

Background: TASK3 potassium channels regulate cellular excitability and intracellular potassium homeostasis.
Results: TNFα enhances current through TASK3 channels by modulating channel gating.
Conclusion: TNFα treatment, together with an increase of potassium efflux through TASK3 channels, promotes apoptosis.
Significance: A novel mechanism for TNFα-mediated changes in cellular homeostasis is described.
Keywords: Apoptosis, Ion Channels, Jun N-terminal Kinase (JNK), p38, Patch Clamp Electrophysiology, Potassium Channels, Tumor Necrosis Factor (TNF)
Abstract
TASK3 two-pore domain potassium (K2P) channels are responsible for native leak K channels in many cell types which regulate cell resting membrane potential and excitability. In addition, TASK3 channels contribute to the regulation of cellular potassium homeostasis. Because TASK3 channels are important for cell viability, having putative roles in both neuronal apoptosis and oncogenesis, we sought to determine their behavior under inflammatory conditions by investigating the effect of TNFα on TASK3 channel current. TASK3 channels were expressed in tsA-201 cells, and the current through them was measured using whole cell voltage clamp recordings. We show that THP-1 human myeloid leukemia monocytes, co-cultured with hTASK3-transfected tsA-201 cells, can be activated by the specific Toll-like receptor 7/8 activator, R848, to release TNFα that subsequently enhances hTASK3 current. Both hTASK3 and mTASK3 channel activity is increased by incubation with recombinant TNFα (10 ng/ml for 2–15 h), but other K2P channels (hTASK1, hTASK2, hTREK1, and hTRESK) are unaffected. This enhancement by TNFα is not due to alterations in levels of channel expression at the membrane but rather to an alteration in channel gating. The enhancement by TNFα can be blocked by extracellular acidification but persists for mutated TASK3 (H98A) channels that are no longer acid-sensitive even in an acidic extracellular environment. TNFα action on TASK3 channels is mediated through the intracellular C terminus of the channel. Furthermore, it occurs through the ASK1 pathway and is JNK- and p38-dependent. In combination, TNFα activation and TASK3 channel activity can promote cellular apoptosis.
Introduction
TASK2 (TWIK-related acid-sensitive K) channels are members of the two-pore domain potassium (K2P) channel family (1–3). These channels are widely distributed throughout the body, with notable expression in the central nervous system (CNS) (4). They are responsible for native leak K currents found in many cell types including neurons such as cerebellar granule neurons (CGNs), where they underlie the background leak K current IKSO (5, 6). TASK channels are known to play a role in various pathological conditions such as epilepsy, brain ischemia, cancer, and inflammation (7, 8). TASK channels are regulated by anesthetics such as isoflurane and halothane that, by enhancing the K current, hyperpolarize neurons (9). Other agents such as methanandamide, a synthetic cannabinoid, extracellular acidification, activation of G protein- coupled receptors linked to Gαq such as M3 muscarinic acetylcholine receptors, zinc, and ruthenium red block TASK3 channels (10–15) and consequently the native currents that they encode (e.g. 16). Because TASK3 channels regulate cellular potassium homeostasis, a role for these channels in other cellular processes such as oncogenesis and neuronal apoptosis has been proposed (7, 17, 18).
Inflammation is a response of the innate immune system that generates a stress signal mediated by proinflammatory cytokines (such as tumor necrosis factor α, TNFα) released to counteract homeostatic disturbances. This can often lead to deleterious outcomes as observed in chronic inflammatory disorders (19). TNFα activates the TNFR1 receptor to trigger various phenomena such as inflammation, apoptotic cellular death, or tumorigenesis (20). TNFα can also be released in the CNS under normal physiological conditions; for example, constitutive release of TNFα from glial cells is known to regulate synaptic strength (21).
K homeostasis, which controls neuronal activity and is involved in the regulation of cell viability (22), is modified in neurons affected by inflammation (23). A number of studies have shown that TNFα may be a mediator of this change of K homeostasis in neurons leading to a K efflux. For example, TNFα enhances an A-type current in cortical neurons contributing to neuronal protection against NMDA excitotoxicity (24) whereas TNFα up-regulates KCa2.2 channel activity in primary cortical neurons, an effect that is neuroprotective against glutamate-induced excitotoxicity (25). In the dorsal root ganglion, a voltage-independent K channel is increased in cells treated with TNFα (26).
Because TASK3 is a K channel important for cell viability, we sought to determine its behavior under inflammatory conditions. In this study, we show that TASK3 channel activity is enhanced by TNFα and that the combination of TNFα expression and TASK3 channel activity can promote cellular apoptosis.
EXPERIMENTAL PROCEDURES
In Vitro tsA-201 Cell and THP-1 Cell Co-culture
tsA-201 cells, modified human embryonic kidney 293 cells, were grown in a monolayer tissue culture flask maintained in a growth medium. The growth medium was composed of 88% minimum essential media with Earle's salts and l-glutamine, 10% heat-inactivated fetal bovine serum, 1% penicillin and streptomycin, and 1% nonessential amino acids. The cells were placed in an incubator at 37 °C with a humidified atmosphere of 95% oxygen and 5% carbon dioxide. After 2 or 3 days, when the cells were 70–90% confluent, they were split and resuspended in a 4-well plate containing 13-mm-diameter coverslips (poly-d-lysine-coated) at a density of 1.5 × 105 cells/ml in 0.5 ml of medium, ready to be transfected the next day. The THP-1 cells, a monocytic cell line, were grown in a similar medium to tsA-201 cells described above, to facilitate the cell co-culture. Added to wells containing the transiently transfected tsA-201 cells in a proportion of 2:1, THP-1 cells were activated using Toll-like receptor 7/8 activator, R848, at 0.1 μg/ml.
Transfection
For the electrophysiological experiments, pcDNA 3.1 vector was cloned with the gene of interest (TASK3 wild-type or mutated or other K2P channels of interest), and a similar vector containing GFP was incorporated into the cells (1 μg/μl for each plasmid) using the calcium phosphate method. For the confocal imaging experiments, TASK3-GFP, a TASK3 channel cloned into the pAcGFP1-N1 vector (500 ng/μl), was added with a pdsRED monomer membrane marker (Clontech) (250 ng/μl) that consists of a fusion protein of dsRED monomer fluorescent protein and neuromodulin (a plasma membrane-specific protein) to the cells. The cells were incubated for 6 h with the transfection solution that includes hTASK3 cDNA (1 mg/liter) and GFP cDNA (1 mg/liter). Then, cells were washed using a phosphate-buffered saline solution (PBS), and new medium was added to each well. The cells were used for experiments after 24 h.
Mutations and Truncations
To generate the mutant H98A and the truncated channel TASK3-stop, complementary oligonucleotide primers with the incorporated mutation were sequenced (MWG-Biotech, Ebersberg, Germany), and we used the QuikChange site-directed mutagenesis kit (Stratagene) to generate the mutated channel. Mutant DNA constructs were sequenced (MWG-Biotech) to confirm the introduction of the correct mutated bases. All mutagenesis experiments were carried out by Emma Veale.
