

Abstract
Bacterial cell division is a highly coordinated process that begins with the polymerization of the tubulin-like protein FtsZ at midcell. FtsZ polymerization is regulated by a set of conserved cell division proteins, including ZapA. However, a zapA mutation does not result in a clear phenotype in Bacillus subtilis. In this study, we used a synthetic-lethal screen to find genes that become essential when ZapA is mutated. Three transposon insertions were found in yvcL. The deletion of yvcL in a wild-type background had only a mild effect on growth, but a yvcL zapA double mutant is very filamentous and sick. This filamentation is caused by a strong reduction in FtsZ-ring assembly, suggesting that YvcL is involved in an early stage of cell division. YvcL is 25% identical and 50% similar to the Streptomyces coelicolor transcription factor WhiA, which induces ftsZ and is required for septation of aerial hyphae during sporulation. Using green fluorescent protein fusions, we show that YvcL localizes at the nucleoid. Surprisingly, transcriptome analyses in combination with a ChIP-on-chip assay gave no indication that YvcL functions as a transcription factor. To gain more insight into the function of YvcL, we searched for suppressors of the filamentous phenotype of a yvcL zapA double mutant. Transposon insertions in gtaB and pgcA restored normal cell division of the double mutant. The corresponding proteins have been implicated in the metabolic sensing of cell division. We conclude that YvcL (WhiA) is involved in cell division in B. subtilis through an as-yet-unknown mechanism.
INTRODUCTION
In most bacteria, cell division begins with the polymerization of the tubulin-like protein FtsZ into a ring-like structure at midcell. This Z-ring serves as a scaffold for other proteins that are required for septum biosynthesis. Several proteins support the assembly of the Z-ring. The protein FtsA contains a membrane anchor and anchors the Z-ring to the cell membrane (1, 2). The protein ZapA cross-links FtsZ-polymers and promotes polymer bundling (3, 4). In Gram-positive bacteria and cyanobacteria the protein SepF supports the bundling of FtsZ polymers, as well (5–8). The absence of SepF results in irregular division septa (9). EzrA is another conserved protein that interacts directly with FtsZ. This protein contains a transmembrane anchor at its N terminus. It can inhibit bundling of FtsZ polymers (10), but it is also involved in shuttling of the penicillin-binding protein PBP1, which is involved in cell wall synthesis, between the lateral wall and the division site (11). Assembly of the Z-ring at midcell is regulated in part by the Min and Noc systems (12). MinCD prevent polymerization of FtsZ close to cell poles, and mutations in minC or minD lead to minicell formation (13). MinC interacts with FtsZ and inhibits FtsZ polymerization (14). MinC is activated by MinD, which is anchored to the membrane through its amphipathic C terminus (15, 16). The polar localization of MinCD in Bacillus subtilis is determined by the proteins MinJ and DivIVA (17–19). The Z-ring does not mature in the area of the cell that is occupied by the nucleoid. In B. subtilis, this nucleoid occlusion mechanism is regulated by Noc that binds DNA and prevents FtsZ polymerization (20, 21). Finally, the frequency of cell division is related to cell mass and the glycosyltransferase UgtP has been shown to inhibit FtsZ assembly and to function as a metabolic regulator of Z-ring assembly (22). The activity of UgtP is determined by the concentration of UDP-glucose, which is abundantly produced in cells grown in rich media.
ZapA is conserved and present in most bacterial species (4). A zapA mutant is very sensitive to reduced FtsZ levels. High levels of ZapA counteract the division inhibition caused by overexpression of MinCD (4). The crystal structure of ZapA from Pseudomonas aeruginosa revealed a tetramer formed by two antiparallel dimers (23), and several biochemical studies have shown that ZapA is capable of promoting the lateral bundling of FtsZ protofilaments (4, 23, 24). A deletion of zapA does not result in a clear phenotype in B. subtilis, and it was postulated that ZapA is only required under circumstances when cells have difficulties forming a Z-ring (25). To find new cell division proteins that become essential when ZapA is absent, we used a synthetic-lethal screening and identified the protein YvcL.
Deletion of yvcL in a zapA mutant background results in very filamentous cells that are blocked in proper Z-ring formation. YvcL is a DNA-binding protein and shows strong homology with the protein WhiA from Streptomyces coelicolor. WhiA regulates the expression of several genes during sporulation, including ftsZ (26, 27). Surprisingly, transcriptome analyses of a yvcL mutant did not show a transcriptional effect on known cell division genes in B. subtilis, and the localization of YvcL binding sites on the genome gave no clear indication that YvcL functions as a transcription factor. Finally, a transposon screen revealed that the cell division defect of a yvcL zapA double mutant can be suppressed by inactivating the genes gtaB, pgcA, or ugtP. These genes are part of a metabolic sensor pathway that couples nutritional availability to cell division (28), providing further evidence that YvcL is involved in cell division in B. subtilis.
MATERIALS AND METHODS
Bacterial strains, plasmids, and oligonucleotides.
The strains and plasmids used in the present study are listed in Table S1 in the supplemental material. The oligonucleotide sequences are listed in Table S2 in the supplemental material. Bacterial cultures were grown at 30°C or 37°C in liquid Luria-Bertani (LB) medium, in competence medium, or on solid nutrient agar (Oxoid). DNA manipulations were carried out by standard techniques, and chromosomal DNA was purified by using phenol-chloroform extraction. Transformations of cells were carried out as described previously (29), and transformants were plated on nutrient agar supplemented with ampicillin (100 μg/ml), chloramphenicol (5 μg/ml), erythromycin (1 μg/ml), glucose (0.4 to 0.8%), kanamycin (5 μg/ml), spectinomycin (50 μg/ml), tetracycline (12 μg/ml), X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; 160 mg/ml), IPTG (isopropyl-β-d-thiogalactopyranoside; 0.5 to 1 mM), or xylose (0.0025 to 1%).
Construction of yvcL mutant strains.
To construct strain KS207, which contains an insertion of pMutin4 in yvcL, thereby disrupting yvcL expression, a 581-bp DNA fragment (primers yvcL-F1 and yvcL-R1) was digested with HindIII and BamHI, and cloned into pMutin4 digested with the same enzymes. The resulting plasmid pMutin4YvcL was integrated into B. subtilis by Campbell integration.
In strain KS400, yvcL is substituted by a kanamycin resistance cassette. DNA fragments outside of yvcL were amplified by using the primer pairs KS80/KS95 and KS84/KS83, resulting in 940- and 984-bp DNA fragments, respectively. The kanamycin resistance cassette was amplified from pBEST501 (30) with the primers km3 and km4. After the fragments were digested with BamHI and EcoRI and ligated, the mixture was transformed into B. subtilis.
Strain KS696 is a markerless yvcL mutant strain. To generate this strain, plasmid pMutin4YvcLKO was constructed. A 1,094-bp fragment, comprising the 3′ end of yvcK and the 5′ end of yvcL, was amplified by using the primers KS94 and yvcL-R1 and subsequently cloned into pMutin4 after digestion with HindIII and BamHI. Site-directed mutagenesis on the plasmid was performed using the primers KS120 and KS121 that introduced an EcoRI site and a stop codon in the beginning of yvcL (32 bp from start). The resulting plasmid pMut4YKO was transformed into B. subtilis cells and selected for erythromycin resistance and blue colonies on X-Gal plates. In order to excise the plasmid, one of the transformants was grown in competence medium for ∼10 generations without any antibiotic pressure and screened for the loss of the plasmid on plates with X-Gal. White and erythromycin-sensitive colonies were cultured, and the chromosomal DNA checked for the EcoRI site in yvcL. The yvcL region was also analyzed by sequencing.
