N, Kaiser UB, Crowley WF, Jr, Seminara SB. Prevalence, phenotypic spectrum, and modes of inheritance of gonadotropin-releasing hormone receptor mutations in idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2001;86:1580–8. doi: 10.1210/jcem.86.4.7395. [DOI] [PubMed] [Google Scholar]
- 22.Pitteloud N, Boepple PA, DeCruz S, Valkenburgh SB, Crowley WF, Jr, Hayes FJ. The fertile eunuch variant of idiopathic hypogonadotropic hypogonadism: spontaneous reversal associated with a homozygous mutation in the gonadotropin-releasing hormone receptor. J Clin Endocrinol Metab. 2001;86:2470–5. doi: 10.1210/jcem.86.6.7542. [DOI] [PubMed] [Google Scholar]
- 23.Costa EM, Bedecarrats GY, Mendonca BB, Arnhold IJ, Kaiser UB, Latronico AC. Two novel mutations in the gonadotropin-releasing hormone receptor gene in Brazilian patients with hypogonadotropic hypogonadism and normal olfaction. J Clin Endocrinol Metab. 2001;86:2680–6. doi: 10.1210/jcem.86.6.7551. [DOI] [PubMed] [Google Scholar]
- 24.Dewailly D, Boucher A, Decanter C, Lagarde JP, Counis R, Kottler ML. Spontaneous pregnancy in a patient who was homozygous for the Q106R mutation in the gonadotropin-releasing hormone receptor gene. Fertil Steril. 2002;77:1288–91. doi: 10.1016/s0015-0282(02)03102-3. [DOI] [PubMed] [Google Scholar]
- 25.Maya-Nunez G, Janovick JA, Ulloa-Aguirre A, Soderlund D, Conn PM, Mendez JP. Molecular basis of hypogonadotropic hypogonadism: restoration of mutant (E(90)K) GnRH receptor function by a deletion at a distant site. J Clin Endocrinol Metab. 2002;87:2144–9. doi: 10.1210/jcem.87.5.8386. [DOI] [PubMed] [Google Scholar]
- 26.Layman LC, Cohen DP, Xie J, Smith GD. Clinical phenotype and infertility treatment in a male with hypogonadotropic hypogonadism due to mutations Ala129Asp/Arg262Gln of the gonadotropin-releasing hormone receptor. Fertil Steril. 2002;78:1317–20. doi: 10.1016/s0015-0282(02)04341-8. [DOI] [PubMed] [Google Scholar]
- 27.Silveira LF, Stewart PM, Thomas M, Clark DA, Bouloux PM, MacColl GS. Novel homozygous splice acceptor site GnRH receptor (GnRHR) mutation: human GnRHR “knockout”. J Clin Endocrinol Metab. 2002;87:2973–7. doi: 10.1210/jcem.87.6.8535. [DOI] [PubMed] [Google Scholar]
- 28.Bedecarrats GY, Kaiser UB. Differential regulation of gonadotropin subunit gene promoter activity by pulsatile gonadotropin-releasing hormone (GnRH) in perifused L beta T2 cells: role of GnRH receptor concentration. Endocrinology. 2003;144:1802–11. doi: 10.1210/en.2002-221140. [DOI] [PubMed] [Google Scholar]
- 29.Bedecarrats GY, Linher KD, Janovick JA, Beranova M, Kada F, Seminara SB, Michael Conn P, Kaiser UB. Four naturally occurring mutations in the human GnRH receptor affect ligand binding and receptor function. Mol Cell Endocrinol. 2003;205:51–64. doi: 10.1016/s0303-7207(03)00201-6. [DOI] [PubMed] [Google Scholar]
- 30.Bedecarrats GY, Linher KD, Kaiser UB. Two common naturally occurring mutations in the human gonadotropin-releasing hormone (GnRH) receptor have differential effects on gonadotropin gene expression and on GnRH-mediated signal transduction. J Clin Endocrinol Metab. 2003;88:834–43. doi: 10.1210/jc.2002-020806. [DOI] [PubMed] [Google Scholar]
- 31.Wolczynski S, Laudanski P, Jarzabek K, Mittre H, Lagarde JP, Kottler ML. A case of complete hypogonadotropic hypogonadism with a mutation in the gonadotropin-releasing hormone receptor gene. Fertil Steril. 2003;79:442–4. doi: 10.1016/s0015-0282(02)04667-8. [DOI] [PubMed] [Google Scholar]
