

Abstract
Background
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia in adults. We hypothesized that gain-of-function KCNQ1 mutations previously associated with familial AF have distinct pharmacological properties that may enable targeted inhibition.
Methods and Results
Wild-type (WT) KCNQ1 or the familial AF mutation KCNQ1-S140G were heterologously co-expressed with KCNE1 to enable electrophysiological recording of the slow delayed rectifier current (IKs) and investigation of pharmacological effects of the IKs selective blocker HMR-1556. Co-expression of KCNQ1-S140G with KCNE1 generated potassium currents (S140G-IKs) that exhibited greater sensitivity to HMR-1556 than WT-IKs. Enhanced HMR-1556 sensitivity was also observed for another gain-of-function AF mutation, KCNQ1-V141M. Heteromeric expression of KCNE1 with both KCNQ1-WT and KCNQ1-S140G generated currents (HET-IKs) with gain-of-function features including larger amplitude, a constitutively active component, hyperpolarized voltage dependence of activation, and extremely slow deactivation. A low concentration of HMR-1556, which had little effect on WT-IKs but was capable of inhibiting the mutant channel, reduced both instantaneous and steady-state HET-IKs to levels that were not significantly different from WT-IKs and attenuated use-dependent accumulation of the current. In cultured adult rabbit left atrial myocytes, expression of S140G-IKs shortened action potential duration (APD) compared to WT-IKs. Application of HMR-1556 mitigated S140G-IKs -induced APD shortening and did not alter APD in cells expressing WT-IKs.
Conclusions
The enhanced sensitivity of KCNQ1 gain-of-function mutations for HMR-1556 suggests the possibility of selective therapeutic targeting and, therefore, our data illustrates a potential proof-of-principal for genotype-specific treatment of this heritable arrhythmia.
Keywords: arrhythmia, atrial fibrillation, antiarrhythmic drug, potassium channels, genetics
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia in adults. The prevalence of AF rises exponentially with age, and because of the aging population, the number of persons with AF in the United States is projected to increase to 12 million by 2050.1 Importantly, AF confers a 6-fold increased risk for thromboembolic disease including stroke, predisposes to heart failure and is associated with premature death.2 The incremental healthcare costs directly related to the diagnosis and management of AF in the United States have been estimated at $6 billion.3
Most often, AF occurs within the context of structural heart disease with onset past the age of 65 years. However, an estimated 10–30% of AF, designated as lone AF, arises in the absence of overt heart disease and has a younger age of onset.4–7 Genetic predisposition to AF has been demonstrated in populations8,9 and in families with monogenic forms of the disease.10 AF-associated mutations have been identified in potassium channels,11–16 sodium channels,17–19 and other genes.20 The mutation KCNQ1-S140G was the first identified mutation and remains the best-studied genetic variant associated with autosomal dominant AF.11,21–23
KCNQ1 encodes a pore-forming voltage-gated potassium channel (Kv7.1 or KCNQ1) that combines with the auxiliary subunit KCNE1 to generate the slow component of the delayed rectifier potassium current (IKs), critical for cardiac action potential repolarization. Co-expression of KCNQ1-S140G with KCNE1 (S140G-IKs) demonstrated a gain-of-function with larger and more instantaneous current activation.11,23 A similar gain-of-function effect occurs with the AF-associated mutation KCNQ1-V141M.12 These in vitro data are consistent with the notion that increased repolarizing potassium current evoked by these mutations cause shortening of atrial action potentials in myocytes and an abbreviated effective refractory period in atrial tissues, resulting in an increased probability of reentry circuits and AF.23
We hypothesized that KCNQ1 gain-of-function mutations have pharmacological properties distinct from the WT channel that may enable selective inhibition of mutant channel complexes. Pharmacological targeting of this gain-of-function behavior would be predicted to decrease AF susceptibility in persons with this dominant mutant allele. We tested this hypothesis using the chromanol 293B derivative HMR-1556, a highly specific IKs blocker when used at low concentrations. Here we present evidence that S140G-IKs and V141M-IKs exhibit enhanced sensitivity to HMR-1556 due to an additional high affinity state. Using a concentration that predominantly inhibits the high affinity state, HMR-1556 effectively suppressed S140G-IKs amplitudes to levels not different from WT-IKs, attenuated the use-dependent accumulation of current without significant effects on WT-IKs, and mitigated action potential shortening in cultured adult rabbit left atrial myocytes without affecting WT-IKs action potential duration. These data suggest a potential opportunity for genotype-specific treatment of familial AF.