The conditional mutant in yvcL-crh-yvcN was constructed by using plasmid pMutiYvcL. An ∼400-bp region upstream of yvcL was amplified using the primers KS94 and KS95. The fragments and plasmids were cut with HindIII and BamHI and cloned into pMutin4 plasmid digested with the corresponding enzymes. Plasmid pMutiYvcL was transformed into B. subtilis and selected for single-crossover events that led to insertion of the IPTG-inducible Pspac promoter upstream of yvcL (strain KS438). To allow tight regulation of yvcL expression, an extra copy of lacI was introduced by transforming plasmid pAPNC213 (31), which integrates into aprE locus, resulting in strain KS891.
To construct a xylose-inducible YvcL overexpression strain, the gfp gene was removed from plasmid pSG1729 by digestion with KpnI and XhoI, followed by insertion of the yvcL gene, which was amplified using the primers KS98 and yvcL-C5, and then digested with the same enzymes. The ensuing plasmid pSG1729YvcLΔGFP was transformed into B. subtilis, resulting in strain KS396.
Construction of the GFP-YvcL fusion protein.
To construct a xylose-inducible GFP-YvcL fusion, plasmid pSG1729YvcL was generated by ligating the amyE-integration vector pSG1729 and a 1,020-bp PCR fragment (primers yvcL-N5 and yvcL-N3), both digested with HindIII and EcoRI. Plasmid pSG1729YvcL was used for a QuikChange mutagenesis reaction with oligonucleotides HS410 and HS411 in order to introduce the A206K mutation in the green fluorescent protein (GFP) coding sequence to reduce protein dimerization (32). The resulting plasmid was verified by sequencing and named pSG1729YvcL(mGFP). Plasmid pSG1729YvcL(mGFP) was transformed into B. subtilis, resulting in strain PG732. The amyE::Pxyl-mgfpmut1-yvcL-spc allele of PG732 was combined with a yvcL mutation (strain KS400) so that mgfp-yvcL is the only copy of yvcL in the cell (strain KS736).
YvcL antibody.
To raise antibodies against YvcL, an expression vector pQE60EYvcL was created, which allows the expression of a C-terminally tagged YvcL-His6 fusion protein. To construct pQE60EYvcL, a 967-bp PCR product (primers KS89 and KS99) was cloned into pQE60E using BamHI and BglII. Escherichia coli XL1-Blue was used as a host for cloning and protein expression. The resultant E. coli XL1-Blue strain containing pQE60EYvcL (KS432) was used for the expression of the fusion protein as follows. Strain KS432 was inoculated into 300 ml of LB medium supplemented with ampicillin and 0.8% glucose (to allow tight repression). When the cell density reached an optical density at 600 nm (OD600) of ∼0.5, expression was induced with 1 mM IPTG for 3.5 h. The pelleted cells were resuspended in 1.2 ml of buffer (100 mM NaCl, 50 mM Tris-Cl [pH 8.0]) and sonicated. The inclusion bodies containing YvcL-His6 were separated by centrifugation and isolated on a 12% SDS-PAA gel. The protein band corresponding to YvcL-His6 was cut from the gel and used to raise rabbit polyclonal antiserum (Eurogentec, Ltd). For Western blotting, a 10,000×-diluted anti-YvcL serum was used.
Synthetic lethal screen.
To perform a synthetic lethal screen with ΔzapA strain, we used the method described by Claessen et al. (11). In brief, the zapA-yshB genes were amplified using yshA-F and yshB-R primers and cloned into the unstable plasmid pLOSS*. pLOSS* contains the lacZ reporter gene, which enables blue-white screening as an indicator for plasmid stability. To prevent possible induction of the endogenous β-galactosidase, the lacA gene was also deleted (33). The resulting plasmid pLOSS-zapA was transformed into cells with a zapA-yshB deletion (strain KS6) and subsequently a lacA::cat deletion was introduced to prevent transposon insertions that would activate the native B. subtilis β-galactosidase (strain KS50). This strain was transformed by pMarB, which carries the transposon TnYLB-1 (34), and the transposon mutant library was screened on nutrient agar plates supplemented with X-Gal and 1 mM MgSO4. Magnesium was added to the media, since it enhanced blue colony formation. Loss of pLOSS-zapA was further stimulated by incubating the plates at 50°C. After selection for blue colonies, chromosomal DNA of positive clones was backcrossed into strain KS50 to check whether the transposon stabilized pLOSS-zapA in cells. To map the transposon insertions, DNA fragments were TaqI digested, religated, and amplified by inverse PCR using the primers OIPCR1 and OIPCR2. The chromosomal position of the transposon was mapped by sequencing using primer OIPCR3 (34).
Suppressor screen for yvcL zapA double mutant.
To select transposon mutants that suppress the lethal phenotype of a yvcL zapA double deletion, the following procedure was developed. First, the instable plasmid pLOSS-YvcL was constructed by cloning yvcL (amplified by KS128 and KS97) into pLOSS*. Leaky transcription from the Pspac promoter gave sufficient levels of YvcL to prevent cell death in the yvcL zapA double-mutant background (strain KS742). Nevertheless, 0.1 mM IPTG and spectinomycin was used during construction of the strains. This strain also contained a lacA deletion.
Strain KS742 was used for transposon mutagenesis using pMarB (34), and the transposon mutant library was screened on nutrient agar plates supplemented with X-Gal, 0.1 mM IPTG, and 1 mM MgSO4, followed by incubation at 37°C. After screening and selection for white colonies, the chromosomal DNA of 82 positive clones was purified and transformed into the conditional yvcL zapA mutant strain (KS859). Suppressor mutations that were able to recover growth of KS859 in the absence of IPTG were mapped as described for the synthetic lethal screen.
Microscopic imaging.
Before the cells were mounted onto microscopic slides, the slides were covered with a thin layer of 1.5% agarose solution. For fluorescence microscopy, also 1 mM MgSO4 and 0.5% glucose were added to the agarose. Zeiss Axiovert 200M microscope was used to capture images. For analyses of all microscopic pictures, ImageJ software (http://rsb.info.nih.gov/ij/) was used.
Microarray analysis.
To identify differences in gene expression between wild-type B. subtilis (strain 168), the KS400 strain, and the KS696 strain, microarray analyses were performed using a 135K tiling array that was designed using the National Center for Biotechnology Information Bacillus subtilis 168 uid57675 Fasta sequence (26 January 2011, Refseq NC_000964.3, gi|255767013). Probes (60 nucleotides and a Blatbitscore threshold of 80) were designed with a tile step of 32 bases with an overlap of 28 bases between probes on opposite strands.
To isolate RNA, cell pellets were flash frozen in liquid nitrogen immediately after harvesting and stored at −80°C prior to RNA extraction. Frozen pellets were grounded by using a mortar and pestle before immersion in 300 μl of Qiazol reagent (Qiagen). RNA was isolated using the Qiazol protocol and further purified using an E.Z.N.A. MicroElute RNA cleanup kit (Omega Biotek), including an on-column treatment with the RNase-free DNase set (Life Technologies). RNA was quantified on a NanoDrop ND-1000 (Thermo Scientific), and RNA integrity was measured with a BioAnalyzer (Agilent Technologies) using an RNA Nano-6000 kit (Agilent Technologies), yielding RIN values of ≥9.7. Labeling was performed by reverse transcription using random octamers, incorporating Cy3 for the test samples and Cy5 for the common reference, as described previously (35). Hybridization, washing, and scanning was performed as described elsewhere (36).
