

For a wide breadth of human noncoding RNAs, including intergenic, intronic, and antisense RNAs, the authors investigate connections between tissue specificity and level of expression, evolutionary conservation, and locus chromatin signature. Uncharacterized ncRNAs are benchmarked against ncRNAs with known cellular roles. It will be of interest to test the authors’ prediction that as yet poorly characterized ncRNAs with profiles of expression, conservation, and locus chromatin similar to known functional ncRNAs are a set enriched for ncRNAs with biological function.
Keywords: noncoding RNA, comparative genomics, ultraconserved elements, EvoFold predictions, lncRNA
Abstract
The past decade has shown mammalian genomes to be pervasively transcribed and identified thousands of noncoding (nc) transcripts. It is currently unclear to what extent these transcripts are of functional importance, as experimental functional evidence exists for only a small fraction. Here, we characterize the expression and evolutionary conservation properties of 12,115 known and novel nc transcripts, including structural RNAs, long nc RNAs (lncRNAs), antisense RNAs, EvoFold predictions, ultraconserved elements, and expressed nc regions. Expression levels are evaluated across 12 human tissues using a custom-designed microarray, supplemented with RNAseq. Conservation levels are evaluated at both the base level and at the syntenic level. We combine these measures with epigenetic mark annotations to identify subsets of novel nc transcripts that show characteristics similar to known functional ncRNAs. Few novel nc transcripts show both high expression and conservation levels. However, overall, we observe a positive correlation between expression and both conservation and epigenetic annotations, suggesting that a subset of the expressed transcripts are under purifying selection and likely functional. The identified subsets of expressed and conserved novel nc transcripts may form the basis for further functional characterization.
INTRODUCTION
Thousands of short nc transcripts have been discovered over the past decade, primarily driven by different types of direct expression evidence (Kapranov et al. 2002; Bertone et al. 2004; Kampa et al. 2004; Carninci et al. 2005). More recently, first chromatin structure analysis and later sequencing provided evidence for many intergenic lncRNA (Ponjavic et al. 2007; Guttman et al. 2009; Khalil et al. 2009). Collectively, such efforts have led to the definition of numerous large sets of putative nc transcripts scattered throughout the human genome, which now exceed the number of annotated protein-coding (pc) genes (Mattick and Makunin 2006).
It is debatable to what extent pervasive transcription of noncoding genomic regions has biological function or represents “noisy” transcription from active chromatin regions. Ravasi et al. (2006) have estimated that up to one-third of entries in the FANTOM database may represent fragments of unprocessed transcription. On the other hand, the potential for functional discoveries exists, as shown by nc transcripts with roles in basic biological functions such as chromosome X inactivation for XIST and p53-mediated apoptosis for lincRNA-p21 (Brockdorff et al. 1991; Huarte et al. 2010). However, much work remains in pinpointing the genuinely functional nc transcripts among the many novel nc transcripts.
Comparative genomics contributes important layers of evolutionary understanding and functional evidence and is a powerful tool for analyzing poorly characterized nc transcripts. Ponjavic et al. (2007) found that long nc transcripts defined in the FANTOM consortium (Okazaki et al. 2002) had lower nucleotide substitution rates, higher promoter conservation, and higher conservation of splice sites than proximal control regions. This is underscored by similar findings for intergenic lncRNAs that show exon conservation exceeding background intergenic regions and promoter conservation comparable to pc genes (Guttman et al. 2009). Recent analysis of the large set of GENCODE annotated human lncRNAs; Derrien et al. (2012) found only 0.7% to be specific to the human lineage and 30% appeared primate specific. These results indicate that far more nc transcripts are subject to purifying selection, and thus potentially functional, than the relatively few currently assigned specific functions.
This study aims to define sets of nc transcripts with an increased chance of being functional based on a combined analysis of features indicative of functionality. We use three types of functional evidence criteria: expression, conservation, and overlap, with chromatin modifications indicative of active transcription. We have designed an expression microarray and profile a set of 12,115 nc transcripts, including both database annotations and putatively transcribed regions such as ultraconserved elements (UCEs), EvoFold predictions, and regions with expression evidence in ENCODE data sets. This set forms the input set for all the analysis. Throughout the analysis, we compare the results against three sets of functional transcripts: a set of known functional lncRNAs, a set of structural RNAs, and a subset of disease-related pc mRNAs, which were specifically included for later use of the array on clinical cancer samples.
We found that expressed nc transcripts as well as pc transcripts have increased conservation profiles in mammals and vertebrates, suggesting that a subset of the uncharacterized expressed nc transcripts are functional. Expressed transcripts were enriched for overlap with both tissue-specific chromatin marks associated with expression and, more generally, expressed sequence tags (ESTs) in both mouse and human. Based on our annotations and combined analyses, we define sets of uncharacterized nc transcripts that show similar expression, conservation, and chromatin mark overlap properties as the well-characterized functional nc transcript sets, with the hope that these can help focus experimental screens for biological function.
RESULTS
Expression platform and transcript definition
To characterize the expression of human nc transcripts, we profiled 12 human tissues (bladder, brain, breast, colon, heart, kidney, liver, lung, muscle, ovary, prostate, and skin) in triplicates using a custom-designed microarray with both nc transcripts (n = 12,115) and pc transcripts (n = 6856). The set of nc transcripts was designed to be comprehensive, encompassing different classes of nc transcripts and only constrained by the transcripts being longer than the array probes (60 nt). Collectively, we refer to these features as nc transcripts, although we did not have existing expression evidence for all. The nc transcript annotations were collected from public databases and literature sources (for sources and filtering steps, see Materials and Methods). Briefly, the set includes lncRNAs (n = 4740) from UCSC (Pruitt et al. 2009b) and RefSeq (Hsu et al. 2006) and intergenic lncRNAs (Cabili et al. 2011); antisense RNAs (n = 625) (Chen et al. 2004; Engström et al. 2006; Ge et al. 2008); structural RNAs (n = 131) (Griffiths-Jones et al. 2003; Schattner et al. 2005; Xie et al. 2007); regions with consistent expression evidence in ENCODE RNAseq data sets (n = 4340) (Birney et al. 2007); nonexonic ultra conserved elements (n = 331) (Bejerano et al. 2004); and long (>60 nt) EvoFold predictions of regions with evolutionarily conserved RNA secondary structure (n = 1599) (Pedersen et al. 2006). Given that the array would also be applied in a cancer setting, we included transcripts of cancer-associated pc genes (n = 6856), primarily from the Cancer Gene Index (Suh et al. 2010). See Figure 1 for relative transcript abundance and Supplemental Table S1 for the complete annotated set of nc transcripts. A UCSC Genome Browser mirror including all transcripts is available at http://moma.ki.au.dk/genome-mirror-mmn.
FIGURE 1.
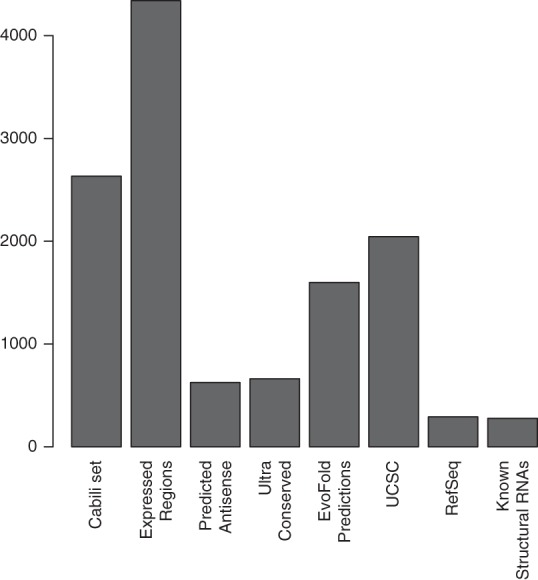
Sources of noncoding transcripts on the expression microarray. Bars represent number of transcripts from each source (see text for details).
