

Abstract
Autosomal dominant mutations in PTH1R segregate with primary failure of eruption (PFE), marked by clinical eruption failure of adult teeth without mechanical obstruction. While the diagnosis of PFE conveys a poor dental prognosis, there are no reports of PFE patients who carry PTH1R mutations and exhibit any other skeletal problems. We performed polymerase chain reaction–based mutational analysis of the PTH1R gene to determine the genetic contribution of PTH1R in 10 families with PFE. Sequence analysis of the coding regions and intron-exon boundaries of the PTH1R gene in 10 families (n = 54) and 7 isolated individuals revealed 2 novel autosomal dominant mutations in PTH1R (c.996_997insC and C.572delA) that occur in the coding region and result in a truncated protein. One family showed incomplete penetrance. Of 10 families diagnosed with PFE, 8 did not reveal functional (nonsynonymous) mutations in PTH1R; furthermore, 4 families and 1 sporadic case carried synonymous single-nucleotide polymorphisms. Five PFE patients in 2 families carried PTH1R mutations and presented with osteoarthritis. We propose that the autosomal dominant mutations of PTH1R that cause PFE may also be associated with osteoarthritis; a dose-dependent model may explain isolated PFE and osteoarthritis in the absence of other known symptoms in the skeletal system.
Keywords: tooth eruption, arthritis, genetics, incomplete penetrance, orthodontics, polymorphism
Introduction
Recent molecular studies have revealed that tooth eruption requires a precise and coordinated signaling process regulated by a series of signaling events between the dental apparatus and the surrounding osseous and soft tissues. Tooth eruption anomalies occur in a significantly large segment of the population causing functional and aesthetic problems that affect the quality of life for affected individuals. A disruption in this eruption process, termed primary failure of eruption (PFE; Proffit and Vig, 1981), represents the most severe end of the spectrum for eruption anomalies. PFE is characterized by eruption failure of permanent teeth in the absence of mechanical obstruction (i.e., dental cyst or adjacent tooth) and affected teeth that do not respond to orthodontic force. It was speculated to be familial in nature (Frazier-Bowers et al., 2007), but the cause was unknown until recently, where the causative gene was identified as PTHR1 (Decker et al., 2008). While the true incidence of PFE is unknown, it has been estimated to occur in approximately 0.6% of the population (Stellzig-Eisenhauer et al., 2010).
Autosomal recessive mutations in the parathyroid hormone receptor gene (PTH1R or PTHR1) cause lethal conditions, including Blomstrand osteochondrodysplasia (BOCD) (OMIM: 215045), a genetic disorder characterized by advanced endochondral bone maturation and lethality. In addition, Jansen metaphyseal chondrodysplasia (OMIM: 156400) enchondromatosis (OMIM: 166000), and Eiken syndrome (OMIM: 600002)—all of which include a cartilage or skeletal dysplasia—are caused by autosomal recessive or dominant mutations in PTHR1 (Jobert et al, 1998; Duchatelet et al., 2005). Our group and others have found that autosomal dominant mutations in PTH1R cause PFE (Decker et al., 2008; Frazier-Bowers et al., 2010). The phenotype is thought to be due to haploinsufficiency of the gene product. The precise mechanisms by which PTH1R leads to the PFE phenotype are poorly understood.
PTH1R is a 7-helical-transmembrane G-protein-coupled receptor that is activated upon binding to 2 distinct ligands: parathyroid hormone (PTH) and parathyroid hormone–related peptide (PTHrP) (Datta and Abou-Samra, 2009). Both PTH and PTHrP are known to regulate calcium homeostasis and bone metabolism, but they also have other diverse functions (Strewler, 2000). While PTH is produced solely from parathyroid glands, PTHrP is produced by several tissues, including skin, endothelium, smooth muscle, growth plate chondrocytes, bone, kidney, neuronal/glial tissues, and developing tooth buds. Reciprocally, PTH1R is expressed where PTH and PTHrP are biologically active, in particular, osteoblasts and renal tubular cells (Lavi-Moshayoff et al., 2010). PTH and PTHrP share only 16% sequence homology, and the homologous region is in the N-terminal where receptor binding is thought to occur. While PTH regulates systemic calcium and bone metabolism, PTHrP likely acts on the same pathways but at the local level (Cornish, 2010). Tight coordination of PTH/PTHrP/PTH1R expression is therefore required for normal skeletal development.