All arrays were subjected to a set of quality control checks, such as visual inspection of the scans, checking for spatial effects through pseudo-color plots, and inspection of pre- and post-normalized data with box plots, density plots, ratio-intensity plots, and principal component analysis. Expression values were calculated by using the robust multi-array average (RMA) algorithm (37), where probe-sets were defined based on the coding sequences with a BSU locus tag. Differences in gene expression between wild-type B. subtilis, the KS400 strain, and the KS696 strain, were statistically analyzed using the Limma package in R 2.14.1 (http://cran.r-project.org/). Empirical Bayes test statistics were used for hypothesis testing (38), and all P values were corrected for false discoveries (39). Gene expression data and array design have been deposited at the public Gene Expression Omnibus, accession number GSE45824. The processed array data are listed in Table S4 in the supplemental material.
ChIP-on chip.
Chromatin immunoprecipitation (ChIP) and ChIP-on-chip analysis of YvcL was performed as described previously (40). ChIP was performed with YvcL antibody bound to protein A-coated magnetic Dynabeads (Invitrogen). The input (whole DNA) and immunoprecipitated DNA was amplified, labeled, and applied onto Nimblegen custom-made chip (∼54,000 50-bp oligonucleotides in array) containing genomic-wide probes. The oligonucleotides used for quantitative PCRs (qPCRs) are listed in Table S2 in the supplemental material.
RESULTS
Synthetic lethal screen.
The cell division protein ZapA is conserved in Gram-positive and Gram-negative bacteria, and yet a deletion of zapA does not result in a clear cell division defect (4, 41). To find new cell division proteins that become essential when ZapA is absent, we applied a synthetic lethal screen using the unstable plasmid pLOSS* (11). zapA was cloned into pLOSS*, resulting in pLOSS-zapA, and the plasmid was introduced into a zapA mutant. pLOSS* contains the lacZ reporter gene which enables blue-white screening as an indicator for plasmid stability. Transposon mutagenesis was performed using the mariner transposon (34). We isolated three mutants that formed blue colonies, indicating that they maintained pLOSS-zapA and required ZapA for growth (Fig. 1A and B). When chromosomal DNA from these mutants was backcrossed into a zapA mutant, only very small colonies appeared (Fig. 1C). Cells in these colonies were very filamentous and lysed easily (Fig. 1E). Mapping of the transposons by reversed PCR revealed three independent transposon insertions into the gene yvcL (Fig. 1F).
Fig 1.
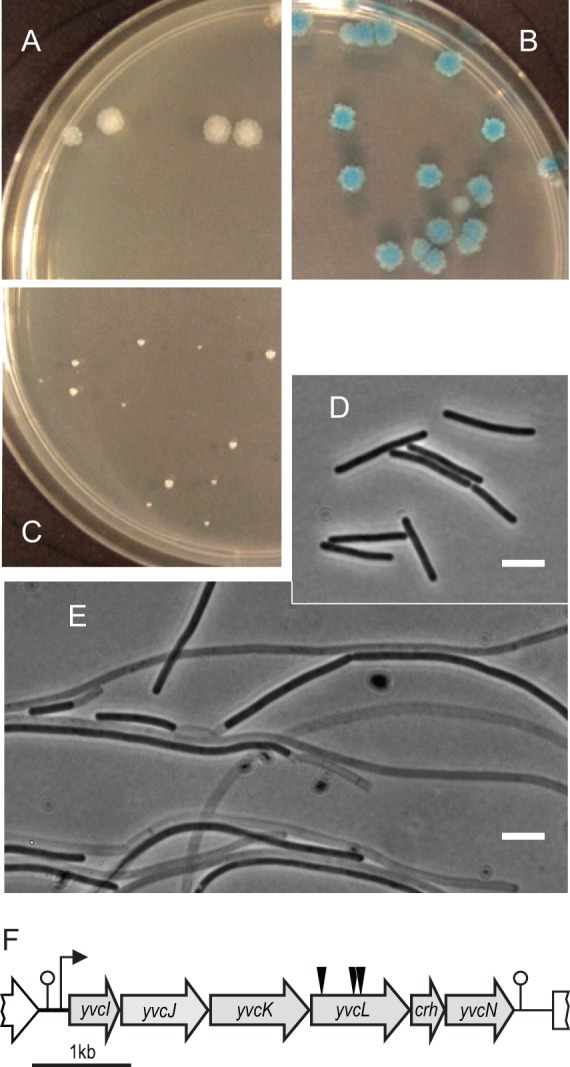
Synthetic lethal screen with zapA. Chromosomal DNA from a transposon mutant was transformed into wild-type B. subtilis strain 168 (A), zapA mutant containing the instable plasmid pLOSS*-zapA (KS50) (B), and zapA mutant (KS6) (C). Plates contained X-Gal, and maintenance of pLOSS*-zapA results in blue colonies. (D and E) Phase-contrast image of cells from plates A and C, respectively. Scale bar, 5 μm. (F) Schematic presentation of the yvcI-yvcN operon. Black triangles indicate the transposon insertion positions in yvcL.
Analysis of the yvcL operon.
The yvcL gene is part of the yvcI-yvcN operon (Fig. 1F) (42). YvcI and yvcN encode proteins of unknown function. YvcJ is a GTPase required for full induction of genetic competence, although the mechanism of this regulation is unclear (43). YvcK is involved in cell wall synthesis under gluconeogenic growth conditions and is required for the correct localization of penicillin-binding protein PBP1 (44, 45). crh, located downstream of yvcL, is involved in the control of carbon flux (42, 46). The inactivation of neither crh nor yvcN resulted in a clear growth defect, but to exclude any polar effects of the transposon insertions, an IPTG-inducible Pspac promoter was integrated either up- or downstream of yvcL and introduced into a zapA mutant. To obtain full repression of the Pspac promoter, an extra copy of lacI was introduced, as well. IPTG was only required for normal growth in the construct that contained Pspac upstream of yvcL (data not shown), thus confirming that the synthetic lethal phenotype is indeed linked to yvcL.
YvcL shows a high homology with WhiA from Streptomyces coelicolor with 25% identity and 50% similarity (see Fig. S1 in the supplemental material). The name was derived from the fact that a whiA mutant forms white colonies as a result of its inability to form gray-pigmented spores (47). The sporulation defect appears to be a consequence of the inability of whiA mutants to form division septa in aerial hyphae. Despite its homology with WhiA, a yvcL mutation has a relatively mild effect on the sporulation in B. subtilis and reduces the sporulation efficiency by only 30 to 40% (see Table S3 in the supplemental material).
Growth defect of the yvcL mutant.
We noticed that in a wild-type background the transposon insertions resulted in slightly smaller colonies. This reduction in growth may be caused by a polar effect, and therefore a markerless mutation was constructed by introducing a stop codon at the beginning of yvcL (strain KS696). However, even this mutant grows slower compared to the wild-type strain (Fig. 2A). Microscopic analyses revealed that the mutant cells are longer (Fig. 2B and C). Deletion of the upstream yvcK gene is known to affect growth and cell shape, and this can be compensated by the addition of an excess of Mg2+ (44). However, addition of Mg2+did not abrogate the lower growth rate or increased cell length of a yvcL mutant (not shown).
Fig 2.

Phenotype of a yvcL mutant. (A) Growth curve of wild type and the markerless yvcL mutant (KS696, ΔyvcL) in LB medium at 37°C. (B) Phase-contrast images (upper panels) and fluorescent membrane stain (lower panels) of wild-type B. subtilis cells and yvcL mutant cells (ΔyvcL). Scale bar, 5 μm. (C) Cell length measurements of exponentially growing wild-type and yvcL mutant (ΔyvcL) cells. The y-axis indicates frequency (%) within the population. (D) Western blot analysis of YvcL levels at different ODs. Cells were grown in LB medium, and cells from an overnight culture (on) were analyzed in the first lane. Transition to the stationary growth phase is indicated (T0). Immunodetection of FtsZ was used as loading control.
