

Abstract
In mice, three major B-cell subsets have been identified with distinct functionalities: B1 B cells, marginal zone B cells and follicular B2 B cells. Here, we used the growing body of publicly available transcriptomics data to create an expression atlas of 84 gene expression microarray data sets of distinct mouse B-cell subsets. These data were subjected to network-based cluster analysis using BioLayout Express3D. Using this analysis tool, genes with related functions clustered together in discrete regions of the network graph and enabled the identification of transcriptional networks that underpinned the functional activity of distinct cell populations. Some gene clusters were expressed highly by most of the cell populations included in this analysis (such as those with activity related to house-keeping functions). Others contained genes with expression patterns specific to distinct B-cell subsets. While these clusters contained many genes typically associated with the activity of the cells they were specifically expressed in, many novel B-cell-subset-specific candidate genes were identified. A large number of uncharacterized genes were also represented in these B-cell lineage-specific clusters. Further analysis of the activities of these uncharacterized candidate genes will lead to the identification of novel B-cell lineage-specific transcription factors and regulators of B-cell function. We also analysed 36 microarray data sets from distinct human B-cell populations. These data showed that mouse and human germinal centre B cells shared similar transcriptional features, whereas mouse B1 B cells were distinct from proposed human B1 B cells.
Keywords: B cells, meta-analysis, microarray, transcriptomics
Introduction
In mice, B cells are considered to comprise three distinct subsets: B1 B cells; marginal zone (MZ) B cells; B2 follicular (FO) B cells. The B2 B cells produce antibodies with high affinity and specificity to T-cell-dependent antigens. B1 B cells and MZ B cells, in contrast, respond rapidly to T-cell-independent antigen and produce natural antibody with low-affinity and wide antigen-reactivity, typically recognizing conserved structures on pathogens. Some B-cell populations, including B1 B cells and MZ B cells, can also act as antigen-presenting cells and secrete regulatory cytokines such as interleukin-10.
To gain further insights into the transcriptomes of distinct tissues and cell populations we have previously used the novel network tool BioLayout Express3D to perform detailed cluster analyses on large collections of publicly available micro-array data sets.1,2 This tool identifies co-regulated genes based on the construction of correlation networks, where genes (probe sets) are represented as nodes, and edges represent the similarity (above a given threshold) between the expression profiles. The clusters are then defined using the Markov Clustering algorithm (MCL) and both the network and clusters are visualized using a powerful three-dimensional (3D) network rendering engine. These meta-analyses show that clusters of genes with correlated expression across these data collections are associated with specific tissues (e.g. intestine, bone marrow), cell lineages (e.g. mononuclear phagocytes, follicular dendritic cells, M cells) or cellular functions (e.g. extracellular matrix).3–5 These analyses enable assessments to be made on the transcriptional relations between distinct cell populations.6,7 Furthermore, predictions can also be made on the functions of novel candidate genes based on the common activities and expression patterns of the majority of the other genes within particular clusters.
In the current study we analysed the transcriptional profiles of multiple B-cell populations. A large collection of publicly available mouse gene expression data was assembled comprising a total of 84 individual microarray data sets, representing 16 different B-cell subsets isolated from distinct tissues. These data were then subjected to network-based cluster analysis using BioLayout Express3D. Several co-expressed gene clusters were identified with expression restricted to B1 B cells, MZ B cells and B2 B cells. Many of these clusters contained genes related to the characteristic activities of the B-cell subsets by which they were expressed. However, these clusters often contained many uncharacterized genes as well as many potentially novel B-cell-subset-specific candidate genes. We also analysed 36 microarray data sets from distinct human B-cell populations. While these data showed that mouse and human germinal centre (GC) B cells shared similar transcriptional features, mouse B1 B cells were distinct from proposed human B1 B cells. Further characterization of the contents of these co-expressed gene expression clusters will lead to the identification of novel B-cell lineage-specific transcription factors and regulators of B-cell function. To enable readers to explore this interactive expression atlas of distinct mouse B-cell subsets in greater detail the network graph is freely available on the author's institutional website.
Materials and methods
Selection of gene expression data sets
The Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) database was searched for mouse B-cell subset expression data sets. Data sets were selected based on the following three criteria: (i) chip platform (Affymetrix mouse gene 1.0 ST expression arrays), (ii) B-cell subsets, and (3) availability of raw data (.cel) files. A large selection of mouse gene expression data was initially obtained comprising a total of 96 individual microarray data sets, including many from a large collection of publicly available gene expression data from different immune cell populations8 (Immunological Genome Project; http://www.immgen.org; GEO data sets accession number, GSE15907). Raw data (.cel) files were downloaded and the quality of the raw data from each data set was reanalysed using the arrayQualityMetrics package in Bioconductor (http://www.bioconductor.org) and scored on the basis of five metrics, namely maplot, spatial, boxplot, heatmap and rle. Any array failing on more than one quality control (QC) metric was removed. The remaining 84 data sets represented 16 different B-cell subsets isolated from distinct tissues (see Supplementary material, Table S1). Data sets were normalized using Robust Multichip Analysis (RMA Express; http://rmaexpress.bmbolstad.com/), and annotated using the latest library available from Affymetrix (http://www.affymetrix.com/). Samples were given a standard annotation (chip no.: cell class: description: replicate no.) and arranged according to cell-type grouping to ease interpretation of these data. Full details on each data set are provided in Table S1.
We also analysed 24 data sets from three independent studies of human B-cell subsets performed on Affymetrix Human Genome U133A plus 2.0 expression arrays (Ref. 9; GSE12366, Ref. 10; GSE15271, Ref. 11), and 12 data sets including samples of ‘proposed’ human B1 B cells performed on Affymetrix Human Gene 1.0 ST expression arrays (GSE42724, Ref. 12).
Network analysis
The analysis pipeline used in the current study is presented in Fig. 1. All the 84 combined, normalized and annotated data sets were saved as an ‘.expression’ file and imported into the tool BioLayout Express3D. This file format contains a unique identifier for each probe set on the array (gene symbol:probe set ID), followed by columns of gene annotation information and finally the non-log-transformed data values (normalized probe-set expression levels) for each sample (each column of data being derived from a different sample). First, a sample-to-sample correlation matrix was calculated from the normalized and non-log-transformed gene expression data. A pairwise Pearson correlation matrix was calculated, which comprised an all versus all comparison of the expression profile of each probe set on the array. A graph was then plotted using all sample-to-sample relationships ≥ 0·95 (Fig. 2). In this graph all the nodes represent individual data sets (cells) and the edges that link these data sets represent Pearson correlation coefficients ≥ 0·95.
Figure 1.
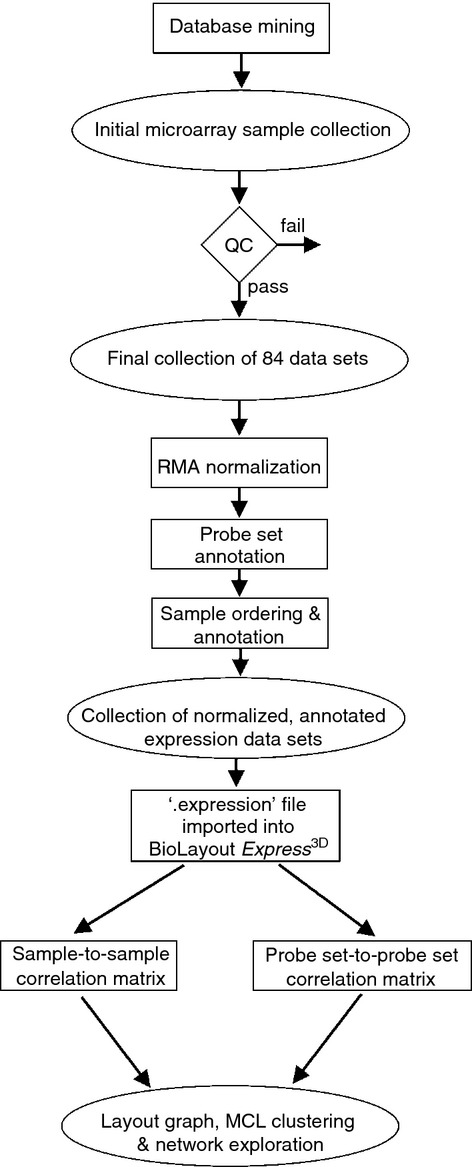
Data analysis pipeline used in the current study.
Figure 2.