Both human and animal studies have documented the genetic contribution of PTHrP in the developing tooth and in the skeletal system (Philbrick et al., 1998). Clinical evaluation of human fetuses affected with BOCD illustrates that severe defects in endochondral bone formation are accompanied by an absence of breasts and unusually impacted teeth within the alveolar bone. In rodent studies, PTHrP expression is found in the bell stage (Liu et al., 1998) and the secretory stage (of enamel formation) (Philbrick et al., 1998). These studies suggest that elevated expression of PTH1R and PTHrP and perhaps the PTH1R-PTHrP complex are important during tooth development. Additional studies in mice also support the significant role for PTHrP and PTH1R in the developing skeletal system and their association with osteoarthritis (Chen et al., 2008; Chang et al., 2009).
Although PFE is reported to be a nonsyndromic condition, we seek to refine the clinical characteristics associated with the PFE phenotype and accompanying systemic conditions to determine the relative contribution of the PTH1R gene to the development of PFE and related disorders. We report here that genetic mutations in familial PFE can be inherited in an incompletely penetrant fashion and may be associated with early onset of osteoarthritis.
Materials & Methods
Ascertainment of Families and Diagnosis
This study was approved by the Biomedical Institutional Review Board at the University of North Carolina at Chapel Hill. Consent was obtained from every adult participant or from a parental guardian in the case of minors. Typically, the index case was identified through a referring orthodontist; through subsequent interviews, the pedigrees were extended for a total of 54 individuals from 10 families (Figure 1) and 7 isolated cases with an age range from 6 to 68 years. Forty-four individuals were available for pretreatment clinical photographs and panoramic and cephalometric radiographs following the initial clinical evaluation described here.
Figure 1.
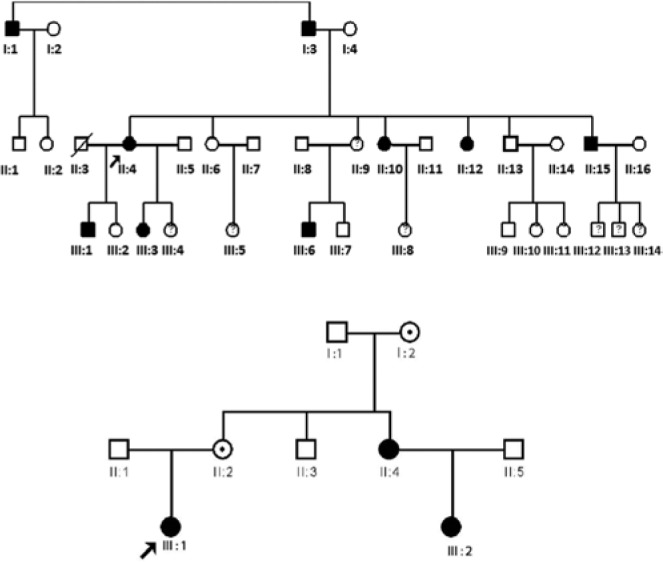
(A, B) Pedigree analysis by inspection for 2 families segregating primary failure of eruption showing an autosomal dominant inheritance pattern. Both families 1 and 2 segregate functional mutations in PTH1R (shown in Figure 3).
A positive diagnosis of PFE was based on at least 1 infraoccluded first molar according to clinical data (i.e., radiographs and/or examination at minimum). A clinical interview was completed for each affected individual and/or one’s family members to determine general health status (including the presence of other medical disorders diagnosed by an internist; e.g., arthritis).
Radiographic Analysis
A detailed clinical analysis was performed for affected individuals; unaffected individuals provided documentation of normal occlusion through the family dentist. Characterization of PFE was carried out via 2 criteria: (1) eruption potential in the anteroposterior gradient and (2) vertical gradients. Affected individuals were categorized as either type I, marked by a consistently progressive open bite from the anterior to the posterior of the dental arches, or type II (Appendix Figure), also presenting as a progressive open bite from the anterior to the posterior but with greater, albeit inadequate, eruption of a second molar. The PFE phenotype was also characterized with respect to the extent of eruption—that is, whether the infraoccluded teeth are intraosseous (completely submerged within bone) or supraosseous (partially erupted through the bone) and whether affection was bilateral or unilateral.
