

Abstract
Human rhinovirus (HRV) is a major causative agent of the common cold, and thus has several important health implications. As a member of the picornavirus family, HRV has a small genomic RNA that utilizes several host cell proteins for RNA replication. Host proteins poly(rC) binding protein 2 (PCBP2) and polypyrimidine tract binding protein (PTB) are cleaved by a viral proteinase during the course of infection by the related picornavirus, poliovirus. The cleavage of PCBP2 and PTB inhibits poliovirus translation and has been proposed to mediate a switch in poliovirus template usage from translation to RNA replication. HRV RNA replication also requires a switch in template usage from translation to RNA replication; however, the mechanism is not yet known. We demonstrate that PCBP2 and PTB are differentially cleaved during HRV infection in different cell lines, suggesting that HRV utilizes a mechanism distinct from PCBP2 or PTB cleavage to mediate a switch in template usage.
Keywords: human rhinovirus, host cell protein cleavage, PCBP2, PTB, coxsackievirus, poliovirus, picornavirus, viral proteinases
Introduction
Human rhinovirus (HRV) is the most prevalent causative agent of the common cold, with 80% of common cold cases caused by HRV (Arruda et al., 1997). Although the common cold is not known to cause a severe disease phenotype, it costs billions of dollars per year and contributes to lost wages (McErlean et al., 2008). Additionally, HRV infections can be severe in individuals with chronic lung illnesses such as COPD, asthma, or allergies. HRV is a major cause of asthma exacerbations, causing up to 70% of asthma exacerbations in children and can increase the rate of morbidity from decreased lung function due to asthma (Johnston et al., 1996; Rakes et al., 1999). Therefore, a better understanding of HRV will allow for antiviral drugs to be developed to help those with chronic lung illnesses. A potential impediment to development of an antiviral drug is that there are over 100 antigenically distinct serotypes of HRV. These serotypes can be divided into three groups, group A, group B, or group C. Additionally, serotypes are grouped based on the receptor used for host cell entry. Most HRV types use ICAM-I as a receptor and are known as the major group serotypes, while the remaining minor group types utilize a low-density lipoprotein (LDL) receptor.
HRV is a member of the Picornaviridae family of viruses, which also includes coxsackievirus, enterovirus 71, and poliovirus, the prototypic picornavirus, among others. Picornaviruses are a family of small, single-stranded, positive-sense RNA viruses that replicate in the cytoplasm of infected host cells. Unlike cellular mRNAs, picornavirus genomes lack a cap on the 5′ end and have a highly structured 5′ noncoding region (NCR) that precludes ribosome scanning (Fernandez-Munoz and Darnell, 1976; Fitzgerald and Semler, 2009; Hewlett et al., 1976; Nomoto et al., 1976). Therefore, translation is initiated in a cap-independent manner. The 5′ NCR is composed of six stem-loop structures, where stem-loops II – VI make up the internal ribosome entry site (IRES) that mediates cap-independent, IRES-driven translation (Belsham and Sonenberg, 1996; Borman and Jackson, 1992; Jang et al., 1988; Pelletier and Sonenberg, 1988). As a positive-sense RNA virus, the genome can serve as a template for both viral translation and RNA replication. Therefore, upon entry into the host cell, the viral genome is first used as a template for translation into a single polyprotein that is subsequently processed by viral proteinases, including 3CD, to produce viral proteins. Picornaviruses utilize host cell proteins, referred to as IRES trans-acting factors (ITAFs), to mediate non-canonical translation. Several host proteins have been shown to be important for poliovirus or HRV translation, including poly(rC) binding protein 2 (PCBP2), polypyrimidine tract binding protein (PTB), lupus autoantigen (La), and upstream of N-ras (unr) (Blyn et al., 1996; Blyn et al., 1997; Gamarnik and Andino, 1997; Gosert et al., 2000; Hellen et al., 1993; Meerovitch et al., 1993; Sawicka et al., 2008; Svitkin et al., 1994).
Following translation, the genomic RNA can be used as a template for synthesis of negative-strand RNA, followed by the subsequent synthesis of positive-strand RNA for further rounds of translation and RNA replication, or packaging into progeny virions. Previous studies have shown that although the viral genome can be used as a template for both translation and RNA replication, RNA that is actively being translated cannot function as a template for RNA replication (Barton et al., 1999; Gamarnik and Andino, 1998). This suggests that there must be a mechanism to mediate a switch in template usage from translation to RNA replication.
Multiple candidates have been proposed to play a role in the switch from viral translation to RNA synthesis, including PCBP2 and PTB. PCBP2 binds to stem-loop IV of the poliovirus IRES to form a complex that is required for translation of the polyprotein (Blyn et al., 1996; Blyn et al., 1997; Gamarnik and Andino, 1997). Additionally, PCBP2 binds to a stem-loop I structure upstream of the IRES and forms a ternary complex with viral proteinase 3CD that is required for initiation of negative-strand RNA synthesis (Andino et al., 1993; Andino et al., 1990; Gamarnik and Andino, 1997, 1998; Parsley et al., 1997). At peak times of viral RNA synthesis, PCBP2 is cleaved by poliovirus 3CD proteinase, disrupting the interaction of PCBP2 with stem-loop IV and inhibiting translation. However, the cleaved form of PCBP2 is still able to bind to stem-loop I and form a functional ternary complex, remaining active in RNA replication (Perera et al., 2007). Additional studies have shown that PCBP2 is also cleaved during coxsackievirus infection and can be cleaved in vitro by HRV type 16 (HRV16) 3CD proteinase (A. J. Chase, S. Daijogo, and B. L. Semler, submitted for publication). Therefore, these data suggest that cleavage of PCBP2 by viral 3CD proteinase could be important for mediating a switch in template usage for multiple picornaviruses.
PTB is an additional ITAF that stimulates poliovirus and HRV translation (Gosert et al., 2000; Hellen et al., 1993; Hunt et al., 1999; Hunt and Jackson, 1999; Sawicka et al., 2008). It has been shown that, like PCBP2, PTB is also cleaved during poliovirus or HRV type 14 (HRV14) infection, and the cleavage is mediated by the viral proteinase 3C (Back et al., 2002). PTB is a member of the hnRNP family and shuttles from the nucleus to the cytoplasm (Michael et al., 1995). Cleavage of PTB results in the redistribution of the cleaved fragments to the cytoplasm and the inhibition of poliovirus translation. While the mechanism of how PTB cleavage inhibits translation is not known, it has been proposed that the cleaved fragments bind viral RNA to preclude binding of full-length, functional PTB (Back et al., 2002). Thus, it has been proposed that cleavage of cellular protein PTB by viral proteinase 3C could also play a role in mediating a switch in template usage.
