

Abstract
Many biological processes, including differentiation, reprogramming, and disease transformations, involve transitions of cells through distinct states. Direct, unbiased investigation of cell states and their transitions is challenging due to several factors, including limitations of single-cell assays. Here we present a stochastic model of cellular transitions that allows underlying single-cell information, including cell-state-specific parameters and rates governing transitions between states, to be estimated from genome-wide, population-averaged time-course data. The key novelty of our approach lies in specifying latent stochastic models at the single-cell level, and then aggregating these models to give a likelihood that links parameters at the single-cell level to observables at the population level. We apply our approach in the context of reprogramming to pluripotency. This yields new insights, including profiles of two intermediate cell states, that are supported by independent single-cell studies. Our model provides a general conceptual framework for the study of cell transitions, including epigenetic transformations.
A number of biologically important processes involve transitions through distinct cell states. Differentiation1,2,3,4,5,6,7,8,9, reprogramming10,11 and disease initiation and progression12,13,14 are among the many examples of this kind. State changes in such processes are in general stochastic, as reflected in experimentally observed variation in transition latency even in the setting where transitions arise in homogenous cell cultures subjected to defined driving events (e.g. Hanna et al.17).
Stochasticity of transitions at the single-cell level (Fig. 1a) imply that during such a process a cell population is a mixture of cells in different states, with the state composition of the cell population itself time-varying (Fig. 1b). Studying single-cell events in heterogenous, time-varying populations is challenging and the global changes in single-cell transcriptional, metabolic, and epigenetic state that are involved in these processes remain incompletely understood. High-throughput assays based on homogenates provide only population-averaged data; in transition processes such data represent averages over heterogenous states (Fig. 1c). Genome-wide single-cell protocols are now emerging2,4, but their efficiency, availability and depth remain limited. Furthermore, these are not live cell assays, so cannot be used to directly track genome-wide molecular profiles of single cells undergoing state transitions.
Figure 1. Stochastic cell state transitions and population-averaged molecular data (illustrated, without loss of generality, with reference to reprogramming and gene expression data).

(a), Coarse schematic showing a cell changing state (to a pluripotent state) via intermediate single-cell states. (b), Since cells stochastically change state during the transition process (horizontal lines), at any given time the population is heterogeneous. (c), Gene expression levels, as measured in mainstream high-throughput assays based on homogenates, represent averages over cells that may be in different states. (d), Our model specifies a latent stochastic process that describes state transitions at the single-cell level. Aggregation of these latent processes gives a likelihood (i.e. a data model) at the level of population-averaged data as depicted in part c. Estimation of model parameters gives information regarding state-to-state transition rates, wi,i+1 as well as molecular profiles that are specific to cell states (part e). Here, estimated state-specific expression profiles βij (where j indexes genes and i indexes states) are represented as an illustrative, genome-wide heat map (with genes in columns and states in rows). (f), Estimated transition rates w give information on the cell population dynamics, specifically the fraction of cells pi(t) in each state i as a function of time t.
Here we present a general stochastic model of transition processes that links parameters at the single cell level to time-course data at the cell population level, as obtained for example in conventional expression, proteomic or epigenetic assays based on homogenates. The key novelty of our approach is to specify latent stochastic models at the single-cell level and then (mathematically) aggregate the models to give a likelihood at the level of homogenate data. As we show below, this allows parameters specific to single-cell states and transitions between them to be estimated from homogenate, time-course data. To facilitate analysis of data collected at non-uniform time points we use continuous-time Markov processes as the single-cell models.
Estimation of model parameters from population-averaged time-course data then gives information on several aspects of the single-cell states and transitions, including:
Single-cell state profiles (e.g. state-specific expression, protein or epigenetic profiles);
State markers (e.g. genes, proteins or marks that are highly specific to individual states); and,
Dynamical information concerning transition rates, cell residence times, and population composition through time.
To fix ideas and illustrate our approach, we develop and apply our model in the context of reprogramming of mouse embryonic fibroblasts (MEFs) to a state of pluripotency10,15,16. This is a process that has been widely studied in recent years, and where a number of advanced experimental approaches have been brought to bear. Recent studies have shown that reprogramming has a substantial stochastic component. Subclones derived from the same transduced somatic cells activate pluripotency markers, such as Nanog-GFP, at very different times, over a range of a few weeks10,15,16. Further, there is evidence that the entire cell population has the potential to give rise to pluripotent cells during direct reprogramming, i.e., there is not an “elite” group of cells that are uniquely able to do so17. Thus, current evidence suggests reprogramming is an inherently stochastic process17 in which individual cells change from an initial differentiated state to an induced pluripotent stem cell (iPSC) state. Single-cell studies using pre-selected sets of genes have begun to elucidate cellular events in reprogramming19,20,21,22. However, at the genome-wide level many questions remain open and our understanding of the state transitions, including the number of traversed states, their marker genes and transition rates, remains limited.