We categorized all transcripts into either intergenic (not overlapping pc genes, n = 6309); intronic (no overlap with either pc exons or intergenic regions, n = 4616); or antisense (overlapping pc exons, n = 1190), as exemplified in Figure 2A–C. When a transcript overlapped multiple regions the following ranking was used: exonic > intronic > intergenic. In addition, we defined two sets of known nc transcripts for use as reference points in the later analysis. (1) A set of lncRNAs with functional evidence in the literature (Amaral et al. 2011) and a length over 500 nt, with at least 80% mutual overlap with array transcripts (n = 35). (2) A set of known structural RNAs (n = 321) defined as all array entries from the Rfam, snoRNA, and tRNAscan databases combined with all other database entries annotated as snoRNAs or scaRNAs. In addition, we compare against the pc transcripts on the array (n = 6856).
FIGURE 2.

Expression profiles of differentially expressed transcripts. (A–C) Examples of three different categories of noncoding transcripts. (A) Intergenic lncRNA located between the protein coding genes ITGA5 and GTSF1. (B) Intronic transcript, uc002xlx, located in intron of HNF4A. (C) Five antisense transcripts overlapping exons of the protein-coding gene CIDEB. (D) Fraction of genes differentially expressed in the 12 tissues for each category. Number of genes displayed on top of bars. (E) Violin plots of expression correlation of subsets of ncRNA with their protein-coding counterparts. Within each category, correlations were calculated for the subset with protein-coding counterparts present on the array. Intergenic 3′ proximal regions are defined as 5 kb downstream from a protein-coding gene. (F–I) Expression heatmaps of differentially expressed genes. Gene profiles were normalized across tissues and hierarchically clustered within categories of coding, intergenic, intronic, and antisense transcripts.
Array validation
As we have designed and used a new expression platform, we compared our array results with RNAseq profiling on the same 12 tissue samples. In agreement with numerous reports, both RNAseq and the array platform showed that nc transcripts generally are lower expressed than pc transcripts (Supplemental Fig. S1A,B). We found that pc transcripts generally agreed well on the two platforms (tissue-wise correlation test, median Spearman's rho [msr] = 0.70; P for all tissues <2 × 10−16) as did highly expressed nc transcripts (expression > 5.5 in all tissues; n = 235; msr = 0.44; P for all tissues <4 × 10−5). However, nc transcripts with low expression (<5.5 in all tissues, n = 5,201) correlated less (msr = 0.16; P for all tissues <2 × 10−9) (Supplemental Fig. S1E–G). Short transcripts (<200 nt) were particularly problematic on the RNAseq platform because FPKM expression is overestimated for short transcripts. This is indicated by a clear correlation between transcript length and expression for short transcripts in the RNAseq platform, not present for the array platform (Supplemental Fig. S1C,D). We conclude that the platforms agree for highly expressed transcripts. However, for lowly expressed transcripts, as most nc transcripts in our setting, the benefits of replicates and the absence of length bias for short transcripts makes the array platform better suited, which has also been concluded by others (Derrien et al. 2012). In the subsequent expression analysis we thus use the array platform.
Expression analysis
We identified expressed nc transcripts using two strategies. First, we tested all transcripts for differential expression between the tissues by fitting data to a linear model testing for equal means among the tissues. Second, because we were also interested in absolute expression across tissues, we tested transcript expression against a set of artificial nonexpressed genes based on a set of nontarget probes (see Materials and Methods).
Differential expression was found much more frequent for pc transcripts (33%) than for nc transcripts (3%). Although we see a tendency for highly expressed transcripts to be more differentially expressed, this difference persists when comparing similarly expressed transcripts, showing that lack of power for lowly expressed nc transcripts is not the main reason (Supplemental Fig. S4A). Slightly more antisense transcripts (5.3%) than intronic (3.1%) and intergenic transcripts (2.7%) were differentially expressed (Fig. 2D). In comparison, the degree of differential expression was higher for both the known structural RNAs (11%) and the known lncRNAs (28%). Also, the different sources of transcripts show some variation in differential expression frequency, with the UCSC set (5.9%) being higher than ENCODE-defined expressed regions (4.4%), RefSeq (1.3%), UCEs (1.2%), and EvoFold predictions (0.8%).
We next used heatmaps to visualize the expression profiles of the differentially expressed nc transcripts and noted that some tissues stood out (Fig. 2F–I). First, remarkably many intergenic transcripts are expressed predominantly in brain tissue. Out of 173 differentially expressed intergenic transcripts, 59 (34%) show highest expression in brain. This fraction dramatically exceeds that of intronic transcripts (5%) and antisense transcripts (9%) (Fig. 2G–I), showing that intergenic nc transcripts are more frequently expressed in brain than in the other tissues. Second, a major fraction of antisense transcripts are expressed predominantly in heart and muscle tissues (Fig. 2I). Out of 65 differentially expressed antisense transcripts, 25 (38%) show the highest expression in heart and muscle, contrasting with 4% for intergenic and intronic transcripts. However, out of these 25 transcripts, 23 are antisense to the titin pc gene (TTN), coding for TITIN, a well-studied structural component of contractile machinery and expressed in striated muscle tissue. The gene has more than 300 exons and spans over 280 kb, with numerous alternatively spliced transcript isoforms, and encodes the largest known protein. There are 36 putative transcripts antisense to titin present on the array, out of which the 23 differentially expressed correlate with titin expression. In general, we found that the differentially expressed antisense transcripts correlate positively with the pc transcript of the opposite strand (Fig. 2E). Negative correlation, which could be expected if an antisense transcript suppressed expression of the pc transcript, was seen only for one transcript (collapsin response mediator protein 1) and with modest correlation (Spearman's rho = −0.37; P = 0.025).
We next compared the expression signal of transcripts with those of background probes, not targeting any transcripts, and used this to decide whether a transcript is significantly expressed in a tissue. A transcript was considered expressed if it scored above a tissue-specific expression threshold, which was defined so that only 5% of artificial transcripts with randomly assigned background probes were expressed in any tissue (see Materials and Methods). Using this approach, we found that 3720 of all 12,115 nc transcripts included in the analysis (∼30%) are expressed in at least one tissue. This number includes 85% of the differentially expressed nc transcripts. By comparison, a much higher fraction of pc transcripts are expressed in one or more tissues (n = 6,138; 90%), including all of the differentially expressed pc transcripts. The use of separate analysis of expression and differential expression allows for cases where the expression analysis lacks power to call significance despite significant differential expression. This happened for some of the lowly expressed, differentially expressed nc transcripts (15%), but not for any of the pc transcripts, showing the two types of analysis to be overall consistent. The known sets of nc transcripts are expressed at intermediate levels, with 56% of the structural RNAs and 77% of the known lncRNAs being expressed. Again, the fraction of expressed transcripts vary between sources, with the highest fraction seen for the UCSC (57%) and RefSeq (58%) transcripts, followed by the Cabili set (48%), ENCODE expressed regions (19%), UCEs (6.0%), and, finally, EvoFold predictions (4.6%).