Cleavage of PCBP2 is required for efficient poliovirus RNA replication, and plays an important role in mediating a switch in template usage from translation to RNA replication (A. J. Chase, S. Daijogo, and B. L. Semler, submitted for publication). Since PCBP2 is cleaved by HRV16 viral proteinase 3CD, this suggests that cleavage of PCBP2 could also be important for HRV RNA replication and the switch in template usage. However, the role host cell proteins play in HRV infection and the importance of PCBP2 or PTB cleavage during HRV infection is not yet understood. To more fully explore these mechanisms, PCBP2 cleavage was investigated during infection with three different HRV serotypes, types 14, 16, and 1A, which includes serotypes from groups A or B and serotypes that use either the major or minor group receptor. HRV14 and HRV16 utilize the major ICAM-1 receptor, while HRV1A uses the minor group LDL receptor. In addition, HRV14 is grouped into group B, while HRV16 and HRV1A are group A serotypes. We found that PCBP2 and PTB are cleaved during infection of HeLa cells by all three HRV serotypes investigated; however, neither protein was cleaved during infection of a human lung fibroblast cell line, suggesting differential cleavage of host cell proteins. In addition, unlike poliovirus, PTB and PCBP2 cleavage does not appear to be required for efficient human rhinovirus infection.
Materials and Methods
HRV16 3CD Construct
Synthetic oligonucleotide primers used were: HRV16-3CD-Nde(+) (5′ agccatatgggtccagaagaagaattt 3′), HRV16-3CD-BamHI(−) (5′ gccggatccctattaagaatttttcatacatt 3′), HRV16-3CDμ10(+) (5′ atcatacttcactgaacaagcagcccaaattcaaatctctaaac 3′), HRV16-3CDμ10(−) (5′ gtttagagatttgaatttgggctgcttgttcagtgaagtatgat 3′). Synthetic oligonucleotide primers HRV16-3CD-Nde (+) and HRV16-3CD-BamHI(−) were used to generate PCR fragments containing HRV16 amino acids 5155 to 7085, flanked by NdeI or BamHI upstream or downstream, respectively. The restriction sites were then used to clone HRV16 3CD into the pET15b vector through ligation. Site-directed mutagenesis of the newly synthesized pET15b HRV16 3CD plasmid was then used as a template for site-directed mutagenesis to alter the 3C/D self-cleavage site such that recombinant 3CD could not be self-cleaved into 3C and 3D. The synthetic oligonucleotide primers used were HRV16-3CDμ10(+) and the reverse complement, HRV16-3CDμ10(−). The resulting plasmid, pET15b HRV16 3CDμ10, was used for protein expression.
Protein purification
Purification of hexahistidine-tagged pET22b PCBP2 was carried out as previously described (Parsley et al., 1997; Perera et al., 2007). Briefly, PCBP2 was expressed in BL21DE3 (Rosetta) cells. The cells were grown at 37°C to an A600 of 0.6 and then induced with isopropyl-β-D-thiogalactopyranoside (IPTG; 1 mM concentration) for 3 hours at 25°C. Cells were resuspended in lysozyme buffer without lysozyme (10 mM EDTA, 25 mM Tris-HCl [pH 8.0], 4.5% glucose) and lysed by sonication, 15 seconds followed by 1 minute on ice repeated five times. The soluble fraction following sonication was precipitated with ammonium sulfate (20% wt/vol). The resulting pellet fraction was dialyzed overnight in I-60 buffer (20 mM Tris-HCl [pH 8.0], 250 mM NaCl, 60 mM imidazole, 10% glycerol) with 0.5% NP-40. The fraction was then subjected to Ni2+ ion-based affinity chromatography (GE Healthcare). The column was washed with I-60 buffer, and the protein was eluted with I-200 buffer (20 mM Tris-HCl [pH 8.0], 250 mM NaCl, 200 mM imidazole, 10% glycerol).
Hexahistidine-tagged 3CD viral proteinase (poliovirus or HRV16) was purified as previously described, with minor modifications (Parsley et al., 1999; Parsley et al., 1997). Briefly, 3CD was expressed in BL21DE3 (Rosetta) cells, and the cells were grown at 37°C to an A600 of 0.4. The protein expression was induced by the addition of IPTG (1 mM) for 3 hours at 25°C. Cells were resuspended in Buffer A (20 mM Tris-HCl [pH 8.0], 25 mM NaCl, 5% glycerol, 1 mM DTT, added fresh) and lysed by one passage through a French pressure cell (Amicon) at 8,000 psi. The lysate was then subjected to centrifugation for 20 minutes at 13,800×g using a Beckman Coulter centrifuge and JA-17 rotor and the resulting supernatant was discarded. The pellet was washed three times in Buffer A, the last wash without DTT. To extract protein from the pellet fraction, the pellet was resuspended in high salt I-30 buffer (20 mM Tris-HCl [pH 8.0], 1M NaCl, 30 mM imidazole, 10% glycerol) and incubated on ice for 30 minutes. The resuspended pellet was then subjected to centrifugation for 20 minutes at 13,800×g as above. The resulting supernatant was subjected to Ni2+ ion-based affinity chromatography (GE Healthcare). The column was washed with high salt I-30 buffer, and the protein was eluted with high salt I-200 buffer (20 mM Tris-HCl [pH 8.0], 1M NaCl, 200 mM imidazole, 10% glycerol).
In vitro cleavage assay
The in vitro cleavage assay was carried out as previously described, with some minor changes (Perera et al., 2007). Briefly, recombinant purified PCBP2 and poliovirus or HRV16 3CD proteinase were incubated at a 1:1 molar ratio in cleavage buffer (20 mM HEPES [pH 7.4], 1 mM DTT, 150 mM KOAc) for 1 to 6 hours at 30°C. The cleavage products were analyzed by SDS-PAGE and Western blot.
Western blot analysis
Following SDS-PAGE, proteins were electroblotted to a PVDF membrane. The membranes were blocked in 5% milk in phosphate buffered saline (PBS) with 0.1% Tween-20 for 1 hour at room temperature. Mouse monoclonal antibody against PCBP2 (1:2000 in PBS with 0.1% Tween-20), as described in Sean et al., was used to detect full-length and cleaved forms of PCBP2 (Sean et al., 2008). To detect PTB protein, mouse monoclonal antibody against PTB (1:2000 in PBS with 0.1% Tween-20) was used. Poliovirus viral proteins were analyzed with a rabbit polyclonal antibody against poliovirus 3CD proteinase (Cathcart et al., 2013). HRV16 viral proteins were detected with a rabbit polyclonal antibody against HRV16 3D RNA-dependent RNA polymerase. A synthetic HRV16 3D peptide was generated and used to induce antibody production in rabbits (Bethyl). The PVDF membrane was incubated with anti-PCBP2, anti-PTB, anti-poliovirus 3CD, or anti-HRV16 3D antibody (1:2000) for 1 hour. This was followed by incubation with goat anti-mouse horseradish peroxidase (HRP) secondary antibody (Millipore) or goat anti-rabbit HRP antibody (Millipore) for 1 hour. Protein bands were visualized using chemiluminescence (Thermo Scientific).
Virus infection and cell culture
HeLa cells were grown as monolayers in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 8% newborn calf serum (NCS). WisL human fetal lung fibroblast cells (Bochkov et al., 2011) were grown as monolayers in DMEM supplemented with 10% fetal bovine serum (FBS). Cells (HeLa or WisL) were infected with poliovirus, coxsackievirus B3 (CVB3), HRV14, HRV16, or HRV1A at a multiplicity of infection (MOI) of 20, and adsorption was carried out at room temperature for 60 minutes for HRV or 30 minutes for poliovirus and coxsackievirus. HRV1A virus stock was kindly provided by Dr. Yury Bochkov (University of Wisconsin – Madison). DMEM with 8% NCS (HeLa cells) or 10% FBS (WisL cells) was added, and the cells were incubated at 34°C or 37°C, as specified, until the indicated times. At specific times post-infection, cells were washed twice with phosphate buffered saline (PBS), scraped, and collected to generate cytoplasmic lysates. Cells were lysed in NP-40 lysis buffer (50 mM Tris-HCl [pH 8.0], 5 nM EDTA [pH 8.0], 150 mM NaCl, 0.5% NP-40) on ice for 30 minutes. After incubation on ice, cells debris was pelleted, the resulting supernatant was collected, and total protein concentration was determined by Bradford assay. Total protein (100 μg) from cytoplasmic lysates were analyzed by Western blot. Where indicated, four times total protein concentration (400 ug) or 2.5 times total protein concentration (250 ug) was analyzed.