In our model, we suppose that a cell can stochastically visit a set of n states during the transition process (Fig. 1d). Transitions between these states are described by a latent continuous-time Markov process whose discrete state space is identified with cell states (see Methods and SI for details). The model parameters are (i) the transition rates wi,i′ between states i and i′ and (ii) state-specific parameters βij that represent the mean expression level for gene j in state i (we focus on transcriptomic data here, but the analysis could be readily applied to e.g. proteomic or epigenomic data). We refer to the β's as state-specific signatures (Fig. 1e). The population dynamics are characterized solely by the transition rates: given the rates wi,i′, the Markov model yields the probability pi(t) of being in state i at time t. For a large number of cells, the population-averaged expression xj(t) of gene j at time t is then a combination of state-specific expression levels weighted by the probability of being in each state (Fig. 1f):
 |
Both wi,i′ and βij can be estimated from time-course data. Complicated transition networks may require ancillary data to ensure identifiability. Here, for simplicity, here we limit ourselves to consider only linear forward-transition models (i.e., no reverse arrows in Fig. 1d); this constraint allows direct application to conventional, time-course data. In the reprogramming context, we note that recent data17 support the idea that almost all donor cells eventually give rise to iPS cells. These results were determined from single cell assays that observed the appearance of one marker for the final state, expression of the Nanog pluripotency gene, and indicated an irreversible switch to pluripotency during reprogramming11. For the reprogramming application we present, as discussed in detail below, we further assume that all cells in the starting population are in an initial (differentiated) state.
We put forward a computationally efficient approach for estimation, as implemented in software called STAMM (State Transitions using Aggregated Markov Models; see Methods and SI for details). As we illustrate below, STAMM can be readily applied to full-genome studies. Furthermore, since STAMM is rooted in a probabilistic model, model selection methods allow exploration of the likely number of single-cell states in a transition process of interest.
Hidden Markov models (HMMs) are widely used to describe latent processes in biological applications and have previously been used to describe cell populations26 and model the cell cycle27,28. It is interesting to contrast our model with a classical HMM. The key differences are twofold. First, our model involves aggregation of single-cell level Markov chains, thus it deals with states that are not only hidden, but whose connection to population-level observables necessarily involves averaging over multiple instances of the latent process. In contrast, a HMM applied to time-course data from a transition process does not provide a model at the single-cell level. Second, our model operates in continuous time and applies naturally to non-uniformly sampled data. In contrast, in a HMM the underlying Markov process operates in discrete time, such that the probability of a state transition is the same between successive time points regardless of the intervening time period. This assumption generally will not hold at all under uneven time sampling of a heterogenous population. Due to these reasons, in our view HMMs are intrinsically ill-suited to the study of transition processes of the type we consider here.
An alternative approach to using HMMs is to attempt direct deconvolution based on a model of single-cell expression profiles, e.g.29,30,31. These approaches have greater deconvolution power but are hindered by the upfront requirement for an expression model. For example, Siegal-Gaskins et al.29 established a model for the progression of Caulobacter cells through their own cell cycle. Similarly, Rowicka et al.31 measured the distribution of cell cycle time-shifts based on well-known cell cycle regulated genes. While these methods can in principle be adapted for other organisms and systems, the STAMM method presented here is immediately and directly applicable to time-course data from general transition processes. In particular, it is not necessary to find genes following regular expression profiles, nor is it necessary to have an a priori knowledge of the phases of the process of interest, which in many applications, including reprogramming as considered here, remain incompletely understood.
Differential expression analysis is widely used to highlight potentially important players in high-throughput studies. Approaches have also been proposed for time course data that rank genes (or proteins) according to whether they show evidence of change over time or relative to a control time course23,24,32 or that cluster together genes that show similar temporal profiles25. However, these approaches do not attempt to model single-cell state transitions nor account for cellular heterogeneity.
Results
Our main results were obtained by application of STAMM to genome-wide gene expression time-course data due to Samavarchi-Tehrani et al.18 (hereafter referred to as “the Samavarchi-Tehrani data”). These data were obtained during reprogramming of a “secondary” mouse embryonic fibroblast (MEF) system that expresses Oct4, Sox2, Klf4, and cMyc for 30 days. The starting MEF culture was isolated from chimeric mice and maintained for less than 5 passages; under these conditions the simplifying assumption of an initially homogeneous cell population is arguably a reasonable one, since substantial long-term changes are unlikely. Below we describe the results we obtained from analysis of these data, including detailed profiles of intermediate states, and insights regarding transition rates and population dynamics. Furthermore, we compare these results with recent single-cell data19,20,21,22 from related secondary MEF reprogramming systems.
Number of intermediate single-cell states
We explored the number of model states using several criteria (Fig. 2). Model fit (as captured by the squared difference between output of the fitted model and observed expression, i.e. the residual sum-of-squares) improves with number n of states; this is unsurprising as the number of model parameters increases with n. However, models with too many states may overfit. Overfitting can occur by introduction of artifactual states that are not transcriptionally or biologically distinct (e.g., splitting of a state into two; Supplementary Fig. S3b). Alongside fit-to-data, we therefore also monitored the extent to which state signatures were mutually distinct (Fig. 2b) quantified using a standard linear algebraic quantity called the condition number (see SI). We find that already with just five states the condition number is sharply increased and signatures are no longer distinct (Fig. 2b), suggesting that the improved fit is simply due to artifactual splitting of states. In addition, we carried out a Bayesian model selection, computing a probability score over number of states that takes account of both fit-to-data and model complexity in a principled way (see Methods and SI for details and discussion). This analysis supports the existence of intermediate states, with highest posterior probability associated with a four-state model (Fig. 2c).