Whole Cell Patch Clamp Recording
Currents were recorded using the whole cell patch clamp in a voltage clamp configuration in tsA-201 cells transiently transfected with the channel of interest. The coverslip with the cells was placed in a recording chamber filled with an external medium composed of 145 mm NaCl, 2.5 mm KCl, 3 mm MgCl2, 1 mm CaCl2, and 10 mm HEPES (pH 7.4). The internal medium used in the glass pipette comprised 150 mm KCl, 3 mm MgCl2, 5 mm EGTA, and 10 mm HEPES (pH 7.4). All of the data presented were collected at room temperature (19–22 °C). The transfected cells were detected using a fluorescent microscope with UV light. To study the potassium leak current, a step of voltage from −80 mV to −40 mV and then a ramp from −120 mV to +20 mV were imposed to the cell membrane. This protocol composed of sweeps lasting 1.8 s. For all quantitative current analysis in this study, we measured the current difference between the −80-mV and −40-mV steps. The current-voltage graphs were obtained from the ramp change in voltage between −120 mV and +20 mV. For each set of experiments, matched control cell recordings (usually cells transfected with WT hTASK3) were obtained on the same experimental days from cells grown at the same time as the treated cells, to facilitate direct comparisons.
The currents obtained with the imposed voltage protocol were recorded and analyzed using pCLAMP 10.2 software and Microsoft Excel. For each cell, the current amplitude (pA) was normalized to the cell capacitance (pF).
Confocal Microscopy and Co-localization Analysis
After treatment with recombinant human TNFα as required, cells were fixed with 4% paraformaldehyde and the DNA subsequently stained with 5 μg/ml Hoechst 33258 dye (Invitrogen). Coverslips were then rinsed with PBS and water before mounting onto glass slides using Mowiol mounting medium with 0.05% p-phenylenediamine. Images were taken using a Leica TCSSP2 confocal microscope and images processed using ImageJ software. For analysis of channel and membrane co-localization, the Pearson's correlation coefficient (Rr) was calculated using ImageJ and the WCIF co-localization plug-in (co-localization test plug-in, Tony Collins and Wayne Rasband). Coefficients were calculated by taking the average of five regions on the cell membrane, minimum of five cells used, and values are displayed as the mean ± S.E.
Western Blot Analysis
We assessed the ASK1, TLR7, and TLR8 proteins in the cell lysates by Western blot analysis as described previously (27).
Measurement of TNFα Production
To measure the production of TNFα by THP-1 cells, co-cultured with tsA-201 cells, we used an ELISA R&D kit according to the manufacturer's protocol.
Measurement of ASK1 Kinase Activity and Detection of ROS Generation
The kinase activity of ASK1 was assessed by immunoprecipitation followed by analysis of phosphorylation of an exogenous substrate, myelin basic protein (28). ROS generation was analyzed as described previously (29).
Annexin V Assay
Cell apoptosis was determined using an annexin V-Fluos staining kit (Roche Applied Science; 11 585 777 001). Cells were grown in a 4-well plate on 13-mm-diameter coverslips (poly-d-lysine-coated). After observing a 70% confluence, cells were transfected with either dsRED endoplasmic protein gene alone or together with the TASK3 channel gene and used the following day for the annexin V assay. The protocol for adherent cells was performed using the annexin V-Fluos labeling reagent. Annexin V binding to externalized phophatidylserine was detected by the green fluorescence of fluorescein linked to annexin V. Fluorescein was excited at 488 nm and emitted at a wavelength of 518 nm and visualized using a fluorescent microscope. Images showing green fluorescence representing the apoptotic cells and red fluorescence for all transfected cells were captured digitally and analyzed using ImageJ software and visual cell counting.
Measurement of Caspase-3 Activity
The enzymatic activity of the caspase-3 proteases in apoptotic tsA-201 cells was determined using a caspase-3 colorimetric assay (R&D Systems; BF3100). tsA-201 cells were grown and transfected in tissue culture dishes of 35-mm diameter for 24 h, followed by a 15-h TNFα treatment or no treatment. After transfection and treatment, tsA-201 cells were lysed to collect their intracellular contents. The samples were prepared according to the manufacturer's instructions. The assay was performed in a 96-well plate that could be read with a microplate reader. The cell lysate was then tested for caspase-3 protease activity by addition of a caspase-specific peptide that contained a specific DEVD amino acid sequence conjugated to a color reporter molecule p-nitroaniline (pNA). The cleavage of the peptide by the caspase released the chromophore pNA, which could be quantified spectrophotometrically at a wavelength of 405 nm. The level of caspase enzymatic activity in the cell lysate was directly proportional to the color reaction. A recombinant caspase-3 enzyme was used as a positive control or standard.
Data Analysis
Data were expressed as means ± S.E., and the statistical analyses used either Student's t test or a one-way ANOVA with a post hoc test, the Bonferroni's comparison of all variation test. The differences were considered as significant for p < 0.05 (*) or p < 0.01 (**) with p the probability to obtain the score randomly. The n represents the number of cells used for the experiment.
Chemicals
Human recombinant TNFα (T6676), SP600125 (S5567), and Bay11-7082 (B5556) were purchased from Sigma-Aldrich. SB203580 was from Ascent Scientific (Bristol, UK), R848 from Alexis Biochemicals (Nottingham, UK), and TNFα neutralizing antibody and TNFR1 neutralizing antibody (mAb225) from R&D Systems (Abingdon, UK).
RESULTS
Activated THP-1 Cells Release TNFα Which Enhances hTASK3 Current
To investigate the role of inflammatory mediators on hTASK3 channels, we co-cultured THP-1 human myeloid leukemia monocytes with tsA-201 cells, the latter transiently transfected with hTASK3. A specific Toll-like receptor 7/8 activator resiquimod (R848), involved in the innate immune system response to infection, was used to activate THP-1 cells (tsA-201 cells do not express these Toll-like receptors, which was confirmed by Western blot analysis; data not shown). Co-cultured cells were treated with R848 (0.1 μg/ml) for 15 h, and hTASK3 current was measured with and without treatment. hTASK3 current was 84 ± 4 pA/pF (n = 14) in the absence of treatment but significantly larger at 109 ± 5 pA/pF (n = 20) following treatment with R848 (Fig. 1, b and c), showing that TASK3 channels are a target of a released inflammatory mediator. Activated THP-1 cells release many inflammatory mediators including TNFα. We measured levels of TNFα in the culture medium, directly, following stimulation with R848 and found it increased from 72 ± 11 to 919 ± 89 pg/ml (Fig. 1a). Furthermore, a TNFα-neutralizing antibody, applied during R848 treatment, completely abolished the enhancement of hTASK3 channel current in tsA-201 cells (86 ± 5 pA/pF, n = 16) (Fig. 1, b and c).
FIGURE 1.