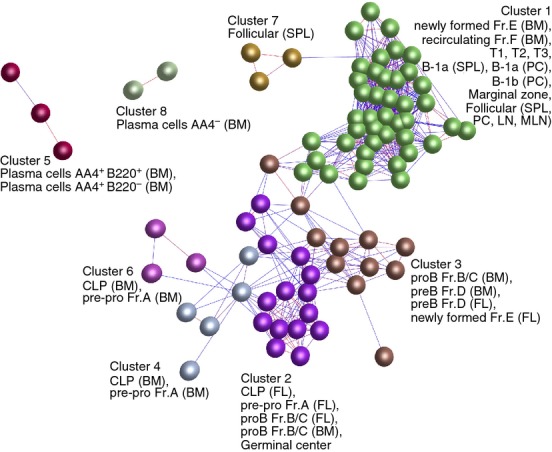
Clustering of samples based on their global gene expression profile. A Pearson correlation matrix was prepared by comparing data derived from all 84 samples. A network graph was then constructed using sample-to-sample relationships greater than r ≥ 0·85, and clustered using a Markov Clustering inflation value of 2·2. Here, the nodes represent samples (individual micro-array data sets) and each cluster of nodes is assigned a different colour. The edges represent the connections between data sets and are coloured according to the strength of the correlation (red, r = 1·0; blue, r ≥ 0·85). Full descriptions of the sources of each data set are given in Table S1. BM, bone marrow; FL, fetal liver; LN, lymph node; MLN, mesenteric lymph node; PC, peritoneal cavity; SPL, spleen.
Next, a pairwise probe set-to-probe set Pearson correlation matrix was calculated based on each probe set's profile across each of the samples. A Pearson correlation coefficient cut-off threshold of r ≥ 0·85 was selected and an undirected network graph of these data was generated. In this graph the nodes represent individual probe sets (genes/transcripts) and the edges between them represent Pearson correlation coefficients ≥ 0·85. The network was then clustered into groups of probe sets (genes) sharing similar profiles using the built-in MCL algorithm using an inflation value (which controls the granularity of clustering) set to 2·2.
Cluster analysis
The probe set-to-probe set network graph (Fig. 3) was then explored extensively to understand the significance of the gene clusterings and the functional activities of the cell populations were investigated. Genes in the clusters of interest were assessed for cellular functions and activities using a combination of literature review and bioinformatics. Significantly over-represented gene ontologies (GO) within clusters of interest were identified using GOstat (http://gostat.wehi.edu.au). For each GO term, the probability was calculated that the observed counts occurred by the random distribution of this GO term between the cluster of interest and the reference group (all genes on the microarray). The Benjamini and Hochberg correction was used to control the false discovery rate of errors expected from multiple testing. Over-represented gene ontologies with P values < 0·05 were accepted as significant (see Supplementary material, Table S2). Groups of genes often shared several GO terms that were indicative of the same biological process, molecular function or cellular compartment. In these instances the most informative GO terms within the top 10 identified are presented.
Figure 3.
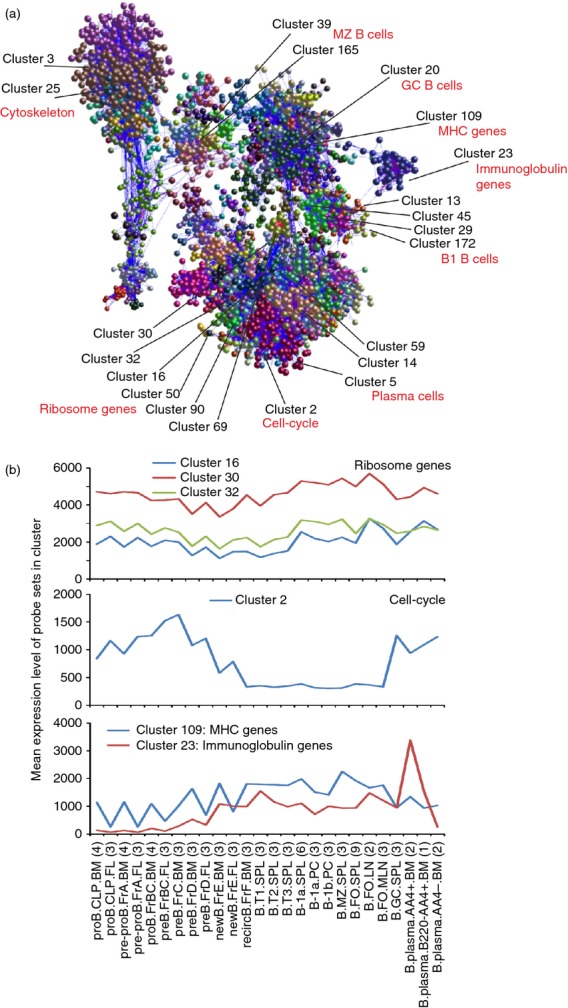
Network analysis of mouse B-cell subset transcriptomics data. (a) Main component of the network graph derived from 84 micro-array data sets of distinct mouse B-cell subsets. Here, the nodes represent probe sets (genes) and the edges represent correlations between individual expression profiles above r ≥ 0·85. (b) The mean expression profiles of the genes in selected clusters across the 84 samples. x-axis shows the samples, grouped according to cell type (in order of presentation in Table S1). For each cell population mean expression levels are presented and the number of replicates indicated in parenthesis on the x-axis. y-axis shows the mean signal expression intensity for the cluster (probe set intensity).
Availability of supporting data
The entire data set used here is available on a dedicated page on the authors’ institutional website (http://www.roslin.ed.ac.uk/neil-mabbott/b-cells). Included are the ‘.expression’ file containing all the combined, normalized and annotated expression data, and a webstart version of BioLayout Express3D. This interactive expression atlas of mouse B-cell subsets enables the reader to explore the network graph in 3D, visualize the mean expression profile of each cluster and the specific expression patterns of individual genes across the entire data collection.
Results and discussion
Comparison of the global gene expression profiles of distinct mouse B-cell subsets
First a graph was created of the sample-to-sample correlations across all the 84 data sets using Pearson correlation relationships r ≥ 0·85 to define edges. The graph was then clustered into groups of data sets (samples) sharing similar expression profiles using the MCL algorithm and individual clusters were assigned a different colour (Fig. 2). Different progenitor and differentiated B-cell subsets clustered together like-with-like and were situated in specific regions of the graph. For example, all the progenitor stages used in this analysis up to the pre-B Fr.D stage clustered in a distinct region of the graph (clusters 2, 3, 4 and 6; Fig. 2). Data sets within these clusters were mostly distributed in order of developmental stage. Those in cluster 3 were connected by a number of edges to the newly formed Fr.E data sets within the largest cluster (cluster 1; Fig. 2), which contained most of the differentiated B-cell subsets from the newly formed Fr.E stage. Exceptions to this were three FO B-cell samples that were located in a separate cluster (cluster 7), but directly connect by an edge to the other FO B-cell data sets in cluster 1. The plasma cell data sets were also located in distinct clusters based on their expression of AA4 (CD93; AA4+, cluster 5; AA4−, cluster 8) suggesting distinct expression profiles.
Creation of the probe set-to-probe set correlation network graph
Next, a full probe set-to-probe set Pearson correlation matrix was calculated whereby the similarity in the expression profile of each probe set represented on the array was compared across each of the 84 data sets. A network graph was constructed using a correlation threshold of r ≥ 0·85. Here, each node represents an individual Affymetrix probe set (representing a specific gene) and correlations between probe sets greater than the threshold value were represented by graph edges. The network graph contained 12 149 nodes representing individual probe sets connected by 385 142 edges, indicating Pearson correlations between probe sets of r ≥ 0·85. After clustering using the MCL algorithm, 315 clusters of six or more nodes were obtained. An image of the 3D network graph is shown in Fig. 3(a) with the locations of some example clusters highlighted. Table S2 lists the contents of each of the 315 clusters. To enable readers to explore the network graph in greater detail the entire data set and a webstart version of the network graph are available on the authors’ institutional website (http://www.roslin.ed.ac.uk/neil-mabbott/b-cells).
The network graph comprises cliques of genes that are co-expressed in a specific manner (correlated in their expression profiles ≥ 0·85) and connected by a large number of edges (Fig. 3a). Clusters containing genes with similar functions or cellular activities typically occupied similar regions of the network graph. For example, clusters 3 and 25 were significantly enriched with genes encoding components of the cytoskeleton/extracellular matrix (e.g. cluster 25, GO:0005201 extracellular matrix structural constituent, P < 0·000646; Table S2) and were situated adjacent to each other in the same region of the graph. Similarly, clusters 16, 30, 32, 50, 69, 90 and 92 were significantly enriched in genes encoding ribosomal components (e.g. cluster 16, GO:0003735, structural component of ribosome, P < 2·09 × 10−8; Table S2). All of these clusters were located within specific regions of the 3D network graph (Fig. 3a). Other examples showed that clusters of genes that were expressed highly by specific cell populations such as plasma cells (clusters 5, 14 and 59), B1 lineage B cells (clusters 13, 29, 45 and 172) and MZ B cells (cluster 39 and 165) were similarly located in specific regions of the graph, like-with-like (Fig. 3a).