Mutational Analysis of PTH1R
Extraction and purification of DNA was carried out with buccal cells (PureGene kit, Gentra Systems, Minneapolis, MN) or saliva with Oragene kits (DNA Genotek, Ottawa, Canada) for all individuals in this study. We amplified and sequenced all coding exons of PTH1R (exons 3-14) from 10 families (n = 54) and 7 isolated cases using primer sets as described previously (Decker et al., 2008). Primer sets were designed to delineate splice junctions and included a minimum of 25 bases of intron sequence in addition to the exon sequence. Amplification was performed with Accuprime polymerase chain reaction buffer and enzyme mix (Life Technologies/Invitrogen, Bethesda, MD) under previously published conditions (Frazier-Bowers et al., 2010). Sequences were compared to wild-type PTH1R (accession NM_000316.2) from Genbank release GRCh37.
Results
Phenotype: Genotype Correlation
The clinical presentations of PFE varied in terms of severity and type within and between families (see Appendix Table 1). Of 10 families, 2 were classified as type II PFE, while the majority (8 families) were classified as type I. All the affected individuals who provided records (n = 32) were found to have an eruption failure that manifested during the supraosseous phase (i.e., the teeth had emerged partially through the bone). A unilateral versus bilateral eruption failure (i.e., manifesting on one side of the dental arches) occurred equally.
In family 1, of 10 affected individuals who participated, 3 were classified as type I PFE (Figure 2A) and segregated a c.996_997insC mutation (Figure 3A) that leads to a frameshift (p.A333fsX397). One individual had a unilateral manifestation, and 2 had a bilateral manifestation. Unaffected individuals (n = 4) did not carry the c.996_997insC mutation nor reveal any signs of the dental eruption phenotype, indicating an autosomal dominant inheritance with complete penetrance. Notably, systemic and/or skeletal problems were detected in family 1. A total of 5 individuals with PFE (see Appendix Table 1) in 2 families had a previous diagnosis of early-onset arthritis by a medical doctor, manifesting in the back and/or hip region around the second or third decade of life (2 in family 1 and 3 in family 2).
Figure 2.
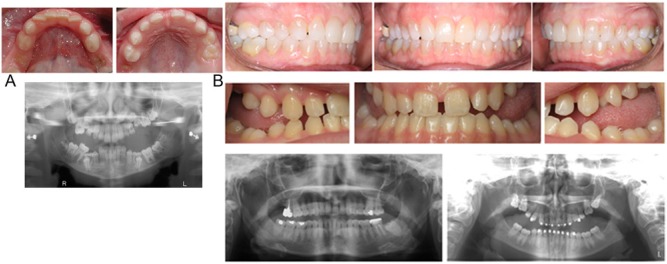
(A) Clinical representation of primary failure of eruption-affected child from family 1 showing type I primary failure of eruption. Her affected family members reported similar clinical patterns (not shown since multiple extractions and restorations have altered their dentitions significantly). (B) Individual I:1 and II:2 (above) do not show clinical expressions of the disorder but have passed the alteration on to their offspring who exhibit clinical signs (III:1 above).
Figure 3.
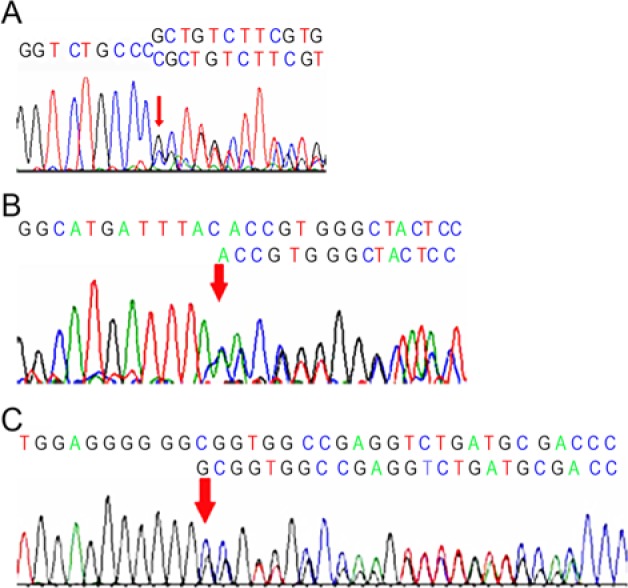
(A) Chromatogram showing the c.996_997insC mutation that segregates in family 1. (B) Chromatogram representative of c.572delA mutation identified in family 2. The mutation segregates in an autosomal dominant fashion but exhibits incomplete penetrance in this family. (C) Chromatogram of PTH1R mutation in an isolated patient with a severe case of primary failure of eruption and Charcot-Marie tooth syndrome. The alteration is an intronic insertion mutation, c.841+46_841+47, in exon 8.