S. coelicolor WhiA is upregulated when sporulation is initiated (26). To examine whether synthesis of YvcL may be growth phase dependent, we purified the protein and raised antibodies. Western blot analysis with YvcL-specific antibodies indicated that the protein is constitutively expressed throughout the growth phase (Fig. 2D). This is in agreement with a recent comprehensive transcriptome study that demonstrated the constitutive transcription of yvcL (48). Apparently, the function of YvcL is not restricted to a certain developmental stage in B. subtilis.
Effect with other cell division mutants.
The synthetic sick phenotype when a yvcL mutation is combined with a zapA deletion suggests that YvcL might affect the activity of FtsZ. If this is the case, then it is likely that the introduction of a yvcL mutation in other cell division mutants will also result in a cell division phenotype. Like ZapA, the cell division protein SepF stimulates bundling of FtsZ protofilaments. However, when a yvcL deletion was introduced into a sepF mutant no effect on cell division was observed, and the double knockout grew fine (Fig. 3). Interestingly, when a yvcL deletion was combined with a mutation in either ezrA, minCD, or noc, the resulting transformants grew poorly and formed very filamentous cells (Fig. 3). These findings further support the suggestion that YvcL influences the activity of FtsZ.
Fig 3.

Transformation of a yvcL deletion into either ezrA, minCD or noc mutant strains results in filamentation. (A) Colony formation of the different mutants: ΔyvcL (KS267), ΔyvcL ΔsepF (KS341), ΔyvcL ΔezrA (KS344), ΔyvcL ΔminCD (KS356), ΔyvcL Δnoc (KS354), ΔzapA (KS6), ΔzapA ΔminCD (PG740), and ΔzapA Δnoc (PG739) strains. (B) Phase-contrast image of ΔyvcL ΔezrA (left panel) and ΔyvcL ΔminCD (right panel) cells. The ΔyvcL Δnoc double mutant shows comparable filamentous cells (not shown). Insets show cells from ΔezrA and ΔminCD single mutants (strains KS44 and KS338, respectively). Scale bar, 5 μm.
It has been shown that a zapA ezrA double mutation is sick and grows filamentous (4, 9). However, deleting zapA in either a minCD or noc mutant does not result in impaired growth or excessive filamentation (Fig. 3). Thus, YvcL and ZapA are not functionally redundant.
The ΔyvcL mutant is sensitive to reduced FtsZ concentrations.
If YvcL supports the assembly of the Z-ring, it is likely that a yvcL mutant is sensitive for a reduction of intracellular FtsZ levels. To test this, an IPTG-inducible ftsZ allele was introduced into the ΔyvcL mutant as the only copy of ftsZ (strain KS748). Serial dilutions were spotted onto plates containing low (30 μm) or high (500 μm) IPTG concentrations. As shown in Fig. 4A, the yvcL mutant shows reduced growth at low FtsZ concentrations (low IPTG), although the effect is much less compared to a zapA mutant (middle panel). This raises the question whether overexpression of FtsZ might rescue the synthetic lethal phenotype of a yvcL zapA double mutant. Both the yvcL and the zapA mutations were introduced into a strain carrying an extra copy of ftsZ driven by the strong xylose-inducible Pxyl promoter (strain PG738). In the absence of xylose, this strain forms very small colonies and filamentous cells (Fig. 4B and C). The induction of FtsZ (0.025 to 0.05% xylose induction) clearly stimulated colony formation of the double mutant (Fig. 4B), and microscopic analyses indicated that cell division was restored (Fig. 4C).
Fig 4.
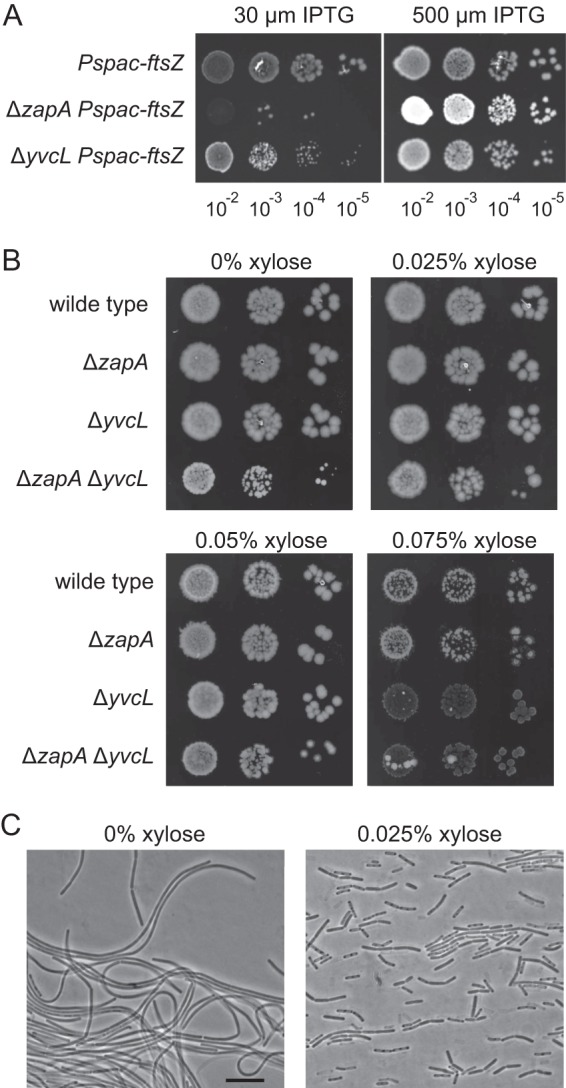
Sensitivity of yvcL mutants for altered FtsZ levels. (A) yvcL mutant is sensitive to reduced cellular FtsZ levels. Serial dilutions of ΔyvcL and ΔzapA mutant strains containing an IPTG-inducible ftsZ gene (KS268, Pspac-ftsZ; KS162, ΔzapA Pspac-ftsZ; and KS748, ΔyvcL Pspac-ftsZ). Dilutions were spotted onto plates with 30 or 500 μM IPTG. (B) FtsZ overexpression restores growth and cell division of a yvcL zapA double mutant. FtsZ overexpression was accomplished by the introduction of an ectopic Pxyl-driven ftsZ copy. Serial dilutions of strains PG8 (Pxyl-ftsZ), PG735 (ΔzapA Pxyl-ftsZ), KS737 (ΔyvcL Pxyl-ftsZ), and PG738 (ΔzapA ΔyvcL Pxyl-ftsZ) were spotted onto plates with 0, 0.025, 0.05, and 0.075% xylose. (C) Phase-contrast images of strain PG738 (ΔzapA ΔyvcL Pxyl-ftsZ) grown in the absence or presence of 0.025% xylose, indicating that overexpression of FtsZ restores cell division. Scale bar, 5 μm.
Although increased FtsZ levels do stimulate growth of the yvcL zapA double mutant, the colonies are still slightly smaller compared to the single mutants (Fig. 4B). In fact, detailed growth analyses in microtiter plates revealed that FtsZ overexpression neither restores the growth rate reduction of the yvcL single mutant nor that of a the yvcL zapA double mutant (see Fig. S2 in the supplemental material). In addition, Fig. 4B shows that high levels of FtsZ (0.075% xylose) causes lyses of the yvcL mutants, which is not observed with wild-type cells or the zapA mutant. These data suggest that YvcL is not simply a regulator of FtsZ activity.
Reduced Z-ring formation in a yvcL zapA double mutant.