Identification of B-cell subset-specific gene expression signatures
The average expression profile of the probe sets, and the genes they represent, within each cluster can help to provide insights into their biological activities. While some clusters contained genes that were expressed highly by almost all the cell subsets included in this analysis (such as those related to house-keeping functions) (Fig. 3b), others were restricted to individual B-cell subsets, or groupings of B-cell lineages. Below we discuss examples of the tightly associated clusters with expression restricted to the major B-cell subsets identified in mice: B1 B cells, MZ B cells and B2 B cells.
B1 B cells
Two clusters were identified (cluster 13 and 172) with expression restricted to B1a and B1b B cells, especially those from the peritoneal cavity (Fig. 4). B1 B cells are long-lived, self-renewing cells that produce high levels of low-affinity, serum poly-reactive and weakly autoreactive natural IgM. Due to their broad antigen reactivity, B1 B cells play important roles in the early response to a wide range of pathogens and auto-antigens such as apoptotic cells. Across this data collection, the CD28 family receptor CTLA4 (CD152), CD30 (encoded by Tnfrsf8) and two probe sets encoding CD80 were highly and specifically expressed only by B1 B cells (Fig. 4c). While generally considered to display inhibitory effects on T cells, a role for CTLA4-signalling in the inhibition of B-cell effector functions in response to T-cell-dependent antigen has been described.13 B1 B cells characteristically play an important role in the rapid induction of antibody responses against T-cell-independent antigen. The specific expression of Ctla4 by B1 B cells, coupled with the demonstration that specific antibody responses to T-cell-dependent antigen were down-regulated on CTLA4-expressing B cells,13 suggests a potential mechanism through which this specificity is mediated.
Figure 4.

Analysis of the genes within clusters 13 and 172 which were expressed specifically by B1 B cells. (a) The mean expression profile of all the probe set intensities within clusters 13 (blue) and 172 (red) over the 84 samples. (b) Heat map showing the mean expression levels of probe sets of interest in clusters 13 and 172. Each column represents the mean (log2) probe set intensity for all samples from each source. Significant differences between groups were sought by analysis of variance. P-values for those genes that were expressed significantly (P < 0·05) by B1 B cells at levels > 2·0 fold when compared with the other cell populations. (c) The mean expression profile of probe sets representing Ctla4 (green), Cd80 (red and blue) and Tnfrsf8 (purple) across the 84 samples. (d) Comparison of the mean expression profile of probe sets representing Ciita (blue) and Zbtb32 (red) across the 84 samples. (a–d) Samples are grouped according to cell type and are arranged in order of presentation as listed in Table S1. For each cell population mean expression levels are presented and the number of replicates is indicated in parenthesis on the x-axis. Red-boxed area indicates the B1 B-cell data sets. (e) Cartoon illustrating the putative functions of all the genes represented in cluster 13 (black font) and 172 (blue font) in B1 B cells. These genes were then classified into groupings of related cellular function based on published data from literature searches and bioinformatics data bases.
Although the direct role of many of the genes in these clusters in relation to the activity of B1 B cells was uncertain, based on their descriptions or published evidence it was evident that several were involved in cell signalling, transcription and cytoskeletal regulation (Fig. 4e). For example, lymphocyte-expressed A2A adenosine receptor (Adora2ar) regulates the inflammatory response in experimental allergic encephalomyelitis,14 and guanylate-binding protein-2 (Gbp2) can inhibit nuclear factor-κB activity.15 The presence of probe sets encoding Gbp2 and two related genes, Gbp6 and Gnb3, suggests similar activity in B1 B cells.
Among the genes in cluster 13 associated with cytoskeletal regulation, cell adhesion and spreading was Itgb1, which encodes integrin β1, and has an important role in promoting B-cell transit from the MZ into the splenic white pulp cords.16,17 The co-expression of Nrp2 (which encodes neuropilin 2) was also interesting as this has been shown to activate the α6β1 integrin, enabling it to form focal adhesions.18 Three genes related to tubulin polymerization were also present (Tppp, Tppp3 and Tubb6). Across this data collection these genes were expressed highly and specifically by B1 B cells (Fig. 4b), implying a role in the reorganization of the cytoskeleton during cell spreading and/or migration.19
Several transcriptional regulators were also represented in cluster 13. The Zbtb32 gene encodes the zinc finger, broad complex, tram track, bric-a-brac (BTB) domain containing 32 transcriptional repressor. Across this data collection Zbtb32 was expressed highly and specifically only by B1 B cells, implying an important role in regulating gene expression in these cell populations (Fig. 4d). Using an ex vivo model, data suggest that Zbtb32 together with Blimp-1 (encoded by Prdm1) regulate plasma cell differentiation by repressing CIITA and MHC class II expression.20 However, the significant expression of high levels of Zbtb32 across this data collection only by differentiated B1 B cells coincident with high expression of Ciita (Fig. 4d) suggests an alternative regulatory activity in these populations. Interestingly, two other transcription factors, Sox5 and Fbxw13, were expressed highly by B1 B cells derived from the peritoneal cavity when compared with those from the spleen (Fig. 4b).
Many un-annotated probe sets were also present in cluster 13. Among these, some were expressed highly and significantly only by B1 B cells (cDNA sequence BC005685; hypothetical protein LOC641050; Affymetrix probe set IDs 10344590, 10401933, 10504755, 10504759 and 10549219; Fig. 4e). Our experience shows that the principle of guilt-by-association works well in the meta-analysis of large and diverse collections of microarray data sets.3–6 For example, our analysis of a large collection of mouse lymphocytes and leucocytes (304 data sets) identified a small cluster of genes expressed highly by tissue-derived classical dendritic cells (Cluster 79 in Ref. 4). Among the 12 annotated genes within this cluster was the known classical dendritic cell transcriptional regulator BATF3.21 Also present was the transcription factor Zbtb46, which was later shown in independent studies to be a Toll-like receptor-responsive transcriptional repressor in classical dendritic cells.22 In the current study many of the clusters were significantly enriched with genes associated with common cellular activities such as those encoding cytoskeleton/extracellular matrix components (clusters 3 and 25), ribosome components (clusters 16, 30, 32, 50, 69, 90 and 92), cell-cycle (cluster 2), immunoglobulin (cluster 23, GO:0006959, humoral immune response, P < 0·000551; Table S2) and MHC class I (cluster 109, GO:0042612, MHC class I protein complex, P < 1·03 × 10−11; Table S2). For example, cluster 2 was a large cluster of 610 probe sets and was highly enriched in genes related to the cell-cycle and mitosis (GO:0007049, cell cycle; P = 0; Table S2). Indeed, almost all of the 497 annotated genes represented in this cluster have known cell-cycle functions including many cyclins, histones, kinesins, centromere and kinetochore complex components and genes involved in DNA replication and repair. This cluster also contained many cell-cycle-related transcription factors, including Foxm1, which is critical for DNA replication and mitosis,23 and several E2F family members 24 such as E2f1. The mean expression profile of all the probe sets in cluster 2 showed that these genes were expressed highly by all the proliferating cell populations included in this analysis, but not by recirculating, transitional, B1, MZ and FO B cells (Fig. 3b). Hence, by using the concept of guilt-by-association it is reasonable to speculate that due to their restricted cellular expression across this large data collection the uncharacterized genes in cluster 13 may have important functions in the B1 B cells.
B1a B cells
Cluster 29 contained 45 probe sets encoding 34 annotated genes. The mean expression profile of this cluster indicated that the genes in it were co-expressed highly by B1a B cells (Fig. 5a). B1a cells are distinguished from other B-cell subsets by their expression of the pan-T-cell surface glycoprotein, CD5.25 The co-expression of Cd5 in cluster 29 is consistent with the suggestion that the genes within this cluster are related to the activity of B1a B cells. These cells reside in the peritoneal and pleural cavities, to a lesser extent in the spleen, and produce high levels of natural antibody. B1a B cells also play an important role in the induction of antibody responses against T-cell-independent type II antigen, and aid the clearance of apoptotic cells and auto-antigen. Although the immediate roles of many of the co-expressed genes in cluster 29 in relation to the function of B1a B cells were uncertain, some had been reported to possess immunoregulatory activity. For example, aryl-hydrocarbon receptor (encoded by Ahr) agonists can suppress IgM, IgG1 and IgE expression and plasma cell differentiation.26 Folate receptor 4 (Folr4) may play a role in the maintenance of regulatory T cells,27 implying a similar requirement for B1a B cells to incorporate folic acid. Stimulation via hypocretin receptor 2 (Hcrtr2) has also been proposed to regulate the function of myeloid cells.28
Figure 5.