For single cycle growth analysis, HeLa cells or WisL cells were infected with HRV16 at an MOI of 20 as above. Cells were washed three times with phosphate buffered saline (PBS) following the adsorption prior to adding media. At designated times post-infection (0–12 hours), supernatant and cells were collected and subjected to four rounds of freeze-thawing. To determine the titer of virus at each time point, the collected virus was then diluted in serum-free media and the dilutions were used to infect HeLa cell monolayers (as above). Following adsorption, the cells were overlaid with DMEM plus 8% NCS and 0.45% agarose. Seven days after infection, the cells were treated with 10% trichloroacetic acid (TCA) and stained with crystal violet (0.1% crystal violet, 25% ethanol). The plaques were counted to determine the virus titer.
Results
Cleavage of purified recombinant PCBP2 in vitro by poliovirus or HRV 3CD proteinases
Previous studies have shown that PCPB2 is cleaved by poliovirus proteinase 3C or precursor 3CD (Perera et al., 2007). To determine if PCBP2 is also cleaved by HRV 3CD proteinase, recombinant poliovirus or HRV16 3CD proteinase was incubated with purified PCBP2, and the electrophoretic mobilities of PCBP2 and cleavage products were analyzed by Western blot (Fig. 1). PCBP2 and HRV16 or poliovirus 3CD proteinase recombinant proteins were incubated for varying amounts of time, as indicated. The level of cleaved PCBP2 increases during the first two hours and then remains consistent during the remainder of the incubation period. Comparing PCBP2 cleavage by poliovirus 3CD (lanes 2–5) to cleavage by HRV16 3CD (lanes 6–9), the levels of cleaved PCBP2 are similar. We have also shown that HRV14 3CD can cleave PCBP2 with a similar efficiency (data not shown). These data suggest that HRV 3CD can cleave PCBP2 as efficiently as poliovirus 3CD, further suggesting that PCBP2 cleavage could be important during HRV infection.
Fig. 1. Cleavage of recombinant PCBP2 by 3CD proteinase.
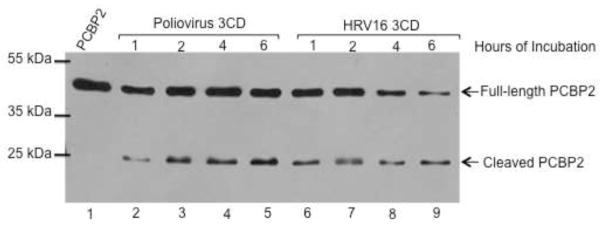
Recombinant PCBP2 was incubated with purified poliovirus or HRV16 3CD for 1 to 6 hours, as indicated. PCBP2 alone is in lane 1. PCBP2 was incubated with recombinant poliovirus 3CD (lanes 2–5) or recombinant HRV16 3CD (lanes 6–9). The arrows to the right of the gel image indicate full-length or cleaved PCBP2. Molecular weight markers are indicated to the left of the gel image.
Cleavage of PCBP2 during HRV infection
Although HRV 3CD can cleave PCBP2 in vitro, the question remains as to whether PCBP2 cleavage occurs during the course of HRV infection. To address this question, HeLa cells were infected with different serotypes of HRV, and cleavage of PCBP2 was analyzed by Western blot, using antibodies specific for PCBP2. HRV has slower growth kinetics than poliovirus, with peak titers reached at 10 hours post-infection compared to 4 hours for poliovirus (data not shown) (Lee and Wang, 2003). Therefore, extracts were harvested out to 16 hours post-infection to account for the protracted time course. As expected, no cleavage of PCBP2 was detected in mock-infected HeLa cells (Fig. 2A). In HRV14-infected HeLa cells, PCBP2 cleavage was not detected until 14 hours post-infection (Fig. 2B). Additionally, cleaved PCBP2 could only be observed when four times the total protein concentration was analyzed compared to poliovirus. Human rhinovirus grows optimally in tissue culture at 34°C, whereas poliovirus grows and replicates optimally at 37°C, although our work and the work of others has recently shown that HRV16 and HRV14 can also replicate efficiently at 37°C (data not shown) (Papadopoulos et al., 1999). To determine if the lower growth temperature contributes to the cleavage of PCBP2 seen during HRV14 infection, poliovirus infection was carried out in HeLa cells at 34°C as a comparison. When HeLa cell lysates from poliovirus-infected cells were analyzed by Western blot, PCBP2 cleavage was detected at 6 hours post-infection (Fig. 2C). While this is slightly delayed relative to cleavage of PCBP2 during poliovirus infection at 37°C, which is detected at 4 hours post-infection, it still occurs much earlier than what was observed during HRV14 infection (A. J. Chase, S. Daijogo, B. L. Semler, submitted for publication) (Perera et al., 2007). These results suggest that cleavage of PCBP2 during HRV14 infection is less efficient than during poliovirus infection. To determine if other rhinovirus serotypes cleave PCBP2 more efficiently than HRV14, HeLa cells were infected with a group A HRV serotype, type 16 (HRV16), which is more similar in sequence to circulating HRV serotypes than HRV14. Again, in mock-infected cells, only full-length PCBP2 is detected (Fig. 3A). PCBP2 cleavage during HRV16 infection was observed by 10–12 hours post-infection, delayed relative to cleavage during poliovirus infection, but not as delayed as cleavage during HRV14 infection (Fig. 3B). Although cleavage of PCBP2 is delayed until 10 hours post-infection during HRV16 infection, it corresponds to the delayed growth kinetics compared to the poliovirus growth kinetics. These data confirm that PCBP2 can be cleaved during human rhinovirus infection of HeLa cells.
Fig. 2. PCBP2 cleavage during HRV14 or poliovirus infection at 34°C.

To determine cleavage during infection at a lower temperature, HeLa cells were mock-infected (A) or infected with HRV14 (B) or poliovirus (C). Cytoplasmic lysates from infected cells were collected at indicated times post-infection and were analyzed by Western blot with an antibody specific to PCBP2. The arrows to the right of the gel image indicate full-length or cleaved PCBP2, and molecular weight markers are indicated to the left of the gel image.
Fig. 3. Cleavage of PCBP2 during HRV16 infection of HeLa cells or WisL cells.

Cytoplasmic lysates were harvested from infected cells and analyzed by Western blot with a monoclonal antibody against PCBP2. HeLa cells were mock-infected (A) or infected with HRV16 (B), and cytoplasmic lysates were harvested from 0 – 16 hours post-infection, as indicated. (C) WisL cells derived from human fetal lung fibroblast cells were mock-infected or infected with HRV16, and cytoplasmic extracts were collected out to 42 hours post-infection and analyzed for PCBP2 cleavage as described for (A) and (B). Arrows to the right of the gel image indicate full-length or cleaved PCBP2, and molecular weight markers are indicated on the left of the gel image.