In family 2, the proband (III:1) had a severe form of type I PFE similar to her maternal aunt and cousin (data not available). The proband and 2 other family members—the mother (II:2) and maternal grandmother (I:1) of the proband—harbored a c.572delA mutation (Figure 3B) that creates a frameshift and premature stop codon, p.Y191fsX203. While the proband (III:1) and her maternal aunt (II:4) and cousin (III:2) were severely affected, the mother and maternal grandmother were normal with respect to the eruption of the permanent dentition (Figure 2B). The finding that individuals carrying functional mutations in the PTH1R gene were unaffected indicates an autosomal dominant inheritance with incomplete penetrance associated with PFE. However, all individuals who harbored the genetic mutation also reported symptoms and diagnosis of arthritic changes during their 20s and 30s except for II:2, suggesting that variable expressivity may explain the absence of the dental feature and the concordance with respect to the skeletal feature (arthritis). If the presence of arthritis is included, then the classification of variable expressivity (affecting tooth eruption, skeletal changes, or both) is another reasonable possibility.
One isolated case (case 3) of severe PFE occurred in a 16-year-old male who was also diagnosed with Charcot-Marie tooth syndrome, a neurologic disorder causing slow degeneration of nerves to the extremities and concomitant muscle weakening. Clinical evaluation of his dental arches revealed that he had bilateral type I PFE with both intraosseous and supraosseous eruption failure. Subsequent mutational analysis of PTH1R in case 3 led to the detection of 3 alterations—including an intronic nonfunctional single-nucleotide polymorphism (SNP) also found in other individuals (described later)—and an intronic insertion mutation, c.841+46_841+47, adjacent to exon 8 (Figure 3C).
Mutational analysis of 8 remaining families (data not shown) revealed the presence of synonymous/nonfunctional SNPs in PTH1R, with PFE for 4 families and the absence of any alterations in PTH1R for 4 families (see Appendix Table 2). Additionally, 1 SNP, c.1116+58 T>C (rs113602108), appeared as a heterozygous allele in 13 affected individuals but did not faithfully segregate with the PFE phenotype. There was no specific correlation with affection status (i.e., type I vs. type II or bilateral vs. unilateral) and allele variant. In our study, the most common allele of the c.1116+58 T>C SNP was the heterozygous T/C variant.
Discussion
Previous studies in our laboratory (Frazier-Bowers et al., 2007; Proffit and Frazier-Bowers, 2009; Frazier-Bowers et al., 2010) and others (Decker et al., 2008) have confirmed the heritability of PFE and the association with mutations in PTH1R. Our analysis of a PFE cohort reveals that there are multiple families and individuals with PFE who do not carry functional mutations in the PTH1R gene, suggesting either that additional genes are responsible for PFE or that regulatory noncoding regions may be altered. However, in 2 families with PFE, we observed cosegregation of autosomal dominant mutations with clinical presence of osteoarthritis in some individuals with PTH1R mutations and, in one family, incomplete penetrance of the autosomal dominant mutation. The phenomenon of incomplete penetrance is poorly understood, but recent studies in Caenorhabditis elegans indicate that incomplete penetrance can be due to “random fluctuations” in gene expression (Raj et al., 2010). Hence, our novel finding that incomplete penetrance is observed in 1 family with PFE may indicate that the c.572delA mutation leads to a partially inactive protein product and/or variability in the PTH1R gene expression. This may explain the inconsistent appearance of osteoarthritis and/or PFE in those individuals who harbor the c.572delA mutation in family 2.