Methods
Voltage clamp experiments were performed in Chinese hamster ovary (CHO) cells transiently transfected with plasmids containing potassium channel subunits (KCNQ1, KCNE1) constructed in plasmids co-expressing a transfection marker (dsRedMST, eGFP, or CD8). Recordings were done in the absence or presence of HMR-1556. Current clamp recordings were performed with cultured adult rabbit left atrial myocytes infected with adenoviruses encoding WT or mutant KCNQ1 subunits in combination with a separate adenovirus encoding KCNE1. Action potentials were elicited from transduced myocytes using whole cell patch clamp 48–72 hours post-isolation in the absence or presence of HMR-1556.
Differences between two groups were assessed using unpaired Student’s t test. When comparing more than two groups, one-way ANOVA followed by a Tukey post hoc test was performed on values obtained for a given membrane voltage. Statistical tests were performed using SigmaStat 2.03 (Systat Software, Inc., Chicago, IL). Significance levels are reported as two-sided p-values.
A complete description of all experimental methods is presented in the Supplementary Material.
Results
S140G-IKs exhibits enhanced sensitivity to HMR-1556
We tested whether heterologously expressed IKs channel complexes consisting of either wildtype (WT) or mutant (S140G) KCNQ1 subunits in combination with the auxiliary subunit KCNE1 are inhibited by HMR-1556. Whole-cell recordings of CHO cells transfected with S140G and KCNE1 (S140G-IKs) demonstrated nearly instantaneous activation of outward current in contrast to the slowly activating current observed in cells expressing WT-IKs (Figure 1A, B). Because KCNQ1-S140G mutation-positive subjects were reported to be heterozygous in familial AF and because WT and mutant KCNQ1 subunits can co-assemble in heteromeric channels, we examined channel complexes consisting of both WT and mutant subunits co-expressed with KCNE1 (HET-IKs), which exhibited larger amplitudes with a large fraction of instantaneous current (Figure 1C). Superfusion of 1μM HMR-1556 completely and rapidly inhibited all channel complexes activated by low frequency pulsing (10 s interpulse duration) to +40 mV (Figure 1D–F). However, inhibition of S140G-IKs and HET-IKs was more pronounced than WT-IKs at lower concentrations.
Figure 1.

S140G-IKs and HET-IKs exhibit enhanced sensitivity to HMR-1556. A, B and C, Representative current recordings from cells expressing WT-IKs (A), S140G-IKs (B), or HET-IKs (C). Recordings illustrated in A, B and C were obtained using the activation protocol described in the Methods. D, E, and F, Average current densities (current normalized to cell capacitance) elicited by a 2 s voltage step to +40 mV followed by a 10 s interpulse during application of vehicle or various concentration of HMR-1556 from cells expressing WT-IKs (D), S140G-IKs (E), or HET-IKs (F). Current density traces in D, E, and F are averages from 9–11 cells.
We assessed concentration-response relationships for WT-IKs, S140G-IKs, and HET-IKs to determine whether the channel complexes have different affinities for HMR-1556 (Figure 2). Cells expressing WT-IKs exhibited a concentration-response curve that was fit by the Hill equation yielding an IC50 of 214nM and Hill coefficient of 1.2. By contrast, S140G-IKs exhibited a complex concentration-response that suggested two affinity states. The IC50 of the high affinity state was 3.7nM, whereas the lower affinity state had an IC50 of 97.7nM. Both states were significantly different from the IC50 for WT-IKs (p<0.001). Hill coefficients for the high (2.2) and low (2.5) affinity states on S140G-IKs suggested positive cooperative binding of the drug. The gating kinetics of the current sensitive to 30nM HMR-1556, a concentration near the crux between the two affinity states on the concentration-response curve, was not overtly different than drug-insensitive current (Figure S1) suggesting that the two affinity states do not emerge from distinct populations of channels. The HET-IKs complex demonstrated an intermediate pharmacologic phenotype with a complex concentration-response curve. The high affinity state had an IC50 of 5.1nM (Hill coefficient 1.7) whereas the low affinity state IC50 was 240nM (Hill coefficient 2.4).