Figure 2. Number of model states.
Models having n states were fitted to gene expression time-course data from a secondary MEF-based reprogramming system due to Samavarchi-Tehrani et al.18. (a), Model fitting error (quantified as the squared difference between output of the fitted model and the observed data) decreases with n but at the cost of introducing artifactual, non-distinct states as shown, (b), by an increase in the condition number that quantifies linear dependence between the state-specific gene expression signatures, i.e., mutual similarity of the n state-specific profiles. (c), A Bayesian model selection approach was used to score models with different numbers of states in terms of their posterior probability. The four-state model has the highest posterior probability, while there is negligible support for a 2-state, or single step, model. Taken together these results suggest that a four-state model strikes a good balance between fit-to-data and model complexity.
Thus, a four-state model appears to strike a good balance between parsimony and fit-to-data. The model shows clearly distinct state signatures (Fig. 2b and Fig. 3e and f) yet, despite having only three dynamical parameters, fits diverse time-courses well (see, e.g., Fig. 3d where some of the genes considered by Samavarchi-Tehrani et al.18 are shown). Taken together, these results suggest that a total of four single-cell states with distinct expression profiles, including two, new intermediate states, are visited during reprogramming of secondary MEFs to pluripotency. Below we explore the four-state model in detail.
Figure 3. A stochastic, multi-state model for reprogramming.

(a), We fitted a four-state model to gene expression time-course data from a secondary MEF-based reprogramming system due to Samavarchi-Tehrani et al.18. (b), Estimated rates for each transition. (c), The probability of a cell to be in a particular state as a function of time during reprogramming. (d), Observed time-course data (blue with crosses) and output of the fitted model (green) for selected genes. (e), Genome-wide, state-specific gene expression signatures. The cell states have markedly different global transcriptional profiles. (f), Gene expression signatures for selected genes that are discussed in the text. Red genes have state-specific expression that is below and green genes above the state average (dotted vertical lines). Genes are shown by category, from top to bottom: pluripotency, epithelial, signaling, mesenchymal, and growth. The model highlights major transcriptional changes between states, including a mesenchymal-to-epithelial transition from state 2 to 3, and the establishment of pluripotency marker genes in the transition from state 3 to 4.
Cell state-specific transcriptional profiles
We identify a total of four cell states as visited in the transition from MEF to iPSC. Genome-wide transcriptional profiles for these four states are shown in Figure 3e (full list in Supplementary Table S2; genes were filtered as described in Methods and SI and a total of 4383 genes were fit; parameter estimation and checks of robustness appear in Methods and SI). Figure 3f shows signatures for a subset of core reprogramming-related genes (listed in Supplementary Table S1). The sets of genes that characterize individual states broadly recapitulate known functional groups and the order in which the states appear are consistent with specific roles. The initial state (S1, triangle) is marked by high expression of MEF marker genes such as Cdh2 and Thy1 and mesenchymal genes including Snai2 (also known as Slug) and Zeb1. Many of these genes remain on in the first intermediate state (S2, square), but Jag1, Notch1 and Cdh2 have been switched off. Correspondingly, expression of proliferation genes, such as Ccnd1, start increasing. The second intermediate state (S3, diamond) is marked by epithelial-associated genes such as Epcam, Ocln, Cdh1, and the loss of the MEF markers. Thus, our model identifies a mesenchymal-to-epithethial transition (MET) of a group of genes from state S2 to S3 that is consistent with previous observations18. However, our results reveal that the MET is just one aspect of a much broader change of state involving a dramatic, global reconfiguration of the transcriptional program of a substantial fraction of genes (more than 70% of the genes show two-fold or greater change in state-resolved expression between successive states, as illustrated by the heat-map of Fig. 3e). The final state (S4, star) is negative for genes such as Tgfb1, and positive for ESC markers such as Nanog, Zfp42 (also known as Rex1), Esrrb, Dppa5a, Utf1, Dppa3, consistent with its iPSC nature.
Functional enrichment in individual states
To further characterize the functional nature of the states identified by STAMM we carried out a Gene Ontology (GO) analysis using the estimated state-specific expression profiles. Specifically, we identified GO terms that are over-represented among genes up- or down-regulated in individual states: the over-representation p-values are shown in Supplementary Figure S4 (see Supplementary Methods for details). The broad categories highlighted reveal the progression of reprogramming seen in the single-cell states identified by STAMM. Overall, GO analysis of the states traversed during reprogramming of secondary MEF into iPSC in the Samavarchi-Tehrani system illustrates that after the typical MEF signature of state S1, in state S2 reprogramming factors seem to trigger a broad range of cell activities including signaling, morphogenesis, differentiation and transcription regulation; in S3 a narrowing seems to occur around fewer key activities in preparation for state S4 where convergence towards the iPSC state occurs. The latter state is high in activities related, among other processes, with cycle regulation and chromatin organization.