The sensitivity of a yvcL mutant for reduced FtsZ concentrations suggests that the filamentous phenotype of a yvcL zapA double mutant is caused by a defect in Z-ring assembly. To examine this, a GFP-ftsZ reporter fusion was introduced into a strain containing an IPTG-inducible yvcL and a zapA deletion (strain 754). When grown in the presence of IPTG, normal cells were formed with clear fluorescent bands indicative of Z-rings (Fig. 5, left panel). However, in the filamentous cells that are formed when IPTG is omitted, no clear Z-rings could be observed (Fig. 5, right panel). Thus, a yvcL zapA double mutant has difficulties to complete the first step in cell division; the assembly of a Z-ring.
Fig 5.
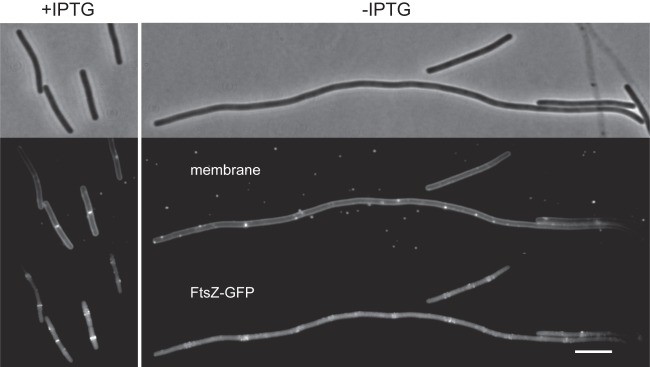
Localization of FtsZ in a yvcL zapA double mutant. A conditional yvcL zapA double mutant (Pspac-yvcL ΔzapA) expressing FtsZ-GFP (strain KS754) was grown in the presence (left panels) or absence of IPTG (right panels) at 30°C. Phase-contrast (top), Nile red membrane stain (middle), and GFP fluorescence (bottom) images are shown. Xylose (0.05%) was used for induction of FtsZ-GFP. Scale bar, 5 μm.
YvcL localizes at the nucleoid.
The crystal structure of a WhiA homolog from Thermatoga maritima indicated that the conserved C-terminal region comprises a typical DNA-binding helix-turn-helix domain (49). To examine whether YvcL binds to DNA in B. subtilis, an N-terminal mGFP fusion (monomeric GFP) was constructed under the control of the xylose-inducible Pxyl promoter (strain PG736). The mGFP-YvcL fusion appears to be active since it restored growth of a yvcL zapA double mutant (data not shown). Western blot analyses indicated that induction with 0.01% xylose resulted in mGFP-YvcL levels that are comparable to wild-type YvcL (see Fig. S3 in the supplemental material). Figure 6 shows the localization of induced mGFP-YvcL in exponentially growing cells. The GFP signal clearly localizes at the nucleoid. This is even more apparent at higher xylose concentrations (see Fig. S3 in the supplemental material) and supports the assumption that YvcL binds to DNA.
Fig 6.

Localization of mGFP-YvcL in B. subtilis cells. Fluorescence microscopy images of strain PG736 containing a Pxyl-mgfp-yvcL fusion and yvcL deletion, grown in LB medium at 30°C in the presence of 0.01% xylose, are shown. Scale bar, 5 μm.
Transcriptome analysis of yvcL mutants.
S. coelicolor WhiA binds to its own promoter and is required for its own expression (26, 50), the sporulation-dependent expression of ftsZ, and the expression of other sporulation genes (27, 51, 52). The nucleoid-binding activity of YvcL and its homology to WhiA suggest that YvcL functions as a transcription factor, possibly regulating ftsZ expression. To identify genes that are regulated by YvcL, a genome-wide transcriptome analysis was performed by using tiling arrays. Wild-type B. subtilis and the yvcL::kan deletion strain (KS400) were grown in LB medium to an OD600 of ∼0.5, followed by the isolation of RNA. The microarray results indicated that the downstream located yvcN and crh genes were significantly upregulated (Table 1). This is likely a consequence of the kanamycin marker that deletes yvcL and reads into crh and yvcN. Therefore, a new transcriptome analysis was performed, this time using the markerless yvcL mutant (KS696). In this case, there was no difference in yvcN and crh expression between ΔyvcL cells and wild-type cells (Table 1). The data sets of both transcriptome analyses were combined. A total of 49 genes showed 2-fold downregulation and 92 genes showed 2-fold upregulation in both yvcL mutants. The significance cutoff was set at an adjusted P value of <1.0E−5. Table 1 lists 46 genes that show a >4-fold difference in expression in both data sets. Surprisingly, we found no significant difference in the expression of known cell division genes, including ftsZ. Western blot analyses confirmed that FtsZ levels are not markedly affected in a yvcL mutant (see Fig. S4 in the supplemental material). The overexpression of YvcL has also no effect on cellular FtsZ levels (see Fig. S4 in the supplemental material) and does not suppress the synthetic cell division defect of a zapA ezrA double mutant (4). Finally, the presence of many up- and downregulated genes in the transcriptome profiles might be related to the fact that a yvcL mutant grows slower (Fig. 2).
Table 1.
Transcriptome analysis of yvcL mutantsa
| Gene | KS400/wt | KS696/wt | Product |
|---|---|---|---|
| yvcL | 0.01 | 1.07 | Putative morphogen |
| mtnA | 0.02 | 0.02 | Methylthioribose-1-phosphate isomerase (methionine salvage pathway) |
| mtnK | 0.02 | 0.02 | Methylthioribose kinase (methionine salvage pathway) |
| cidA | 0.09 | 0.15 | Holin regulator of murein hydrolases |
| lrgB | 0.10 | 0.11 | Anti-holin factor controlling activity of murein hydrolases |
| yxiM | 0.16 | 0.14 | Putative esterase (lipoprotein) |
| yxiK | 0.14 | 0.10 | Putative phage head maturation protein |
| yxiJ | 0.11 | 0.07 | Conserved hypothetical protein |
| yxiI | 0.09 | 0.07 | Conserved hypothetical protein |
| yxzG | 0.10 | 0.07 | Putative nucleic acid binding protein |
| yxiG | 0.10 | 0.06 | Conserved hypothetical protein |
| yxzC | 0.10 | 0.06 | Putative nucleic acid binding protein |
| yxiF | 0.09 | 0.05 | Putative phage reverse transcriptase or polymerase |
| yxxG | 0.12 | 0.07 | Hypothetical protein |
| wapA | 0.12 | 0.07 | Cell wall-associated protein precursor |
| ydaK | 0.18 | 0.15 | Putative membrane protein with diguanylate cyclase domain |
| ydaL | 0.23 | 0.16 | Conserved hypothetical protein |
| ydaM | 0.19 | 0.13 | Putative glycosyl transferase associated to biofilm formation |
| ydaN | 0.20 | 0.11 | Putative regulator |
| opuE | 0.20 | 0.21 | Proline transporter |
| pyrF | 0.22 | 0.19 | Orotidine 5′-phosphate decarboxylase |
| pyrE | 0.25 | 0.21 | Orotatephosphoribosyl transferase |
| ydcF | 54.3 | 46.1 | Hypothetical protein |
| ydcG | 21.0 | 17.8 | Conserved hypothetical protein |
| ydcH | 22.7 | 25.1 | Putative transcriptional regulator |
| clpE | 13.3 | 11.7 | ATP-dependent Clp protease (class III stress gene) |
| ycdA | 11.7 | 16.6 | Putative lipoprotein |
| bmrC | 9.9 | 7.4 | ABC transporter involved in the signaling pathway that activates KinA |
| bmrD | 7.9 | 5.8 | ABC transporter involved in the signaling pathway that activates KinA |
| yvcN | 6.6 | 1.0 | Putative acetyltransferase |
| crh | 6.2 | 1.0 | Catabolite repression HPr-like protein |
| yvcA | 5.4 | 8.7 | Putative lipoprotein |
| lrgA | 5.4 | 25.7 | Antiholin factor |
| ywdA | 4.2 | 6.4 | Hypothetical protein |
| sacA | 5.0 | 7.8 | Sucrase-6-phosphate hydrolase |
| sacP | 5.4 | 7.8 | Phosphotransferase system (PTS) sucrose-specific enzyme |
| yjhA | 5.3 | 7.1 | Putative lipoprotein |
| yjhB | 4.1 | 5.9 | Putative ADP-ribose pyrophosphatase |
| yfjC | 4.1 | 5.8 | Hypothetical protein |
| yfjB | 4.5 | 8.6 | Hypothetical protein |
| dhbF | 4.1 | 11.8 | Siderophore bacillibactin synthetase |
| dhbB | 4.4 | 12.5 | Isochorismatase |
| dhbE | 4.1 | 12.8 | 2,3-Dihydroxybenzoate-AMP ligase (enterobactin synthetase component E) |
| dhbA | 4.0 | 11.4 | 2,3-Dihydro-2,3-dihydroxybenzoate dehydrogenase |
| bslA | 4.2 | 5.1 | Protein involved in biofilm formation |
| tasA | 4.1 | 4.7 | Major biofilm matrix component |
| sacB | 4.0 | 6.3 | Levansucrase |
Genes with a 4-fold expression difference and an adjusted P value of <1.0E–5 present in both transcriptome experiments are listed (except for yvcL, yvcN, and crh). KS400 contains a kanamycin marker in yvcL, and KS696 is the markerless yvcL mutation. Fold differences are shown, and genes shifted to the right in column 1 indicate operons. wt, wild type.