Figure 2.

HMR-1556 concentration-response curves for WT-IKs (○), S140G-IKs (△), and HET-IKs (●). Solid lines represents fits of the averaged data to either monophasic (WT-IKs) or biphasic (S140G-IKs and HET-IKs) Hill function (see Supplemental Material). IC50 values and Hill coefficients are provided in the text.
There were also substantial differences in the kinetics of HMR-1556 inhibition. Specifically, on- and off-rates observed for suppression of S140G-IKs were significantly slower than WT-IKs (Figure S2). The dramatically slower off-rate for S140G-IKs suggested a stronger interaction between HMR-1556 and the channel consistent with our finding that the mutant subunit confers an enhanced affinity for the drug. The intermediate phenotype of HET-IKs exhibited an on-rate comparable to WT-IKs but a significantly slower off-rate that was more similar to S140G-IKs. This enhanced sensitivity suggested an opportunity to selectively suppress the mutant current with minimal effects on WT-IKs.
V141M-IKs exhibits enhanced sensitivity to HMR-1556
We also determined the pharmacologic effects of HMR-1556 on another previously reported gain-of-function KCNQ1 mutation, V141M, associated with early onset AF.12 V141M-IKs exhibited enhanced sensitivity to HMR-1556 and a complex IC50 binding curve with values similar to S140G-IKs (Figure 3). The off-rate was significantly slower than WT-IKs (Figure S2). These data demonstrate that increased HMR-1556 sensitivity was not specific to S140G-IKs. All further experiments were conducted with S140G-IKs as a prototypic familial AF mutation.
Figure 3.
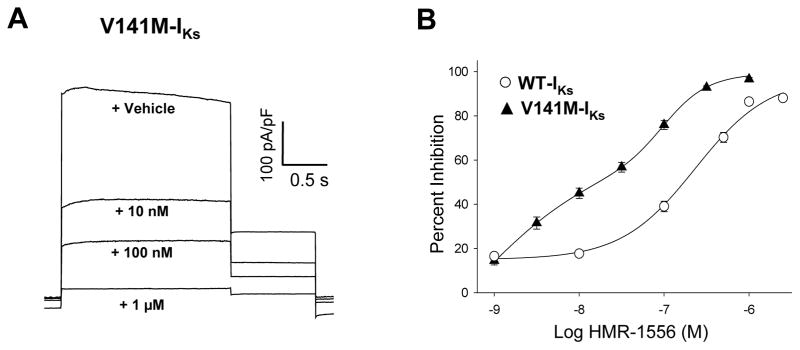
V141M-IKs exhibits enhanced sensitivity to HMR-1556. A, Representative current densities (current normalized to cell capacitance) recorded from cells expressing WT-IKs that were elicited by a 2 s voltage step to +40 mV followed by a 10 s interpulse during application of vehicle or various concentration of HMR-1556. B, HMR-1556 concentration-response curves for V141M-IKs (▲) and WT-IKs (○). Solid lines represents fits of the averaged data (9–11 cells) to a biphasic Hill function (see Supplemental Material). For V141M-IKs, the high affinity state had an IC50 of 0.72 nM and Hill coefficient of 0.6; the low affinity state had an IC50 of 204 nM and Hill coefficient of 1.7.
Properties of heteromeric S140G-IKs and WT-IKs
We elucidated the functional properties of HET-IKs. Compared to WT-IKs, cells expressing HET-IKs exhibited larger amplitudes with a large fraction of instantaneous current between −80 and −20 mV (Figure S3). At more positive voltage steps (−20 to +60 mV), HET-IKs exhibits both time-dependent and constitutive activation with significantly greater current density than WT-IKs (Figure S3). The voltage dependence of activation was shifted significantly in the hyperpolarized direction for HET-IKs (V1/2: HET-IKs, 1.4±8.1 mV; WT-IKs, 30.1±9.3 mV; p<0.001) without any difference in slope factor (Figure S3). The time course of deactivation was extremely slow for HET-IKs as compared to WT-IKs (Figure S3).