Cell population dynamics
STAMM gives estimates of transition rates in the latent stochastic process (that describes changes in cell state) that give information on the dynamics of the changing cell population. Figure 3c shows the distribution of the cell population across states as a function of time; the changing fraction of cells in each state result from single cell transitions between states (Fig. 3b). The transition from S1 to S2 takes on average about 4 days, and the transition between S2 and S3 has a similar timescale. We find, however, that the final step of the process, from second intermediate (S3) to final state (S4), is the slowest to occur, taking on average 15 days. Our model predicts therefore that the last transition is the process bottleneck, implying that it would be natural to act on it to try to increase reprogramming speed and efficiency. The average transition rates that we determine shed light on the strong stochasticity of reprogramming, whereby even identical subclones reach the iPSC state with very different latency times16. In fact, each intermediate transition is a Poisson process and the variance of its transition time is proportional to its transition rate. Since the overall process comprises multiple steps, its overall variance is roughly the sum of the single step variances. Thus, our model predicts a variance of the total reprogramming time of the order of 3 weeks, a value consistent with experimental findings16.
Cell state markers and the molecular circuitry of reprogramming
STAMM allows ranking of genes according to state-specific expression, and can be used to provide insights and hypotheses concerning how specific genes are modulated during state transitions. We focus, in particular, on:
Switch genes, which are expressed in a particular state and persist at moderate-to-high expression levels in subsequent states; and
Pulse genes, which are switched on in a particular state, but turned off in all other states (see also Methods; full ranked lists of state genes are reported in Supplementary Table S2).
Gene lists for each pulse and switch were further analyzed using the Gene Set Enrichment Analysis tool (GSEA; www.broadinstitute.org/gsea/msigdb/; see also Methods and SI) to investigate overlap with published gene sets (Supplementary Table S4).
We sought to investigate whether STAMM is able to identify genes that are known to play a role in reprogramming and further to identify new insights, including potentially novel players in the process, from our genome-wide analysis. We focused on a selected group of the top-5% high ranked switch and pulse genes (see Supplementary Fig. S7); these are shown in Figure 4, which also highlights micro-RNAs (MIRs) and transcription factors whose DNA binding motifs and known occupancy of promoters through ChIP data show targeting of those genes. Switches identify genes which can be turned on to drive reprogramming via the induced factors (OSKM), and genes which can serve as reprogramming factors. The histone demethylase Utx, for instance, turns on quickly in S2 and remains active: it has been recently shown to be indispensable for reprogramming33. The S3 switch contains Wdr5, which mediates reprogramming34. Expression of Gnl3 is specific to the S4 switch and promotes reprogramming35. Myc and MIR-302 regulate subsets of genes in these switches and are have been shown to promote reprogramming36.
Figure 4. Cell state markers and the molecular circuitry of reprogramming.

Application of our model to data from the the reprogramming system of Samavarchi-Tehrani et al.18 yields a gene ranking that, unlike conventional differential expression and related analyses, is based on cell state-specific estimates. We used our approach to identify state marker genes and to explore the molecular circuitry underpinning reprogramming. Five state-resolved expression profiles are shown: “switches” occur when a gene is expressed in a particular cell state and remains on in subsequent states, while “pulses” occur when a gene is switched on in only one state, and is off in all other states. Estimated state-specific expression levels were used to rank genes in each profile; genes shown are selected from the top 5%, genome-wide, under each profile. Highlighted are transcription factors whose DNA binding motifs and known occupancy of promoters through ChIP data show targeting of genes in each switch or pulse, as well as micro-RNAs (MIRs) targeting the genes.
Intriguingly, several commonly accepted pluripotent marker genes, such as Nanog and Sall4 are already activated in the S3 switch, despite the fact that cells in S3 are on average 15 days away from the final state. In contrast, Klf5 and Sox2 are highly specific to S4 alone (Fig. 4). These observations are supported by recent independent single-cell experiments that indicate activation of Klf5 and Sox2 are late steps in reprogramming19. Other factors specifically marking iPS cells in these single-cell studies were Dppa4, Utf1 and Esrrb; all of these genes are highly specific to S4 in our analysis. The single cell analysis also indicated that Gdf3 was activated in partially reprogrammed cells while Sox2 was not, consistent with the putative pre-pluripotent nature of that state and with our observation that Gdf3 is switched on in S3. Other poor markers of iPS cells in the single-cell assays were Sall4 and Kdm1 which are promiscuously turned on in S3 or earlier.
Pulses identify genes that we hypothesize must be tightly controlled during reprogramming, as they are turned on in only one state. Indeed, constitutive over-expression of these factors has a complex relationship with reprogramming. Notably the Tcf3 binding motif is highly represented in the S3 pulse group (p = 1.83 × 10−12, hypergeometric test), and expression of these genes is lost in S4. Tcf3 forms an interconnected autoregulatory loop with Oct4, Sox2, and Nanog in pluripotent stem cells, and is mainly in a repressive complex promoting differentiation, although some Tcf3 associates with β-catenin to activate target genes and promote pluripotency37. Tcf3 deletion increases reprogramming38 and Wnt signaling is known to accelerate reprogramming39. Our results suggest that Tcf3 needs to be recruited to form a repressive complex on the differentiation genes in S3, so that proper programming of the pluripotent state can occur. The S3 pulse further reveals that other master regulators of differentiated lineages - Pax4, Zeb1, Foxo4, and Sox11 - also need to be turned off during this final transition. Several annotated sets of known pluripotency genes have significant overlap with the top 5% genes of the S4 switch (p < 10−15; see Methods and Supplementary Table S4 for gene lists). The top 5% genes of the S3 switch also have significant overlap with pluripotency gene lists (p ≤ 1.35 × 10−6) indicating that S3 may represent a pre-pluripotent state.