Similarly to the differential expression analysis, the expression analysis shows a higher fraction of intergenic transcripts with tissue-specific expression patterns, i.e., expressed in few tissues, when compared with both intronic and antisense transcripts and, in particular, pc transcripts (Fig. 3). A similar finding has been observed for intergenic lncRNAs (Cabili et al. 2011). Conversely, intronic transcripts appear to be more broadly expressed, with a larger fraction expressed in all tissues and a profile more reminiscent of pc transcripts (Fig. 3A,C). We note that a similar expression profile was found among the tissues when considering all pc transcripts based on the RNAseq data (Supplemental Fig. S4B). The pattern with numerous antisense transcripts expressed in both heart and muscle can also be observed here (Fig. 3D). By comparison, a higher fraction of known structural RNAs are ubiquitously expressed (25%, Supplemental Fig. S7), and few (2.5%) are expressed tissue specifically. For all sources of transcripts except EvoFold, the highest number of tissue-specific transcripts was found in brain (Supplemental Fig. S7), thereby extending earlier findings for intergenic lncRNAs to a broader class of nc transcripts. For EvoFold, brain had the second highest number of tissue-specific transcripts, exceeded by breast tissue.
FIGURE 3.

Tissue-specific expression estimates. (A–D) For each transcript category, the number of genes expressed above the background level is given for each tissue (see Materials and Methods). The expressed genes are further stratified by their tissue specificity (gray scale), which shows that nc transcripts are more often tissue specific (white) than pc transcripts, which have the highest fraction of ubiquitously expressed transcripts (black).
Conservation analysis
Functional genomic regions are generally under purifying selection, which slows down the rate of substitution, resulting in conserved elements. Base-level conservation and the presence of conserved elements thus reveal regions of biological function. The function may reside in the DNA (e.g., a transcription-factor binding site), in RNA (e.g., an RNA–RNA interaction), or at the protein level (e.g., a catalytic domain). Function may also reside at several levels at the same time, as would be the case for functional antisense transcript overlapping pc regions. It is worth noting that recent evolutionary innovations, such as an nc transcript recently acquiring a function, may not have significantly influenced the overall substitution rate, and therefore not be predicted as conserved despite being functional. In contrast, regions that recently lost their function may still be designated as conserved.
In our effort to determine the functional evidence for nc transcripts, we evaluate the fraction of transcript bases found in PhastCons vertebrate conserved elements (Siepel et al. 2005), thus assigning a score to each between zero and one. Generally, the nc transcripts are less conserved (mean = 0.38) than the protein-coding transcripts of the array (mean = 0.54). There is a slight bias for higher conservation of the array pc transcripts over all pc transcripts (mean = 0.51, Supplemental Fig. S4C). The different classes of nc transcripts show considerable variation, with antisense (mean = 0.47) and intronic transcripts (mean = 0.47) showing the highest level of conservation, followed by intergenic (mean = 0.30). There is also much variation between the different sources of nc transcripts. The UCEs and the EvoFold predictions are by construction fully conserved. The UCSC (mean = 0.14) and RefSeq lncRNAs (mean = 0.13) are more conserved than the Cabili set (mean = 0.06). In comparison, the known functional lncRNAs are more conserved (mean = 0.15), albeit much less than structural RNAs (mean = 0.55). We note that the structural RNAs have a bimodal conservation profile because many snoRNAs have zero overlap with conserved elements. These nonconserved snoRNAs are generally intergenic or found in large introns and thus far away from pc exons (median exon distance = 2178 nucleotides), which is in contrast to the conserved snoRNAs (median exon distance = 113 nt). This pattern could either be explained by pc exons anchoring and improving genomic alignments, and hence bias conservation measures, or by snoRNA pseudogenes being wrongly included in the known sets.
Selection to maintain function, not only slows down the rate of substitution, it also slows down the fixation rate for other mutational processes, such as the rate of chromosomal rearrangements. Rearrangements that displace one exon from another will likely disrupt function, as will rearrangements that displace regulatory elements from a gene locus. The absence of rearrangements, and hence the conservation of presence and order along the genomic sequence, is often called syntenic conservation. In contrast to base-level conservation, syntenic conservation may act on nonfunctional regions between functional elements, if they must stay together to maintain functionality overall. This is likely often the case for lncRNAs, which appear to often have a modular structure with functional, conserved domains interspersed by neutrally evolving regions (Guttman and Rinn 2012). Similarly, host genes that encode snoRNAs or other ncRNAs in their introns comprise another class of nc transcripts likely to be syntenically conserved, yet show little base-level conservation in their exons. We therefore consider the presence of syntenic conservation a relevant measure for nc transcripts. One caveat is that syntenic conservation does not provide the same resolution as base-level conservation, as chromosomal rearrangements are infrequent compared with base-level mutations.
As a measure of syntenic conservation, we evaluate the fraction of bases in a transcript that are syntenically preserved in other species of mammals and vertebrates. The analysis was based on the pairwise genomic alignments between human and chimpanzee, mouse, rat, dog, chicken, and pufferfish (Tetraodonnigroviridis), as given by the Chains and Nets of the UCSC Genome Browser (Kent et al. 2003). As a control set, the analysis was also carried out on transcripts with shuffled coordinates.
Overall, pc transcripts show high levels of syntenic conservation, with 88% (n = 6002) fulfilling that at least half of the human bases are syntenically conserved (found in a single pairwise alignment chain) in each of the other mammals. This drops to 23% (n = 1566) when including fish and pufferfish in the analysis. By comparison, in the shuffled set of pc transcripts only 19% (n = 1308) fulfill this criterion in mammals and 0% (n = 5) across all species. For the nc transcripts, we leave out the UCE and EvoFold predictions, which are deeply conserved by construction (95% fulfill the criterion across the mammals and 33% in all the species). Of the remaining nc transcripts, 55% (n = 5457) fulfill the criterion for mammals and 4.1% across all species, which is well above the rates for shuffled transcripts (23% for mammals and 0.6% across all species). When requiring syntenic conservation of 90% of the human bases across mammals, nearly half of pc transcripts (45%) and 27% of nc transcripts (omitting EvoFold and UCE) still fulfill this.
The number of transcripts passing a criterion for syntenic conservation should be regarded as a lower bound, given that incomplete assemblies will cause some transcripts to falsely fail the criterion. In addition, a criterion is easier to pass for short than for long transcripts, which introduce a bias against pc genes. However, for a given transcript, deep syntenic conservation is an indication of functional preservation.
It is interesting to note that for the set of known lncRNAs, 60% (n = 21) overlap with more than half of the bases in all mammals, and 17% (n = 6) also at a threshold of 90%, yet none are syntenically conserved to chicken or pufferfish. This may be explained by many known lncRNAs being mammalian-specific innovations. The structural RNAs are more syntenically conserved with 77% (n = 248) fulfilling that at least 50% of the bases are conserved across the mammals and 34 (10%) across all the species, which is also expected given that these are largely composed of short single exon RNAs.
Presence of antisense transcripts correlate with excess conservation in protein-coding regions
In protein-coding regions, changes between synonymous codons, coding for the same amino acid, are often assumed to be neutral, as they cannot directly affect the protein product. However, other levels of functionality may be encoded by the same DNA, such as exonic splicing enhancers/silencers or transcriptional enhancers, which may constrain the evolution of synonymous base positions and lead to an overall excess in base-level conservation compared with regions only coding for protein.
To evaluate whether selection acts on the subset of antisense nc transcripts, we annotate them with the base fraction that overlaps a recently published set of mammalian synonymous constrained elements (SCEs; n = 10,575) within all consensus coding sequences (CCDS) (Pruitt et al. 2009a; Lin et al. 2011). We found 72 of 706 (10.2%) antisense transcripts overlapped SCEs (and CCDS), with an overall base-overlap of 3.2% compared with 2.8% (P < 2 × 10−16, Fisher's exact test) for the background set of all CCDS exons that the SCEs were based on (Fig. 4C). When extending this analysis to the set of GENCODE antisense transcripts (Harrow et al. 2006), we found a similar level of overlap with SCEs (3.1%, P = 2 × 10−13, Fisher's exact test), supporting the generality of the finding. There is much variation in SCE conservation of individual antisense transcripts, with 19 being >50% overlapped. Seven of these were among 16 antisense transcripts both expressed and overlapping a SCE (Supplemental Table S2). Though the included antisense transcripts are from different sources with variable amounts of evidence (Chen et al. 2004; Engström et al. 2006; Ge et al. 2008), these results suggest that some are under purifying selection, and hence functional. An example of a conserved antisense transcript with expression evidence is found in POU4F2 (Fig. 6F,E, below). It is highly expressed in skin, whereas the pc gene is not differentially expressed, showing that the antisense expression is not merely a noisy byproduct of regional transcription.