Identification of YvcL binding sites on the genome.
The GFP-YvcL fusion indicated that the protein accumulates at the nucleoid but the transcriptome analysis did not identify any YvcL-regulated gene that could explain why this protein becomes important when ZapA, EzrA, MinCD, or Noc are absent. To determine whether the transcriptome profile can be linked to specific YvcL operator sites on the genome, the chromosomal YvcL-binding sites were determined using ChIP combined with microarrays (ChIP-on-chip assay). After cross-linking, chromosomal DNA was isolated, and YvcL-DNA fragments were immunoprecipitated with YvcL antibodies. The DNA fragments were amplified, fluorescently labeled, and hybridized to Nimblegen tiling arrays, as previously described (40). The intensity plot of YvcL-enriched genomic regions is shown in Fig. 7A. We noticed that several of the peaks are also present in the published ChIP-on-chip profiles with Noc and Smc (21, 40). These peaks might therefore indicate unspecific amplification. The peaks unique for YvcL are marked in red (Fig. 7A). The strongest peaks were verified with a ChIP experiment, followed by qPCR, whereby DNA from a yvcL mutant served as a negative control (Fig. 7B). The YvcL peaks do not seem to reveal a special binding pattern and could not be assigned to genes that showed up in the transcriptome analysis. The protein is enriched at an actively transcribed region that encompasses the ribosomal genes rpsL, rplB, and rplN, but in fact a closer inspection of peaks indicated that YvcL binds within coding regions instead of promoter regions (Fig. 7C). Thus, the ChIP-on-chip data do not support the assumption that YvcL functions as a transcription factor.
Fig 7.

ChIP-on-chip analysis of YvcL binding sites on the B. subtilis genome. (A) Ratios of immunoprecipitated DNA versus total DNA (IP/IN) are depicted on a linear genome map. Peaks that were unique for YvcL and that are not present in previous published ChIP-on-chip data obtained with SMC or Noc are highlighted in red. (B) Verification of ChIP-on-chip data using qPCR. (C) Detailed distribution of WhiA binding sites around acoC, spoVID, and xynD.
Isolation of suppressor mutants.
Possibly, the identification of mutants that suppress the filamentous phenotype of a yvcL zapA double mutant might shed light on the function of YvcL. To find such suppressors, we again made use of the instable pLOSS* plasmid, but this time yvcL was cloned into the plasmid. When the plasmid was introduced into a yvcL zapA double mutant, the resulting transformants formed blue colonies of normal size on X-Gal-containing plates. After transposon mutagenesis, a few white colonies of normal size appeared. These colonies had lost the plasmid and contained a transposon insertion that suppressed the sick phenotype caused by the combined deletion of yvcL and zapA. Eventually, three suppressors were identified. Two clones had a transposon insertion in gtaB, and one clone contained a transposon in pgcA. Interestingly, mutations in gtaB and pgcA have been shown to increase the frequency of cell division (28). GtaB and PgcA provide the UDP-glucose substrate for the glucosyltransferase UgtP, and this protein binds directly to FtsZ and inhibits the assembly of FtsZ (28). Indeed, when a ugtP deletion was introduced into a conditional yvcL zapA double mutant, the transformants formed normally sized colonies without IPTG (Fig. 8). Microscopic analyses also showed that the disruption of gtaB, pgcA, or ugtP suppresses the cell division defect of the yvcL zapA double mutant (Fig. 8).
Fig 8.

Suppressors of yvcL zapA double mutant. The different suppressor mutations were introduced into a conditional yvcL zapA double mutant (KS859, ΔzapA Pspac-yvcL aprE::lacI) and tested for colony formation in diluted cultures on plates in the presence or absence of IPTG. The effect of suppressor mutations on cell length of a yvcL mutant is indicated in the table. The average cell lengths and standard deviations (SD) of at least 80 cells were determined. Strains: 168, KS696, KS902, KS903, and KS1015.
DISCUSSION
Using a synthetic lethal screen, we have identified a new protein involved in cell division in B. subtilis. This protein, YvcL, is required for normal growth and cell division when the FtsZ regulators ZapA, EzrA, MinCD, or Noc are absent. In the filamentous yvcL zapA double mutant, no clear Z-rings are observed. This phenotype can be suppressed by blocking the activity of UgtP, the metabolic regulator that inhibits Z-ring formation. Together, these data suggest that YvcL acts at the level of Z-ring formation, which is supported by the finding that a yvcL mutant is sensitive for reduced FtsZ concentrations, and that increased FtsZ levels suppress the cell division defect in the yvcL zapA double mutant.
YvcL belongs to the conserved protein family DUF199 whose members are assumed to act as transcriptional regulators (53). The crystal structure of Thermatoga maritima WhiA revealed a typical helix-turn-helix fold related to bacterial sigma-70 factors (49). This domain is required for binding of S. coelicolor WhiA to its own promoter (50). WhiA is essential for the induction of ftsZ during sporulation in S. coelicolor (37, 54), and constitutive ftsZ expression rescues sporulation in a whiA mutant (55). Because of these structural and functional homologies, we propose to use the name WhiA instead of YvcL.
Considering the homologies between B. subtilis and S. coelicolor WhiA, it was surprising to find that B. subtilis WhiA does not regulate ftsZ or other known cell division genes. S. coelicolor WhiA is also required for the expression of parAB, whiB, and hupS during sporulation (26, 51, 52). We could not detect transcriptional changes in soj/spo0J, which are the B. subtilis equivalents of parA/B, in strains lacking WhiA. B. subtilis does not encode a whiB homologue, and hupS encodes a histone-like protein typical for Actinomycetes. The expression of B. subtilis chromosome architectural proteins Hbs, ScpA/B, and Smc was also not altered in a whiA mutant. Thus far, any possible transcriptional activity of WhiA fails to explain why this protein is required for cell division in B. subtilis.
The exact function of B. subtilis WhiA remains elusive. Because the protein binds DNA, it might play a role in nucleoid occlusion. However, it is unlikely that WhiA regulates Noc directly since a whiA noc double mutant shows a severe cell division phenotype that is not observed with the single mutants. We have examined whether the nucleoid localization of Noc is affected in a whiA mutant, but that is not the case (see Fig. S5 in the supplemental material). Moreover, we failed to notice the classical FtsZ rings and spirals that overlap the nucleoid or chromosome bisection, which are typical for noc mutants (21).