STAMM differs fundamentally from existing gene-ranking approaches because it estimates expression profiles that are specific to cell states. Thus, although existing gene ranking approaches based on differential expression are certainly informative, STAMM offers complementary insights, rooted in a state-specific view. Indeed, genes highly ranked under STAMM that are implicated in reprogramming and shown in Figure 4 are not highly ranked under conventional differential expression or temporal change criteria (Supplementary Fig. S7). STAMM can also be extended to identify pairs of genes that can jointly act as state markers (see SI and Supplementary Fig. S9).
Testing model predictions against single-cell data
The foregoing results were obtained from analysis of homogenate time-course data only (the microarray data due to Samavarchi-Tehrani et al.18). To test the ability of our approach to uncover information on cell states we compared results with recent independent single-cell datasets on reprogramming.
We focused first on a single-cell mRNA expression dataset due to Buganim et al.19 that considered a different secondary MEF system under reprogramming by the transduction of Oct4, Sox2, Klf4, and cMyc. The data were obtained using the Fluidigm assay, and included single-cell gene expression data for 48 genes in up to 96 cells, in a variety of populations, ranging from starting MEFs, to cells at the 2-to-6 days stage of reprogramming, to iPS cells (see Fig. 5).
Figure 5. Testing model predictions against independent single-cell data.

Results obtained from application of our model to the microarray time-course data were tested against independent single-cell gene expression data from a secondary reprogramming system due to Buganim et al.19. (a), Number of clusters in the single-cell data. A score called the Bayesian Information Criterion (BIC) is shown as a function of number of clusters (see text for details). (b), Matching individual cells to predicted states. Single cells within different experimental populations in Buganim et al. were assigned to each of the four states in our model (all parameters were estimated using the microarray data only). Matching was done by similarity between single-cell expression profiles and the state signatures. The heatmap shows the fraction of cells in each experimental population that were assigned to each state. Assignments for different experiments show clear preference for certain states, in a manner consistent with the nature of the states, and in line with a progression towards iPS via the intermediate states defined by our model. For example, the MEF populations have a marked peak in state 1, while cell lines at the 2-to-6 days stage of reprogramming have an heterogeneous population spread over the first three states, with a peak at state 3. Finally, the populations of dox-independent colonies and iPS cells belong to state 3 and 4, peaking respectively at 3 and 4.
Since these data are single-cell readouts, clustering of the data can be used to identify cell states that are distinct with respect to expression patterns, as individual cells belonging to the same state with similar single-cell expression profiles should lie close to each other in gene expression space. We carried out a cluster analysis of the data (using a widely-used multi-variate clustering tool called mclust, see SI) and selected the number of clusters using a score known as the Bayesian Information Criterion (BIC). Although single cell technologies are still not fully mature and remain affected by relevant experimental errors, we find that the BIC from the single-cell data (see Fig. 5a) has a large increase above two clusters but plateaus above three clusters. This does not support a single-step (two clusters) model but is consistent with the existence of one or two intermediate states, in line with our predictions using independent microarray data.
Next, we asked whether the state-specific expression profiles we identified in the four-state model (as estimated from microarray time-course data only) were consistent with single-cell expression profiles. To this end, we computed the distance of the gene expression profile of each single cell in the Buganim et al. data from each of the four state signatures that we estimated, and assigned each individual cell to the state that was closest to it in expression space. The fraction of single cells assigned to each state in the various experimental populations is shown in Figure 5b. In contrast to a random assignment that would populate each state roughly equally, we find that specific states are highly represented in specific cell populations. The MEFs show a clear population peak in state 1, while MEFs at the 2-to-6 days stage of reprogramming have an heterogeneous population spread over the first three states, with a peak at state 3. Finally, dox-independent colonies and iPS cells have a population distributed over state 3 and 4, peaking respectively at 3 and 4. The state assignment is consistent with the nature of the states and the progression through the reprogramming process. Thus we find that individual cells from an independent single-cell study of a different secondary MEF system project consistently onto the four states we identified using microarray time-course data only.
We also briefly discuss three very recent comprehensive single-cell studies of secondary reprogramming systems20,21,22. A key result in those studies is the discovery of two intermediate “transcriptional waves” that occur in the first 12 days of reprogramming and mark the transition from initial MEF to two subsequent “stages”, which are later followed by a “DNA methylation wave” when cells acquire stable pluripotency. These observations strongly support our independent, model-based prediction of two intermediate states and their appearance within the first 10 days of reprogramming, followed by the establishment of a fourth (iPSC-like) state. Thus, several recent single-cell studies appear to support the results we obtain from application of STAMM to homogenate time-course data.