FIGURE 4.

Conservation of expressed noncoding RNA transcripts. (A,B) Box plots of transcript conservation for intergenic (A) and protein-coding transcripts (B). Each box represents transcripts expressed in the given number of tissues. (C) Distribution of bases overlapping synonomous constraint elements (SCE) among the background set of bases overlapping the human consensus coding sequences (CCDS). The fraction that overlaps with antisense (AS) ncRNAs is indicated. (D,E) Average overlap with chained alignments in seven species for intergenic (D) and protein-coding transcripts (E). Transcripts were stratified by tissue specificity of expression (gray scale). Red lines indicate similar overlap values for corresponding transcripts with shuffled genome coordinates. UCE and EvoFold predictions were omitted from this analysis (see text for details). (F–I) Gene body (F,G) and promoter (H,I) overlap with ChromHMM epigenetically derived functional elements. Fraction of transcripts overlapping regions with epigenetic marks associated with either transcription (TXN) or transcription start sites (TSS) are given for each tissue for both intergenic nc transcripts (F,H) and pc transcripts (G,I). The overlap is evaluated for both expressed (red) and nonexpressed (cyan) transcripts as well as shuffled control sets (orange and blue) (see Materials and Methods). Over-representation P-values (Fisher's exact test) for expression and overlap are given for each tissue.
FIGURE 6.

Examples of expressed noncoding transcripts. (A) Genomic context of EvoFold prediction 36852, with breast tissue RNAseq read depth given below. (B) Structure of EvoFold 36852. (C,D) Expression profiles and genomic context of transcripts located in and near the DACH1 locus including UCE 352 (red). Bars represent triplicate microarray expression means for each tissue (legend at bottom) with standard error bars. (E,F) Antisense transcript located in exon of POU4F2. Syntenic conservation in seven species is shown below (chained alignments in black) together with base-level conservation (gray). (F) Microarray expression profiles as in D. (G) Intergenic lncRNA transcript with syntenic alignments for cow, mouse, and rat shown below. (H) Expression profile of transcripts from F.
We note that it is plausible that some antisense transcripts function through complete base-complementarity to pc sense transcript, i.e., functionally similar to endogenous siRNAs (Watanabe et al. 2008). However, such complementarity does not cause additional constraint by itself, as an antisense transcript is complementary to its sense counterpart by definition.
Expression of nc transcripts correlate with conservation
We next compared the expression and conservation evidence. The EvoFold and UCE classes, which are fully conserved by construction, were omitted as they would introduce a bias and are instead analyzed separately below. When focusing on the top 25% highest expressed nc transcripts, we found a higher overlap with conserved elements (mean = 0.42) compared with the full set (mean = 0.25), and in particular, compared with the lowest expression quartile (mean = 0.09). Generally, we found base-level conservation to correlate positively with expression levels for both pc transcripts (Spearman's rho = 0.20; P < 2 × 10−16) and nc transcripts (Spearman's rho = 0.52; P < 2 × 10−16) (Supplemental Fig. S2A,B). We did see a weaker correlation also for the structural set (Spearman's rho = 0.27; P = 1 × 10−6), but for the 35 known lncRNAs, we did not observe it.
A correlation between pc transcript expression and base-level conservation is well supported and has been reported for several species (Pál et al. 2001; Krylov et al. 2003; Subramanian and Kumar 2004; Drummond et al. 2006). For nc transcripts, this has only recently been observed (Managadze et al. 2011). In addition, we also observed a positive correlation between the number of expressed tissues (tissue specificity) and the conservation level for both pc and nc transcripts (Fig. 4A,B). To exclude that the correlation is an artifact of transcript size, e.g., if highly expressed transcripts have higher probability of overlapping conserved elements by chance because of length or exon structure, we calculated P-values for base-level conservations by shuffling the location of each transcript in the genome up to 1,000,000 times, which confirmed the trend for both pc and nc transcripts (Supplemental Fig. S5). Furthermore, the trend was also observed for all pc transcripts based on RNAseq data (Supplemental Fig. S4D).
Given that spliced lncRNAs have a particularly low conservation profile and show little correlation between expression and conservation, we looked in more detail at the conservation profile of known, functional cases. We found that there is often much variation in conservation levels between exons, with a single or a few exons explaining the overall conservation levels (Fig. 5A). Inspired by this, we focused on exon-level conservation for spliced ncRNAs as well as pc transcripts. In both cases we saw a similar trend of broadly expressed transcripts being more conserved (Fig. 5B).
FIGURE 5.

Overview of transcript annotations and exon conservation. (A) Table of exon conservation of four known, functional lncRNAs. (B) Mean maximal exon conservation across all exons as well as mean conservation for exon number one, and two for nc transcripts (left) and pc transcripts (right) stratified by tissue specificity. (C) Table summarizing annotations statistics for pc transcripts, the three classes of nc transcripts and two known nc sets. The statistics are divided into three main types: expression (red), conservation (blue), and epigenetic marks (green). Numbers represent counts of transcripts overlapping a given annotation (ESTs and epigenetic marks) or fulfilling the criteria of max exon conservation >0.5 and syntenic P-value measure <0.01 (see text for details). Fraction of transcripts in a given category fulfilling the given annotation criteria is given in parenthesis, followed by FDR estimates either defined by the inference method (array expression) or by genomic shuffling (all other annotations). (D) Venn diagrams showing number of transcripts fulfilling at least one criteria within each of the three main types. Colored as in C.
As for base-level conservation, the top 25% highest expressed nc transcripts are more syntenically conserved compared with both the full set and the lowest expressed quartile. This was found in all species except in chimp, where the level is equal because of the high overall syntenic conservation to human. Moreover, the level of syntenic conservation increased consistently across expression quartiles in all species (except chimp), with the strongest trend for the most distant species (Supplemental Fig. S2C,D). We made a similar observation when transcripts were stratified by number of tissues they were expressed in (Fig. 4D,E). Because of the bias for long transcript mentioned above, we assigned a P-value to the observed measure of syntenic conservation for each transcript in each species. The P-value records the number of shuffled transcripts with a measure of syntenic conservation that is equal or higher than the true transcript, thus normalizing for the effect of transcript length and structure. For a given transcript, the minimum P-value across species was used to summarize the level of syntenic conservation. When using these P-values to quantify the level of syntenic conservation, we again saw a positive correlation with expression (0.09, P = 4 × 10−10, Pearson, Supplemental Fig. S5C,D). Insignificant P-values may merely reflect lack of power to reject nonsyntenic evolution, which may often be the case for short transcripts. For this reason, we use this measure only as a positive selection mark when defining sets of transcripts below.
We finally evaluated the degree of conserved expression by calculating overlaps of human and mouse expressed sequence tags (ESTs) for individual transcripts (Supplemental Fig. S6A,B). For the mouse EST overlap, transcripts were first lifted to mouse coordinates when possible. The overlap was higher for expressed transcripts compared with nonexpressed transcripts for both human (P = 4.4 × 10−16) and mouse (P = 2.4 × 10−7, Fisher's exact test).