Depletion of FtsZ, although causing filamentation that makes cells prone to lysis, does not reduce cell growth (elongation) (56). However, a whiA mutant clearly grows slower, and the additional expression of FtsZ does not suppress this growth defect. It is unclear how the activity of WhiA links cell growth with cell division. Interestingly, inactivation of UgtP, the metabolic sensor of cell division, suppresses the severe cell growth defect of the whiA zapA double mutant. UgtP interacts with FtsZ and inhibits assembly of FtsZ when cells grow in rich medium (28). Possibly, WhiA represses directly or indirectly the activity of UgtP. The transcriptome data indicate that the expression levels of ugtP or other genes involved in the activity of UgtP (pgcA, gtaB) are unaffected in a whiA knockout strain. We also could not detect increased septal localization of UgtP in such backgrounds (data not shown). Further research will be necessary to determine whether WhiA controls UgtP activity.
Supplementary Material
ACKNOWLEDGMENTS
We thank Yoshi Kawai and Ling Wu for strains and plasmids, and we thank other members of the Centre for Bacterial Cell Biology for helpful discussions. In addition, we thank Nigel Saunders and Richard Capper for help with the initial microarray analysis, Stephan Gruber for assistance with the ChIP-on-chip experiment, Rob Dekker of the University of Amsterdam MicroArray Department for transcriptome analyses, and Richard Daniel and Alexander TerBeek for providing the microarrays.
This study was supported by Marie Curie ITN EST grant ATPBCT (L.W.H., J.E.), and STW Vici grant 12128 (L.W.H.).
Footnotes
Published ahead of print 4 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00507-13.
REFERENCES
- 1.Pichoff S, Lutkenhaus J. 2005. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol. Microbiol. 55:1722–1734 [DOI] [PubMed] [Google Scholar]
- 2.Szwedziak P, Wang Q, Freund SM, Lowe J. 2012. FtsA forms actin-like protofilaments. EMBO J. 31:2249–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dajkovic A, Pichoff S, Lutkenhaus J, Wirtz D. 2010. Cross-linking FtsZ polymers into coherent Z rings. Mol. Microbiol. 78:651–668 [DOI] [PubMed] [Google Scholar]
- 4.Gueiros-Filho FJ, Losick R. 2002. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev. 16:2544–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gundogdu ME, Kawai Y, Pavlendova N, Ogasawara N, Errington J, Scheffers DJ, Hamoen LW. 2011. Large ring polymers align FtsZ polymers for normal septum formation. EMBO J. 30:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishikawa S, Kawai Y, Hiramatsu K, Kuwano M, Ogasawara N. 2006. A new FtsZ-interacting protein, YlmF, complements the activity of FtsA during progression of cell division in Bacillus subtilis. Mol. Microbiol. 60:1364–1380 [DOI] [PubMed] [Google Scholar]
- 7.Marbouty M, Saguez C, Cassier-Chauvat C, Chauvat F. 2009. Characterization of the FtsZ-interacting septal proteins SepF and Ftn6 in the spherical-celled cyanobacterium Synechocystis strain PCC 6803. J. Bacteriol. 191:6178–6185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh JK, Makde RD, Kumar V, Panda D. 2008. SepF increases the assembly and bundling of FtsZ polymers and stabilizes FtsZ protofilaments by binding along its length. J. Biol. Chem. 283:31116–31124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamoen LW, Meile JC, de Jong W, Noirot P, Errington J. 2006. SepF, a novel FtsZ-interacting protein required for a late step in cell division. Mol. Microbiol. 59:989–999 [DOI] [PubMed] [Google Scholar]
- 10.Haeusser DP, Schwartz RL, Smith AM, Oates ME, Levin PA. 2004. EzrA prevents aberrant cell division by modulating assembly of the cytoskeletal protein FtsZ. Mol. Microbiol. 52:801–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Claessen D, Emmins R, Hamoen LW, Daniel RA, Errington J, Edwards DH. 2008. Control of the cell elongation-division cycle by shuttling of PBP1 protein in Bacillus subtilis. Mol. Microbiol. 68:1029–1046 [DOI] [PubMed] [Google Scholar]
- 12.Rodrigues CD, Harry EJ. 2012. The Min system and nucleoid occlusion are not required for identifying the division site in Bacillus subtilis but ensure its efficient utilization. PLoS Genet. 8:e1002561. 10.1371/journal.pgen.1002561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Boer PA, Crossley RE, Rothfield LI. 1989. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in Escherichia coli. Cell 56:641–649 [DOI] [PubMed] [Google Scholar]
- 14.Hu Z, Mukherjee A, Pichoff S, Lutkenhaus J. 1999. The MinC component of the division site selection system in Escherichia coli interacts with FtsZ to prevent polymerization. Proc. Natl. Acad. Sci. U. S. A. 96:14819–14824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Boer PA, Crossley RE, Hand AR, Rothfield LI. 1991. The MinD protein is a membrane ATPase required for the correct placement of the Escherichia coli division site. EMBO J. 10:4371–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu Z, Lutkenhaus J. 2003. A conserved sequence at the C terminus of MinD is required for binding to the membrane and targeting MinC to the septum. Mol. Microbiol. 47:345–355 [DOI] [PubMed] [Google Scholar]
- 17.Edwards DH, Errington J. 1997. The Bacillus subtilis DivIVA protein targets to the division septum and controls the site specificity of cell division. Mol. Microbiol. 24:905–915 [DOI] [PubMed] [Google Scholar]
- 18.Patrick JE, Kearns DB. 2008. MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol. Microbiol. 70:1166–1179 [DOI] [PubMed] [Google Scholar]
- 19.Bramkamp M, Emmins R, Weston L, Donovan C, Daniel RA, Errington J. 2008. A novel component of the division-site selection system of Bacillus subtilis and a new mode of action for the division inhibitor MinCD. Mol. Microbiol. 70:1556–1569 [DOI] [PubMed] [Google Scholar]
- 20.Wu LJ, Errington J. 2004. Coordination of cell division and chromosome segregation by a nucleoid occlusion protein in Bacillus subtilis. Cell 117:915–925 [DOI] [PubMed] [Google Scholar]
- 21.Wu LJ, Ishikawa S, Kawai Y, Oshima T, Ogasawara N, Errington J. 2009. Noc protein binds to specific DNA sequences to coordinate cell division with chromosome segregation. EMBO J. 28:1940–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weart RB, Levin PA. 2003. Growth rate-dependent regulation of medial FtsZ ring formation. J. Bacteriol. 185:2826–2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Low HH, Moncrieffe MC, Lowe J. 2004. The crystal structure of ZapA and its modulation of FtsZ polymerisation. J. Mol. Biol. 341:839–852 [DOI] [PubMed] [Google Scholar]
- 24.Small E, Marrington R, Rodger A, Scott DJ, Sloan K, Roper D, Dafforn TR, Addinall SG. 2007. FtsZ polymer-bundling by the Escherichia coli ZapA orthologue, YgfE, involves a conformational change in bound GTP. J. Mol. Biol. 369:210–221 [DOI] [PubMed] [Google Scholar]
- 25.Monahan LG, Robinson A, Harry EJ. 2009. Lateral FtsZ association and the assembly of the cytokinetic Z ring in bacteria. Mol. Microbiol. 74:1004–1017 [DOI] [PubMed] [Google Scholar]