- 32.Karges B, Karges W, Mine M, Ludwig L, Kuhne R, Milgrom E, de Roux N. Mutation Ala(171)Thr stabilizes the gonadotropin-releasing hormone receptor in its inactive conformation, causing familial hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2003;88:1873–9. doi: 10.1210/jc.2002-020005. [DOI] [PubMed] [Google Scholar]
- 33.Meysing AU, Kanasaki H, Bedecarrats GY, Acierno JS, Jr, Conn PM, Martin KA, Seminara SB, Hall JE, Crowley WF, Jr, Kaiser UB. GNRHR mutations in a woman with idiopathic hypogonadotropic hypogonadism highlight the differential sensitivity of luteinizing hormone and follicle-stimulating hormone to gonadotropin-releasing hormone. J Clin Endocrinol Metab. 2004;89:3189–98. doi: 10.1210/jc.2003-031808. [DOI] [PubMed] [Google Scholar]
- 34.Topaloglu AK, Lu ZL, Farooqi IS, Mungan NO, Yuksel B, O’Rahilly S, Millar RP. Molecular genetic analysis of normosmic hypogonadotropic hypogonadism in a Turkish population: identification and detailed functional characterization of a novel mutation in the gonadotropin-releasing hormone receptor gene. Neuroendocrinology. 2006;84:301–8. doi: 10.1159/000098147. [DOI] [PubMed] [Google Scholar]
- 35.Lin L, Conway GS, Hill NR, Dattani MT, Hindmarsh PC, Achermann JC. A homozygous R262Q mutation in the gonadotropin-releasing hormone receptor presenting as constitutional delay of growth and puberty with subsequent borderline oligospermia. J Clin Endocrinol Metab. 2006;91:5117–21. doi: 10.1210/jc.2006-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antelli A, Baldazzi L, Balsamo A, Pirazzoli P, Nicoletti A, Gennari M, Cicognani A. Two novel GnRHR gene mutations in two siblings with hypogonadotropic hypogonadism. Eur J Endocrinol. 2006;155:201–5. doi: 10.1530/eje.1.02198. [DOI] [PubMed] [Google Scholar]
- 37.Cerrato F, Shagoury J, Kralickova M, Dwyer A, Falardeau J, Ozata M, Van Vliet G, Bouloux P, Hall JE, Hayes FJ, Pitteloud N, Martin KA, Welt C, Seminara SB. Coding sequence analysis of GNRHR and GPR54 in patients with congenital and adult-onset forms of hypogonadotropic hypogonadism. Eur J Endocrinol. 2006;155(Suppl 1):S3–S10. doi: 10.1530/eje.1.02235. [DOI] [PubMed] [Google Scholar]
- 38.Vagenakis GA, Sgourou A, Papachatzopoulou A, Kourounis G, Papavassiliou AG, Georgopoulos NA. The gonadotropin-releasing hormone (GnRH)-1 gene, the GnRH receptor gene, and their promoters in patients with idiopathic hypogonadotropic hypogonadism with or without resistance to GnRH action. Fertil Steril. 2005;84:1762–5. doi: 10.1016/j.fertnstert.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 39.Pedersen-White JR, Chorich LP, Bick DP, Sherins RJ, Layman LC. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol Hum Reprod. 2008;14:367–70. doi: 10.1093/molehr/gan027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brothers SP, Cornea A, Janovick JA, Conn PM. Human loss-of-function gonadotropin-releasing hormone receptor mutants retain wild-type receptors in the endoplasmic reticulum: molecular basis of the dominant-negative effect. Mol Endocrinol. 2004;18:1787–97. doi: 10.1210/me.2004-0091. [DOI] [PubMed] [Google Scholar]
- 41.Janovick JA, Maya-Nunez G, Conn PM. Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J Clin Endocrinol Metab. 2002;87:3255–62. doi: 10.1210/jcem.87.7.8582. [DOI] [PubMed] [Google Scholar]
- 42.Leanos-Miranda A, Janovick JA, Conn PM. Receptor-misrouting: an unexpectedly prevalent and rescuable etiology in gonadotropin-releasing hormone receptor-mediated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002;87:4825–8. doi: 10.1210/jc.2002-020961. [DOI] [PubMed] [Google Scholar]