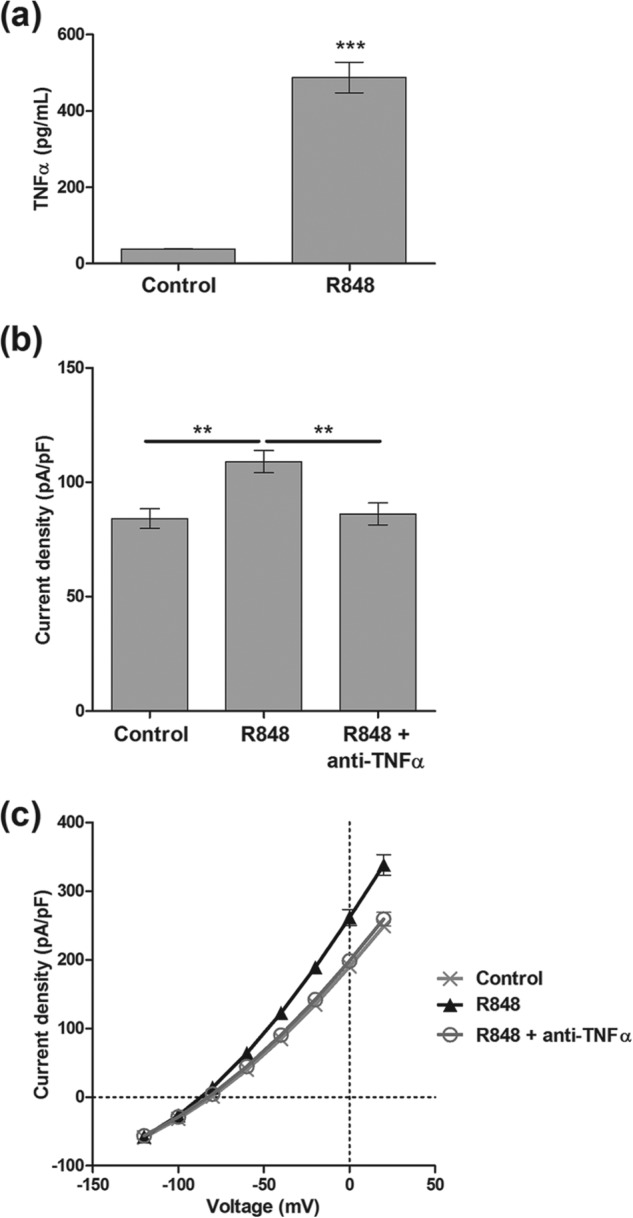
Activated THP-1 cells release TNFα which increases the K+ current in tsA-201 cells transfected with wild-type hTASK3. a, quantity of TNFα produced by THP-1 cells activated or not by R848 in presence of tsA-201 cells (unpaired t test; ***, p ≤ 0.001). b, histogram of current density (mean ± S.E. (error bars)) after co-culture of transiently transfected tsA-201 cells with THP-1 cells. THP-1 cells (none activated; control) are compared with R848 (15-h incubation) activated THP-1 cells incubated with or without TNFα-neutralizing antibody (one-way ANOVA; **, p ≤ 0.01). c, current-voltage plot of hTASK3 current density in tsA-201 cells co-culture with THP-1 cells.
Direct Incubation with TNFα Enhances hTASK3 Current
To understand the mechanism of TNFα action on hTASK3, tsA-201 cells transiently transfected with hTASK3 channel were exposed to a recombinant TNFα (10 ng/ml) for various time intervals: 1, 2, and 15 h (Fig. 2, a and b). Although no significant effects were found after a 1-h incubation, TASK3 current density increased after a period of just a 2-h incubation from 68 ± 6 pA/pF (n = 9) using matched control cells (or 64 ± 1 pA/pF (n = 187) for all control cells) to 103 ± 6 pA/pF (n = 10). This effect was maintained after a 15-h TNFα incubation (65% increase) (from 58 ± 9 pA/pF, n = 14, to 93 ± 10 pA/pF, n = 18). The increase of current induced by TNFα occurred across the voltage range examined (Fig. 2, c and d). TNFα did not significantly enhance current in the presence of a neutralizing antibody against TNFR1, mAb225 (30). After treatment with mAb225 (1 μg/ml for 30 min), current density was 69 ± 4 pA/pF, n = 7 in the absence of TNFα and 78 ± 6 pA/pF, n = 15, after 2-h TNFα treatment.
FIGURE 2.

TNFα (10 ng/ml) increases K+ current in tsA-201 cells transfected with wild-type hTASK3. a, traces of current obtained using the voltage protocol (see “Experimental Procedures”) on cells treated for 1, 2, and 15 h with TNFα (10 ng/ml) compared with untreated cells. b, histogram of current density (mean ± S.E. (error bars)) in control cells and cells incubated with TNFα at different times: 1, 2, and 15 h (unpaired t test; *, p ≤ 0.05; **, p ≤ 0.01). c, current-voltage plot of hTASK3 current density in untreated tsA-201 cells (control) or cells treated with TNFα 10 ng/ml, 2-h incubation, and transfected with hTASK3 gene.
TNFα action on a murine TASK3 channel (mTASK3) was investigated. The amino acid similarity between mTASK3 and hTASK3 channel is only approximately 68% with most of the differences concentrated in the C-terminal domain. Nevertheless, TNFα was able to enhance, similarly, mTASK3 current by approximately 70% (from 78 ± 7 pA/pF, n = 7, to 138 ± 8 pA/pF, n = 6; Student's t test, p < 0.001).
The Effect of TNFα Is Not Seen for hTREK1, hTASK1, hTASK2, or hTRESK K2P Channels
Despite the fact that K2P channels have similar function, their distribution in the body and their biophysical and pharmacological properties are different. We determined the specificity of TNFα effect on TASK3 channels by observing other K2P channel activity in the presence of TNFα. Of other K2P channels, TASK1 is the closest channel in term of sequence similarity. However, our results showed no effect of TNFα on TASK1 channels (control, 19 ± 2 pA/pF, n = 11; TNFα, 21 ± 4 pA/pF, n = 12; Student's t test p > 0.05) (Fig. 3). Three other representative K2P channels, hTASK2, hTREK1, and hTRESK, were considered, but currents through these channels were also not modified by TNFα (Fig. 3).
FIGURE 3.

TNFα does not affect hTASK1, hTREK1, hTASK2, and hTRESK1 currents. Graphs show various K2P channel current densities in untreated cells and cells treated for 2 h with TNFα (10 ng/ml). The K2P current densities between −80 and −40 mV are on the left and the density of current-voltage plot on the right.
TNFα Does Not Influence Surface Expression of hTASK3 Channels
To visualize the channel distribution in cells, a fluorescent-tagged TASK3-GFP channel was created with the tag present on the M1P1 loop (therefore located on the extracellular face of the channel). To measure the effect of TNFα on TASK3 channel surface expression, cells were co-transfected with hTASK3-GFP and a dsRED membrane marker. Cells were incubated with or without TNFα (10 ng/ml) for 2 and 15 h and subsequently fixed and DNA-stained (blue) (see “Experimental Procedures”). Confocal imaging showed a large overlap between hTASK3-GFP (green) and dsRED-membrane marker (red), as denoted by yellow co-localized pixels, confirming a high expression of hTASK3 channel at the plasma membrane (Fig. 4, a and b). Quantification of the overlap using the Pearson's correlation coefficient showed that hTASK3-GFP alone compared with hTASK3-GFP incubated with TNFα for 2 h (0.66 ± 0.06 AU compared with 0.68 ± 0.05 AU, Fig. 4c) or 15 h (0.52 ± 0.07 AU compared with 0.62 ± 0.04 AU) were not significantly different.
FIGURE 4.

TNFα does not affect the surface expression of hTASK3 channels. a, hTASK3-GFP- (green) and dsRED membrane marker- (red) transfected cells. b, cells co-transfected with hTASK3-GFP and dsRED membrane marker as above then treatment with TNFα (10 ng/ml) for 2 h. White scale bars represent 10 μm. c, histogram showing co-localization analysis using the Pearson's correlation coefficient comparing membrane expression of TNFα-treated (2 h) and control untreated cells (Student's t test; NS, p > 0.5). d, histogram of current density (mean ± S.E. (error bars)) in control cells and cells treated with TNFα for 2 h.
The ability of TNFα to enhance the GFP-tagged TASK3 channels after 2-h application was investigated, and, despite a reduced overall current density of the tagged channel, these channels remained functional, and an enhancement by TNFα of 50% was observed (from 32 ± 6 pA/pF, n = 10–50 ± 6 pA/pF, n = 10; Student's t test p < 0.05, Fig. 4d). TNFα was again effective over all voltages measured. Thus, addition of a GFP tag did not alter the action of TNFα on TASK3 channels. This result suggested that the amount of channel at the membrane is not changed by TNFα and, furthermore, that the effect seen with TNFα treatment was not due to changes in the trafficking of hTASK3 channel.