- 26.Ainsa JA, Ryding NJ, Hartley N, Findlay KC, Bruton CJ, Chater KF. 2000. WhiA, a protein of unknown function conserved among gram-positive bacteria, is essential for sporulation in Streptomyces coelicolor A3(2). J. Bacteriol. 182:5470–5478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flardh K, Leibovitz E, Buttner MJ, Chater KF. 2000. Generation of a non-sporulating strain of Streptomyces coelicolor A3(2) by the manipulation of a developmentally controlled ftsZ promoter. Mol. Microbiol. 38:737–749 [DOI] [PubMed] [Google Scholar]
- 28.Weart RB, Lee AH, Chien AC, Haeusser DP, Hill NS, Levin PA. 2007. A metabolic sensor governing cell size in bacteria. Cell 130:335–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamoen LW, Smits WK, de Jong A, Holsappel S, Kuipers OP. 2002. Improving the predictive value of the competence transcription factor (ComK) binding site in Bacillus subtilis using a genomic approach. Nucleic Acids Res. 30:5517–5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itaya M, Kondo K, Tanaka T. 1989. A neomycin resistance gene cassette selectable in a single copy state in the Bacillus subtilis chromosome. Nucleic Acids Res. 17:4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morimoto T, Loh PC, Hirai T, Asai K, Kobayashi K, Moriya S, Ogasawara N. 2002. Six GTP-binding proteins of the Era/Obg family are essential for cell growth in Bacillus subtilis. Microbiology 148:3539–3552 [DOI] [PubMed] [Google Scholar]
- 32.Landgraf D, Okumus B, Chien P, Baker TA, Paulsson J. 2012. Segregation of molecules at cell division reveals native protein localization. Nat. Methods 9:480–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daniel RA, Haiech J, Denizot F, Errington J. 1997. Isolation and characterization of the lacA gene encoding beta-galactosidase in Bacillus subtilis and a regulator gene, lacR. J. Bacteriol. 179:5636–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Breton Y, Mohapatra NP, Haldenwang WG. 2006. In vivo random mutagenesis of Bacillus subtilis by use of TnYLB-1, a mariner-based transposon. Appl. Environ. Microbiol. 72:327–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Knegt GJ, Bruning O, ten Kate MT, de Jong M, van Belkum A, Endtz HP, Breit TM, Bakker-Woudenberg IA, de Steenwinkel JE. 2013. Rifampicin-induced transcriptome response in rifampicin-resistant Mycobacterium tuberculosis. Tuberculosis 93:96–101 [DOI] [PubMed] [Google Scholar]
- 36.Pennings JL, Rodenburg W, Imholz S, Koster MP, van Oostrom CT, Breit TM, Schielen PC, de Vries A. 2011. Gene expression profiling in a mouse model identifies fetal liver- and placenta-derived potential biomarkers for Down Syndrome screening. PLoS One 6:e18866. 10.1371/journal.pone.0018866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264 [DOI] [PubMed] [Google Scholar]
- 38.Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3:Article3 [DOI] [PubMed] [Google Scholar]
- 39.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. R. Stat. Soc. Ser. B 57:289–300 [Google Scholar]
- 40.Gruber S, Errington J. 2009. Recruitment of condensin to replication origin regions by ParB/SpoOJ promotes chromosome segregation in Bacillus subtilis. Cell 137:685–696 [DOI] [PubMed] [Google Scholar]
- 41.Johnson JE, Lackner LL, Hale CA, de Boer PA. 2004. ZipA is required for targeting of DMinC/DicB, but not DMinC/MinD, complexes to septal ring assemblies in Escherichia coli. J. Bacteriol. 186:2418–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galinier A, Haiech J, Kilhoffer MC, Jaquinod M, Stulke J, Deutscher J, Martin-Verstraete I. 1997. The Bacillus subtilis crh gene encodes a HPr-like protein involved in carbon catabolite repression. Proc. Natl. Acad. Sci. U. S. A. 94:8439–8444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luciano J, Foulquier E, Fantino JR, Galinier A, Pompeo F. 2009. Characterization of YvcJ, a conserved P-loop-containing protein, and its implication in competence in Bacillus subtilis. J. Bacteriol. 191:1556–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorke B, Foulquier E, Galinier A. 2005. YvcK of Bacillus subtilis is required for a normal cell shape and for growth on Krebs cycle intermediates and substrates of the pentose phosphate pathway. Microbiology 151:3777–3791 [DOI] [PubMed] [Google Scholar]
- 45.Foulquier E, Pompeo F, Bernadac A, Espinosa L, Galinier A. 2011. The YvcK protein is required for morphogenesis via localization of PBP1 under gluconeogenic growth conditions in Bacillus subtilis. Mol. Microbiol. 80:309–318 [DOI] [PubMed] [Google Scholar]
- 46.Landmann JJ, Busse RA, Latz JH, Singh KD, Stulke J, Gorke B. 2011. Crh, the paralogue of the phosphocarrier protein HPr, controls the methylglyoxal bypass of glycolysis in Bacillus subtilis. Mol. Microbiol. 82:770–787 [DOI] [PubMed] [Google Scholar]
- 47.Hopwood DA, Wildermuth H, Palmer HM. 1970. Mutants of Streptomyces coelicolor defective in sporulation. J. Gen. Microbiol. 61:397–408 [DOI] [PubMed] [Google Scholar]
- 48.Nicolas P, Mader U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S, Becher D, Bisicchia P, Botella E, Delumeau O, Doherty G, Denham EL, Fogg MJ, Fromion V, Goelzer A, Hansen A, Hartig E, Harwood CR, Homuth G, Jarmer H, Jules M, Klipp E, Le Chat L, Lecointe F, Lewis P, Liebermeister W, March A, Mars RA, Nannapaneni P, Noone D, Pohl S, Rinn B, Rugheimer F, Sappa PK, Samson F, Schaffer M, Schwikowski B, Steil L, Stulke J, Wiegert T, Devine KM, Wilkinson AJ, van Dijl JM, Hecker M, Volker U, Bessieres P, Noirot P. 2012. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335:1103–1106 [DOI] [PubMed] [Google Scholar]
- 49.Kaiser BK, Clifton MC, Shen BW, Stoddard BL. 2009. The structure of a bacterial DUF199/WhiA protein: domestication of an invasive endonuclease. Structure 17:1368–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaiser BK, Stoddard BL. 2011. DNA recognition and transcriptional regulation by the WhiA sporulation factor. Sci. Rep. 1:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jakimowicz D, Mouz S, Zakrzewska-Czerwinska J, Chater KF. 2006. Developmental control of a parAB promoter leads to formation of sporulation-associated ParB complexes in Streptomyces coelicolor. J. Bacteriol. 188:1710–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salerno P, Larsson J, Bucca G, Laing E, Smith CP, Flardh K. 2009. One of the two genes encoding nucleoid-associated HU proteins in Streptomyces coelicolor is developmentally regulated and specifically involved in spore maturation. J. Bacteriol. 191:6489–6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knizewski L, Ginalski K. 2007. Bacterial DUF199/COG1481 proteins including sporulation regulator WhiA are distant homologs of LAGLIDADG homing endonucleases that retained only DNA binding. Cell Cycle 6:1666–1670 [DOI] [PubMed] [Google Scholar]
- 54.Ryding NJ, Bibb MJ, Molle V, Findlay KC, Chater KF, Buttner MJ. 1999. New sporulation loci in Streptomyces coelicolor A3(2). J. Bacteriol. 181:5419–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Willemse J, Mommaas AM, van Wezel GP. 2012. Constitutive expression of ftsZ overrides the Whi developmental genes to initiate sporulation of Streptomyces coelicolor. Antonie Van Leeuwenhoek 101:619–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beall B, Lutkenhaus J. 1991. FtsZ in Bacillus subtilis is required for vegetative septation and for asymmetric septation during sporulation. Genes Dev. 5:447–455 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.