PCBP2 remains intact during HRV infection of lung fibroblast cells
To further determine the importance of PCBP2 cleavage during rhinovirus infection, a human lung fibroblast-derived WisL cell line was utilized (Bochkov et al., 2011). As HRV infects the respiratory tract, this cell line may allow for analysis of HRV infection of cells more relevant than the standard HeLa cell model. Cytoplasmic extracts from WisL cells infected with HRV16 were collected and analyzed at indicated times post-infection. As seen in Fig. 3C, cleavage products of PCBP2 cannot be detected out to 42 hours (lane 10), and this was consistent when four times the total protein concentration was analyzed (data not shown).
To ensure that a productive infection was occurring, cytoplasmic lysates from HRV16-infected HeLa cells or WisL cells were analyzed by Western blot, using an antibody specific for HRV16 3D RNA-dependent RNA polymerase. Viral proteins 3D and 3CD precursor were detected at 8 hours post-infection of WisL cells (Fig. 4A). During HRV16 infection of HeLa cells, viral proteins were detected at 6 hours post-infection (Fig. 4B). The production of viral proteins suggesting productive HRV infection of WisL cells was further supported by single cycle growth analysis showing similar growth during HRV16 infection of WisL cells compared to growth kinetics during HRV16 infection of HeLa cells (Fig. 4C). Both the growth analysis and production of viral proteins indicate that HRV16 can productively infect WisL lung fibroblast cells, although PCBP2 is not cleaved. Therefore, we conclude that HRV16 can productively infect WisL cells in the absence of PCBP2 cleavage.
Fig. 4. Detection of HRV16 proteins during infection of HeLa or WisL cells.


To ensure that a progressive infection was occurring, we analyzed cytoplasmic lysates from infected WisL lung fibroblast cells (A) or HeLa cells (B) by Western blot with an antibody specific for HRV16 3D polymerase. Lane 1 [and lane 2 in (B)] is extract from mock-infected cells as a control. The arrows to the right of the gel image indicate 3CD, 3D, or 3D′ viral protein products. Molecular weight markers are indicated on the left of the gel image. (C) Single cycle growth analysis of HeLa cells (blue) or WisL cells (green) infected with HRV16 was analyzed by determining plaque-forming units per ml (pfu/ml).
To determine if PCBP2 can be cleaved in WisL cells, a poliovirus infection of WisL cells was carried out. Following infection, cytoplasmic lysate was harvested at indicated times post-infection, and PCBP2 cleavage was analyzed by Western blot. In contrast to HRV16 infection, cleavage of PCBP2 during poliovirus infection of WisL cells is readily detected at 6 hours post-infection, indicating that PCBP2 can be cleaved during infection of WisL cells (Fig. 5A). While the cleavage of PCBP2 during poliovirus infection of WisL cells was slightly delayed relative to HeLa cells (detected at 4 hours post-infection), it corresponds to a slight delay in growth kinetics, as indicated by the slightly delayed detection of viral proteins (Fig. 5B). During infection of WisL cells, poliovirus 3CD proteinase and 3D polymerase are detected at 4 and 6 hours post-infection, respectively, which is slightly delayed relative to detection during infection of HeLa cells, when viral proteins are detected by 3 hours post-infection. These data demonstrate that poliovirus productively infects WisL cells, and that PCBP2 can be cleaved in WisL cells during poliovirus infection. Therefore, the lack of cleavage observed during HRV16 infection appears to be unique to HRV infection.
Fig. 5. Poliovirus infection of WisL cells.

Human lung fibroblast WisL cells were infected with poliovirus and cytoplasmic lysates were collected from 0 – 12 hours post-infection, as indicated at the top of the gel image, and analyzed by Western blot. (A) Western blot analysis with an antibody specific for PCBP2, with the cleaved or full-length forms of the protein indicated to the right of the gel image. (B) Western blot analysis with poliovirus 3CD specific antibodies to verify a productive viral infection. 3CD precursor protein and 3D or 3D′ products are indicated to the right of the gel image. Lane 6 displays extract from mock-infected cells as a control. Molecular weight markers are indicated to the left of the gel image.
To further characterize the cleavage state of PCBP2 during HRV infection, WisL cells were infected with a group B serotype (HRV14), which also utilizes the major group receptor, similar to group A serotype HRV16, or with minor group HRV1A (Fig. 6). During HRV14 infection of HeLa cells, cleaved PCBP2 is barely detected at 12 hours post-infection, but is readily detected by 24 hours post-infection (lanes 5 and 6). Similar to HRV16 infection, PCBP2 is not cleaved during HRV14 infection of WisL cells (Fig. 6A, lanes 3 and 4). These data demonstrate that major group HRV serotypes 14 and 16 are able to cleave PCBP2 in HeLa cells, but this cleavage event does not appear to be required for efficient infection of WisL cells. To determine if the observed lack of cleavage in WisL cells was unique to major group rhinovirus types, WisL cells were infected with minor group HRV1A. Similar to HRV14 and HRV16, at 24 hours post HRV1A infection of HeLa cells, cleaved PCBP2 is readily detected (Fig. 6B, lane 7). Additional studies have shown that PCBP2 cleavage during HRV1A infection of HeLa cells can also be detected at 16 hours post-infection, similar to both HRV14 and HRV16 (data not shown). However, during HRV1A infection of WisL cells, cleaved PCBP2 is not detected (Fig. 6B, lanes 4 and 5), consistent with what we observed for HRV16 or HRV14 infection of WisL cells. Collectively, these data suggest that PCBP2 cleavage is not necessary for efficient HRV infection of WisL cells.
Fig. 6. PCBP2 cleavage during human rhinovirus infection.

HeLa or WisL cells were infected with HRV and cytoplasmic lysates were collected at indicated times post-infection. The collected lysates were then analyzed by Western blot with an antibody specific for PCBP2. (A) Cells were infected with HRV14. Extracts from mock-infected WisL or HeLa cells are shown in lanes 1 and 2, respectively. Lysates from infected HeLa cells are shown in lanes 3 and 4, and lysates from infected WisL cells are analyzed in lanes 5 and 6. (B) WisL (lanes 3–5) or HeLa cells (lanes 6 and 7) were infected with HRV1A. Extracts from mock-infected WisL or HeLa cells are analyzed in lanes 1 or 2, respectively. Arrows on the right of the gel image indicate full-length or cleaved PCBP2, and molecular weight markers are on the left of the gel image.