TNFα Modulates hTASK3 Channel Gating
pH is an important regulator of TASK3 current. We examined the correlation between pH sensitivity of the channel and TNFα effect (Fig. 5). An acidic pH of 6.4 decreased hTASK3 current observed at a physiological pH of 7.4 by approximately 60% (from 83 ± 4 pA/pF, n = 13, to 32 ± 5 pA/pF, n = 14). Interestingly, the action of TNFα was abolished at pH 6.4 (34 ± 5 pA/pF, n = 15).
FIGURE 5.

TNFα (10 ng/ml) has no effect on K+ current at an external acidic pH of 6.4 in tsA-201 cells transfected with wild-type hTASK3. a, traces of current obtained using the voltage protocol on cells incubated in an external solution of pH 6.4 treated with TNFα (10 ng/ml) compared with untreated cells. b, current-voltage plot of hTASK3 current density in tsA-201 cells untreated or treated with TNFα for 2 h and incubated in an external solution of pH 6.4. c, histogram of current density (mean ± S.E. (error bars)) in control cells and cells treated with TNFα at an external pH of 6.4 or 7.4 (one-way ANOVA; *, p ≤ 0.05; NS, p > 0.5). Significant inhibition of current induced by pH 6.4 is compared with pH 7.4 for control and TNFα-treated cells (one-way ANOVA; +, p ≤ 0.05; ++, p ≤ 0.01).
Histidine at position 98 (His-98) in the pore region of the channel is known to play an important role in the pH sensitivity of the channel (10). As expected, the hTASK3 mutant H98A lost its pH sensitivity (Fig. 6); at pH 7.4 the density of current was 53 ± 10 pA/pF (n = 10) and at pH 6.4, 56 ± 4 pA/pF (n = 11). However, TNFα was now able to enhance current through this mutated, pH-insensitive, TASK3 channel at either pH 7.4 or pH 6.4 (respectively, 86 ± 8 pA/pF, n = 10, and 82 ± 9 pA/pF, n = 12).
FIGURE 6.

TNFα (10 ng/ml) similarly increases K+ current at external pH 6.4 and pH 7.4 in tsA-201 cells transfected with mutant H98A hTASK3. a, traces of current obtained using the voltage protocol on cells incubated in an external solution of pH 6.4 or pH 7.4 treated with TNFα compared with untreated cells. (b) current-voltage plot of hTASK3 current density in tsA-201 cells untreated or treated with TNFα for 2 h incubated in an external solution of pH 6.4 and pH 7.4. (c) Histogram of current density (mean ± S.E.) in control cells and cells treated with TNFα at an external pH of 6.4 or 7.4 (one-way ANOVA *: p ≤ 0.05).
The C Terminus of hTASK3 Is a Major Contributor to TNFα Effect
The large intracellular C terminus region of hTASK3 contains many regulatory sites. We used a truncated hTASK3 channel (hTASK3-stop) with the last 124 amino acids deleted. This mutant was still functional and pH-sensitive. An inhibition of 50% was observed when the pH was decreased to 6.4 (control at pH 6.4, 66 ± 6 pA/pF, n = 9; control at pH 7.4, 120 ± 6 pA/pF, n = 8). However, the truncated channel was not affected by TNFα at either pH 6.4 or 7.4, suggesting that the C terminus of hTASK3 is an important region involved in its modulation by TNFα (at pH 7.4, control, 120 ± 6 pA/pF, n = 8; TNFα, 127 ± 6 pA/pF, n = 8) (Fig. 7).
FIGURE 7.

TNFα (10 ng/ml) has no effect on K+ current at external pH 6. 4 and pH 7.4 in tsA-201 cells transfected with truncated mutant hTASK3. (a) Traces of current obtained using the voltage protocol on cells incubated in an external solution of pH 6.4 or pH 7.4 treated with TNFα compare with untreated cells. b, current-voltage plot of hTASK3 current density in tsA-201 cells untreated or treated with TNFα for 2 h and incubated in an external solution of pH 6.4 and pH 7.4. c, histogram of current density (mean ± S.E. (error bars)) in control cells and cells treated with TNFα at an external pH of 6.4 or 7.4 (one-way ANOVA; NS, p > 0.05). Significant inhibition of current induced by pH 6.4 is compared with pH 7.4 for control and TNFα-treated cells (one-way ANOVA; ++, p ≤ 0.01)
TNFα Enhances TASK3 Current via the ASK1 Pathway
Having established that TNFα enhances the hTASK3 channel via a modulation of its C-terminal region, we sought to characterize the signaling pathway underlying this effect in more detail. The role of ASK1, a second messenger mediator of TNFα, and the role of reactive oxygen species were both investigated. A plasmid containing either wild-type ASK1 or a dominant negative isoform (ASK1-KM) was co-transfected into the tsA-201 cells with hTASK3. Western blotting, using an ASK1 protein antibody, showed a small increase in ASK1 protein expression induced by 15-h TNFα treatment (137 ± 6% of control value). When tsA-201 cells co-transfected with either the dominant negative (165 ± 9% of control value) or the wild-type ASK1 gene (153 ± 9% of control value) were treated with TNFα, a greater enhancement of ASK1 protein expression was observed (Fig. 8). TNFα increased significantly ASK1 activity from 0.058 nmol/min mg of protein to 0.073 nmol/min mg of protein. The dominant negative ASK1-KM gene blocked ASK1 activity (0.053 nmol/min mg of protein) compared with the wild-type ASK1 gene (0.084 nmol/min mg of protein) (Fig. 8). Because the TNFα effect is similar with or without wild-type ASK1 transfection, it seems that there is sufficient endogenous ASK1 activity to fully mediate the effect of TNFα. This increase in ASK1 activity induced by TNFα was related to an enhancement of hTASK3 current: cells co-transfected with the wild-type ASK1 gene had a current density significantly higher (98 ± 6 pA/pF, n = 17) compared with cells co-transfected with ASK1-KM dominant negative gene (76 ± 6 pA/pF, n = 20). Consequently, ASK1 seems to be involved in the pathway that activates hTASK3 (Fig. 8, a and b).
FIGURE 8.

TNFα enhances K+ current via ROS-ASK1 pathway in tsA-201 cells transfected with wild-type hTASK3. a, current-voltage plot of hTASK3 current density in tsA-201 cells co-transfected with either ASK1-wt gene or ASK1-KM gene and treated with TNFα for 2 h. b, histogram of current density (mean ± S.E. (error bars)) in control cells and cells co-transfected with either ASK1-wt gene or ASK1-KM gene, treated with TNFα. c, reactive oxygen species produced in comparison with control (100%) after treatment in TNFα in cells co-transfected with either ASK1-wt gene, ASK1-KM gene or without co-transfection. d, activity of ASK1 protein in control cells treated or untreated with TNFα and in cells co-transfected with either ASK1-wt gene or ASK1-KM gene treated with TNFα (one-way ANOVA; *, p < 0.05; **, p < 0.01; NS, p > 0.05). e, Western blot analyses of ASK1 protein levels.
To determine the relation between ROS and ASK1 in a TNFα-activated pathway, ROS formation induced by TNFα after a 15-h treatment was quantified. TNFα induced a significant increase of ROS compared with control (195 ± 13% of control, n = 3). A similar enhancement was obtained in the presence of either ASK1-wt or ASK1-KM gene (respectively, 201 ± 14%, n = 3 and 204 ± 22%, n = 3) (Fig. 8c). Because in the presence of the ASK1-KM gene an increase of ROS production was observed and yet no enhancement of TASK3 current was seen, the hTASK3 current enhancement has no direct link with the production of ROS.