During normal signaling, the PTH1R receptors dimerize upon activation by the ligand (PTH or PTHrP), stimulating the downstream signaling cascades (Figure 4A). Two classical G-protein signaling cascades are activated: adenylate cyclase and phospholipase C (PLC) (Abou-Samra et al., 1992; Takasu & Bringhurst 1998; Takasu et al., 1999; Miedlich & Abou-Samra, 2008; Datta & Abou-Samra, 2009). While activation of adenylate cyclase occurs at the physiologic concentration of agonist (subnanomolar), the activation of PLC requires micromolar agonist concentration (Bringhurst et al., 1993). It is possible that the PLC activation is a result of an extremely high local concentration of PTHrP in reciprocation with areas abundant in PTH1R, as in a growth plate or developing tooth bud. This putative dose-dependent model explained by the second signaling pathway (PLC) (Becher et al., 2010) may be important in PFE and osteoarthritis development.
Figure 4.
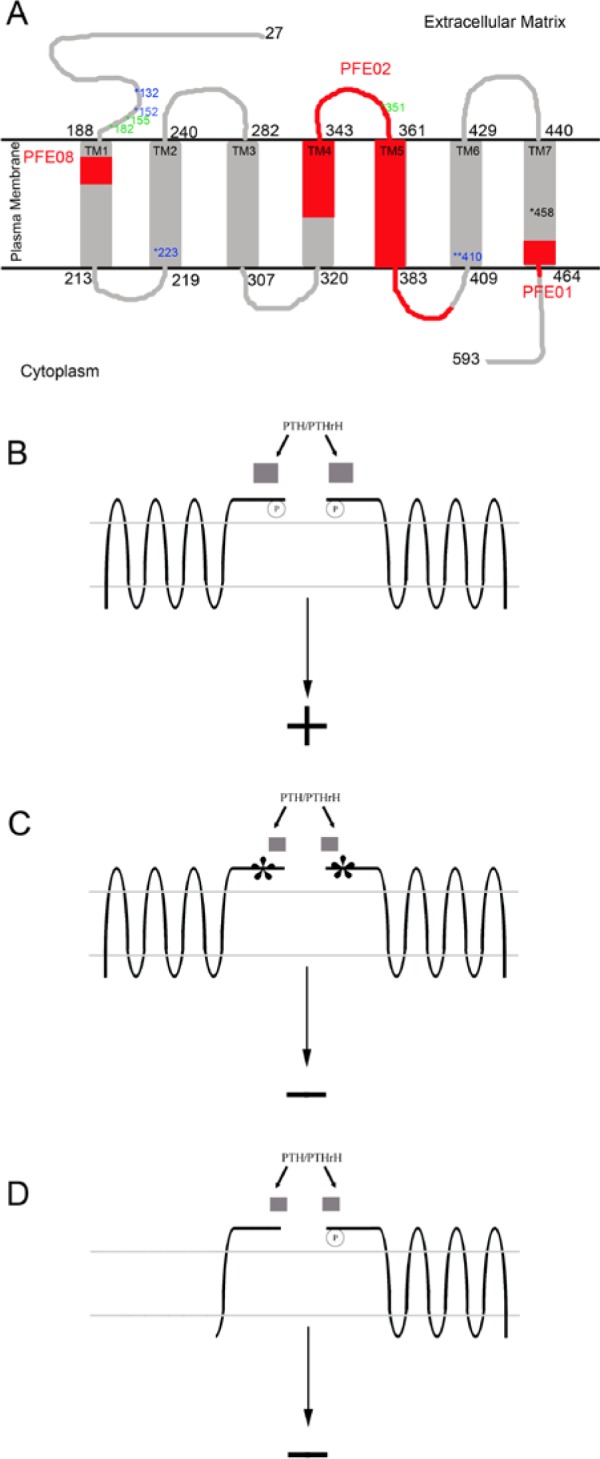
(A) The membrane-associated structure of mature PTH1R is shown here between the extracellular matrix and the cytoplasm. The N-terminal extracellular domain starts from residue 27 to 188. Residues 189 to 463 form 7-helical transmembrane domain. Selected mutations are shown in blue, green, and red. Residues in blue are the point mutations; 132 P to L in Blomstrand osteochondrodysplasia (Zhang et al. 1998), 152 R to C in enchondromatosis-Ollier type (Hopyan et al. 2002), 223 H to R, 410 T to P or R, and 458 I to R. Of these, the last 3 mutations are related to Jansen’s metaphyseal chondrodysplasia (Schipani et al. 1995, Schipani et al. 1996, Schipani et al. 1997, and Bastepe et al. 2004). Residues in green are the truncation mutations of primary failure of eruption (PFE) in previous publications. The regions in red represent mutations identified by the laboratory in this report (PFE-02 [family 1; II:4] and PFE-08 [family 2: III:1]) and previously (PFE-01; Frazier-Bowers et al., 2010). Human PTH1R is composed of 14 exons, and the structure is divided into 3 domains: the extracellular N-terminal domain, the J domain (composed of the transmembrane helices and connected loop), and the intracellular C-terminal domain. The extracelluar N-terminal domain of PTH1R, functioning as a regulatory domain, interacts with the carboxy-terminal portion of parathyroid hormone, and parathyroid hormone–related peptide binds to enhance the ligand interaction with the J domain. The J domain, the main functional domain, interacts with the N-terminal of parathyroid hormone and parathyroid hormone–related peptide. Once PTH1R is activated, the ligand receptor is inactivated by phosphorylation of PTH1R. The receptor is then internalized and recycled. The crystal structures and biochemistry/cell biology studies of the extracellular N-terminal domain of PTH1R suggest that PTH1R may function in a dimeric form and that only the extracellular N-terminal domain is required for this dimeric interaction. However, the C-terminal domain is necessary for appropriate intracellular signaling. (B-D) The proposed mechanism of PTH1R activation: We propose here that the dimeric form of PTH1R and its ligands are required for PTH1R activation (B). The mutations in the N-terminal peptide can disrupt either PTH1R dimeric interaction or the PTH1R-ligand interaction (C). Truncated version of PTH1R may allow PTH1R dimerization and PTH1R-ligand interaction, and the mutants may disrupt the downstream PTH1R signaling (D).