Cleavage of polypyrimidine tract binding protein during HRV infection
Previous studies have shown that cellular protein polypyrimidine tract binding protein (PTB) enhances both poliovirus and HRV IRES-mediated translation (Gosert et al., 2000; Hellen et al., 1993; Hunt and Jackson, 1999; Sawicka et al., 2008). Additionally, PTB can be cleaved during poliovirus infection by 3C/3CD proteinase, and it has been proposed that the cleavage event can help mediate a switch in template usage from translation to RNA replication (Back et al., 2002). It has been reported that all three PTB isoforms (PTB-1, -2, and -4) can be cleaved by 3CD, producing two major cleavage products at 41 kDa and 39 kDa, and a minor cleavage product at 16 kDa (Back et al., 2002). Therefore, this protein may also be cleaved during HRV infection to help mediate initiation of RNA replication by inhibiting translation to allow a switch in template usage. To determine the cleavage state of PTB during HRV infection, lysates from HRV16-infected HeLa cells were analyzed by Western blot with an antibody specific for PTB. During infection of HeLa cells at 34°C, cleavage was detected by 12 hours post-infection (Fig. 7A, lane 7), and cleavage can also be detected as early as 10 hours post-infection (data not shown). Additionally, PTB cleavage was detected by 12 hours post-infection during HRV14 infection of HeLa cells (Fig. 7A, lane 11). During HRV16 or HR14 infection, three cleavage products were detected, two products with molecular masses of approximately 27–30 kDa, and a product with a molecular mass of approximately 13–14 kDa. To further determine if PTB cleavage occurs during HRV infection, WisL lung fibroblast cells were infected with HRV16 or HRV14 and lysates were collected at the indicated times post-infection and analyzed by Western blot. Similar to what we observed for PCBP2, only trace levels of PTB cleavage were detected in HRV16-infected WisL cells (Fig. 7A, lanes 3–4). To confirm the lack of cleavage, 2.5 times the total protein concentration in lysates from WisL cells infected with HRV16 was analyzed, and again only trace levels of PTB cleavage were detected (Fig. 7A, lanes 5 and 6, indicated by the asterisk). However, during HRV14 infection, PTB cleavage products were detected by 12 hours post-infection; however, only low levels of the largest PTB cleavage product PTB could be detected at 24 hours post-infection (Fig. 7A, compare lanes 9 and 10). This suggests that PTB cleavage is not required for efficient HRV16 infection, but PTB cleavage may play a role in HRV14 infection. However, the cleavage of PTB appears to be inefficient in HRV14-infected cells, perhaps limiting the possible role of this event in the HRV14 replication cycle.
Fig. 7. PTB cleavage during human rhinovirus infection.
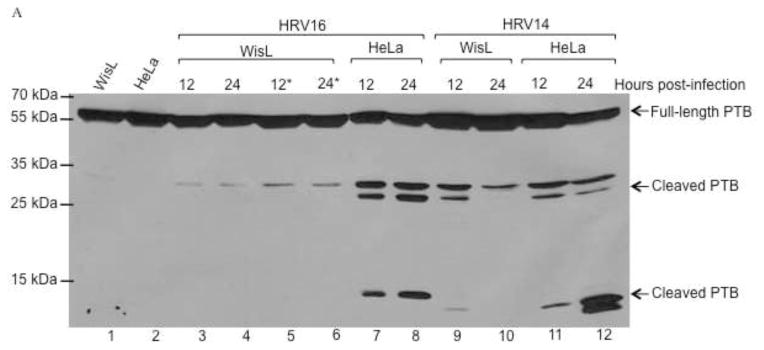

HeLa or WisL cells were infected with HRV14, 16, or 1A, as described for Fig. 6. Lysates were analyzed by Western blot utilizing an antibody specific for PTB. (A) Cells were infected with HRV16 or HRV14. Lanes 1 and 2 display extracts from mock-infected WisL or HeLa cells, respectively. Extracts from HRV16-infected WisL cells are shown in lanes 3–6, with 100 μg total protein in lanes 3 and 4, and 250 μg total protein in lanes 5 and 6 (indicated by the asterisks). Lanes 7 and 8 show lysates from HRV16-infected HeLa cells, with 100 μg total protein analyzed. Lanes 9–12 analyze WisL cells (lanes 9 and 10) or HeLa cells (lanes 11 and 12) infected with HRV14. Arrows to the right of the gel image indicate full-length or cleaved PTB. (B) Cells were infected with HRV1A, and cytoplasmic lysates were collected at 12 or 24 hours post-infection. Mock-infected HeLa cell or WisL cell lysates (lanes 1 and 4, respectively) confirm full-length PTB. Lysates from HRV1A-infected HeLa cells are in lanes 2 and 3, and lysates from HRV1A-infected WisL cells are analyzed in lanes 5 and 6. Full-length and cleaved forms of PTB are indicated by the arrows to the right of the gel image and molecular weight markers are indicated to the left of the gel image.
To further analyze cleavage of PTB during HRV infection, cytoplasmic lysates from HeLa or WisL cells infected with minor receptor group serotype HRV1A were analyzed, as shown in Fig. 7B. During infection of HeLa cells, cleaved PTB was detected by 12 hours post-infection (lane 2), and at 24 hours post-infection both higher and lower molecular weight PTB cleavage products were detected (lane 3). However, during infection of WisL cells by HRV1A, only a trace amount of PTB cleavage product is detected at 12 or 24 hours post-infection (lanes 5 and 6). From these data, it can be concluded that while PTB cleavage occurs during HRV infection of HeLa cells, it is not detected during HRV infection of WisL cells and does not appear to be necessary for HRV replication in these cells.
Differential cleavage of host proteins during coxsackievirus infection of different cell types
Coxsackievirus, also a member of the picornavirus family of viruses, has similar growth properties to poliovirus. Coxsackievirus grows and replicates optimally at 37°C and has similar replication kinetics, with peak titer being reached at 5 hours post-infection. PCBP2 is cleaved during coxsackievirus infection at a similar time post-infection to PCBP2 cleavage during poliovirus infection (A. J. Chase, S. Daijogo, B. L. Semler, submitted for publication). To determine if coxsackievirus cleaves PCBP2 differentially in HeLa versus WisL cells, both cell lines were infected with coxsackievirus and the resulting cytoplasmic lysates from infected cells were analyzed by Western blot. During coxsackievirus infection of HeLa cells, PCBP2 is cleaved beginning at 4–6 hours post-infection (Fig. 8A), further demonstrating that PCBP2 cleavage during coxsackievirus infection is similar relative to cleavage during poliovirus infection. However, during coxsackievirus infection of WisL human lung fibroblast cells, only a small amount of cleaved PCBP2 is detected at 12 hours post-infection (Fig. 8B). This is delayed relative to observed PCBP2 cleavage at 6 hours following poliovirus infection of WisL cells, indicating that coxsackievirus may cleave PCBP2 differentially based on cell type.
Fig. 8. Cleavage of PCBP2 and PTB during coxsackievirus infection.


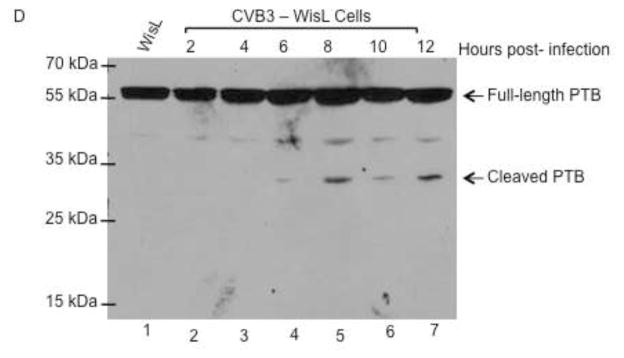
HeLa or WisL cells were infected with coxsackievirus B3 (CVB3), and cytoplasmic lysates were analyzed by Western blot with antibodies specific for host cell proteins PCBP2 or PTB. (A) HeLa cells or (B) WisL cells were infected with CVB3 and PCBP2 cleavage was analyzed. Lysates from mock-infected cells were analyzed in lane 1. Arrows to the right of the gel image indicate full-length or cleaved PCBP2. To determine the status of PTB cleavage, cytoplasmic lysates from CVB3-infected HeLa cells (C) or WisL cells (D) were analyzed by Western blot with an antibody specific for PTB. Lysate from mock-infected cells is shown in lane 1. Arrows to the right of the gel image indicate full-length or cleaved PTB. The molecular weight standards are shown on the left of the gel image.