During repetitive depolarization to +40 mV with a short recovery period, both WT-IKs and HET-IKs exhibited a use-dependent accumulation of instantaneous and steady-state current over time, but HET-IKs current density was significantly greater than WT-IKs at each successive pulse (Figure S4). The ratio of instantaneous to steady-state current at the end of this protocol, a proxy for the degree of constitutive activation, was much greater for HET-IKs (84±3%) than WT-IKs (38±4%; p<0.001). These findings illustrate the dynamic nature of IKs and further emphasize the biophysical consequences of the gain-of-function mutation, KCNQ1-S140G.
Selective inhibition of HET-IKs with HMR-1556
Given the enhanced sensitivity of S140G-IKs to HMR-1556, we hypothesized that HET-IKs could be selectively suppressed by using a concentration HMR-1556 that predominantly inhibits the high affinity state. To test this hypothesis, we applied 20nM HMR-1556 or vehicle to heterologously expressed channels and assessed the effects of drug on gating kinetics and current amplitudes.
Vehicle had no effects on the behavior of WT-IKs and HET-IKs (Figure 4A,B). Further, 20nM HMR-1556 had no appreciable effect on current levels or gating behavior of WT-IKs (Figure 4C), but the drug exerted notable effects on HET-IKs including suppression of both instantaneous and steady-state current amplitude and attenuation of use-dependent current accumulation (Figure 4D). The effects of 20nM HMR-1556 on WT-IKs and HET-IKs are quantified in Figure 5A–D. Importantly, 20nM HMR-1556 did not inhibit WT-IKs, but did reduce the amplitude of HET-IKs to levels that were not significantly different (p=0.32) from WT-IKs. Additionally, the ratio of instantaneous to steady-state current for HET-IKs was also modified by the drug to a value that was not significantly different from WT-IKs (p=0.06, Figure 5E). These findings demonstrated the selective suppression of HET-IKs and the normalization of this mutant current to WT-IKs levels.
Figure 4.

Selective inhibition of HET-IKs by HMR-1556. A and B, Effects of vehicle on whole-cell currents during an activation voltage clamp protocol (middle panel) and during repetitive stimulation (right panel) from cells expressing WT-IKs (A) or HET-IKs (B). C and D, Effect of HMR-1556 (20 nM) on whole-cell currents during activation (middle panel) and repetitive stimulation (right panel) protocols from cells expressing WT-IKs (C) or HET-IKs (D). In A–D, traces in each row are from the same cell.
Figure 5.

Selective inhibition of HET-IKs by HMR-1556. A, Voltage dependence of instantaneous current density for vehicle-treated WT-IKs (○, n = 10), vehicle-treated HET-IKs (●, n = 11), HMR-1556 (20 nM) treated WT-IKs (□, n = 10), and HMR-1556 (20 nM) treated HET-IKs (■, n = 9). Differences between vehicle-treated HET-IKs and other groups were significant at the p<0.001 level for voltages between −20 and +60 mV. B, Voltage dependence of steady-state current density for vehicle or HEM-1556 treated WT-IKs or HET-IKs (symbols defined in A). Differences between vehicle-treated HET-IKs and other groups were significant at the p<0.02 level for voltages between −40 and +60 mV. C, Use dependence of instantaneous current density for vehicle or HEM-1556 treated WT-IKs or HET-IKs (symbols defined in A). Differences between vehicle-treated HET-IKs and other groups were significant at the p<0.001 level at all tested potentials. D, Use dependence of steady-state current density for vehicle or HEM-1556 treated WT-IKs or HET-IKs (symbols defined in A). Differences between vehicle-treated HET-IKs and other groups were significant at the p<0.02 level at all tested potentials. In A–D, there were no significant differences (p=0.09–0.74) among vehicle-treated WT-IKs, HMR-1556 treated WT-IKs, and HMR-1556 treated HET-IKs at any voltage. E, Ratios of instantaneous current density to steady-state current density. Differences between vehicle-treated WT-IKs (open black bar) or HET-IKs (solid black bar) was significant at p<0.001, whereas there was no significant difference (p=0.06) between HMR-1556 treated WT-IKs (open red bar) and HET-IKs (solid red bar).