- 43.Pask AJ, Kanasaki H, Kaiser UB, Conn PM, Janovick JA, Stockton DW, Hess DL, Justice MJ, Behringer RR. A novel mouse model of hypogonadotrophic hypogonadism: N-ethyl-N-nitrosourea-induced gonadotropin-releasing hormone receptor gene mutation. Mol Endocrinol. 2005;19:972–81. doi: 10.1210/me.2004-0192. [DOI] [PubMed] [Google Scholar]
- 44.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, Kang GB, Rosenberger G, Tekin M, Ozata M, Bick DP, Sherins RJ, Walker SL, Shi Y, Gusella JF, Layman LC. Mutations in CHD7, Encoding a Chromatin-Remodeling Protein, Cause Idiopathic Hypogonadotropic Hypogonadism and Kallmann Syndrome. Am J Hum Genet. 2008 doi: 10.1016/j.ajhg.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raevaara TE, Korhonen MK, Lohi H, Hampel H, Lynch E, Lonnqvist KE, Holinski-Feder E, Sutter C, McKinnon W, Duraisamy S, Gerdes AM, Peltomaki P, Kohonen-Ccorish M, Mangold E, Macrae F, Greenblatt M, de la Chapelle A, Nystrom M. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129:537–49. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 47.Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, Plummer L, Hughes V, Seminara S, Cheng YZ, Li WP, Maccoll G, Eliseenkova AV, Olsen SK, Ibrahimi OA, Hayes FJ, Boepple P, Hall JE, Bouloux P, Mohammadi M, Crowley W. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest. 2007;117:457–63. doi: 10.1172/JCI29884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raivio T, Falardeau J, Dwyer A, Quinton R, Hayes FJ, Hughes VA, Cole LW, Pearce SH, Lee H, Boepple P, Crowley WF, Jr, Pitteloud N. Reversal of Idiopathic Hypogonadotropic Hypogonadism. N Engl J Med. 2007;357:863–73. doi: 10.1056/NEJMoa066494. [DOI] [PubMed] [Google Scholar]
- 49.Pitteloud N, Acierno JS, Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, Yialamas M, Hall JE, Grant E, Mohammadi M, Crowley WF., Jr Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2006;103:6281–6. doi: 10.1073/pnas.0600962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pitteloud N, Meysing A, Quinton R, Acierno JS, Jr, Dwyer AA, Plummer L, Fliers E, Boepple P, Hayes F, Seminara S, Hughes VA, Ma J, Bouloux P, Mohammadi M, Crowley WF., Jr Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol. 2006;254-255:60–9. doi: 10.1016/j.mce.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 51.Layman LC. Human gene mutations causing infertility. J Med Genet. 2002;39:153–61. doi: 10.1136/jmg.39.3.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beier DR, Dluhy RG. Bench and bedside--the G protein-coupled receptor GPR54 and puberty. N Engl J Med. 2003;349:1589–92. doi: 10.1056/NEJMp038155. [DOI] [PubMed] [Google Scholar]
- 53.Tusnady GE, Simon I. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J Mol Biol. 1998;283:489–506. doi: 10.1006/jmbi.1998.2107. [DOI] [PubMed] [Google Scholar]
- 54.Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849–50. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 55.Lanfranco F, Gromoll J, von Eckardstein S, Herding EM, Nieschlag E, Simoni M. Role of sequence variations of the GnRH receptor and G protein-coupled receptor 54 gene in male idiopathic hypogonadotropic hypogonadism. Eur J Endocrinol. 2005;153:845–52. doi: 10.1530/eje.1.02031. [DOI] [PubMed] [Google Scholar]
- 56.Trarbach EB, Baptista MT, Garmes HM, Hackel C. Molecular analysis of KAL-1, GnRH-R, NELF and EBF2 genes in a series of Kallmann syndrome and normosmic hypogonadotropic hypogonadism patients. J Endocrinol. 2005;187:361–8. doi: 10.1677/joe.1.06103. [DOI] [PubMed] [Google Scholar]