To further characterize differential cleavage of host proteins during coxsackievirus infection, PTB cleavage was analyzed in WisL or HeLa cells infected with coxsackievirus. Cytoplasmic lysates from HeLa- or WisL-infected cells were analyzed by Western blot for PTB cleavage. As shown in Fig. 8C, the higher molecular weight PTB cleavage products are detected at 4 hours post-infection in HeLa cells, and the lower molecular weight product is seen by 8 hours post-infection. During coxsackievirus infection of WisL cells, only one of the major PTB cleavage products is detected at 6 hours post-infection, and is more readily detected at 8 hours post-infection (Fig. 8D). Similar to PCBP2 cleavage, a very low level of cleaved product is observed relative to infection of HeLa cells, further suggesting that there is a significant difference in cleavage of host cell proteins PTB and PCBP2 during coxsackievirus infection, based on cell type being infected. Taken together, these data suggest that, unlike poliovirus infection, cleavage of host cell proteins PCBP2 and PTB are not required for efficient HRV or coxsackievirus infection.
Discussion
Human rhinovirus translation is driven in a cap-independent, IRES-dependent manner. Several host cell proteins help mediate this non-canonical translation, known as IRES trans-acting factors (ITAFs), including PCBP2 and PTB (Blyn et al., 1996; Blyn et al., 1997; Gamarnik and Andino, 1997; Gosert et al., 2000; Hellen et al., 1993; Hunt and Jackson, 1999; Sawicka et al., 2008). PCBP2 is cleaved during poliovirus infection by the 3CD proteinase, and it has been proposed that the cleavage event helps mediate a switch in template usage by inhibiting translation (Perera et al., 2007). Additionally, PTB is known to stimulate both poliovirus and HRV translation. Cleavage of PTB by poliovirus 3C proteinase also leads to an inhibition in translation, and thus has also been proposed to be involved in mediating a switch in template usage (Back et al., 2002; Hunt and Jackson, 1999). The picornavirus genome can be used as a template for both translation and RNA replication, and it has been shown that a template actively being translated cannot function as a template for RNA replication (Barton et al., 1999; Gamarnik and Andino, 1998). A switch in template usage is essential for efficient viral RNA synthesis, and could therefore be important for the progression of the replication cycle.
HRV serotypes can be divided into group A, group B, or group C, and also by the receptor used to mediate cell entry; major group serotypes use ICAM-I and minor group serotypes utilize low-density lipoprotein (LDL) receptor. To provide a more comprehensive study of the cleavage or use of host cell proteins PCBP2 and PTB during human rhinovirus infection, serotypes representing different groups were studied. HRV16, group A, and HRV14, group B, are both major group serotypes, and HRV1A, group A, is a minor group serotype. PCBP2 and PTB were both shown to be cleaved during the course of HRV14, HRV16, or HRV1A infection of HeLa cells, suggesting that PCBP2 might also be important for a switch in template usage during HRV infection of HeLa cells. During HRV16 or HRV14 infection of HeLa cells, PCBP2 cleavage is detected at 10 or 14 hours post-infection, respectively, while cleavage is also detected prior to 24 hours during HRV1A infection (Fig. 2, 3, and 6, summarized in Table 1). In contrast, HRV infected WisL human lung fibroblast cells showed a lack of PCBP2 cleavage out to 24 hours post-infection during HRV16, HRV14, or HRV1A infection (Fig. 3 and 6, Table 1). However, single cycle growth analysis and Western blot analysis with an antibody specific for HRV16 3D polymerase indicated that there was a productive HRV16 infection occurring in WisL cells (Fig. 4). Additionally, although slightly delayed, PCBP2 cleavage was observed in WisL cells during poliovirus infection. The observed delay is consistent with the delay in viral protein accumulation, suggesting that poliovirus infection may be slightly delayed in WisL cells, but that cleavage of PCBP2 is still necessary for infection (Fig. 5). This demonstrates that PCBP2 can be cleaved in WisL cells, and that the lack of observed cleavage in this cell line appears to be unique to HRV infection.
Table 1.
Summary of when cleavage of host cell proteins PCBP2 and PTB is detected during picornavirus infections
| Virus | Cell Type | PCBP2 | PTB |
|---|---|---|---|
| HRV16 – 34°C | HeLa | 10 hpi | 10 hpi |
| HRV16 – 37°C | HeLa | 4 hpi | 8 hpi |
| HRV16 – 34°C | WisL | Not detected up to 42 hpi | Not detected at 32 hpi |
| HRV14 – 34°C hpi | HeLa | 14 hpi (with 4× total protein) | 10 hpi; 12 significantly more detected |
| HRV14 – 34°C | WisL | Not detected up to 32 hpi | Not detected up to 32 hpi |
| HRV1A – 34°C | HeLa | 16 hpi | 12 hpi |
| HRV1A – 34°C | WisL | Not detected out to 24 hpi | Not detected out to 24 hpi |
| Coxsackievirus – 37°C | HeLa | 4 hpi | 4 hpi |
| Coxsackievirus – 37°C | WisL | 12 hpi and at significantly lower levels levels | 6–8 hpi, and significantly lower |
| Poliovirus – 37°C | HeLa | 4 hpi | 3 hpi* |
| Poliovirus – 37°C | WisL | 6 hpi | 4 hpi |
As it is known that other cellular proteins such as PTB can be cleaved by 3C proteinase, and the cleavage could also help mediate a switch in template usage, further analysis was carried out on cleavage of PTB during HRV infection of WisL cells. The data showed that PTB was cleaved during HRV14, HRV16, or HRV1A infection of HeLa cells, but similar to PCBP2, PTB was also not cleaved efficiently during HRV infection of WisL cells (Fig. 7, Table 1). Because HRV can establish a productive infection of WisL cells, our data suggest that cleavage of PCBP2 or PTB is not necessary for efficient HRV infection of these cells.
Coxsackievirus has similar growth kinetics to poliovirus, and it has been shown that PCBP2 is cleaved during coxsackievirus infection of HeLa cells with similar kinetics to cleavage during poliovirus infection (A. J. Chase, S. Daijogo, and B. L. Semler, submitted for publication). Therefore, we hypothesized that PCBP2 cleavage would also occur during coxsackievirus infection of WisL cells, similar to what was observed during poliovirus infection of WisL cells (Fig. 5). However, PCBP2 cleavage was significantly delayed and less efficient during coxsackievirus infection of WisL cells compared to infection of HeLa cells. When cleavage of PTB was analyzed, there was a similar occurrence in which PTB cleavage was both less efficient and delayed. Taken together, these data suggest that efficient coxsackievirus infection of WisL cells does not require PCBP2 or PTB cleavage.