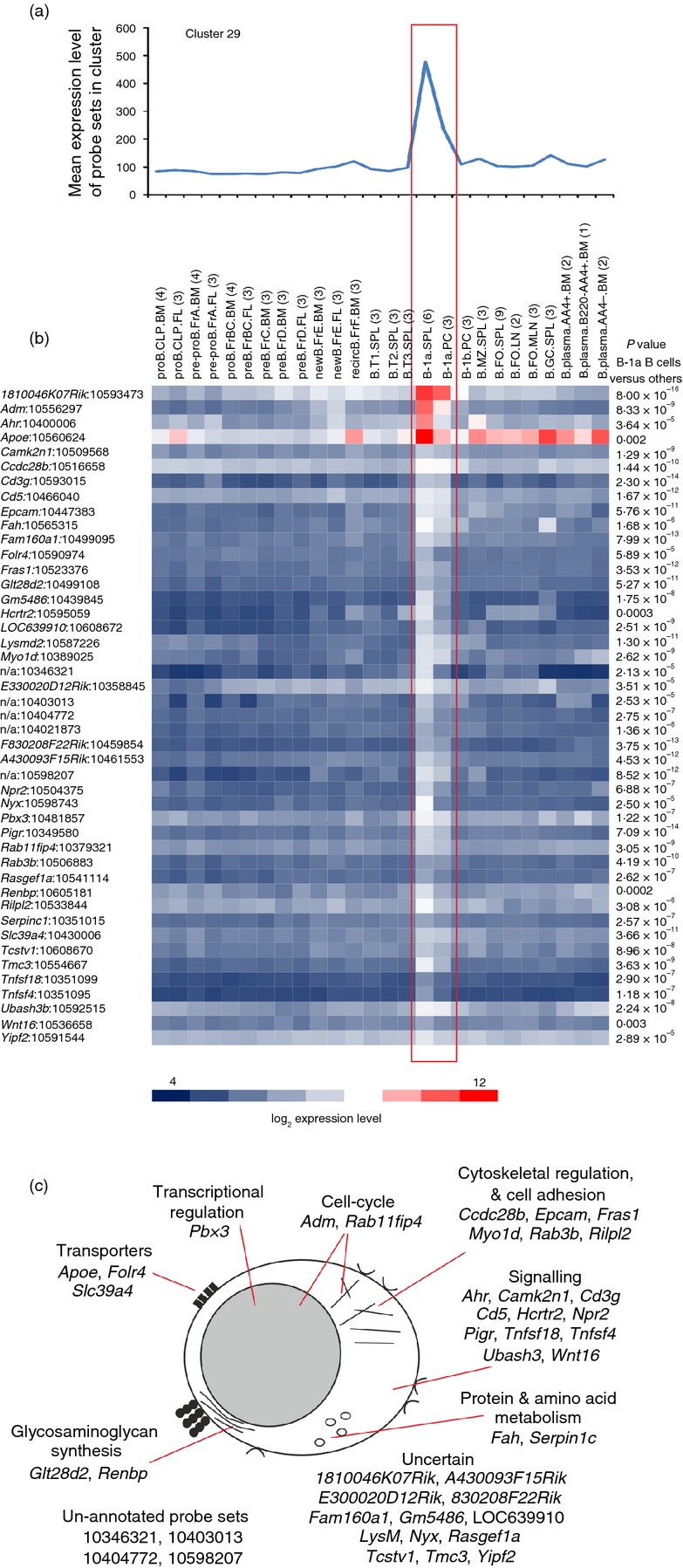
Analysis of the genes within cluster 29 which were expressed specifically by B1a B cells. (a) The mean expression profile of all the probe set intensities within cluster 29 across the 84 samples. (b) Heat map showing the mean expression intensity of each probe set in cluster 29. Each column represents the mean (log2) probe set intensity for all samples from each source. Significant differences between groups were sought by analysis of variance. P-values for those genes that were expressed significantly (P < 0·05) by B1a B cells at levels > 2·0-fold when compared with the other cell populations. (a, b) Samples are grouped according to cell type and are arranged in order of presentation as listed in Table S1. For each cell population mean expression levels are presented and the number of replicates is indicated in parenthesis on the x-axis. Red-boxed area indicates the B1a B-cell data sets. (c) Cartoon illustrating the putative functions of all the genes represented in cluster 29 in B1a B cells.
The expression of at least two of the genes within this cluster may influence the activity of T cells. Glucocorticoid-induced tumour necrosis factor receptor ligand (GITRL, encoded by Tnfsf18) expression by interleukin-10-expressing regulatory B cells is important for the maintenance of regulatory T cells and suppression of autoimmunity.29 Whereas the engagement of OX40L (encoded by Tnfsf4) on B cells was important for the induction of T helper type 2 responses.30
Cluster 29 also contained many uncharacterized genes or un-annotated probe sets that were expressed highly and specifically only by B1a B cells (Fig. 5c) and that may have important roles in B1a B-cell function.
B1 B cells and MZ B cells
Cluster 45 contained 34 probe sets encoding 25 annotated genes. In addition to producing antibody, B1 and MZ B cells share many properties with cells of the innate immune system by acting as antigen-presenting cells and providing cytokines. As a consequence, these B-cell populations are also referred to as innate-like B cells.31 Data in this study indicate that these cell populations also share transcriptional similarities because the mean probe set expression profile of cluster 45 indicated that the genes in this cluster were co-expressed by B1 cells and MZ B cells (Fig. 6a). Two probe sets were present specific for ataxin 1 (encoded by Atxn1), otherwise known as the product of the spinocerebellar ataxia type 1 (SCA1) gene. Whereas SCA1 expression is low in bone marrow common lymphoid progenitors, expression by MZ B cells differentiated from these progenitors is much higher 32 (Fig. 6c). Follicular B cells, in contrast, express much lower levels of SCA1. Data here indicate that Atxn1 was additionally expressed highly by B1 B-cells (Fig. 6c). Whether the expression of high levels of SCA1 is indicative of the activation status of these cells,33 or is a reflection of the precursor cells from which they differentiate is uncertain.32
Figure 6.

Analysis of the genes within cluster 45 with expression enriched in B1 B cells and marginal zone (MZ) B cells. (a) The mean expression profile of all the probe set intensities within cluster 45 over the 84 samples. (b) Heat map showing the mean expression intensity of each probe set in cluster 45. Each column represents the mean (log2) probe set intensity for all samples from each source. Significant differences between groups were sought by analysis of variance. P-values for those genes that were expressed significantly (P < 0·05) by B1 B cells and MZ B cells at levels > 2·0-fold when compared with the other cell populations. (c) The mean expression profile of probe sets representing Atxn1 (blue and red) and Bhlhe41 (green) across the 84 samples. (d) Comparison of the mean expression profile of probe sets representing Cnr2 (blue), Gpr55 (red) and Tbc1d9 (green) across the 84 samples. (a–d) Samples are grouped according to cell type and are arranged in order of presentation as listed in Table S1. For each cell population mean expression levels are presented and the number of replicates is indicated in parenthesis on the x-axis. Red-boxed area indicates the B1 B-cell and MZ B-cell data sets. (e) Cartoon illustrating the putative functions of all the genes represented in cluster 45 in B1 B cells and MZ B cells.
Cluster 45 contained the basic helix-loop-helix family transcription factor BHLHE41 and was specifically expressed at high levels by B1 B cells, implying an important role in the regulation of gene expression by these cells (Fig. 6c).
The co-expression of Gpr55, which encodes for the G protein-coupled receptor 55, was also intriguing. Expression of cannabinoid receptor 2 (CB2) by B cells promotes their chemoattraction towards the endocannabinoid 2-archionoylglycerol (2-AG), and is important for the homing of MZ B cells and their precursors to the MZ.34,35 Data suggest that GPR55 can also act as a cannabinoid receptor,36 and regulate the CB2-mediated responses of neutrophils to endocannabinoids such as 2-AG.37 Whereas Cnr2 (which encodes CB2) was expressed by all B-cell subsets from the newly formed Fr.E stage, Cnr2 and Gpr55 were co-expressed only by B1 B cells and MZ B cells (Fig. 6d). This suggests that cross-talk between CB2 and GPR55 may play an important role in regulating the chemotactic responses of these populations to endocannabinoids. The inclusion of TBC1 domain family member 9 (Tbc1d9) was also interesting (Fig. 6d). Although the putative function of Tbc1d9 in B cells is unknown, this gene was reported to be up-regulated along with that encoding cannabinoid receptor 1 in mantle cell lymphoma (considered to derive from B cells from the follicular mantle zone).38
Marginal zone B cells
Cluster 39 (GO:0045727, positive regulation of transcription, P < 0·0376; GO:0042221, response to chemical stimulus, P < 0·0376) comprised a cluster of 39 probe sets encoding 29 annotated genes with expression significantly enriched in MZ B cells (Fig. 7a). Within this cluster were several genes known to be expressed highly by MZ B cells including Cd1d, S1pr3 and Tlr3 (Fig. 7b),39–43 but it was interesting in that it appeared to lack an obvious transcriptional regulator. The MZ B cells are situated on the exterior of the marginal sinus. These specialized, non-recirculatory B cells express B-cell receptors specific for microbial polysaccharides, Toll-like receptors, complement receptors (CD21/CD35; CR2/CR1) and can self-renew. These features, and their MZ positioning, enable them to trap and concentrate blood-borne antigen on their surfaces and rapidly mount T-cell-independent type-2 antibody responses to polysaccharide antigen such as those on encapsulated bacteria.44 The MZ B cells also capture blood-borne immune complexes and rapidly shuttle into B-cell follicles,43 and can act as regulatory cells that control the function of other cells involved in the innate immune response.31
Figure 7.