HMR-1556 mitigates S140G-IKs-induced atrial action potential duration shortening
We examined the effects of HMR-1556 on action potentials in cultured adult rabbit left atrial myocytes expressing WT-IKs or S140G-IKs channel complexes. Action potentials were elicited at 1 Hz during whole-cell current clamp recording of adenovirus transduced atrial myocytes (Figure S5). Expression of S140G-IKs in left atrial myocytes hyperpolarized the resting membrane potential and significantly reduced 90% action potential duration (APD90) compared to WT-IKs expression (APD90: WT-IKs, 177.4±21.0 msec; S140G-IKs, 68.9±19.2 msec; p<0.001) (Figure 6A). Application of 1μM HMR-1556 did not alter APD90 of WT-IKs expressing myocytes (185.2±27.3 msec, p=0.59), whereas application of 1μM HMR-1556 significantly lengthened APD90 of S140G-IKs expressing myocytes (117.1±12.7 msec, p<0.04) (Figure 6B). These findings illustrate that HMR-1556 can selectively suppress S140G-IKs effects on atrial action potential duration without altering action potentials in myocytes expressing WT-IKs.
Figure 6.
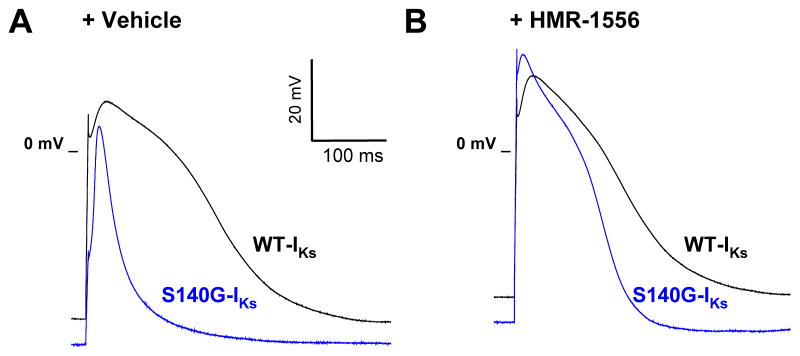
HMR-1556 mitigates atrial action potential shortening by S140G-IKs. Representative averages of 10 sequential action potentials from cultured rabbit left atrial myocytes expressing either WT-IKs (black line, n=6) or S140G-IKs (blue line, n=6). A, Action potentials elicited after application of vehicle. B, Action potentials elicited after application of 1 μM HMR-1556. APD90 values are provided in the text.
Discussion
The discovery of mutations in familial AF illustrated the contribution of specific genetic factors to AF susceptibility and suggested molecular mechanisms for some heritable forms of this common arrhythmia. For gain-of-function KCNQ1 mutations in particular, we sought to exploit this knowledge to explore a potential targeted therapy. Specifically, we hypothesized that KCNQ1 mutations predisposing to AF encode potassium channels with distinct pharmacological properties that could render them susceptible to selective inhibition. Genotype-specific therapies for inherited arrhythmia syndromes such as congenital long-QT syndrome and catecholaminergic polymorphic ventricular tachycardia are emerging.24,25 Further, a precedent for mutation-specific pharmacology of a rare, inherited disorder was established by the approval of ivacaftor for treatment of cystic fibrosis caused by CFTR-G551D.26
In this study, we investigated the utility of the selective and high affinity IKs blocker HMR-1556 to inhibit gain-of-function KCNQ1 mutations S140G and V141M. Chromanol 293B was the first identified selective IKs blocker, which exerts its effect at low micromolar concentrations.27 The chromanol derivative HMR-1556 was developed to increase potency and improve the selectivity of IKs inhibition.28 This derivative was initially demonstrated to have an IC50 of 120 nM against IKs expressed in Xenopus oocytes with little effect on other recombinant potassium channels at 10 μM consistent with a high level of specificity.28 In isolated canine ventricular myocytes, HMR-1556 inhibits IKs with a nanomolar IC50 whereas inhibition of other ionic currents (e.g., IKr, IK1, Ito, ICa,L) required much higher concentrations.29
Consistent with our hypothesis, we observed that S140G-IKs exhibits enhanced sensitivity to HMR-1556. This enhancement was correlated with the emergence of an additional high affinity state, which was also observed with V141M-IKs. Importantly, using a concentration that predominantly inhibits the high affinity state, we demonstrated that HMR-1556 effectively suppressed HET-IKs amplitude to a level that was not significantly different from WT-IKs. Further, this drug concentration attenuated the use-dependent accumulation of HET-IKs that occurs during repetitive pulsing. Importantly, we demonstrated that HMR-1556 can mitigate the S140G-IKs induced APD shortening in cultured adult rabbit atrial myocytes without affecting action potentials in myocytes expressing WT-IKs. These findings offer evidence supporting the potential for genotype-specific therapy of familial AF.