We propose 3 possible mechanisms for PTH1R-PTH/PTHrP signaling that distinguish among normal, heterozygous, and homozygous mutant protein (Figure 4B). PTH1R point mutations—most of which occur in the extracellular domain, where dimerization and ligand interaction occur (Mierke et al., 2007)—interrupt downstream signaling because of faulty ligand binding or dimerization. When the mutations are autosomal recessive (2 defective copies of the PTH1R protein), a severe skeletal phenotype results. However, in some cases, a single mutation can cause a severe autosomal dominant condition if the mutation (1) disrupts the dimerization (extracellular domain), (2) alters ligand activation (transmembrane domain), or (3) alters other G-protein subunit interaction (intracellular domain). Finally, as observed in our study, 2 families with PFE harbor autosomal dominant mutations that lead to truncation of the protein. Since the resultant mutant protein will not allow normal signaling, it is likely that the defective protein interacts with the normal copy of the protein and functions as a competitive inhibitor for PTH1R activation (Figure 4B). Biochemical data and x-ray crystallographic structures reveal that only the extracellular domain is required for receptor dimerization (Mierke et al., 2007). Hence, we speculate that 1 normal copy of PTH1R is necessary and sufficient in most tissues, but in the developing tooth (and perhaps developing joints), significant quantities of activated PTH1R are physiologically necessary (i.e., for PTH1R-PLC pathway). Accordingly, the PFE phenotype (preferentially affecting large multirooted posterior teeth) may be the result of dose-dependent inactivation of PTH1R. Normal activation of PTH1R requires sufficient interaction of ligand (PTH or PTHrP) and PTH1R, as well as dimerization of PTH1R. This may explain why some individuals who carry a PTH1R mutation do not show the clinical phenotype and others do. Moreover, a consistent observation is that PFE due to autosomal dominant mutations in PTH1R does not affect anterior teeth and other skeletal systems, except for some reports of osteoarthritis.
Although the mechanism for PTH1R in tooth eruption may not be fully understood, we know that a failure of osteoclast formation is shown to cause eruption failure in the mouse model (Wise and King, 2008). Hence, in patients with PFE due to PTH1R mutations, there may be an as-yet-undetermined osteoclast defect. While our observations in 2 families associate PTH1R mutations with tooth eruption and the development of osteoarthritis, we do not historically find osteoarthritis in all individuals who carry a PTH1R mutation. However, recent evidence confirms the association of osteoarthritis and a decrease in PTH1R expression in rat chondrocytes (Becher et al., 2010). Larger cohort studies examining the causal relationship of PTH1R with osteoarthritis will fully test this hypothesis since osteoarthritis otherwise occurs frequently in the population (Felson et al., 2000). Additional studies of PFE also should include functional studies to characterize the mechanism of specific mutations and mutational analysis to understand the role of high-priority candidate genes in the tooth eruption and development. Taken together, these studies using the tooth organ as a model for broader systemic processes will undoubtedly contribute to the gaps of knowledge in our understanding of normal versus abnormal bone turnover and the pathogenesis of arthritis.