Analysis of the genes within cluster 39 with expression enriched in marginal zone (MZ) B cells. (a) The mean expression profile of all probe set intensities within cluster 39 over the 84 samples. (b) Heat map showing the mean expression intensity of each probe set in cluster 39. Each column represents the mean (log2) probe set intensity for all samples from each source. Significant differences between groups were sought by analysis of variance. P-values for those genes that were expressed significantly (P < 0·05) by MZ B cells at levels > 2·0 fold when compared with the other cell populations. (c) The mean expression profile of probe sets representing S1pr1 (blue), S1pr2 (red) and S1pr3 (green) across the 85 samples. (d) Treatment of mice with the S1P receptor modulator FTY720 rapidly displaces MZ B cells (CD1d+ cells, red) from the splenic MZ. In control mice many MZ B cells (left-hand panel, arrow) are situated within the MZ adjacent to the ring of MADCAM1-expressing sinus-lining cells (green). Following treatment with FTY720 MZ B cells are displaced from the MZ and retained in the follicles (right-hand panel, arrow-heads). FO, B-cell follicle. (e) Comparison of the mean expression profile of probe sets representing Adam28 (light blue), Asb2 (red), Tspan15 (green) and Zc3h12c (dark blue) across the 85 samples. (a, b, c and e) Samples are grouped according to cell type and are arranged in order of presentation as listed in Table S1. For each cell population mean expression levels are presented and the number of replicates is indicated in parenthesis on the x-axis. Red-boxed area indicates the MZ B-cell data sets. Blue-boxed area in (e) indicates the germinal centre (GC) B-cell data sets. (f) Cartoon illustrating the putative functions of all the genes represented in cluster 39 in MZ B cells.
Stimulation through various G protein-coupled receptors is important for the positioning of MZ B cells in the MZ. Cells in the MZ are continually exposed to sphingosine-1-phosphate (S1P) in the bloodstream. MZ B cells strongly express the S1P receptors S1P1 and S1P3 (encoded by S1pr1 and S1rp3, respectively; Fig. 7c), which aid the S1P-mediated positioning of MZ B cells in the MZ.42,43,45 When S1P-stimulation is blocked, for example by treatment with the pharmacological S1P receptor modulator FTY720, MZ B cells are rapidly displaced from the MZ and migrate into the follicles in response to CXCL13–CXCR5 stimulation42,43,45,46 (Fig. 7d). Across this data collection, S1pr3 was expressed highly only by MZ B cells, whereas S1pr2 was restricted to GC B cells (see cluster 29 below). In contrast S1pr1 was expressed by all B-cell populations from the pre-B Fr.D stage, except GC B cells and plasma cells (Fig. 7c), consistent with its role in controlling the egress of mature B cells from secondary lymphoid organs.47 In addition to S1P-mediated stimulation, the chemokine receptor CXCR7 (encoded by Cxcr7) can act as a scavenger receptor for CXCL12 on MZ B cells and promote their retention in the splenic MZ.48
Among the other genes present was Adam28, which encodes a disintegrin and metalloprotease 28 (ADAM28) and was specifically and significantly expressed only by MZ B cells (Fig. 7e). This distribution was consistent with immunohistochemistry data elsewhere suggesting that ADAM28 was predominantly expressed in the splenic MZ.49 Marginal zone B cells express high levels of integrins α4β1 and αLβ2, which by binding to stromal cell adhesion molecules intercellular adhesion molecule 1 and vascular cell adhesion molecule 1 aids their retention in the MZ.17 Data show that ADAM28 modulates the binding of integrin α4β1 to vascular cell adhesion molecule 1,49 implying an important role in the integrin-mediated retention of MZ B cells to stromal cells within the MZ.
The specific co-expression of Tspan15 is also intriguing (Fig. 7e). This gene encodes tetraspanin 15, which can regulate the activity of ADAM10.50 Whether TSPAN15 shares a similar activity towards ADAM28 is uncertain, but ADAM10 has been shown to be essential for the development of MZ B cells.51 NOTCH2-activation induced by stromal-derived Delta-like-1 is an essential regulator of MZ B-cell development.52–54 NOTCH2 signalling is impaired in the absence of ADAM10 and as a consequence MZ B-cell development is blocked. As ADAM10 critically regulates MZ B-cell development by initiating NOTCH2 signalling,51 these data suggest that TSPAN15 may also play an important role by modulating ADAM10 activity in B cells. In cells of the B lineage NOTCH signalling leads to the degradation of Janus kinase (JAK3), implying that NOTCH signalling influences lymphopoiesis through the modulation of JAK3. NOTCH signalling transcriptionally activates ankyrin-repeat SOCS box containing protein 2 (ASB2) and S-phase kinase-associated protein 2 (SKP2), and each has been shown to interact with and degrade JAK3.55 The specific and significant expression of Asb2 only by MZ B cells (Fig. 7e) suggests an important role in NOTCH-mediated JAK3 turnover.
The gene encoding zinc finger CCCH-type-containing 12c (Zc3h12c) was expressed highly by MZ B cells and to a lesser extent by cells of the B1a lineage (Fig. 7e). Expression of ZC3H12C has been shown to repress nuclear factor-κB activation and pro-inflammatory gene expression.56,57 Although a range of TLR agonists stimulate the rapid proliferation of MZ B cells and the production of immunoglobulins, their expression of pro-inflammatory cytokines such as tumour necrosis factor-α and interleukin-12p40 is negligible.40,41 Hence, it is plausible that ZC3H12C may play a similar anti-inflammatory function in MZ B cells to that observed in macrophages and endothelial cells.56,57
Recirculating, transitional and B2 B cells
Cluster 37 contained 39 probe sets encoding 33 annotated genes. The mean expression profile of this cluster indicated that the genes within it were co-expressed highly by recirculating, transitional and B2 B cells (Fig. 8a,b). The FO B2 cells are characterized as CD5− CD21lo CD23+ CD45R+ IgMlo IgDhi cells, reside within the B-cell follicle, circulate throughout the bloodstream and produce high-affinity antibodies to T-cell-dependent antigen. The low-affinity IgE receptor (CD23) is encoded by Fcer2a, and across this data set expression was restricted to recirculating, transitional and B2 B cells (Fig. 8c).
Figure 8.

Analysis of the genes within cluster 37 with expression enriched in recirculating, transitional and B2 B cells. (a) The mean expression profile of all the probe set intensities within cluster 37 over the 84 samples. (b) Heat map showing the mean expression intensity of each probe set in cluster 37. Each column represents the mean (log2) probe set intensity for all samples from each source. Significant differences between groups were sought by analysis of variance. P-values for those genes that were expressed significantly (P < 0·05) by recirculating, transitional and B2 B cells at levels > 2·0-fold when compared with the other cell populations. (c) The mean expression profile of Fcer2a across the 84 samples. (d) Comparison of the mean expression profile of probe sets representing Gdf11 (red), Icosl (green), Lrrk2 (dark blue) and Mef2c (light blue) across the 84 samples. (a–d) Samples are grouped according to cell type and are arranged in order of presentation as listed in Table S1. For each cell population mean expression levels are presented and the number of replicates is indicated in parenthesis on the x-axis. Red-boxed areas indicate the recirculating, transitional and B2 B cells data sets. (e) Cartoon illustrating the putative functions of all the genes represented in cluster 45 in recirculating, transitional and B2 B cells.
Many of the other genes in this cluster had activities associated with signalling (Fig. 8e; GO:0035591, signalling adaptor activity, P < 0·039). Among them was Lrrk2, which encodes leucine-rich repeat kinase 2 and has been shown to be differentially expressed by resting B2 B cells58 (Fig 8d). The expression profiles of Gdf11 (which encodes growth factor differentiation factor 11) and Icosl (which encodes icos ligand) also indicated they were restricted to B2 B cells (Fig. 8d).
An interesting feature of this cluster was the inclusion of the myocyte enhancer factor-2 family transcription factor, MEF2C (Fig. 8d). In B cells the Fcer2a gene has been reported to be a direct target of MEF2C.59 Our retrospective analysis of published microarray data from the spleens of Mef2c−/− and wild-type mice59 revealed that in addition to Fcer2a, the expression of many of the other genes within cluster 37 were also affected by Mef2c-deficiency (see Supplementary material, Table S3). These data suggest that the expression of many of the genes in cluster 37 is influenced by MEF2C.