Collectively, these results reveal positive correlations between expression and conservation. For pc transcripts, this pattern has been explained by a restricted codon usage to avoid translation errors and resulting toxicity for highly expressed transcripts (Drummond and Wilke 2008, 2009). We note that this explanation cannot be used for nc transcripts, which are not translated. Likewise, the increased syntenic conservation of more expressed pc transcripts cannot be explained by restricted codon usage only. Instead, we speculate that the expression in multiple tissues, with each their own unique molecular environment, could be coupled to more molecular interactions, and hence, a higher accumulated selection pressure.
Expression of ultraconserved elements and predicted structural RNAs
In contrast to the majority of the nc transcripts (regions) included on the array, the UCEs and the EvoFold structural RNA predictions were included based on their evolutionary signatures instead of prior expression evidence. We therefore separately analyzed the expression evidence for these.
The UCEs were defined as at least 200-bp long regions that were completely conserved between human, mouse, and rat (Bejerano et al. 2004). As such, they are examples of regions of extreme conservation, explained by overlapping levels of functionality in some cases (Bejerano et al. 2004). The array includes representations of both strands of all UCEs not overlapping pc exons (n = 2×332). We found that 40 were significantly expressed (Supplemental Table S3), with only one strand being expressed in most cases. An example is an UCE (uc.352) specifically expressed in skin from one strand only and located in a gene desert ∼200 kb upstream of the DACH1 gene (Fig. 6C,D). This UCE has previously been found to be transcribed and to be differentially expressed in leukemia (Nobrega et al. 2003; Calin et al. 2007). We note that DACH1 is on the same strand as the UCEs, but it is not differentially expressed. The elements were generally (28/40) expressed in one tissue, although seven were expressed in six or more tissues, which contrasts with previous findings of UCEs being ubiquitously expressed (Calin et al. 2007).
The EvoFold structural RNAs were predicted by comparative analysis of 31 vertebrate genomes (Parker et al. 2011). We here included only the small subset of long structures (>60 bp) and only the predicted strand. We found 67 of 1599 predictions to be expressed (Supplemental Table S4), which should be considered a lower bound given that the EvoFold strand prediction is uncertain (Pedersen et al. 2006). A few (n = 5) are expressed across most tissues, though most are only expressed in one tissue (n = 46).
The substitution evidence for the EvoFold predictions, as measured by the EvoP measure (Parker et al. 2011), correlates significantly with expression evidence (P = 0.03, Kolmogorov-Smirnov test; Supplemental Fig. S3). This strongly supports the presence of structurally functional nc transcripts, as substitution and expression evidence are independent. One such expressed EvoFold prediction (EvoFold.36852) with intermediate support (pEvoP = 0.374) is found in the fourth intron of O-linked GlcNAc transferase (OGT), a gene involved in protein glycosidation (Fig. 6A,B). The EvoFold prediction is significantly expressed in breast, colon, and lung, and the same intron also harbors a neighboring expressed UCE (uc.475) 600 bp downstream. Both the EvoFold prediction and the UCE are expressed in the same orientation as the OGT gene. Visual inspection of the mapped RNAseq data reveals demarked boundaries in read depth corresponding closely to the ends of both the EvoFold prediction and the UCE. We note that the UCE also contains two EvoFold predictions (36853 and 36854), both too short (<60 nt) to have been included on the array. This suggests that short nc transcripts potentially originate from the intron of the pc transcript of OGT.
Chromatin state marks as functional indicators
Chromatin state marks provide information on the transcriptional states of genes and have been used successfully to identify intergenic lncRNAs (Guttman et al. 2009). Large consortia have recently made genome-wide maps of chromatin state marks available, facilitating their use in gene analysis. We here used tissue-specific ChromHMM-based functional segmentations of chromatin marks from the Epigenetics Roadmap Project, matching nine of our 12 tissue types (Bernstein et al. 2010; Ernst and Kellis 2012). From these we extracted genomic regions classified as transcribed regions (TXN) or as harboring transcriptional start sites (TSS) based on their epigenetic profile.
We evaluated the overlap with the TXN regions for the full length of all transcripts and between the TSS regions and transcript promoters, which were defined as 500 bp upstream of the transcript start site. For nc transcripts, we focused on intergenic nc transcripts to reduce signal interference from pc genes. Both pc and intergenic nc expressed transcripts were found to have significantly higher overlap with TXN regions than nonexpressed transcripts (Fig. 4F,G). This was more pronounced for pc transcripts and was not observed for a shuffled set of transcripts (see Materials and Methods). For expressed intergenic nc promoters the overlap with the TSS regions was generally even higher than the overlap of their body with the TXN regions (Fig. 4H,I).
We further overlapped the promoter regions with ENCODE tissue-specific maps of DNAse hypersensitivity sites (DHS) (Thurman et al. 2012), which are known to be associated with active promoters, again matching nine of the 12 tissues (Supplemental Fig. S6C,D). Again, for all tissues, expressed intergenic nc transcripts shoved a higher frequency of overlap than nonexpressed transcript. For pc transcripts, the pattern was the same, although the overlapping fraction was about twice as big.
The overlap with both chromatin state marks and DHS were significantly enriched for expressed pc as well as nc transcripts, but not for their respective shuffled counterparts. This signifies a similar regulation of ncRNAs and pc transcripts, and thus indicates that chromatin state marks are useful as functional indicators. The much higher number of overlapping transcripts for pc transcripts may be ascribed to the lower expression levels or a dilution of the signal by nonfunctional nc transcripts.
Sets of expressed and conserved nc transcripts
In the preceding analysis we have annotated all transcripts with statistics for expression, conservation, and overlap with epigenetic marks (Supplemental Table S1). Here we identify sets of ncRNAs for which these statistics lend the strongest functional support. The classes of UCEs, EvoFold predictions, and antisense transcripts are omitted, given their special conservation properties and as they have already been ranked and shortlisted (Supplemental Tables S2–S4).
We divide the statistics into three types: (1) expression, which consists of our microarray expression estimates as well as overlap with human and mouse ESTs; (2) conservation, which consists of base-level conservation and P-values of syntenic conservation measures; (3) epigenetic marks, which consists of overlap with the ChromHMM defined functional TXN and TSS regions (see Materials and Methods). We have summarized these statistics for all analyzed transcripts divided into their main categories (Fig. 5C). For each category we evaluated the fraction of transcripts fulfilling a given annotation criteria as a measure of sensitivity and the FDR estimated by genomic shuffling of transcript locations as a measure of specificity (Fig. 5C). The FDR for microarray expression could not be estimated by genomic shuffling, but was previously fixed at 5% overall (see Materials and Methods). For pc transcripts, 83% fulfill at least one criteria for all the three annotation types, compared with 9% for intergenic nc transcripts, 12% for intronic nc transcripts, and 39% for antisense transcripts (Fig. 5D).
The known functional nc sets generally fulfill more of the annotation criteria than the uncharacterized nc transcripts, however, were still less than the pc transcripts (Fig. 5C). Peculiarly, relatively few known structural transcripts have promoters overlapping the epigenetically inferred TSS regions, which may be explained by the presence of internal RNA polymerase III promoters or multicistronic origin from larger transcripts (Schramm and Hernandez 2002; Li et al. 2010). We observed significant variation in FDR estimates across transcript categories for some annotations (e.g., TXN overlap), illustrating a need for category-specific thresholds to achieve comparable FDRs (Fig. 5C).