TNFα Enhancement of TASK3 Current Is JNK- and p38-dependent
To further study the pathway between TNFα receptor activation and enhancement of TASK3 current, JNK and p38, two downstream targets of ASK1, were investigated. Specific inhibitors of these two kinases were used: respectively, SP600125 (10 μm) and SB203580 (10 μm). These two inhibitors were added independently to TNFα during a 2-h incubation. In the presence of the JNK inhibitor SP600125, there was no significant TNFα-mediated enhancement of TASK3 current (from 75 ± 5 pA/pF, n = 18, to 91 ± 6 pA/pF, n = 17; Fig. 9a). Similarly, in the presence of the p38 inhibitor, SB203580, TNFα action was inhibited (from 60 ± 8 pA/pF, n = 9, to 55 ± 7 pA/pF, n = 9; Fig. 9b). These results reveal a mutual role of JNK and p38 in the TNFα effect on TASK3 channel current.
FIGURE 9.

p38 and JNK are involved in TNFα-induced hTASK3 current enhancement. Current-voltage plot and histogram (mean ± S.E. (error bars)) show hTASK3 current density in tsA-201 cells treated with either SP600125 (a), SB203580 (b), or Bay11-7082 alone (c) and in the presence of TNFα (d) (one-way ANOVA; *, p ≤ 0.05, NS, p > 0.5).
The potential involvement of the NF-κB pathway, which has been shown to be a mediator of TNFα action on some K channels, was investigated. A specific inhibitor of NF-κB, Bay11-7082, was used at a concentration of 2 μm. Added with TNFα, it did not affect TNFα action on hTASK3 current (Bay11-7082, 91 ± 8 pA/pF, n = 6; TNFα + Bay11-7082, 131 ± 17 pA/pF, n = 7; Fig. 9c), and thus the NF-κB pathway is not involved in the TNFα action on hTASK3 channel.
TNFα Needs the Presence of hTASK3 to Induce Death of tsA-201 Cells
To detect any apoptotic action of TNFα on cells expressing hTASK3 channel, we measured translocation of phosphatidylserine from the inner side of the membrane to the outer side, an established marker of early apoptosis. Annexin V interacts with phosphatidylserine externalized during the apoptosis process and generates a green fluorescence at the surface of the cell. Cells were co-transfected with hTASK3 and dsRED endoplasmic reticulum marker (the latter to count total transfected cells), or transfected only with the dsRED marker. These were incubated with or without TNFα (10 ng/ml) for 15 h. TNFα did not induce apoptosis in cells transfected with dsRED only (control, 3.6 ± 1.5%; TNFα, 2.2 ± 0.6%). However, when hTASK3 was present, a significant enhancement of cell death by TNFα, from 7.3 ± 0.8% (control) to 17.0 ± 1.4% (TNFα) was observed (Fig. 10). It is of note that in fields of cells transfected with TASK3 and treated with TNFα there is not always overlap between the annexin V and dsRED signals (Fig. 10iv). Some cells appear to be untransfected but undergoing apoptosis (green but no red signal). Interestingly, there is a significant reduction in the total number of red (transfected) cells and a significant increase in the total number of green cells, only in fields both transfected with TASK3 and treated with TNFα. This suggests that in these fields, apoptosis interferes with the dsRED signal, giving an underestimate of the number of transfected cells and the appearance of untransfected cells undergoing apoptosis.
FIGURE 10.
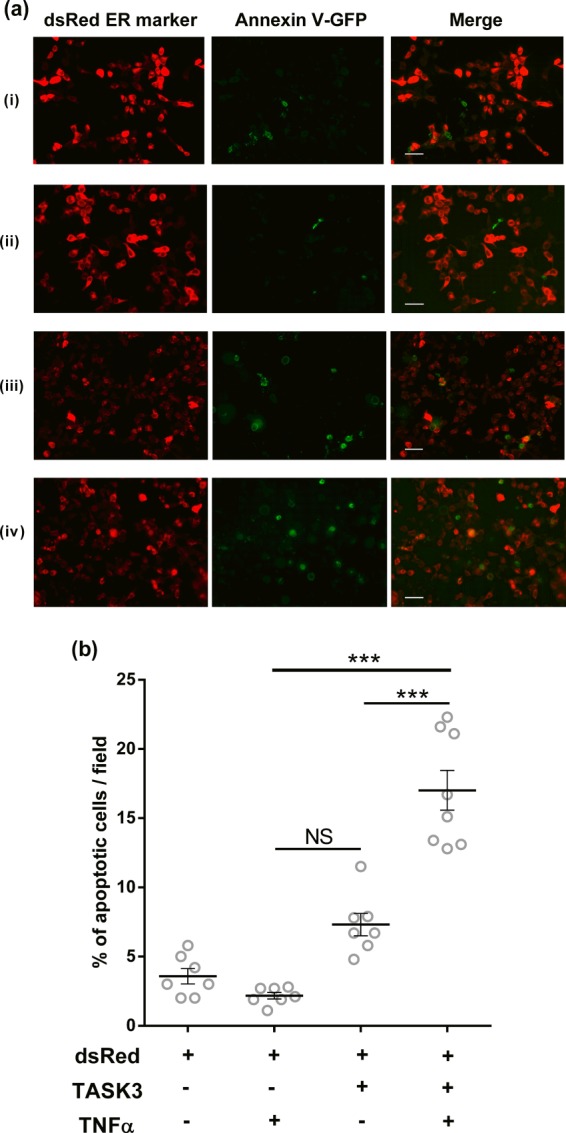
Apoptotic cell death needs the presence of TNFα and hTASK3 potassium channel. a, images representing the annexin V-GFP fluorescence (apoptotic cells; left), the dsRED fluorescence (transfected cells, middle), and the two images merged (right). i, dsRED cell transfection; ii, dsRED cell transfection with 15-h TNFα treatment; iii, dsRED and hTASK3 cells co-transfection; iv, dsRED and hTASK3 cells co-transfection with 15-h TNFα treatment. The scale bar represents 50 μm in all of the images. b, percentage of apoptotic cell death is represented by the annexin V (green) interacting with membrane apoptotic markers. TASK3/dsRED- and dsRED-transfected cells were treated with TNFα (two-way ANOVA; NS, p > 0.05; ***, p < 0.001).
Addition of TNFα did not change significantly the caspase-3 activity in untransfected cells (from 0.11 ± 0.02 to 0.19 ± 0.03 nmol pNA/min per mg of protein, two-way ANOVA, p > 0.05) nor in TASK3/GFP-transfected cells (from 0.13 ± 0.03 to 0.23 ± 0.04 nmol pNA/min per mg of protein, two-way ANOVA, p > 0.05). Thus, the apoptotic event induced by the dual presence of TASK3 and TNFα is caspase-3-independent.
DISCUSSION
In this study, TNFα has been shown to enhance current through TASK3 channels by modulating channel gating. TNFα treatment, combined with an increase of K efflux through TASK3 channels, leads to apoptosis.
TNFα Indirectly Modulates TASK3 Channel Activity via the ASK1-p38/JNK Pathway
The increase of TASK3 current following TNFα treatment did not occur instantaneously and was only seen after an incubation for 2 h or more, suggesting an indirect action of TNFα via second messenger mediators activated downstream. TNFα activates a number of different pathways (see Fig. 11), leading to a range of different effects (32). In this study, activation of ASK1 by TNFα with subsequent stimulation of p38 and JNK kinases emerged as the main mediator of the TASK3 current enhancement.
FIGURE 11.