Germinal centre B cells
Cluster 20 comprised a cluster of 67 probe sets encoding 56 annotated genes with expression enriched in GC B cells (Fig. 9a,b). Several genes within this cluster have previously been reported to be expressed highly by GC B cells including Aicda, Basp1, Fas, Neil1, Plxnb2, Rgs13, S1pr2 and Tnfsf9 (which encodes CD137L/4-1BBL).
Figure 9.

Analysis of the genes within cluster 20 with expression enriched in germinal centre (GC) B cells. (a) The mean expression profile of all probe set intensities within cluster 29 over the 84 samples. (b) Heat map showing the mean expression intensity of each probe set in cluster 29. Each column represents the mean (log2) probe set intensity for all samples from each source. Significant differences between groups were sought by analysis of variance. P-values for those genes that were expressed significantly (P < 0·05) by recirculating, transitional and B2 B cells at levels > 2·0-fold when compared with the other cell populations. (a, b) Samples are grouped according to cell type and are arranged in order of presentation as listed in Table S1. For each cell population mean expression levels are presented and the number of replicates is indicated in parenthesis on the x-axis. Red-boxed areas indicate the GC B-cell data sets. (c) Cartoon illustrating the putative functions of all the genes represented in cluster 20 in recirculating, transitional and B2 B cells.
B2 B cells within secondary lymphoid tissue undergo GC reactions to generate high-affinity antibody with strong antigen-specificity. Expression of the activation-induced cytidine deaminase AID (encoded by Aicda) plays an essential role in initiating somatic hypermutation, gene conversion and class-switch recombination in the immunoglobulin genes of GC B cells. Other genes in this cluster were also related to immunoglobulin production such as Erp44.60 Consistent with the active transcriptional status of GC B cells, many genes with functions associated with transcriptional regulation were also represented in this cluster (Fig. 9c). Of particular interest was the MEF2 family transcription factor Mef2b. Although expression of MEF2C by B cells has been described,61 the expression of MEF2B by GC B cells implies a specific role in their development or function.
Germinal centre B cells also undergo competitive selection to enrich the cells with high antigen affinity, whereas those with low affinity are eliminated by apoptosis. These activities were reflected by the inclusion of genes involved in apoptosis regulation and B-cell selection including Ada, Fas, Fgf11, Neil1, Optn, Ppp4r2, Rassf6, Rgs13, Slc41a2, Stau2 and Zdhhc2. The selected GC B cells then undergo rapid clonal expansion indicated by the presence of genes related to the cell-cycle and cytoskeletal regulation (Fig. 9c).
The expression of S1P receptor type 2 (S1P2, encoded by S1pr2) was restricted to GC B cells (Figs 7c and 9b) and is considered to exert a dual role in these cells by regulating their survival and the positioning at the centre of the B-cell follicle.62 In addition to S1pr2, other genes within this cluster, Arhgap8, Gnaz, Ppap2a and Rgs9 have related activities. The relative concentration of S1P in the B-cell follicle is much lower than that encountered at the perimeter. The low S1P concentration in the centre of the follicle appears to be maintained in part by local degradation by B cells through expression of S1P-degrading lipid phosphate phosphatases such as phosphatidic acid phosphatase type 2 A (PPAP2A, also known as LPP1).62 After GC B cells encounter S1P, S1P2 signals via Gα12-Gα13 and the small GTPase Rho to regulate their survival.62 The co-expression of genes encoding the regulator of G-protein signalling 9 (Rgs9), Rho GTPase activating protein 8 (Arhgap8), and the G protein Gα(z) (encoded by Gnaz), which can modulate Rho signalling, implies similar activities in maintaining GC B-cell homeostasis.
Regulator of G-protein signalling 13 (Rgs13) was also highly expressed across this data collection only by GC B cells (Fig. 9b). RGS13 exerts several roles in GC B cells, including limiting extra-follicular plasma cell generation and the size and number of cells in the GC.63
The function of a number of genes in GC B cells was uncertain, but across this data set 1810010H24Rik, Adhef1, Hbb-bh1 and Vwa3b were highly and specifically expressed only by GC B cells (Fig. 9b). As many of the annotated genes in cluster 20 encode proteins with published or credible functions in GC B cells (Fig. 9c), by using the principle of guilt-by-association it is reasonable to speculate that 1810010H24Rik, Adhef1, Hbb-bh1 and Vwa3b may likewise have similar activities. Also intriguing was the un-annotated gene recognized by Affymetrix probe set ID 10416006. The high and specific expression of this transcript across this data set implies a specific role for this uncharacterized gene in GC B cells.
Comparison with human B cells
We next determined whether GC B cells from human tonsils expressed high levels of the genes in cluster 20 which were enriched in mouse GC B cells. To do so, we analysed 24 microarray data sets from human peripheral blood B cells and tonsil-derived naive B cells, memory B cells, GC B cells, CXCR4+ centrocytes and CXCR4− centroblasts (Table S1). As these data were performed on Affymetrix Human Genome U133A Plus 2.0 expression arrays it was not possible to combine them with the mouse data sets above. These data were normalized and the expression levels of 51 orthologues of the 56 murine annotated genes in cluster 20 were compared. Our analysis showed that the expression of 41% of the mouse GC B-cell-related genes in cluster 20 were significantly higher (> 2·0-fold) in human GC B cells when compared with the other human B-cell populations (see Supplementary material, Table S4). These included S1PR2 and PPAP2A, which in mice help to regulate the survival and the positioning of GC B cells in the centre of the follicle62 (and the related gene GNAZ), and RG13 which limits extra-follicular plasma cell generation and GC size.63 These data clearly show that mouse and human GC B cells share similar transcriptional features.
Whether B cells of the B1 lineage exist in humans is controversial.12,64,65 We therefore analysed 12 microarray data sets from a published study of proposed human B1 B cells, naive B cells, memory B cells and plasmablasts 12 (GSE42724; Table S1). As these data were performed on Affymetrix Human Gene 1.0 ST expression arrays it was not possible to combine them with the human GC B-cell or mouse data sets above. Data were normalized and 23 annotated genes were identified that were expressed significantly more (> 2·0-fold) by proposed human B1 B cells when compared with the other cell populations. We then compared the expression levels of equivalent probe sets across the different mouse B-cell populations. Our analysis showed that orthologues for only three of these genes, CCR1, CD5 and SYT11, were expressed at significant levels (> 2·0-fold) in mouse B1 B cells when compared with the other mouse B-cell populations (see Supplementary material, Table S5). Furthermore, CD5 was the only gene that was represented in any of the mouse B1 and MZ B cell-related clusters identified above (clusters 13, 29, 39, 45 and 172). Studies have proposed that circulating CD20+ CD27+ CD43+ CD70− CD69− B cells might represent the human equivalents of mouse B1 B cells.64 Others consider that these cells might be activated B cells undergoing differentiation into plasma cells.12,65 Data here support this conclusion. Among the 23 annotated genes in Table S5, orthologues for four (CD96, ITGAM, TNFRSF1B and TNFRSF21) were co-expressed in the mouse plasma cell-related clusters (clusters 19, 5, 14 and 10, respectively; Table S2), and the expression levels of 11 genes (48%) were significantly higher (> 2·0-fold) in mouse plasma cells when compared with the other cell populations (Table S5).
Conclusions
This study provides novel insight into the transcriptomes of distinct mouse B-cell populations. A meta-analysis approach was used to compare 84 publicly available gene expression data sets representing 16 different mouse B-cell subsets enriched from various tissues. Several co-expressed gene clusters were identified with expression restricted to the major mouse B-cell subsets: B1 B cells, MZ B cells and B2 B cells. Although these clusters contained many genes typically associated with the activity of the cells they were specifically expressed in, many novel B-cell-subset-specific candidate genes were also identified. Indeed, many clusters contained a large number of un-annotated probe sets or uncharacterized genes. As these genes were expressed highly and specifically by distinct B-cell populations, by using the principal of guilt-by-association it is plausible to speculate that these currently uncharacterized genes may have important activities in the B-cell populations within which they were expressed. We also analysed 36 microarray data sets from distinct human B-cell populations and compared them to our large collection mouse B-cell lineages. Although these data showed that mouse and human GC B cells shared similar transcriptional features, mouse B1 B cells were distinct from proposed human B1 B cells. To enable readers to explore this interactive expression atlas of distinct mouse B-cell subsets in greater detail an interactive webstart version of the network graph is available on the authors’ institutional website (http://www.roslin.ed.ac.uk/neil-mabbott/b-cells). Further characterization of activities of the candidate genes identified above will lead to the identification of novel B-cell lineage-specific transcription factors and regulators of B-cell function.