Our results lead to several questions, including why PCBP2 and PTB are cleaved during infection of HeLa cells but not in WisL cells, and if there are other, as yet undefined, ITAFs that are important for HRV and/or coxsackievirus translation in WisL cells that may be less abundant in HeLa cells. Additional studies are necessary to better understand the intracellular determinants required for HRV translation and RNA replication, and work will need to be done in multiple cell lines including, but not limited to, HeLa cells and WisL cells. Since PCBP2 and PTB are cleaved during poliovirus infection of WisL cells, the reason PCBP2 and PTB are differentially cleaved during HRV infection of WisL cells but not during poliovirus infection is not understood. Perhaps there is a lower concentration of PCBP2 present in replication complexes formed during infection of WisL cells, leading to the decreased detection of cleaved PCBP2. During HRV infection of HeLa cells, PCBP2 cleavage is delayed relative to cleavage during poliovirus infection, suggesting that PCBP2 cleavage may be less efficient during HRV infection, and the lower local concentration of PCBP2 would result in a lack of detectable levels of PCBP2 cleavage. Alternatively, PCBP2 or PTB may be bound to cellular RNA or host cell proteins, making them unavailable as cleavage substrates during HRV infection of WisL cells.
It has been proposed that cleavage of both PCBP2 and PTB helps to mediate a switch in poliovirus template usage from translation to RNA replication to allow the progression of RNA synthesis (Back et al., 2002; Perera et al., 2007). Therefore, the question remains as to what mediates the switch in HRV template usage. The cellular protein upstream of N-ras (unr) has been shown to be important for HRV translation, and can stimulate HRV translation alone and synergistically with PTB (Hunt et al., 1999). Thus, unr could be an important protein involved in template switching (Hunt et al., 1999; Hunt and Jackson, 1999). However, preliminary data suggests that unr is not cleaved in either HeLa cells or WisL cells during HRV16 infection (data not shown).
Previous studies have shown that when 3CD is bound to stem-loop I, levels of translation are decreased while levels of RNA replication increase, and, correspondingly, in the absence of bound 3CD, translation is increased (Gamarnik and Andino, 1998). Subsequent work indicated that in the presence of 3CD, PCBP2 preferentially binds to stem-loop I. The apparent dissociation constant of PCBP2 for poliovirus stem-loop IV is approximately 15 nM, while that of PCBP2 to stem-loop I, in the absence of 3CD, is approximately 95 nM. In the presence of 3CD, the dissociation constant of PCBP2 for stem-loop IV is unaltered, but that of PCBP2 for stem-loop I changes to approximately 1 nM (Gamarnik and Andino, 2000). Additionally, competition assays indicated that PCBP2 bound to poliovirus stem-loop IV can be reduced in the presence of 3CD bound to stem-loop I (Gamarnik and Andino, 2000). As originally proposed by Gamarnik and Andino (1998) for poliovirus, the shift in the binding affinity of PCBP2 for stem-loop I over stem-loop IV in the presence of 3CD could help mediate a switch in template usage from translation to RNA replication independent of the cleavage state of PCBP2 or PTB. As translation of the genomic template proceeds, the concentration of viral proteins, including 3CD, would increase. 3CD interacts with stem-loop I, shifting the occupancy of PCBP2 from stem-loop IV to stem-loop I, and in the process inhibits translation while allowing the initiation of negative-strand RNA synthesis. The change in PCBP2 binding affinity from stem-loop I to stem-loop IV in the presence of 3CD proteinase could be the fundamental mechanism mediating template switching, and could be augmented by cleavage of host cell proteins such as PCBP2 and PTB by viral proteinase 3CD. Since it has been shown that PCBP2 cleavage is important for poliovirus infection, the cleavage of PCBP2 would mediate a more efficient or a faster switch in template usage. As coxsackievirus infection has similar growth properties as poliovirus infection, this would suggest that an alternative mechanism, that is as yet unknown, helps improve the switch in template usage. HRV growth and replication is delayed relative to poliovirus, which could be attributed to a less efficient switch in template usage, leading to a delay in RNA synthesis. During poliovirus infection, the template switch could occur more quickly as cellular protein PCBP2 is cleaved by 3CD to inhibit the RNP complex formed between PCBP2 and stem-loop IV, potentiating the change in PCBP2 binding from stem-loop IV to stem-loop I. Further studies are necessary to identify the mechanism(s) that mediate a switch in HRV template usage.
Research highlights.
Human rhinovirus 3C/3CD proteinase cleaves host cell RNA binding protein PCBP2
Rhinovirus-induced cleavage of IRES trans-acting factors PCBP2 and PTB is host cell dependent
Rhinoviruses employ unique mechanisms to switch from translation to RNA replication
There are distinct intracellular virus-cell requirements during picornavirus replication cycles
Acknowledgments
We are grateful to Andrea Cathcart and Sonya Maciejewski for critical comments on the manuscript. We thank Dr. Yury Bochkov for generously providing the HRV1A virus stock and the WisL cell line. We thank Hung Nguyen and MyPhuong Tran for their expert technical assistance. The research was supported by Public Health Service grant AI026765 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andino R, Rieckhof GE, Achacoso PL, Baltimore D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 1993;12:3587–3598. doi: 10.1002/j.1460-2075.1993.tb06032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andino R, Rieckhof GE, Baltimore D. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell. 1990;63:369–380. doi: 10.1016/0092-8674(90)90170-j. [DOI] [PubMed] [Google Scholar]
- Arruda E, Pitkaranta A, Witek TJ, Jr, Doyle CA, Hayden FG. Frequency and natural history of rhinovirus infections in adults during autumn. J Clin Microbiol. 1997;35:2864–2868. doi: 10.1128/jcm.35.11.2864-2868.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SH, Kim YK, Kim WJ, Cho S, Oh HR, Kim JE, Jang SK. Translation of polioviral mRNA is inhibited by cleavage of polypyrimidine tract-binding proteins executed by polioviral 3C(pro) J Virol. 2002;76:2529–2542. doi: 10.1128/jvi.76.5.2529-2542.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton DJ, Morasco BJ, Flanegan JB. Translating ribosomes inhibit poliovirus negative-strand RNA synthesis. J Virol. 1999;73:10104–10112. doi: 10.1128/jvi.73.12.10104-10112.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsham GJ, Sonenberg N. RNA-protein interactions in regulation of picornavirus RNA translation. Microbiol Rev. 1996;60:499–511. doi: 10.1128/mr.60.3.499-511.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyn LB, Swiderek KM, Richards O, Stahl DC, Semler BL, Ehrenfeld E. Poly(rC) binding protein 2 binds to stem-loop IV of the poliovirus RNA 5′ noncoding region: identification by automated liquid chromatography-tandem mass spectrometry. Proc Natl Acad Sci U S A. 1996;93:11115–11120. doi: 10.1073/pnas.93.20.11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyn LB, Towner JS, Semler BL, Ehrenfeld E. Requirement of poly(rC) binding protein 2 for translation of poliovirus RNA. J Virol. 1997;71:6243–6246. doi: 10.1128/jvi.71.8.6243-6246.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkov YA, Palmenberg AC, Lee WM, Rathe JA, Amineva SP, Sun X, Pasic TR, Jarjour NN, Liggett SB, Gern JE. Molecular modeling, organ culture and reverse genetics for a newly identified human rhinovirus C. Nat Med. 2011;17:627–632. doi: 10.1038/nm.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borman A, Jackson RJ. Initiation of translation of human rhinovirus RNA: mapping the internal ribosome entry site. Virology. 1992;188:685–696. doi: 10.1016/0042-6822(92)90523-r. [DOI] [PubMed] [Google Scholar]