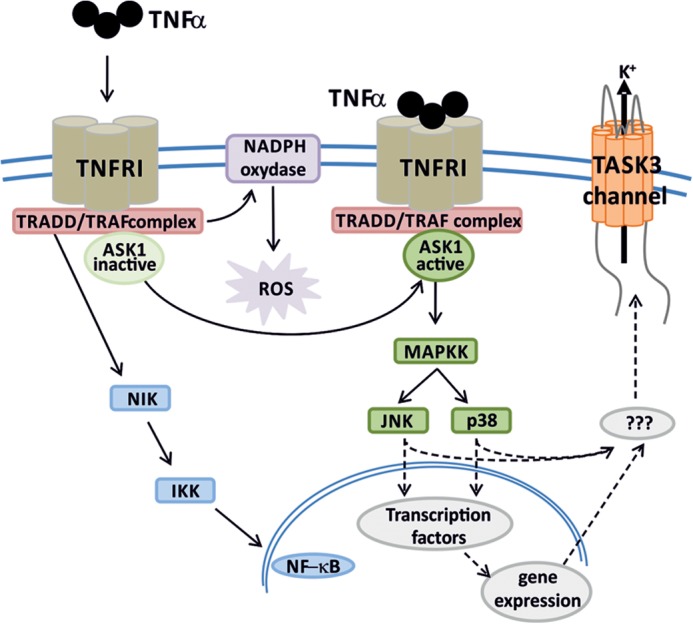
Schematic summarizing TNFα mechanism of action leading to an enhancement of K+ efflux via TASK3 channels.
The p38-JNK pathway is involved in stress signals such as osmotic stress, redox stress, mechanical stress, and radiation in addition to inflammation. In many cases, p38 kinase activation leads to a K efflux and alterations in intracellular K concentration (33–37). This promotes both neurodegeneration and cell death following cellular stress (38). In myocytes, activation of p38 following release of ATP during ischemia increased a TREK1-like K2P channel current (39).
In this study, the dual action of these two kinases on the TASK3 channel could be explained in two ways: either both JNK and p38 directly phosphorylate the TASK3 channel or, perhaps more likely, this occurs indirectly by p38 and JNK activating simultaneously one or more downstream kinases (see Fig. 11).
Gating Modulation of TASK3 Channel C-terminal Domain
TNFα action on TASK3 was sensitive to extracellular acidification. Acidic pH inhibits TASK3 current through a protonation of histidine residue 98, localized in the first pore region of the channel, which modifies channel gating (10). A complete abolition of the TNFα-mediated TASK3 current enhancement was obtained when reducing pH to 6.4 from 7.4, showing an interaction between both TNFα and pH regulation of the channel. Mutation of His-98 to an alanine residue blocked the TASK3 current inhibition induced by the histidine protonation observed at pH 6.4, but TNFα-mediated TASK3 current enhancement at acidic pH was now observed with the H98A-TASK3 mutant channel. This suggests that acidic pH does not interfere with the pathways leading to TNFα enhancement of TASK3 current, but instead acts solely at the level of the TASK3 channel to alter and overcome the TNFα action. In addition, this experiment shows that despite the interaction between TNFα and pH regulation, the two factors regulate the activity of TASK3 channels independently. This is confirmed by experiments showing that C-terminal deletion of the TASK3 channel blocked TNFα action without affecting pH regulation of the channel. Thus, the C terminus of the channel is required for TNFα action, which strongly suggests that TNFα modifies this region of the channel to produce its effect, as seen for many regulators of both TASK3 and other K2P channels (see e.g. 40). Recent studies on the related K2P channel TREK1 have proposed that changes in the C-terminal domain might affect the conformation of the selectivity filter (41–43) rather than influencing an activation gate at the bundle crossing region of the channel (44, 45). Because acidic pH induces a protonation of His-98 which also affects gating at the selectivity filter, the protonation of His-98 may compete with TNFα-mediated regulation at the C-terminal domain to overcome its action at the level of the selectivity filter.
TASK3 Channel Regulation and Apoptosis
The MAPK pathway stimulated by TNFα and, especially, the JNK-p38 pathway is proapoptotic. In this study, we observed that TNFα, in the presence of hTASK3 channels, triggers cell apoptosis in a caspase-3-independent way (Fig. 10).
Changes in intracellular K concentration have been shown to be essential in the regulation of cell activity and viability (46). A loss of cytoplasmic K ion leads to changes in cell osmolarity, which is balanced by an efflux of water, resulting in a pathophysiological cell volume decrease. In addition, K ions are negative regulators of some of the enzymes involved in apoptosis (18). As such, a decrease of intracellular potassium concentration generates a favorable cytoplasmic environment for apoptotic proteolyses and DNA degradation (47–49). A modification of K channel activity and/or of channel membrane expression, resulting from the activation of extrinsic pathways involving death receptor pathways (or apoptotic stimuli), is mainly responsible for this K efflux (46). Indeed, changes of intracellular K concentration have been described as one of the direct instigators of the initial events of this apoptotic cascade (see 50).
Many distinct potassium channels have been linked to this apoptosis process depending on the cell type, the apoptotic stimulus involved, and the environment of the cell (46, 47). K2P channels seem of particular importance in this regard (18). These channels are characterized by voltage-independence and, because they are open at rest, this property leads to them often being the main contributor of the initial intracellular K+ loss. Quinine- and barium-sensitive K+ currents, similar to K2P leak currents, participate in apoptotic volume decrease (50–52). Particular members of the K2P subfamily have been demonstrated to be involved in this loss of K+ and the following apoptosis in a number of different cells, such as TREK1 channels in mouse embryos (53), TALK channels in exocrine pancreatic cells (54), and TASK2 channels in immature B cells (55).
TASK3 channels have been shown to be of particular importance in cultured CGNs (17, 18) and TASK1 and TASK3 channels in evoked inflammation (7, 56, 57). In CGNs, K loss through the native leak current IKSO (5, 6), carried primarily by TASK3 channels (e.g. 58), was found to lead to apoptosis, unless cells were maintained in a high extracellular K environment (17). Decreasing extracellular pH, treatment with ruthenium red, or activation of M3 muscarinic receptors all inhibit IKSO and protect CGNs from K-dependent cell death. Conversely, overexpressing TASK3 channels in hippocampal neurons induced K-dependent cell death (17). In this study, the combination of TASK3 channel activity and TNFα treatment was required to induce apoptotic cell death in tsA-201 cells. Our initial experiments indicate that the native IKSO current in mouse cerebellar granule neurons in culture is enhanced modestly by TNFα consistent with the hypothesis that these native channels are composed, at least in part, of homomeric TASK3 channels. However, it is known that IKSO current is already greatly up-regulated in CGNs maintained in culture. It is of great interest to determine what happens to TASK3-mediated current in native neurons either in vivo or in acute slice preparations when the cells are exposed to inflammatory mediators such as TNFα.
Perhaps paradoxically, an overexpression of TASK3 channels is observed in approximately 44% of breast cancers and in other cancers (59), and a clear correlation between TASK3 channel expression and cell growth has been found (18, 60). Thus, TASK3 channels have the potential to be both proapoptotic or oncogenic (promoting cell proliferation) depending on the cellular environment. It also appears that the level of TASK3 current generated is important, being approximately 10-fold higher in cells where apoptosis occurs (e.g. CGNs) compared with tumor cells where proliferation is promoted (18). This may reflect the importance of the degree of K efflux in a particular circumstance, where modest loss stimulates proliferation and large K efflux stimulates apoptosis. Alternatively, it may reflect a balance between the electrophysiological consequences of K2P channel expression (hyperpolarization of the membrane and decreased input resistance) and the biochemical consequences of decreased intracellular K concentration following increased K efflux (31).