Acknowledgments
This work was supported by project and Institute Strategic Grant funding from the Biotechnological and Biological Sciences Research Council. This work benefitted from expression data assembled by the ImmGen consortium8 (http://www.immgen.org).
Glossary
Abbreviations
- Ab
antibody
- Ag
antigen
- 2-AG
2-archionoylglycerol
- ADAM28
a disintegrin and metalloprotease 28
- CB2
cannabinoid receptor 2
- DC
dendritic cell
- GC
germinal centre
- GO
gene ontology
- FO
follicular
- GPR55
G protein-coupled receptor 55
- IL
interleukin
- JAK3
Janus kinase 3
- MCL
Markov clustering
- MEF2
myocyte enhancer factor-2
- MZ
marginal zone
- S1P
sphingosine-1-phosphate
- SCA1
spinocerebellar ataxia type 1
- TLR
Toll-like receptor
- TSPAN15
tetraspanin 15
- ZBTB32
zinc finger, broad complex, tram track, bric-a-brac domain containing 32
- ZC3H12C
zinc finger CCCH-type-containing 12c
Disclosures
The authors declare no financial or commercial conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Full description of the cellular identities and sources of data sets analysed in this study (in order of presentation from left to right on the cluster profiles). The source of each data set is also provided (Gene Expression Omnibus identifier).
Table S2. Cluster functional enrichment analysis at Pearson r = 0·85, MCL 2.2. A Microsoft Excel file listing all the co-expressed gene clusters derived from the analysis in this study.
Table S3. Effect of Mef2c-deficiency on the expression of the genes in cluster 37 in the spleen.
Table S4. Comparison of the expression of the mouse germinal centre (GC) B-cell related genes in cluster 20 by human GC B cells.
Table S5. Comparison of genes expressed highly in proposed human B1 B-cell by mouse B1 B cells.
References
- 1.Theocharidis A, van Dongen S, Enright AJ, Freeman TC. Network visualisation and analysis of gene expression data using Biolayout Express3D. Nat Protoc. 2009;4:1535–50. doi: 10.1038/nprot.2009.177. [DOI] [PubMed] [Google Scholar]
- 2.Freeman TC, Goldovsky L, Brosch M, et al. Construction, visualisation, and clustering of transcriptional networks from microarray expression data. PLoS Comput Biol. 2007;3:2032–42. doi: 10.1371/journal.pcbi.0030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hume DA, Summers KM, Raza S, Baillie JK, Freeman TC. Functional clustering and lineage markers: insights into cellular differentiation and gene function from large-scale microarray studies of purified primary cell populations. Genomics. 2010;95:328–38. doi: 10.1016/j.ygeno.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Mabbott NA, Baillie JC, Hume DA, Freeman TC. Meta-analysis of co-expressed gene signatures in mouse leukocyte populations. Immunobiology. 2010;215:724–36. doi: 10.1016/j.imbio.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi A, Donaldson DS, Kanaya T, et al. Identification of novel genes selectively expressed in the follicle-associated epithelium from the meta-analysis of transcriptomics data from multiple mouse cell and tissue populations. DNA Res. 2012;19:407–22. doi: 10.1093/dnares/dss022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mabbott NA, Bailie JK, Kobayashi A, et al. Expression of mesenchyme-specific gene signatures by follicular dendritic cells: insights from the meta-analysis of microarray data from multiple mouse cell populations. Immunology. 2011;133:482–98. doi: 10.1111/j.1365-2567.2011.03461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hume DA, Mabbott N, Raza S, Freeman TC. Can DCs be distinguished from macrophages by molecular signatures. Nat Immunol. 2013;14:187–9. doi: 10.1038/ni.2516. [DOI] [PubMed] [Google Scholar]
- 8.Heng TS, Painter MW, Consortium TIGP, et al. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–4. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- 9.Lyons PA, Koukoulaki M, Hatton A, Doggett K, Woffendin HB, Chaudhry AN, Smith KGC. Microarray analysis of human leucocyte subsets: the advantages of positive selection and rapid purification. BMC Genomics. 2007;8:64. doi: 10.1186/1471-2164-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Longo NS, Lugar PL, Yavuz S, et al. Analysis of somatic hypermutation in X-linked hyper-IgM syndrome shows specific deficiencies in mutational targeting. Blood. 2009;113:3706–15. doi: 10.1182/blood-2008-10-183632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caron G, Le Gallou S, Lamy T, Tarte K, Fest T. CXCR4 expression functionally discriminates centroblasts versus centrocytes within human germinal center B cells. J Immunol. 2009;182:7595–602. doi: 10.4049/jimmunol.0804272. [DOI] [PubMed] [Google Scholar]
- 12.Covens K, Verbinnen B, Geukens N, Meyts I, Schuit F, Van Lommel L, Jacquemin M, Bossuyt X. Characterization of proposed human B-1 cells reveals pre-plasmablast phenotype. Blood. 2013;121:5176–83. doi: 10.1182/blood-2012-12-471953. [DOI] [PubMed] [Google Scholar]
- 13.Quandt D, Hoff H, Rudolph M, Fillatreau S, Brunner-Weinzieri MC. A new role for CTLA-4 on B cells in thymus-dependent immune responses in vivo. J Immunol. 2007;179:7316–24. doi: 10.4049/jimmunol.179.11.7316. [DOI] [PubMed] [Google Scholar]
- 14.Mills JH, Kim DG, Krenz A, Chen JF, Bynoe MS. A2A adenosine receptor signaling in lymphocytes and the central nervous system regulates inflammation during experimental autoimmune encephalomyelitis. J Immunol. 2012;188:5713–22. doi: 10.4049/jimmunol.1200545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balasubramanian S, Fan M, Messmer-Blust AF, Yang CH, Trendel JA, Jeyaratnam JA, Pfeffer LM, Vestal DJ. The interferon-γ-induced GTPase, mGTP-2, inhibits tumour necrosis factor α (TNF-α) induction of matrix metalloproteinase-9 (MMP-9) by inhibiting NF-kκB and Rac protein. J Biol Chem. 2011;186:20054–64. doi: 10.1074/jbc.M111.249326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo CG, Lu TT, Cyster JG. Integrin-dependence of lymphocyte entry into the splenic white pulp. J Exp Med. 2003;197:353–61. doi: 10.1084/jem.20021569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu TT, Cyster JG. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science. 2002;297:409–12. doi: 10.1126/science.1071632. [DOI] [PubMed] [Google Scholar]
- 18.Goel HL, Pursell B, Standley C, Fogarty K, Mercurio AM. Neuropilin-2 regulates α6β1 integrin in the formation of focal adhesion and signaling. J Cell Sci. 2012;125:497–506. doi: 10.1242/jcs.094433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sumoza-Toledo A, Santos-Arqumendo L. The spreading of B lymphocytes induced by CD44 cross-linking requires actin, tubulin, and vimentin rearrangements. J Leukoc Biol. 2004;75:233–9. doi: 10.1189/jlb.0803403. [DOI] [PubMed] [Google Scholar]
- 20.Yoon HS, Scharer CD, Majumder P, et al. ZBTB32 is an early repressor of the CIITA and MHC class II gene expression during B cell differentiation to plasma cells. J Immunol. 2012;189:2393–403. doi: 10.4049/jimmunol.1103371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hildner K, Edelson BT, Purtha WE, et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meredith MM, Liu K, Kamphorst AO, et al. Zinc finger transcription factor zDC is a negative regulator required to prevent activation of classical dendritic cells in the steady state. J Exp Med. 2012;209:1583–93. doi: 10.1084/jem.20121003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalin TV, Ustitan V, Kalinchenko VV. Multiple faces of FoxM1 transcription factor. Cell Cycle. 2011;10:396–405. doi: 10.4161/cc.10.3.14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong JV, Dong P, Nevins JR, Mathey-Prevot B, You L. Control of E2F dynamics in cell cycle entry. Cell Cycle. 2011;10:3086–94. doi: 10.4161/cc.10.18.17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan B, Morel L. Role of B-1a cells in autoimmunity. Autoimmun Rev. 2006;5:403–8. doi: 10.1016/j.autrev.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida T, Katsuya K, Oka T, Koizumi S, Wakita D, Kitamura H, Nishimura T. Effects of AhR ligands on the production of immunoglobulins in purified mouse B cells. Biomed Res. 2012;33:67–74. doi: 10.2220/biomedres.33.67. [DOI] [PubMed] [Google Scholar]
- 27.Yamaguchi T, Hirota K, Nagahama K, Ohkawa K, Takahashi T, Nomura T, Sakaguchi S. Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity. 2007;27:145–59. doi: 10.1016/j.immuni.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 28.Wright GJ, Puklavec MJ, Willis AC, Hoek RM, Sedgwick JD, Brown MH, Barclay AN. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity. 2000;13:233–42. doi: 10.1016/s1074-7613(00)00023-6. [DOI] [PubMed] [Google Scholar]