- Cathcart AL, Rozovics JM, Semler BL. Cellular mRNA decay protein AUF1 negatively regulates enterovirus and human rhinovirus infections. J Virol. 2013 doi: 10.1128/JVI.01049-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Munoz R, Darnell JE. Structural difference between the 5′ termini of viral and cellular mRNA in poliovirus-infected cells: possible basis for the inhibition of host protein synthesis. J Virol. 1976;18:719–726. doi: 10.1128/jvi.18.2.719-726.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KD, Semler BL. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim Biophys Acta. 2009;1789:518–528. doi: 10.1016/j.bbagrm.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R. Two functional complexes formed by KH domain containing proteins with the 5′ noncoding region of poliovirus RNA. RNA. 1997;3:882–892. [PMC free article] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998;12:2293–2304. doi: 10.1101/gad.12.15.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R. Interactions of viral protein 3CD and poly(rC) binding protein with the 5′ untranslated region of the poliovirus genome. J Virol. 2000;74:2219–2226. doi: 10.1128/jvi.74.5.2219-2226.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert R, Chang KH, Rijnbrand R, Yi M, Sangar DV, Lemon SM. Transient expression of cellular polypyrimidine-tract binding protein stimulates cap-independent translation directed by both picornaviral and flaviviral internal ribosome entry sites In vivo. Mol Cell Biol. 2000;20:1583–1595. doi: 10.1128/mcb.20.5.1583-1595.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen CU, Witherell GW, Schmid M, Shin SH, Pestova TV, Gil A, Wimmer E. A cytoplasmic 57-kDa protein that is required for translation of picornavirus RNA by internal ribosomal entry is identical to the nuclear pyrimidine tract-binding protein. Proc Natl Acad Sci U S A. 1993;90:7642–7646. doi: 10.1073/pnas.90.16.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewlett MJ, Rose JK, Baltimore D. 5′-terminal structure of poliovirus polyribosomal RNA is pUp. Proc Natl Acad Sci U S A. 1976;73:327–330. doi: 10.1073/pnas.73.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt SL, Hsuan JJ, Totty N, Jackson RJ. unr, a cellular cytoplasmic RNA-binding protein with five cold-shock domains, is required for internal initiation of translation of human rhinovirus RNA. Genes Dev. 1999;13:437–448. doi: 10.1101/gad.13.4.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt SL, Jackson RJ. Polypyrimidine-tract binding protein (PTB) is necessary, but not sufficient, for efficient internal initiation of translation of human rhinovirus-2 RNA. RNA. 1999;5:344–359. doi: 10.1017/s1355838299981414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SK, Krausslich HG, Nicklin MJ, Duke GM, Palmenberg AC, Wimmer E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol. 1988;62:2636–2643. doi: 10.1128/jvi.62.8.2636-2643.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SL, Pattemore PK, Sanderson G, Smith S, Campbell MJ, Josephs LK, Cunningham A, Robinson BS, Myint SH, Ward ME, Tyrrell DA, Holgate ST. The relationship between upper respiratory infections and hospital admissions for asthma: a time-trend analysis. Am J Respir Crit Care Med. 1996;154:654–660. doi: 10.1164/ajrccm.154.3.8810601. [DOI] [PubMed] [Google Scholar]
- Lee WM, Wang W. Human rhinovirus type 16: mutant V1210A requires capsid-binding drug for assembly of pentamers to form virions during morphogenesis. J Virol. 2003;77:6235–6244. doi: 10.1128/JVI.77.11.6235-6244.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McErlean P, Shackelton LA, Andrews E, Webster DR, Lambert SB, Nissen MD, Sloots TP, Mackay IM. Distinguishing molecular features and clinical characteristics of a putative new rhinovirus species, human rhinovirus C (HRV C) PLoS One. 2008;3:e1847. doi: 10.1371/journal.pone.0001847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerovitch K, Svitkin YV, Lee HS, Lejbkowicz F, Kenan DJ, Chan EK, Agol VI, Keene JD, Sonenberg N. La autoantigen enhances and corrects aberrant translation of poliovirus RNA in reticulocyte lysate. J Virol. 1993;67:3798–3807. doi: 10.1128/jvi.67.7.3798-3807.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael WM, Choi M, Dreyfuss G. A nuclear export signal in hnRNP A1: a signal-mediated, temperature-dependent nuclear protein export pathway. Cell. 1995;83:415–422. doi: 10.1016/0092-8674(95)90119-1. [DOI] [PubMed] [Google Scholar]
- Nomoto A, Lee YF, Wimmer E. The 5′ end of poliovirus mRNA is not capped with m7G(5′)ppp(5′)Np. Proc Natl Acad Sci U S A. 1976;73:375–380. doi: 10.1073/pnas.73.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos NG, Sanderson G, Hunter J, Johnston SL. Rhinoviruses replicate effectively at lower airway temperatures. J Med Virol. 1999;58:100–104. doi: 10.1002/(sici)1096-9071(199905)58:1<100::aid-jmv16>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Parsley TB, Cornell CT, Semler BL. Modulation of the RNA binding and protein processing activities of poliovirus polypeptide 3CD by the viral RNA polymerase domain. J Biol Chem. 1999;274:12867–12876. doi: 10.1074/jbc.274.18.12867. [DOI] [PubMed] [Google Scholar]
- Parsley TB, Towner JS, Blyn LB, Ehrenfeld E, Semler BL. Poly (rC) binding protein 2 forms a ternary complex with the 5′-terminal sequences of poliovirus RNA and the viral 3CD proteinase. RNA. 1997;3:1124–1134. [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334:320–325. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- Perera R, Daijogo S, Walter BL, Nguyen JH, Semler BL. Cellular protein modification by poliovirus: the two faces of poly(rC)-binding protein. J Virol. 2007;81:8919–8932. doi: 10.1128/JVI.01013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakes GP, Arruda E, Ingram JM, Hoover GE, Zambrano JC, Hayden FG, Platts-Mills TA, Heymann PW. Rhinovirus and respiratory syncytial virus in wheezing children requiring emergency care. IgE and eosinophil analyses. Am J Respir Crit Care Med. 1999;159:785–790. doi: 10.1164/ajrccm.159.3.9801052. [DOI] [PubMed] [Google Scholar]
- Sawicka K, Bushell M, Spriggs KA, Willis AE. Polypyrimidine-tract-binding protein: a multifunctional RNA-binding protein. Biochem Soc Trans. 2008;36:641–647. doi: 10.1042/BST0360641. [DOI] [PubMed] [Google Scholar]
- Sean P, Nguyen JH, Semler BL. The linker domain of poly(rC) binding protein 2 is a major determinant in poliovirus cap-independent translation. Virology. 2008;378:243–253. doi: 10.1016/j.virol.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkin YV, Meerovitch K, Lee HS, Dholakia JN, Kenan DJ, Agol VI, Sonenberg N. Internal translation initiation on poliovirus RNA: further characterization of La function in poliovirus translation in vitro. J Virol. 1994;68:1544–1550. doi: 10.1128/jvi.68.3.1544-1550.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]