The potential utility of HMR-1556 or a similarly acting drug in the setting of familial AF should be considered in the context of the liabilities of inhibiting IKs in tissues other than atria. Reduction of IKs in ventricular muscle carries the risk of reduced repolarization reserve and predisposition to reentrant arrhythmia as in type 1 congenital long-QT syndrome. In anesthetized dogs receiving continuous intravenous infusions of HMR-1556, there was significant QTc prolongation and reproducible triggering of torsades de points with an isoproterenol bolus.30 Prolongation of QTc during HMR-1556 exposure is accentuated in dogs by co-administration of the IKr blocker dofetilide.31 In Langendorff-perfused rabbit hearts, HMR-1556 alone was not sufficient to prolong monophasic action potential duration (APD) but co-administration of either dofetilide alone or dofetilide with veratridine caused significant lengthening of APD.32,33 These reports emphasize the potential proarrhythmic effects of high concentration HMR-1556 or of concurrent IKs and IKr inhibition. Fortunately, our data indicate that selective inhibition of S140G-IKs can be achieved at HMR-1556 concentrations that do not suppress WT-IKs.
Ototoxicity is another potential concern with HMR-1556. Because IKs expressed in the stria vascularis of inner ear is important in the generation of the K+ rich cochlear endolymph, disruption of IKs has the potential to impair hearing as observed in autosomal recessive Jervell-Lange-Nielson syndrome associated with KCNQ1 or KCNE1 mutations.34–36 Indeed, high concentrations of HMR-1556 exert a reversible ototoxicity in cats.37 Again, our data suggest that there is a potential concentration range that may be free of inner ear adverse effects.
In summary, the AF-associated mutations KCNQ1-S140G and KCNQ1-V141M confer enhanced sensitivity to HMR-1556 in the context of the IKs channel complex. At a concentration that predominantly suppresses current by interacting with a novel high affinity state expressed by the S140G mutant, HMR-1556 normalized current amplitudes to levels that are not significantly different from WT-IKs, and attenuated the use-dependent accumulation of current. In cultured adult rabbit atrial myocytes, HMR-1556 mitigated the shortened APD induced by S140G-IKs expression. Our demonstration of selective targeting of this gain-of-function mutation provides a potential proof-of-principal for genotype-specific treatment of familial AF.
Supplementary Material
Acknowledgments
The authors acknowledge Lauren Manderfield for generating the KCNQ1-S140G mutation, and Richard C. Welch for assisting with isolating rabbit cardiac myocytes.
Funding Sources: This work was supported by Greater Southeast Affiliate Predoctoral Fellowship 11PRE7610043 from the American Heart Association, Ruth L. Kirschstein National Research Service Award for Individual Predoctoral MD/PhD Fellows F30 HL107066 from the National Heart Lung and Blood Institute (NHLBI), Public Health Service Award T32 GM07347 from National Institute for General Medical Studies for the Vanderbilt Medical-Scientist Training Program, Cooperative Research Center grant U19-HL65962-10 from NHLBI, and a Vanderbilt University Graduate School Dissertation Enhancement Grant.