For the shortlisting of intergenic and intronic lncRNA transcripts with the strongest functional support, we use all three types of annotation statistics. We define category-specific thresholds such that the estimated FDR is at most 5% by introducing and adjusting thresholds to overlaps and the synteny P-value measure (see Materials and Methods). By requiring that a lncRNA transcript must fulfill the specified threshold for at least one annotation of each of the three types, we shortlisted 132 from the Cabili set (5%), 151 of the UCSC transcripts (8%), and 12 of the RefSeq transcripts (4%). These are ranked by their sum-of-ranks for the individual annotation statistics and provided with their full set of annotations (Supplemental Table S5). For comparison, we found six transcripts from the known lncRNA set (17%) and nine of the known structural RNAs (3%) that fulfill the same criteria.
For the expressed regions, 451 (11%) fulfill the same set of category-specific selection criteria. We note that this set is particularly challenging to analyze because we do not have the complete transcript structures. Again, we provide a ranked table with the full set of annotations (Supplemental Table S6).
One of the criteria used in the selection is syntenic conservation to other species, which appear stringent and highly dependent on alignment quality, and therefore challenging. This is exemplified by a lncRNA that is syntenic to chimpanzee, dog, and rat, yet fails to fulfill the synteny criterion of all exons falling in the same alignment chain for mouse and cow (Fig. 6G,H). In mouse, however, all exons are conserved in the same order at another locus on chromosome X, which suggests a high-level chromosomal rearrangement preserving synteny. The use of a stringent synteny threshold thus risks introducing false negatives on the basis of such rearrangements.
DISCUSSION
With the aim of defining sets of human nc transcripts enriched for functional potential, we evaluated 12,115 nc transcripts for their levels of expression, conservation, and overlap with epigenetic marks. Based on these results we have narrowed in on concrete sets of nc transcripts that fulfill criteria inspired by known nc transcripts, which may serve as candidates for further functional characterization. The sets are divided into EvoFold predictions, UCEs, antisense transcripts, lncRNAs, and expressed regions, and are provided with all the generated annotations in Supplemental Tables S1–S6. In addition, we provide access to all transcripts through a mirror of the UCSC genome browser (http://mmn.moma.ki.au.dk).
Ideally, it is desirable to translate the evaluated expression, conservation, and epigenetic evidence into functional evidence. However, because there is no simple translation, this leads to both false positives (nc transcripts predicted to be functional, yet are not) and false negatives (true functional nc transcripts failing to be predicted). Expressed pseudogenes, which could represent functional molecules, but could also be rudimentary transcription of regions with lost functionality but apparent conservation, may be a source of false positives. Conversely, false negatives may result from conserved regions failing to be transcribed in a given experimental setting, although transcribed and functional in others, e.g., transcripts expressed only during development. Also, it is possible that a transcript is functional albeit not conserved, e.g., if functionality is based on providing spacing regions without specific structural requirements or if a transcript function has emerged recently. Similarly, a transcript may be located in a region that is transcribed on the opposite strand and thus be associated with epigenetic marks that wrongly support its transcription.
Incomplete annotations and technology-related issues also contribute to false predictions. Most nc transcripts are weekly expressed, which introduces uncertainty of expression estimates. This can, in turn, lead to both false positives and false negatives. Also, hybridization errors may lead to false expression estimates for array platforms. We have tried to minimize false-positive predictions by eliminating nc transcripts with probes that either overlap pseudogenes or that show homology to pc transcripts from the analysis (see Materials and Methods). Our filters reduce the set of analyzed transcripts to 46% of the original amount present on the array, which likely introduce some additional false negatives. Current gene models may not accurately reflect transcript boundaries or exon structure of the expressed isoforms, which may often be the case for expressed regions inferred based on ENCODE expression data. Many of these short transcripts may eventually prove to be part of longer transcripts. In effect, conservation profiles may not be based on the true full-length transcripts and introduce biases in relation to our criteria.
We observed expression of ∼30% of the nc transcripts compared with 90% of the pc transcripts. Although the set of pc transcripts is not complete, and may be biased for differential expression, the finding is in line with pc transcripts having higher expression than nc transcripts, as also reported previously (Ravasi et al. 2006; Nakaya et al. 2007; Dinger et al. 2008; Guttman et al. 2010; Cabili et al. 2011). We found intergenic nc transcripts to have a more tissue-specific expression profile compared with pc transcripts. Notably, far more intergenic nc transcripts are expressed in brain relative to other tissues, in agreement with earlier reports (Mercer et al. 2008, 2010; Ponjavic et al. 2009).
We found a general correlation of expression with both base-level conservation and syntenic conservation. For base-level conservation, this has been observed previously for pc transcripts (Krylov et al. 2003; Subramanian and Kumar 2004; Drummond et al. 2006) and, recently, also for nc transcripts (Managadze et al. 2011). The current model for explaining this phenomenon for pc transcripts is based on minimizing toxic effects of misfolded proteins due to high error rates in the translation process, leaving translation errors in 15% of a median-sized protein (Drummond and Wilke 2008, 2009; Wolf et al. 2010). Thus, translation errors will have higher toxic impact for abundant proteins, and evolutionary analysis has shown that error-minimizing triplet codons are selected for in abundant proteins (Drummond and Wilke 2008). Since nc transcripts are not translated, this model cannot explain the correlation for nc transcripts. Although it is intriguing to speculate that misfolded nc transcripts will lead to cellular toxicity, RNA polymerases have error rates orders of magnitudes lower than translation. Moreover, minimizing this error rate by base-level selection in a transcribed region is hard to imagine.
We also note that this model does not explain the similar correlation between expression and syntenic conservation for either nc or pc transcripts. Based on our findings, we propose that tissue specificity may correlate with functional diversity. A transcript expressed in many tissues is exposed to a more diverse molecular environment, which may lead to functional adaptations as well as added structural constraint, and hence conservation.
EvoFold predictions and UCEs both have high-conservation levels by construction and thus likely represent functional regions in the genome. We found significantly less expressed UCEs (n = 40) than was reported previously in human tissues where 962 UCE were found expressed and 325 (34%) ubiquitously expressed (Calin et al. 2007). We note that apart from platform differences, a lower number of tissues in this study combined with higher stringency from the use of replicates and multiple probes likely contribute to explain this. We found only 67 expressed EvoFold regions out of the 1599 included long predictions (4.2%). Presence of structure may preclude hybridization to array probes, which may contribute to the low-expression rate (Cheng et al. 2005), although we observed a similar expression rate in the RNAseq data. An increase in substitution evidence in this set compared with the background indicates selective pressure for structure conservation among them.
In conclusion, our study provides multiple lines of evidence for correlation between functional indications along with a rich set of annotations for a large set of nc transcripts. Correlation of expression with conservation and epigenetic marks shows that observed expression is not merely random transcriptional noise. We have identified sets of nc transcripts of different classes enriched for functional indications, and it is our hope that these results will be a resource and help in directing focus on functional studies of nc transcripts.
MATERIALS AND METHODS
Array design
The Nimblegene HD2-12 platform (135K 60 mer probes) was used to design an array containing probes against 26,910 nc transcripts and 6856 pc transcripts.
Noncoding transcripts represented on the array were collected from a number of sources:
6614 nc transcripts from the UCSC gene set (Hsu et al. 2006).
2711 additional nonoverlapping nc transcripts from the RefSeq database (Pruitt et al. 2009b).
4004 lncRNAs from an early version of the set defined in Cabili et al. (2011), kindly provided by John Rinn and Aviv Regav.
829 nc transcripts from the Rfam, snoBase, and tRNAscan databases that were at least 60 nucleotides long (Griffiths-Jones et al. 2003; Schattner et al. 2005; Xie et al. 2007).
175 mixed nc transcripts from ncRNAdb (Szymanski et al. 2007).
1425 antisense nc transcripts from the literature (Chen et al. 2004; Engström et al. 2006; Ge et al. 2008).