Footnotes
- TASK
- TWIK-related acid-sensitive potassium
- ANOVA
- analysis of variance
- AU
- absorbance unit
- CGN
- cerebellar granule neuron
- K2P
- two-pore domain potassium
- pA
- picoampere
- pF
- picofarad
- pNA
- p-nitroaniline
- ROS
- reactive oxygen species
- TLR
- Toll-like receptor.
REFERENCES
- 1. Goldstein S. A., Bayliss D. A., Kim D., Lesage F., Plant L. D., Rajan S. (2005) International union of pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol. Rev. 57, 527–540 [DOI] [PubMed] [Google Scholar]
- 2. Lotshaw D. P. (2007) Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell Biochem. Biophys. 47, 209–256 [DOI] [PubMed] [Google Scholar]
- 3. Enyedi P., Czirják G. (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol. Rev. 90, 559–605 [DOI] [PubMed] [Google Scholar]
- 4. Talley E. M., Solorzano G., Lei Q., Kim D., Bayliss D. A. (2001) CNS distribution of members of the two-pore domain (KCNK) potassium channel family. J. Neurosci. 21, 7491–7505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Watkins C. S., Mathie A. (1996) A non-inactivating K+ current sensitive to muscarinic receptor activation in rat cultured cerebellar granule neurons. J. Physiol. 491, 401–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Millar J. A., Barratt L., Southan A. P., Page K. M., Fyffe R. E., Robertson B., Mathie A. (2000) A functional role for the two-pore domain potassium channel, TASK-1, in cerebellar granule neurons. Proc. Natl. Acad. Sci. U.S.A. 97, 3614–3618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bittner S., Budde T., Wiendl H., Meuth S. G. (2010) From the background to the spotlight: TASK channels in pathological conditions. Brain Pathol. 20, 999–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marsh B., Acosta C., Djouhri L., Lawson S. N. (2012) Leak K+ channel mRNAs in dorsal root ganglia: relation to inflammation and spontaneous pain behavior. Mol. Cell. Neurosci. 49, 375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patel A. J., Honoré E., Lesage F., Fink M., Romey G., Lazdunski M. (1999) Inhalational anesthetics activate two-pore domain background K+ channels. Nat. Neurosci. 2, 422–426 [DOI] [PubMed] [Google Scholar]
- 10. Rajan S., Wischmeyer E., Xin Liu G., Preisig-Müller R., Daut J., Karschin A., Derst C. (2000) TASK-3, a novel tandem pore domain acid-sensitive K+ channel: an extracellular histidine as pH sensor. J. Biol. Chem. 275, 16650–16657 [DOI] [PubMed] [Google Scholar]
- 11. Czirják G., Enyedi P. (2003) Ruthenium red inhibits TASK-3 potassium channel by interconnecting glutamate 70 of the two subunits. Mol. Pharmacol. 63, 646–652 [DOI] [PubMed] [Google Scholar]
- 12. Clarke C. E., Veale E. L., Green P. J., Meadows H. J., Mathie A. (2004) Selective block of the human 2-P domain potassium channel, TASK-3, and the native leak potassium current, IKSO, by zinc. J. Physiol. 560, 51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clarke C. E., Veale E. L., Wyse K., Vandenberg J. I., Mathie A. (2008) The M1P1 loop of TASK3 K2P channels apposes the selectivity filter and influences channel function. J. Biol. Chem. 283, 16985–16992 [DOI] [PubMed] [Google Scholar]
- 14. Veale E. L., Buswell R., Clarke C. E., Mathie A. (2007) Identification of a region in the TASK3 two-pore domain potassium channel that is critical for its blockade by methanandamide. Br. J. Pharmacol. 152, 778–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Veale E. L., Kennard L. E., Sutton G. L., MacKenzie G., Sandu C., Mathie A. (2007) Gαq-mediated regulation of TASK3 two-pore domain potassium channels: the role of protein kinase C. Mol. Pharmacol. 71, 1666–1675 [DOI] [PubMed] [Google Scholar]
- 16. Aller M. I., Veale E. L., Linden A. M., Sandu C., Schwaninger M., Evans L. J., Korpi E. R., Mathie A., Wisden W., Brickley S. G. (2005) Modifying the subunit composition of TASK channels alters the modulation of a leak conductance in cerebellar granule neurons. J. Neurosci. 25, 11455–11467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lauritzen I., Zanzouri M., Honoré E., Duprat F., Ehrengruber M. U., Lazdunski M., Patel A. J. (2003) K+-dependent cerebellar granule neuron apoptosis: role of task leak K+ channels. J. Biol. Chem. 278, 32068–32076 [DOI] [PubMed] [Google Scholar]
- 18. Patel A. J., Lazdunski M. (2004) The 2P-domain K+ channels: role in apoptosis and tumorigenesis. Pflugers Arch. 448, 261–273 [DOI] [PubMed] [Google Scholar]
- 19. Tabas I., Glass C. K. (2013) Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science 339, 166–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goetz F. W., Planas J. V., MacKenzie S. (2004) Tumor necrosis factors. Dev. Comp. Immunol. 28, 487–497 [DOI] [PubMed] [Google Scholar]
- 21. Stellwagen D., Malenka R. C. (2006) Synaptic scaling mediated by glial TNF-α. Nature 440, 1054–1059 [DOI] [PubMed] [Google Scholar]
- 22. Burg E. D., Remillard C. V., Yuan J. X. (2006) K+ channels in apoptosis. J. Membr. Biol. 209, 3–20 [DOI] [PubMed] [Google Scholar]
- 23. Knoch M. E., Hartnett K. A., Hara H., Kandler K., Aizenman E. (2008) Microglia induce neurotoxicity via intraneuronal Zn2+ release and a K+ current surge. Glia 56, 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Houzen H., Kikuchi S., Kanno M., Shinpo K., Tashiro K. (1997) Tumor necrosis factor enhancement of transient outward potassium currents in cultured rat cortical neurons. J. Neurosci. Res. 50, 990–999 [DOI] [PubMed] [Google Scholar]
- 25. Dolga A. M., Granic I., Blank T., Knaus H. G., Spiess J., Luiten P. G., Eisel U. L., Nijholt I. M. (2008) TNF-α-mediates neuroprotection against glutamate-induced excitotoxicity via NF-κB-dependent up-regulation of K2.2 channels. J. Neurochem. 107, 1158–1167 [DOI] [PubMed] [Google Scholar]
- 26. Czeschik J. C., Hagenacker T., Schäfers M., Büsselberg D. (2008) TNF-α differentially modulates ion channels of nociceptive neurons. Neurosci. Lett. 434, 293–298 [DOI] [PubMed] [Google Scholar]
- 27. Nicholas S. A., Sumbayev V. V. (2009) The involvement of hypoxia-inducible factor 1α in Toll-like receptor 7/8-mediated inflammatory response. Cell Res. 19, 973–983 [DOI] [PubMed] [Google Scholar]
- 28. Sumbayev V. V. (2008) LPS-induced Toll-like receptor 4 signalling triggers cross-talk of apoptosis signal-regulating kinase 1 (ASK1) and HIF-1α protein. FEBS Lett. 582, 319–326 [DOI] [PubMed] [Google Scholar]
- 29. Kapiszewska M., Cierniak A., Elas M., Lankoff A. (2007) Lifespan of etoposide-treated human neutrophils is affected by antioxidant ability of quercetin. Toxicol. in Vitro 21, 1020–1030 [DOI] [PubMed] [Google Scholar]
- 30. Defer N., Azroyan A., Pecker F., Pavoine C. (2007) TNFR1 and TNFR2 signaling interplay in cardiac myocytes. J. Biol. Chem. 282, 35564–35573 [DOI] [PubMed] [Google Scholar]