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Wolf PA, D’Agostino RB, Silbershatz H, Kannel WB, Levy D. Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation. 1998;98:946–952. doi: 10.1161/01.cir.98.10.946. [DOI] [PubMed] [Google Scholar]
- 3.Kim MH, Johnston SS, Chu BC, Dalal MR, Schulman KL. Estimation of total incremental health care costs in patients with atrial fibrillation in the United States. Circ Cardiovasc Qual Outcomes. 2011;4:313–320. doi: 10.1161/CIRCOUTCOMES.110.958165. [DOI] [PubMed] [Google Scholar]
- 4.Chugh SS, Blackshear JL, Shen WK, Hammill SC, Gersh BJ. Epidemiology and natural history of atrial fibrillation: clinical implications. J Am Coll Cardiol. 2001;37:371–378. doi: 10.1016/s0735-1097(00)01107-4. [DOI] [PubMed] [Google Scholar]
- 5.Kopecky SL, Gersh BJ, McGoon MD, Whisnant JP, Holmes DR, Jr, Ilstrup DM, Frye RL. The natural history of lone atrial fibrillation. A population-based study over three decades. N Engl J Med. 1987;317:669–674. doi: 10.1056/NEJM198709103171104. [DOI] [PubMed] [Google Scholar]
- 6.Brand FN, Abbott RD, Kannel WB, Wolf PA. Characteristics and prognosis of lone atrial fibrillation. 30-year follow-up in the Framingham Study. JAMA. 1985;254:3449–3453. [PubMed] [Google Scholar]
- 7.Scardi S, Mazzone C, Pandullo C, Goldstein D, Poletti A, Humar F. Lone atrial fibrillation: prognostic differences between paroxysmal and chronic forms after 10 years of follow-up. Am Heart J. 1999;137:686–691. doi: 10.1016/s0002-8703(99)70224-3. [DOI] [PubMed] [Google Scholar]
- 8.Fox CS, Parise H, D’Agostino RB, Sr, Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA, Benjamin EJ. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–2855. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- 9.Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, Sigurdsson A, Jonasdottir A, Baker A, Thorleifsson G, Kristjansson K, Palsson A, Blondal T, Sulem P, Backman VM, Hardarson GA, Palsdottir E, Helgason A, Sigurjonsdottir R, Sverrisson JT, Kostulas K, Ng MC, Baum L, So WY, Wong KS, Chan JC, Furie KL, Greenberg SM, Sale M, Kelly P, MacRae CA, Smith EE, Rosand J, Hillert J, Ma RC, Ellinor PT, Thorgeirsson G, Gulcher JR, Kong A, Thorsteinsdottir U, Stefansson K. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353–357. doi: 10.1038/nature06007. [DOI] [PubMed] [Google Scholar]
- 10.Roberts R. Mechanisms of disease: Genetic mechanisms of atrial fibrillation. Nature Clin Pract Cardiovasc Med. 2006;3:276–282. doi: 10.1038/ncpcardio0509. [DOI] [PubMed] [Google Scholar]
- 11.Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J, Huang W. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 12.Hong K, Piper DR, Diaz-Valdecantos A, Brugada J, Oliva A, Burashnikov E, Santos-de-Soto J, Grueso-Montero J, Diaz-Enfante E, Brugada P, Sachse F, Sanguinetti MC, Brugada R. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res. 2005;68:433–440. doi: 10.1016/j.cardiores.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 13.Olson TM, Alekseev AE, Moreau C, Liu XK, Zingman LV, Miki T, Seino S, Asirvatham SJ, Jahangir A, Terzic A. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med. 2007;4:110–116. doi: 10.1038/ncpcardio0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J, Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings P, Barhanin J, Chen Y. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005;332:1012–1019. doi: 10.1016/j.bbrc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R, Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J, Chen Y. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. 2004;75:899–905. doi: 10.1086/425342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinner MF, Pfeufer A, Akyol M, Beckmann BM, Hinterseer M, Wacker A, Perz S, Sauter W, Illig T, Nabauer M, Schmitt C, Wichmann HE, Schomig A, Steinbeck G, Meitinger T, Kaab S. The non-synonymous coding IKr-channel variant KCNH2-K897T is associated with atrial fibrillation: results from a systematic candidate gene-based analysis of KCNH2 (HERG) Eur Heart J. 2008;29:907–914. doi: 10.1093/eurheartj/ehm619. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, Kannankeril PJ, Roden DM. Mutations in sodium channel β1- and β2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2009;2:268–275. doi: 10.1161/CIRCEP.108.779181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makiyama T, Akao M, Shizuta S, Doi T, Nishiyama K, Oka Y, Ohno S, Nishio Y, Tsuji K, Itoh H, Kimura T, Kita T, Horie M. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J Am Coll Cardiol. 2008;52:1326–1334. doi: 10.1016/j.jacc.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 19.Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, George AL, Jr, Roden DM. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117:1927–1935. doi: 10.1161/CIRCULATIONAHA.107.757955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abraham RL, Yang T, Blair M, Roden DM, Darbar D. Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J Mol Cell Cardiol. 2009;48:181–190. doi: 10.1016/j.yjmcc.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bendahhou S, Marionneau C, Haurogne K, Larroque MM, Derand R, Szuts V, Escande D, Demolombe S, Barhanin J. In vitro molecular interactions and distribution of KCNE family with KCNQ1 in the human heart. Cardiovasc Res. 2005;67:529–538. doi: 10.1016/j.cardiores.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, Liu Y, Dong X, Kuang Y, Lin J, Su X, Peng L, Jin Q, He Y, Liu B, Pan Z, Li L, Zhu Q, Lin X, Zhou Q, Pan Q, Eurlings PM, Fei J, Wang Z, Chen YH. Human KCNQ1 S140G mutation is associated with atrioventricular blocks. Heart Rhythm. 2007;4:611–618. doi: 10.1016/j.hrthm.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 23.Restier L, Cheng L, Sanguinetti MC. Mechanisms by which atrial fibrillation-associated mutations in the S1 domain of KCNQ1 slow deactivation of IKs channels. J Physiol. 2008;586:4179–4191. doi: 10.1113/jphysiol.2008.157511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol. 2008;19:1289–1293. doi: 10.1111/j.1540-8167.2008.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380–383. doi: 10.1038/nm.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordonez C, Elborn JS. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bosch RF, Gaspo R, Busch AE, Lang HJ, Li GR, Nattel S. Effects of the chromanol 293B, a selective blocker of the slow, component of the delayed rectifier K+ current, on repolarization in human and guinea pig ventricular myocytes. Cardiovasc Res. 1998;38:441–450. doi: 10.1016/s0008-6363(98)00021-2. [DOI] [PubMed] [Google Scholar]
- 28.Gogelein H, Bruggemann A, Gerlach U, Brendel J, Busch AE. Inhibition of IKs channels by HMR 1556. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:480–488. doi: 10.1007/s002100000284. [DOI] [PubMed] [Google Scholar]
- 29.Thomas GP, Gerlach U, Antzelevitch C. HMR 1556, a potent and selective blocker of slowly activating delayed rectifier potassium current. J Cardiovasc Pharmacol. 2003;41:140–147. doi: 10.1097/00005344-200301000-00018. [DOI] [PubMed] [Google Scholar]
- 30.Gallacher DJ, Van de WA, van der LH, Hermans AN, Lu HR, Towart R, Volders PG. In vivo mechanisms precipitating torsades de pointes in a canine model of drug-induced long-QT1 syndrome. Cardiovasc Res. 2007;76:247–256. doi: 10.1016/j.cardiores.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 31.Nakashima H, Gerlach U, Schmidt D, Nattel S. In vivo electrophysiological effects of a selective slow delayed-rectifier potassium channel blocker in anesthetized dogs: potential insights into class III actions. Cardiovasc Res. 2004;61:705–714. doi: 10.1016/j.cardiores.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 32.So PP, Hu XD, Backx PH, Puglisi JL, Dorian P. Blockade of IKs by HMR 1556 increases the reverse rate-dependence of refractoriness prolongation by dofetilide in isolated rabbit ventricles. Br J Pharmacol. 2006;148:255–263. doi: 10.1038/sj.bjp.0706721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.So PP, Backx PH, Dorian P. Slow delayed rectifier K+ current block by HMR 1556 increases dispersion of repolarization and promotes Torsades de Pointes in rabbit ventricles. Br J Pharmacol. 2008;155:1185–1194. doi: 10.1038/bjp.2008.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Fauré S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15:186–189. doi: 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- 35.Tyson J, Tranebjerg L, Bellman S, Wren C, Taylor JFN, Bathen J, Aslaksen B, Sorland SJ, Lund O, Malcolm S, Pembrey M, Bhattacharya S, Bitner-Glindzicz M. IsK and KVLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum Mol Genet. 1997;6:2179–2185. doi: 10.1093/hmg/6.12.2179. [DOI] [PubMed] [Google Scholar]
- 36.Schulze-Bahr E, Wang Q, Wedekind H, Haverkamp W, Chen Q, Sun Y, Rubie C, Hordt M, Towbin JA, Borggrefe M, Assmann G, Qu X, Somberg JC, Breithardt G, Oberti C, Funke H. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat Genet. 1997;17:267–268. doi: 10.1038/ng1197-267. [DOI] [PubMed] [Google Scholar]
- 37.Hartmann R, Gerlach U, Klinke R. Ototoxic side-effects of the IKs-channel blocker HMR1556. Hear Res. 2002;172:145–150. doi: 10.1016/s0378-5955(02)00576-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.