- 29.Ray A, Basu S, Williams CB, Slazman NH, Dittel BN. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol. 2012;188:3188–98. doi: 10.4049/jimmunol.1103354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linton P-L, Bautista B, Biederman E, Bradley ES, Harbertson J, Kondrack RM, Padrick RC, Bradley LM. Costimulation via OX40L expressed by B cells is sufficient to determine the extent of primary CD4 cell expansion and Th2 cytokine secretion. J Exp Med. 2003;197:875–83. doi: 10.1084/jem.20021290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X. Regulatory functions of innate-like B cells. Cell Mol Immunol. 2013;10:113–21. doi: 10.1038/cmi.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fossati V, Kumar R, Snoeck H-W. Progenitor cell origin plays a role in fate choices of mature B cells. J Immunol. 2010;184:1251–60. doi: 10.4049/jimmunol.0901922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shevach EM, Korty PE. Ly-6: a multigene family in search of a function. Immunol Today. 1989;10:195–200. doi: 10.1016/0167-5699(89)90324-1. [DOI] [PubMed] [Google Scholar]
- 34.Basu S, Ray A, Dittel BN. Cannabinoid receptor 2 is critical for the homing and retention of marginal zone B lineage cells and for efficient T-independent immune responses. J Immunol. 2011;187:5720–32. doi: 10.4049/jimmunol.1102195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muppidi JR, Arnon TI, Bronevetsky Y, Veerapen N, Tanaka M, Besra GS, Cyster JG. Cannabinoid receptor 2 positions and retains marginal zone B cells with the splenic marginal zone. J Exp Med. 2011;208:1941–8. doi: 10.1084/jem.20111083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryberg E, Larsson N, Sjogren S, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balenga NAB, Aflaki E, Kargl J, et al. GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Res. 2011;21:1452–69. doi: 10.1038/cr.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Islam TC, Asplund AC, Lindvall JM, et al. High level of cannabinoid receptor 1, absence of regulator of G protein signalling 13 and differential expression of cyclin D1 in mantle cell lymphoma. Leukemia. 2003;17:1880–90. doi: 10.1038/sj.leu.2403057. [DOI] [PubMed] [Google Scholar]
- 39.Pillai S, Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat Rev Immunol. 2009;9:767–77. doi: 10.1038/nri2656. [DOI] [PubMed] [Google Scholar]
- 40.Gururajan M, Jacob J, Pulendran B. Toll-like receptor expression and responsiveness of distinct murine splenic and mucosal B-cell subsets. PLoS One. 2007;2:e863. doi: 10.1371/journal.pone.0000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bialecki E, Paget C, Fontaine J, Capron M, Trottein F, Faveeuw C. Role of marginal zone B lymphocytes in invariant NKT cell activation. J Immunol. 2009;182:6105–13. doi: 10.4049/jimmunol.0802273. [DOI] [PubMed] [Google Scholar]
- 42.Cinamon G, Matloubian M, Lesneski MJ, Xu Y, Low C, Lu T, Proia RL, Cyster J. Spingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol. 2004;5:713–20. doi: 10.1038/ni1083. [DOI] [PubMed] [Google Scholar]
- 43.Cinamon G, Zachariah MA, Lam OM, Foss FW, Jr, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol. 2008;9:54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Srivastava B, Quin WJ, III, Hazard K, Erikson J, Allman D. Characterization of marginal zone B cell precursors. J Exp Med. 2005;202:1225–34. doi: 10.1084/jem.20051038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Girkontaite I, Sakk V, Wagner M, Borggrefe T, Tedford K, Chun J, Fischer K-D. The sphingosine-1-phosphate (S1P) lysophospholipid receptor S1P3 regulates MAdCAM-1+ endothelial cells in splenic marginal sinus organization. J Exp Med. 2004;200:1491–501. doi: 10.1084/jem.20041483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mok SW, Proia RL, Brinkmann V, Mabbott NA. B cell-specific S1PR1 deficiency blocks prion dissemination between secondary lymphoid organs. J Immunol. 2012;188:5032–40. doi: 10.4049/jimmunol.1200349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–60. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 48.Wang H, Beaty N, Chen S, Qi CF, Masiuk M, Shin DM, Morse HC., 3rd The CXCR7 chemokine receptor promotes B-cell retention in the splenic marginal zone and serves as a sink for CXCL12. Blood. 2012;119:465–8. doi: 10.1182/blood-2011-03-343608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGinn OJ, English WR, Roberts S, Ager A, Newham P, Murphy G. Modulation of integrin α4β1 by ADAM28 promotes lymphocyte ashesion and transendothelial migration. Cell Biol Int. 2011;35:1043–53. doi: 10.1042/CBI20100885. [DOI] [PubMed] [Google Scholar]
- 50.Prox J, Willenbrock M, Weber S, Lehmann T, Schmidt-Arras D, Schwanbeck R, Saftiq P, Schwake M. Tetraspanin 15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cell Mol Life Sci. 2012;69:2919–32. doi: 10.1007/s00018-012-0960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gibb DR, El Shikh M, Kang D-J, et al. ADAM10 is essential for Notch-2-dependent marginal zone B cell development and CD23 cleavage in vivo. J Exp Med. 2010;207:623–35. doi: 10.1084/jem.20091990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tan JB, Xu K, Cretegny K, Visan I, Yuan JS, Egan SE, Guidos CJ. Lunatic and manic fringe cooperatively enhance marginal zone B cell precursor competition for Delta-like 1 in splenic endothelial niches. Immunity. 2009;30:254–63. doi: 10.1016/j.immuni.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 53.Saito T, Chiba S, Ichikawa M, et al. Notch2 is preferentially expressed in mature B cells and indispensible for marginal zone B lineage development. Immunity. 2003;18:675–85. doi: 10.1016/s1074-7613(03)00111-0. [DOI] [PubMed] [Google Scholar]
- 54.Santos MA, Sarmento LM, Rebelo M, et al. Notch1 engagement by Delta-like-1 promotes differentiation of B lymphocytes to antibody-secreting cells. Proc Natl Acad Sci USA. 2007;104:15454–9. doi: 10.1073/pnas.0702891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu W, Sun X-H. A mechanism underlying NOTCH-induced and ubiquitin-mediated JAK3 degradation. J Biol Chem. 2011;286:41153–62. doi: 10.1074/jbc.M111.273755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang J, Wang J, Azfer A, Song W, Tromp G, Kolattudy PE, Fu M. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J Biol Chem. 2008;283:6337–46. doi: 10.1074/jbc.M707861200. [DOI] [PubMed] [Google Scholar]
- 57.Liu L, Zhou Z, Huang S, et al. Zc3h12c inhibits vascular inflammation by repressing NF-κB activation and pro-inflammatory gene expression in endothelial cells. Biochem J. 2013;451:55–60. doi: 10.1042/BJ20130019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kubo M, Kamiya Y, Nagashima R, Maekawa T, Eshima K, Azuma S, Ohta E, Obata F. LRRK2 is expressed by B-2 but not in B-1 B cells, and downregulated by cellular activation. J Neuroimmunol. 2010;229:123–8. doi: 10.1016/j.jneuroim.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 59.Debnath I, Roundy KM, Pioli PD, Weiss JJ, Weiss JH. Bone marow-induced Mef2c deficiency delays B-cell development and alters the expression of key B-cell regulatory proteins. Int Immunol. 2013;25:99–115. doi: 10.1093/intimm/dxs088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cotini M, Sitia R. ERp44 and ERGIC-53 synergise in coupling efficiency and fidelity of IgM polymerisation and secretion. Traffic. 2010;11:651–9. doi: 10.1111/j.1600-0854.2010.01043.x. [DOI] [PubMed] [Google Scholar]
- 61.Swanson BJ, Jack H-M, Lyons GE. Characterization of myocyte enhancer 2 (MEF2) expression in B and T cells: MEF2C is a B cell-restricted transcription factor in lymphocytes. Mol Immunol. 1998;35:445–58. doi: 10.1016/s0161-5890(98)00058-3. [DOI] [PubMed] [Google Scholar]
- 62.Green JA, Suzuki K, Cho B, et al. The sphingosine 1-phosphate receptor S1P2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol. 2011;12:672–80. doi: 10.1038/ni.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hwang I-Y, Hwang K-S, Park C, Harrison KA, Kehrl JH. Rgs13 constrains early B cell responses and limits germinal center sizes. PLoS One. 2013;8:e60139. doi: 10.1371/journal.pone.0060139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+CD27+CD43+CD70−. J Exp Med. 2011;208:67–80. doi: 10.1084/jem.20101499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Descatoire M, Weill J-C, Reynaud CA, Weller S. A human equivalent of mouse B-1 cells? J Exp Med. 2011;208:2563–4. doi: 10.1084/jem.20112232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.