Application to other systems
While note must be taken of the diversity of different reprogramming systems, a four-state model also fits data from the primary Mikkelsen et al.40 system (see Supplementary Methods and Supplementary Fig. S8 for results), with severals genes, including Cdh1, Cdh2, Zeb1 and Nanog having similar state profiles, although others are dissimilar, such as Zeb2, Epcam, Gata4 and Thy1. This highlights the possible existence of common reprogramming mechanisms between primary and secondary systems. However, the small number of time points in the Mikkelsen data currently preclude a fuller comparison of commonalities and differences between state transitions and dynamics in the two systems. The model proposed in Hanna et al. for a B-cell based system17 can be recovered as a simpler case of the model presented here, with exactly two states (one transition). Such a two-state model does not give a good fit to the Samavarchi-Tehrani data (see Supplementary Fig. S3a), indicating that intermediate states are needed to explain the dynamics seen in the secondary MEF system. Also in a single-cell mRNA-seq dataset due to Tang et al.2 obtained during the derivation of embryonic stem cells from the inner cell mass, we found that the number of states seen for gene pairs in the single-cell data mirrors the corresponding discriminatory scores obtained from our analysis of the Samavarchi-Tehrani reprogramming data (see Supplementary Methods and Supplementary Fig. S9).
Discussion
We put forward a new stochastic model for the investigation of cellular transition processes. We showed how the model can be used to explore transition processes in a genome-wide fashion using conventional, population-averaged time-course data, in this way providing new insights as well as detailed guidance for single-cell studies with smaller, selected sets of genes (or other molecular readouts). Application of our approach to stem-cell reprogramming recapitulated a wealth of known biology. Furthermore, the analysis shed new light on the process, including several insights that we found to be consistent with recent single-cell studies and novel hypotheses that could be tested in future experiments. The approach could be similarly applied to differentiation, development or oncogenic transformation, to provide insights and hypotheses concerning cell states visited during these processes.
Our model provides a bioinformatics tool as well as a conceptual framework that should be useful in helping to better understand cell states and their transitions. However, several questions concerning the meaning and interpretation of cell states remain open and were not addressed in our analysis. Our model seeks to identify states that are distinct with respect to the molecular data type used for analysis (here gene expression), but cannot itself determine whether such differences correspond to states that are distinct in a deeper sense, for example in terms of a specific, discrete phenotype of interest. This limitation is analogous to that faced by cluster analysis: identifying groups of samples (e.g. cells, genes or patients) that are distinct with respect to certain measured variables may or may not correspond to a specific functional difference of interest. Our approach provides detailed information regarding putative cell states; such information should be regarded as providing testable hypotheses and guidance for the design of follow-up experiments.
In the case of stem-cell reprogramming we concluded that a four state model can be used to explain transcriptional dynamics observed during reprogramming of MEFs into iPSCs in a secondary system18 (see Fig. 2). This led to detailed molecular profiles of two new, intermediate single-cell states. Our results suggested that the transition between the second intermediate (S3) to the final reprogrammed state (S4) is the process bottleneck. This multiple-state model also explains the variance (over weeks) of subclone reprogramming times, consistent with experimental observations16.
Our results show how state transitions in reprogramming involve global transcriptional changes (see Fig. 3). As one part of these global changes, we observed a mesenchymal-to-epithelial transition that takes place between states S2 and S3, in agreement with previous experimental observations18. Interestingly, our analysis contradicts the view that genes such as Nanog and Sall4 are true markers of pluripotency, since these genes are already expressed in the penultimate state S3 which is 15 days away from the final state S4. Strikingly, this observation is supported by recent single-cell data19.
Output from our model provides immediate and detailed guidance for the design of future single-cell experiments. We showed how state-resolved analysis using STAMM complements and extends available analyses and tools, providing new insights into the molecular circuitry of reprogramming, including lists of single-cell state specific “switch” and “pulse” genes (see Fig. 4). Since the STAMM gene lists and markers were obtained via an unbiased, genome-wide analysis, highly-ranked genes may represent important candidates for future investigation. Moreover, since the model yields mean transition times between states, our results suggest specific times at which to optimally isolate intermediate states. In the reprogramming context, such insights can also help to design strategies to optimally accelerate the transitions.
We tested our model predictions against recent single cell data from a different secondary MEF system19. This analysis suggests that a four state model is consistent with such data, and moreover that individual cells project in a consistent manner onto the states that we identified using the Samavarchi-Tehrani microarray time-course data18 (see Fig. 5). Our predictions are also consistent with, and allow the interpretion of, a number of very recent comprehensive studies20,21,22 which revealed intermediate transcriptional waves in iPSC reprogramming. Overall, the available single-cell experimental data support the picture of the structure of reprogramming that emerges from our genome-wide analysis.
Large-scale epigenetic changes are also observed upon reprogramming11,41. The method we propose should be applicable to time-varying epigenetic data42 to directly identify state-specific epigenetic signatures along with expression patterns. The model we propose and its future extensions can provide a starting point for a comprehensive interpretation of the next-generation of single-cell data on reprogramming and other cellular transition processes in development, differentiation and disease.
Methods
Here, we briefly describe Methods used in the paper. Further information, including full technical details, appear in SI.
The model
Our model describes state changes at the single-cell level using a latent continuous-time Markov process. Here, we restricted attention to forward-only state transitions, such that transitions between n states indexed by i are parameterized by (n − 1) transition rates wi,i+1 (collectively denoted by w). Our general approach could in principle be extended to more general transition topologies, but depending on the specific application and model constraints further data could be required to ensure identifiability. The rates wi,i+1 fully determine the dynamics (assuming rates of cell division and death are independent of state). We assume that the initial population is homogeneous (all cells in an initial state). Under these model assumptions, the master equation for the latent Markov process can be solved fully (see SI for details) to give the probabilities pi(t) that an individual cell is in state i at time t (Fig. 3c).