684 ultraconserved regions outside known gene context (Bejerano et al. 2004).
12,826 consistently expressed nc genomic regions with conserved element overlap of 5% or higher and at least 100 nucleotides long inferred from stranded ENOCDE RNA-seq and CAGE experiments (Birney et al. 2007).
2528 EvoFold predictions of genomic regions with evolutionarily conserved RNA secondary structure longer than 60 nucleotides (Pedersen et al. 2006; Parker et al. 2011).
The 6856 pc genes on the array are from the National Cancer Institute cancer gene index, as well as an additional set of individually selected mostly cancer-associated genes (n = 200).
Transcript filters
The microarray technology is inherently sensitive to cross-hybridization, resulting in a false-positive expression signal. Thus, nc transcripts were applied to various filters to avoid cross-hybridization issues. The following filters were applied to all probes of nc transcripts on the array:
All probes were aligned to all protein-coding mRNAs (UCSC Known Genes) (Hsu et al. 2006) using BLAST and probes with E-scores below 1 × 10−10 failed.
Probes overlapping a genomic region with more than 10 human chained self-alignments (Kent et al. 2003).
Probes overlapping regions with mitochondrial homology.
Probes overlapping repeatMask regions.
The following three filter rules were subsequently applied to all nc transcripts:
Nc transcripts with any probe failing filter 1 were discarded.
Nc transcripts with no probes passing filters 2, 3, and 4 were discarded.
Nc transcripts overlapping pseudogenes defined by GENCODE (V12) were discarded.
Collectively, this reduced the number of analyzed transcripts from 26,910 to 12,115.
Sample preparation
Total RNA (150 ng) from a panel of 12 human tissues (bladder, brain, breast, colon, heart, kidney, liver, lung, muscle, ovary, prostate, and skin, BioChain) was processed using a Low Input Quick Amp WT Labeling Kit (Agilent), and 600 ng labeled product was hybridized to slides using a NimbleGen Hybridization Kit (cat. no. 05 583 934 001), then hybridized for 16 h at 42 degrees. This was done by DTU Multi Assay Core facility at the Center for Biological sequence Analysis, Technical University of Denmark. Slides were scanned using a Roche NimbleGen MS 200 scanner and data extracted with NimbleScan v.2.6.
Expression analysis
Array data was analyzed in R (www.r-project.org). Arrays were normalized using the RMA implementation of the oligo software package (Irizarry et al. 2003) and subsequently analyzed using the LIMMA package (Smyth 2004). For differential transcript expression analysis, a linear model was fitted to the expression data:
Ei,j = µj + ɛi,j, where i is the index for replicates and j is the index for tissues. E is the measured expression (log2), µ is the tissue mean, and ɛ is the error.
P-values were calculated for all transcripts based on the null hypothesis.
H0: µ1 = µ2 = … = µ12, where numbers represents the twelve tissues.
Subsequently, P-values were corrected for multiple testing (Benjamini and Hochberg 1995). Differentially expressed transcripts were defined by a threshold of 0.05 on the adjusted P-value.
For estimation of tissue specific-expression, we constructed a set of 1646 artificial nonexpressed transcripts by randomly distributing 2600 background probes from the array. Arrays including the artificial transcripts were subsequently RMA normalized, which only had a negligible effect on expression values of the original true transcripts. An expression percentile threshold was defined such that 95% of the artificial transcripts fell below it in all tissues, based on the mean of tissue triplicate measurements. Transcripts falling below the threshold percentile were classified as nonexpressed in a given tissue. The data have been deposited in NCBI's Gene Expression Omnibus (Edgar 2002) and are accessible through GEO Series accession number GSE41947.
RNA-Seq library construction and sequencing
Using 500 ng of total-RNA from each of the samples, RNA-Seq libraries were constructed by depletion of rRNA, followed by synthesis of directional, paired-end, and indexed RNA-Seq libraries. The rRNA-depleted RNA was purified using the RNA Clean & Concentrator-5 columns (Zymo Research). Speed-vac was used to reduce the remaining sample volume to 9.5 μL, followed by synthesis of directional, paired-end, and indexed RNA-Seq libraries using the ScriptSeq Kit (Epicentre). Briefly, rRNA-depleted RNA was chemically fragmented, and cDNA was synthesized from a tagged random hexamer. The cDNA was terminal tagged using a 3′-end blocked and tagged oligo, followed by MiniElute (Qiagen) purification. The di-tagged cDNA was then used as template in a 10-cycle PCR reaction. Libraries were purified by agarose gel electrophoresis selecting elements of 200–600 bp in size. The RNA-Seq libraries were denaturated and diluted to 10 pM with pre-chilled Hybridization buffer (Illumina) and loaded into TruSeq PE v3 flowcells (Illumina) on an Illumina cBot, followed by indexed paired-end sequencing (101 + 7 + 101 bp) on a Illumina HiSeq 2000 using TruSeq SBS Kit v3 chemistry.
RNA-seq analysis
Demultiplexed fastq files were generated using Illuminas CASAVA software, followed by analysis using Tophat and Cufflinks (Trapnell et al. 2009, 2010), which were given gtf files of the transcripts on the array. FPKM values per transcript were extracted and used in downstream analysis.
Conservation analysis
Base-level conservation was defined as the fraction of a transcript that overlapped vertebrate-conserved elements defined by PhastCons and downloaded from the UCSC Genome Browser (phasConsElements44way) (Siepel et al. 2005). We calculated P-values for conservation as the fraction of transcripts shuffled in the genome with overlaps equal to or higher than the original case.
The syntenic conservation measure should reveal whether a transcript's exons were preserved and in the same order in another species. We evaluated syntenic conservation of the array transcripts against seven vertebrates (chimpanzee, cow, dog, rat, mouse, chicken, and the pufferfish tetraodon). We calculated the overlap with chained alignments (Kent et al. 2003) between human and the seven species, and calculated a P-value by counting the fraction of shuffled transcript with similar or larger overlap.
Noncoding exon overlaps with SCEs were analyzed by counting all CCDS exons overlapping with both a SCE and nc exons in antisense orientation and applied to a Fisher's exact test.
Chromatin, DHS, and EST overlap statistics
Nine tissue-specific genome-wide maps of chromHMM epigenetically derived functional segmentations were downloaded from the Epigenome Browser at Washington University and overlapped with all transcripts. As a control, all transcripts were shuffled in the genome with the constraint that intergenic transcripts remained intergenic and intronic transcripts remained intronic. Pc and antisense transcripts were shuffled throughout the genome. DHS regions from nine tissues made by the ENCODE consortium were downloaded from UCSC and overlapped with promoter regions defined as 500 bases upstream of transcript start annotation for all transcripts. Human and mouse ESTs were downloaded from UCSC and overlapped with all transcripts and with the shuffled sets. For overlap with mouse ESTs, transcripts were first mapped to the mouse genome, requiring at least 50% base-level mappability.
Transcript selection criteria
Selection of putative functional candidate transcripts was based on a combination of criteria that, in turn, were based on the presented annotations. First, annotations were grouped into three types: expression, conservation, and chromatin marks. Expression annotations include array expression measures as well as overlap statistics with mouse and human ESTs. Conservation annotations include base level and syntenic conservation statistics. Chromatin mark annotations include promoter overlap with chromHMM TSS states and transcript overlap with chromHMM TXN states. For each of these annotations, thresholds were defined so that in each of the transcript categories (e.g., nc intergenic and nc intronic) a FDR of 5% was allowed. For all annotations except expression, the FDR was based on a set of transcripts with similar transcript structure, but shuffled in the genome. For expression annotation, FDR was set to 5% based on background probes (see “Expression analysis” above). Next, transcripts were selected if they passed the given threshold of at least one annotation of each type, i.e., an expression criterion and a conservation criterion and a chromatin mark criterion. For EST overlaps, passing a threshold implied passing both mouse and human EST overlap thresholds.