- 31. Yi B. A., Minor D. L., Jr., Lin Y. F., Jan Y. N., Jan L. Y. (2001) Controlling potassium channel activities: interplay between the membrane and intracellular factors. Proc. Natl. Acad. Sci. U.S.A. 98, 11016–11023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wajant H., Pfizenmaier K., Scheurich P. (2003) Tumor necrosis factor α signaling. Cell Death Differ. 10, 45–65 [DOI] [PubMed] [Google Scholar]
- 33. McLaughlin B., Pal S., Tran M. P., Parsons A. A., Barone F. C., Erhardt J. A., Aizenman E. (2001) p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J. Neurosci. 21, 3303–3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bossy-Wetzel E., Talantova M. V., Lee W. D., Schölzke M. N., Harrop A., Mathews E., Götz T., Han J., Ellisman M. H., Perkins G. A., Lipton S. A. (2004) Cross-talk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron 41, 351–365 [DOI] [PubMed] [Google Scholar]
- 35. Aras M. A., Aizenman E. (2005) Obligatory role of ASK1 in the apoptotic surge of K+ currents. Neurosci. Lett. 387, 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Redman P. T., He K., Hartnett K. A., Jefferson B. S., Hu L., Rosenberg P. A., Levitan E. S., Aizenman E. (2007) Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc. Natl. Acad. Sci. U.S.A. 104, 3568–3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Redman P. T., Hartnett K. A., Aras M. A., Levitan E. S., Aizenman E. (2009) Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. J. Physiol. 587, 4393–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barone F. C., Irving E. A., Ray A. M., Lee J. C., Kassis S., Kumar S., Badger A. M., Legos J. J., Erhardt J. A., Ohlstein E. H., Hunter A. J., Harrison D. C., Philpott K., Smith B. R., Adams J. L., Parsons A. A. (2001) Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med. Res. Rev. 21, 129–145 [DOI] [PubMed] [Google Scholar]
- 39. Aimond F., Rauzier J. M., Bony C., Vassort G. (2000) Simultaneous activation of p38 MAPK and p42/44 MAPK by ATP stimulates the K+ current ITREK in cardiomyocytes. J. Biol. Chem. 275, 39110–39116 [DOI] [PubMed] [Google Scholar]
- 40. Mathie A. (2007) Mammalian K2P channels and their regulation by G protein coupled receptors. J. Physiol. 578, 377–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bagriantsev S. N., Peyronnet R., Clark K. A., Honoré E., Minor D. L., Jr. (2011) Multiple modalities converge on a common gate to control K2P channel function. EMBO J. 30, 3594–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bagriantsev S. N., Clark K. A., Minor D. L., Jr. (2012) Metabolic and thermal stimuli control K(2P_2.1 (TREK-1) through modular sensory and gating domains. EMBO J. 31, 3297–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Piechotta P. L., Rapedius M., Stansfeld P. J., Bollepalli M. K., Erhlich G., Andres-Enguix I., Fritzenschaft H., Decher N., Sansom M. S., Tucker S. J., Baukrowitz T. (2011) The pore structure and gating mechanism of K2P channels. EMBO J. 30, 3607–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cohen A., Ben-Abu Y., Zilberberg N. (2009) Gating the pore of potassium leak channels. Eur. Biophys. J. 39, 61–73 [DOI] [PubMed] [Google Scholar]
- 45. Mathie A., Al-Moubarak E., Veale E. L. (2010) Gating of two pore domain potassium channels. J. Physiol. 588, 3149–3156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu S. P. (2003) Regulation and critical role of potassium homeostasis in apoptosis. Prog. Neurobiol. 70, 363–386 [DOI] [PubMed] [Google Scholar]
- 47. Bortner C. D., Hughes F. M., Jr., Cidlowski J. A. (1997) A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem. 272, 32436–32442 [DOI] [PubMed] [Google Scholar]
- 48. Hughes F. M., Jr., Bortner C. D., Purdy G. D., Cidlowski J. A. (1997) Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J. Biol. Chem. 272, 30567–30576 [DOI] [PubMed] [Google Scholar]
- 49. Montague J. W., Bortner C. D., Hughes F. M., Jr., Cidlowski J. A. (1999) A necessary role for reduced intracellular potassium during the DNA degradation phase of apoptosis. Steroids 64, 563–569 [DOI] [PubMed] [Google Scholar]
- 50. Maeno E., Ishizaki Y., Kanaseki T., Hazama A., Okada Y. (2000) Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. U.S.A. 97, 9487–9492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Séguin D. G., Baltz J. M. (1997) Cell volume regulation by the mouse zygote: mechanism of recovery from a volume increase. Am. J. Physiol. 272, C1854–61 [DOI] [PubMed] [Google Scholar]
- 52. Niemeyer M. I., Hougaard C., Hoffmann E. K., Jorgensen F., Stutzin A., Sepúlveda F. V. (2000) Characterisation of a cell swelling-activated K+-selective conductance of ehrlich mouse ascites tumour cells. J. Physiol. 524, 757–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Trimarchi J. R., Liu L., Smith P. J., Keefe D. L. (2000) Noninvasive measurement of potassium efflux as an early indicator of cell death in mouse embryos. Biol. Reprod. 63, 851–857 [DOI] [PubMed] [Google Scholar]
- 54. Duprat F., Girard C., Jarretou G., Lazdunski M. (2005) Pancreatic two P domain K+ channels TALK-1 and TALK-2 are activated by nitric oxide and reactive oxygen species. J. Physiol. 562, 235–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nam J. H., Shin D. H., Zheng H., Lee D. S., Park S. J., Park K. S., Kim S. J. (2011) Expression of TASK-2 and its up-regulation by B cell receptor stimulation in WEHI-231 mouse immature B cells. Am. J. Physiol. Cell Physiol. 300, C1013–22 [DOI] [PubMed] [Google Scholar]
- 56. Meuth S. G., Herrmann A. M., Ip C. W., Kanyshkova T., Bittner S., Weishaupt A., Budde T., Wiendl H. (2008) The two-pore domain potassium channel TASK3 functionally impacts glioma cell death. J. Neurooncol. 87, 263–270 [DOI] [PubMed] [Google Scholar]
- 57. Bittner S., Meuth S. G., Göbel K., Melzer N., Herrmann A. M., Simon O. J., Weishaupt A., Budde T., Bayliss D. A., Bendszus M., Wiendl H. (2009) TASK1 modulates inflammation and neurodegeneration in autoimmune inflammation of the central nervous system. Brain 132, 2501–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brickley S. G., Aller M. I., Sandu C., Veale E. L., Alder F. G., Sambi H., Mathie A., Wisden W. (2007) TASK-3 two-pore domain potassium channels enable sustained high-frequency firing in cerebellar granule neurons. J. Neurosci. 27, 9329–9340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mu D., Chen L., Zhang X., See L. H., Koch C. M., Yen C., Tong J. J., Spiegel L., Nguyen K. C., Servoss A., Peng Y., Pei L., Marks J. R., Lowe S., Hoey T., Jan L. Y., McCombie W. R., Wigler M. H., Powers S. (2003) Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell 3, 297–302 [DOI] [PubMed] [Google Scholar]
- 60. Pei L., Wiser O., Slavin A., Mu D., Powers S., Jan L. Y., Hoey T. (2003) Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function. Proc. Natl. Acad. Sci. U.S.A. 100, 7803–7807 [DOI] [PMC free article] [PubMed] [Google Scholar]