Each state in the model has state-specific parameters βij that represent the mean expression level for gene j in single-cell state i (“state-specific signatures”). Each cell in the population is associated with its own latent Markov process; we assume the cell-specific processes are stochastically independent. We link the single-cell latent processes to population-level observables by aggregating over individual cells. At any given time t each individual cell in the population is in one of the n states, with the probability of being in state i given by the solution pi(t) to the master equation. For a large number of cells, the fraction of cells in each state i at time t is therefore given by pi(t). Population average expression of gene j at time t can now be written in terms of expression per state weighted by the fraction of cells in each state at the given time; this yields the expression shown in the Introduction and reproduced below
 |
where the dependence of pi(t) on transition rates w is made explicit. The above expression links the parameters of the latent processes at the single-cell level to population-level observables xj(t). Making the noise model explicit we arrive at
 |
where 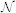 denotes a Normal density and
denotes a Normal density and 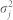 denotes gene-specific variance. This latter expression gives the likelihood. We estimated the parameters βij and wi,i+1 using a
denotes gene-specific variance. This latter expression gives the likelihood. We estimated the parameters βij and wi,i+1 using a 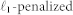 estimator related to the maximum a posteriori estimator for the Bayesian formulation below (see SI for details).
estimator related to the maximum a posteriori estimator for the Bayesian formulation below (see SI for details).
For complex models with many parameters, it is important to check stability of estimation to guard against artifactual results. To check stability of the penalized estimator, we re-estimated parameters following perturbation of the data and compared with estimates obtained from the original data. We perturbed the data in two ways: a) adding Gaussian noise and b) removing an entire time point (see Supplementary Fig. S5 and below). We found that results reported were robust to such perturbations, suggesting that overall estimator variance is well controlled. We also carried out a re-analysis under permutation of the temporal order of the data (Supplementary Fig. S6). We found that both model fit and distinctness of state signatures were systematically worse under such temporal permutation, suggesting that our simple model of transition dynamics captures real temporal structure in the data.
STAMM software, implementing the above model and associated estimators and gene ranking tools (see below) is provided as part of the Supplementary Information and at mukherjeelab.nki.nl/CODE/STAMM.zip.
Model selection
We used a Bayesian model selection procedure to complement the model selection heuristics reported in the Main Text. A full description of the Bayesian formulation, including details of the Markov chain Monte Carlo (MCMC) appear in SI. Let Mn denote the model with n states and y = {yj} denote observed data for all genes. Taking a flat prior over models P(Mn) ∝ 1, the posterior probability over models is P(Mn|y) ∝ p(y|Mn). The marginal likelihood, p(y|Mn), is obtained by integrating out all model parameters (β's, w's and σ's) from the likelihood corresponding to the noise model in Eq. (3) above. This gives a score for each model that takes account of both fit-to-data and model complexity that is then normalized to give the posterior probability over number of states. We further investigated model selection by applying it to the data with time points permuted (Supplementary Fig. S6); while application to the original data showed clear evidence of intermediate states, this is completely lost under temporal permutation, suggesting that the evidence for intermediate states is rooted in real temporal structure in the data.
Gene ranking
STAMM ranks genes by using estimated state-specific signatures β. For each state i and gene j we call the score 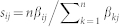 the “state-specific score”, since it indicates state-specific expression relative to all states for that gene (up-regulated genes in a given state are the top scoring genes while down-regulated genes are the lowest scoring). Gene lists for Fig. 4 were then obtained as follows. For profiles with expression switched on in one state only (S2 pulse, S3 pulse and S4 switch) genes were ranked under the respective state-specific scores sij. For the S2 and S3 switch profiles, in which genes are switched on in multiple states, rankings were carried out with respect to fold change in state-specific expression β before and after a switch: this was done using the minimum fold change between β's for any state after the switch with respect to any state before the switch.
the “state-specific score”, since it indicates state-specific expression relative to all states for that gene (up-regulated genes in a given state are the top scoring genes while down-regulated genes are the lowest scoring). Gene lists for Fig. 4 were then obtained as follows. For profiles with expression switched on in one state only (S2 pulse, S3 pulse and S4 switch) genes were ranked under the respective state-specific scores sij. For the S2 and S3 switch profiles, in which genes are switched on in multiple states, rankings were carried out with respect to fold change in state-specific expression β before and after a switch: this was done using the minimum fold change between β's for any state after the switch with respect to any state before the switch.
Gene ontology (GO) analysis
State-specific gene lists were obtained from STAMM rankings as described above. To form a list of genes up-regulated in state i, we retained those genes j with pulse score sij ≥ 2, and to form a list of genes down-regulated we retained those genes j having sij ≤ 0.5. GO analysis was performed using the Cytoscape plugin BINGO43. In Supplementary Fig. S4, for clarity, only the significant GO terms with number of descendants between 200 and 800 are shown.