DATA DEPOSITION
Microarray and NGS data are available at NCBI's Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE41947 and GSE45326.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are grateful to Philippe Lamy and Henrik Hornshøj for helpful technical discussions. This work was supported by The Danish National Advanced Technology Foundation, The John and Birthe Meyer Foundation, and The Lundbeck Foundation.
REFERENCES
- Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick JS 2011. lncRNAdb: A reference database for long noncoding RNAs. Nucleic Acids Res 39: D146–D151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D 2004. Ultraconserved elements in the human genome. Science 304: 1321–1325 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc 57: 289–300 [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. 2010. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 28: 1045–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, et al. 2004. Global identification of human transcribed sequences with genome tiling arrays. Science 306: 2242–2246 [DOI] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff N, Ashworth A, Kay GF, Cooper P, Smith S, McCabe VM, Norris DP, Penny GD, Patel D, Rastan S 1991. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 351: 329–331 [DOI] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL 2011. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25: 1915–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Liu C, Ferracin M, Hyslop T, Spizzo R, Sevignani C, Fabbri M, Cimmino A, Lee EJ, Wojcik SE, et al. 2007. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 12: 215–229 [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. 2005. The transcriptional landscape of the mammalian genome. Science 309: 1559–1563 [DOI] [PubMed] [Google Scholar]
- Chen J, Sun M, Kent WJ, Huang X, Xie H, Wang W, Zhou G, Shi RZ, Rowley JD 2004. Over 20% of human transcripts might form sense–antisense pairs. Nucleic Acids Res 32: 4812–4820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, et al. 2005. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308: 1149–1154 [DOI] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. 2012. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 22: 1775–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinger M, Amaral P, Mercer T 2008. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res 18: 1433–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134: 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO 2009. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet 10: 715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Raval A, Wilke CO 2006. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol 23: 327–337 [DOI] [PubMed] [Google Scholar]
- Edgar R 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engström PG, Suzuki H, Ninomiya N, Akalin A, Sessa L, Lavorgna G, Brozzi A, Luzi L, Tan SL, Yang L, et al. 2006. Complex loci in human and mouse genomes. PLoS Genet 2: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M 2012. ChromHMM: Automating chromatin-state discovery and characterization. Nat Methods 9: 215–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Rubinstein WS, Jung Y-C, Wu Q 2008. Genome-wide analysis of antisense transcription with Affymetrix exon array. BMC Genomics 9: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR 2003. Rfam: An RNA family database. Nucleic Acids Res 31: 439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Rinn JL 2012. Modular regulatory principles of large non-coding RNAs. Nature 482: 339–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. 2009. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458: 223–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. 2010. Ab initio reconstruction of cell type–specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28: 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Denoeud F, Frankish A, Reymond A, Chen C-K, Chrast J, Lagarde J, Gilbert JGR, Storey R, Swarbreck D, et al. 2006. GENCODE: Producing a reference annotation for ENCODE. Genome Biol 7 Suppl 1: S4.1–S4.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D 2006. The UCSC Known Genes. Bioinformatics 22: 1036–1046 [DOI] [PubMed] [Google Scholar]
- Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al. 2010. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142: 409–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP 2003. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa D, Cheng J, Kapranov P, Yamanaka M, Brubaker S, Cawley S, Drenkow J, Piccolboni A, Bekiranov S, Helt G, et al. 2004. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res 14: 331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SPA, Gingeras TR 2002. Large-scale transcriptional activity in chromosomes 21 and 22. Science 296: 916–919 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D 2003. Evolution's cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci 100: 11484–11489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. 2009. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci 106: 11667–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylov DM, Wolf YI, Rogozin IB, Koonin EV 2003. Gene loss, protein sequence divergence, gene dispensability, expression level, and interactivity are correlated in eukaryotic evolution. Genome Res 13: 2229–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Zhou X, Wang X, Zhu D, Zhang Y 2010. Identification and characterization of human snoRNA core promoters. Genomics 96: 50–56 [DOI] [PubMed] [Google Scholar]
- Lin MF, Kheradpour P, Washietl S, Parker BJ, Pedersen JS, Kellis M 2011. Locating protein-coding sequences under selection for additional, overlapping functions in 29 mammalian genomes. Genome Res 21: 1916–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Managadze D, Rogozin IB, Chernikova D, Shabalina SA, Koonin EV 2011. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs. Genome Biol Evol 3: 1390–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS, Makunin IV 2006. Non-coding RNA. Hum Mol Genet 15 Spec No 1: R17–R29 [DOI] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS 2008. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci 105: 716–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Qureshi IA, Gokhan S, Dinger ME, Li G, Mattick JS, Mehler MF 2010. Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci 11: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya HI, Amaral PP, Louro R, Lopes A, Fachel AA, Moreira YB, El-Jundi TA, da Silva AM, Reis EM, Verjovski-Almeida S 2007. Genome mapping and expression analyses of human intronic noncoding RNAs reveal tissue-specific patterns and enrichment in genes related to regulation of transcription. Genome Biol 8: R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobrega MA, Ovcharenko I, Afzal V, Rubin EM 2003. Scanning human gene deserts for long-range enhancers. Science 302: 413. [DOI] [PubMed] [Google Scholar]
- Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, et al. 2002. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420: 563–573 [DOI] [PubMed] [Google Scholar]
- Pál C, Papp B, Hurst LD 2001. Highly expressed genes in yeast evolve slowly. Genetics 158: 927–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BJ, Moltke I, Roth A, Washietl S, Wen J, Kellis M, Breaker R, Pedersen JS 2011. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res 21: 1929–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D 2006. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput Biol 2: e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponjavic J, Ponting CP, Lunter G 2007. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res 17: 556–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponjavic J, Oliver PL, Lunter G, Ponting CP 2009. Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLoS Genet 5: e1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, et al. 2009a. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res 19: 1316–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Klimke W, Maglott DR 2009b. NCBI Reference Sequences: Current status, policy and new initiatives. Nucleic Acids Res 37: D32–D36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasi T, Suzuki H, Pang KC, Katayama S, Furuno M, Okunishi R, Fukuda S, Ru K, Frith MC, Gongora MM, et al. 2006. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res 16: 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattner P, Brooks AN, Lowe TM 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33: W686–W689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm L, Hernandez N 2002. Recruitment of RNA polymerase III to its target promoters. Genes Dev 16: 2593–2620 [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Kumar S 2004. Gene expression intensity shapes evolutionary rates of the proteins encoded by the vertebrate genome. Genetics 168: 373–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh KS, Park SW, Castro A, Patel H, Blake P, Liang M, Goy A 2010. Ovarian cancer biomarkers for molecular biosensors and translational medicine. Expert Rev Mol Diagn 10: 1069–1083 [DOI] [PubMed] [Google Scholar]
- Szymanski M, Erdmann VA, Barciszewski J 2007. Noncoding RNAs database (ncRNAdb). Nucleic Acids Res 35: D162–D164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, et al. 2008. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 453: 539–543 [DOI] [PubMed] [Google Scholar]
- Wolf YI, Gopich IV, Lipman DJ, Koonin EV 2010. Relative contributions of intrinsic structural–functional constraints and translation rate to the evolution of protein-coding genes. Genome Biol Evol 2: 190–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Zhang M, Zhou T, Hua X, Tang L, Wu W 2007. Sno/scaRNAbase: A curated database for small nucleolar RNAs and cajal body-specific RNAs. Nucleic Acids Res 35: D183–D187 [DOI] [PMC free article] [PubMed] [Google Scholar]