Author Contributions
J.W.A., A.A.R. and C.J.O. did computational analyses; they and K.S. performed research; J.W.A., K.S., C.J.O., R.J., M.N. and S.M. wrote the manuscript; M.N. and S.M. conceived and led the project.
Supplementary Material
Table S2
Table S3
Table S4
Supplementary Information
Acknowledgments
This work was supported by UK EPSRC EP/E501311/1 and the Cancer Systems Biology Center grant from the Netherlands Organisation for Scientific Research.
References
- Evans M. J. & Kaufman M. H. Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156 (1981). [DOI] [PubMed] [Google Scholar]
- Tang F. et al. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-seq analysis. Cell Stem Cell 6, 468–478 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T. et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z. P. et al. Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caiazzo M. et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227 (2011). [DOI] [PubMed] [Google Scholar]
- Ieda M. et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efe J. A. et al. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat. Cell Biol. 13, 215–222 (2011). [DOI] [PubMed] [Google Scholar]
- Huang P. et al. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 475, 386–389 (2011). [DOI] [PubMed] [Google Scholar]
- Sekiya S. & Suzuki A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 475, 390–393 (2011). [DOI] [PubMed] [Google Scholar]
- Takahashi K. & Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006). [DOI] [PubMed] [Google Scholar]
- Hanna J. H., Saha K. & Jaenisch R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell 143, 508–525 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K. & Yamanaka S. Induced pluripotent stem cells: opportunities and challenges. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 2198–2207 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmut I., Sullivan G. & Chambers I. The evolving biology of cell reprogramming. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 2183–2197 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel G. Diseases in a dish take off. Science 330, 1172–1173 (2010). [DOI] [PubMed] [Google Scholar]
- Wernig M. et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318–324 (2007). [DOI] [PubMed] [Google Scholar]
- Jaenisch R. & Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 132, 567–582 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J. et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 462, 595–601 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P. et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7, 64–77 (2010). [DOI] [PubMed] [Google Scholar]
- Buganim Y. et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150, 1209–1222 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo J. et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 151, 1617–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson J. et al. Highly coordinated proteome dynamics during reprogramming of somatic cells to pluripotency. Cell Report 2, 1579–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley J. et al. High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature 499, 88–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai Y. & Speed T. A multivariate empirical Bayes statistic for replicated microarray time course data. Ann. Stat. 34, 2387–2412 (2006). [Google Scholar]
- Kalaitzis A. & Lawrence N. A simple approach to ranking differentially expressed gene expression time courses through gaussian process regression. BMC Bioinf. 12, 180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard N., Holmes C., Stephens D., Hand D. & Dimopoulos G. Bayesian coclustering of Anopheles gene expression time series: study of immune defense response to multiple experimental challenges. Proc. Natl. Acad. Sc. USA 102, 16939–16944 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Lane T., Allen C., Aragon A. & Werner-Washburne M. A hidden-state Markov model for cell population deconvolution. J. Comp. Bio. 13, 1749–74 (2006). [DOI] [PubMed] [Google Scholar]
- Bar-Joseph Z., Farkash S., Gifford D., Simon I. & Rosenfeld R. Deconvolving cell cycle expression data with complementary information. Bioinformatics 20, i23–i30 (2004). [DOI] [PubMed] [Google Scholar]
- Bar-Joseph Z. et al. Genome-wide transcriptional analysis of the human cell cycle identifies genes differentially regulated in normal and cancer cells. Proc. Natl. Acad. Sci. USA 105, 955–60 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal-Gaskins D., Ash J. & Crosson S. Model-based deconvolution of cell cycle time-series data reveals gene expression details at high resolution. PLoS Comput. Biol. 5, e1000460 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando D. et al. A probabilistic model for cell cycle distributions in synchrony experiments. Cell Cycle 6, 478–488 (2007). [DOI] [PubMed] [Google Scholar]
- Rowicka M., Kudlicki A., Tu B. P. & Otwinowski Z. High-resolution timing of cell cycle-regulated gene expression. Proc. Natl. Acad. Sci. USA 104, 16892–16897 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa I., Roepcke S., Hafemeister C. & Schliep A. Inferring differentiation pathways from gene expression. Bioinformatics 24, i156–64 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A. et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature 488, 409–413 (2012). [DOI] [PubMed] [Google Scholar]
- Ang Y. et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell 145, 183–197 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J. & Bishop J. M. Nucleostemin maintains self-renewal of embryonic stem cells and promotes reprogramming of somatic cells to pluripotency. J. Cell Biol. 197, 731–745 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanyam D. et al. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 29, 443–448 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole M., Johnstone S., Newman J., Kagey M. & Young R. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev. 22, 746–755 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluis F. et al. T-cell factor 3 (Tcf3) deletion increases somatic cell reprogramming by inducing epigenome modifications. Proc. Natl. Acad. Sc. USA 108, 11912–917 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A. et al. Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell 3, 132–135 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen T. S. et al. Dissecting direct reprogramming through integrative genomic analysis. Nature 454, 49–55 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koche R. P. et al. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell 8, 96–105 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal R. B., Henikoff J. G. & Henikoff S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328, 1161–1164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S., Heymans K. & Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in biological networks. Bioinformatics 21, 3448–3449 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2
Table S3
Table S4
Supplementary Information