Supplementary Material
Acknowledgments
We gratefully acknowledge the support of the families and dentists who participated in this study, especially Drs. Bloyce Britton and Fidel Del Toro; the assistance of Darrin Simmons and Donchuan Guo in the preparation of the data; and Russell Mack in assisting with the manuscript preparation.
Footnotes
This research was supported by the American Association of Orthodontists Faculty Development Award and the National Institutes of Health (grants 1K23RR17442 and M01RR-00046).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Abou-Samra AB, Juppner H, Force T, Freeman MW, Kong XF, Schipani E, et al. (1992). Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc Natl Acad Sci U S A 89:2732-2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastepe M, Raas-Rothschild A, Silver J, Weissman I, Wientroub S, Jüppner H, et al. (2004). A form of Jansen’s metaphyseal chondrodysplasia with limited metabolic and skeletal abnormalities is caused by a novel activating parathyroid hormone (PTH)/PTH-related peptide receptor mutation. J Clin Endocrinol Metab 89:3595-3600. [DOI] [PubMed] [Google Scholar]
- Becher C, Szuwart T, Ronstedt P, Ostermeier S, Skwara A, Fuchs-Winkelmann S, et al. (2010). Decrease in the expression of the type 1 PTH/PTHrP receptor (PTH1R) on chondrocytes in animals with osteoarthritis. J Orthop Surg Res 5:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringhurst FR, Jiippner H, Guo J, Urefia P, Potts JT, Kronenberg HM, et al. (1993). Cloned, stably expressed parathyroid hormone (PTH)/PTH-related peptide receptors activate multiple messenger signals and biological responses in LLC-PK1 kidney cells. Endocrinology 132:2090-2098. [DOI] [PubMed] [Google Scholar]
- Chang JK, Chang LH, Hung SH, Wu SC, Lee HY, Lin YS, et al. (2009). Parathyroid hormone 1-34 inhibits terminal differentiation of human articular chondrocytes and osteoarthritis progression in rats. Arthritis Rheum 60:3049-3060. [DOI] [PubMed] [Google Scholar]
- Chen X, Macica CM, Nasiri A, Broadus AE. (2008). Regulation of articular chondrocyte proliferation and differentiation by indian hedgehog and parathyroid hormone-related protein in mice. Arthritis Rheum 58:3788-3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish J. (2010). Systemic and local regulation of skeletal metabolism: meeting report from the IBMS Davos Workshop. Bone biology and therapeutics. IBMS BoneKey 7:187-189. [Google Scholar]
- Datta NS, Abou-Samra AB. (2009). PTH and PTHrP signaling in osteoblasts. Cell Signal 21:1245-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker E, Stellzig-Eisenhauer A, Fiebig BS, Rau C, Kress W, Saar K, et al. (2008). PTHR1 loss-of-function mutations in familial, nonsyndromic primary failure of tooth eruption. Am J Hum Genet 83:781-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchatelet S, Ostergaard E, Cortes D, Lemainque A, Julier C. (2005). Recessive mutations in PTHR1 cause contrasting skeletal dysplasias in Eiken and Blomstrand syndromes. Hum Mol Genet 14:1-5. [DOI] [PubMed] [Google Scholar]
- Felson DT, Lawrence RC, Dieppe PA, Hirsch R, Helmick CG, Jordan JM, et al. (2000). Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann Intern Med 133:635-646. [DOI] [PubMed] [Google Scholar]
- Frazier-Bowers SA, Koehler KE, Ackerman JL, Proffit WR. (2007). Primary failure of eruption: further characterization of a rare eruption disorder. Am J Orthod Dentofacial Orthop 131:578.e1-578.e11. [DOI] [PubMed] [Google Scholar]
- Frazier-Bowers SA, Simmons D, Wright JT, Proffit WR, Ackerman JL. (2010). Primary failure of eruption and PTH1R: the importance of a genetic diagnosis for orthodontic treatment planning. Am J Orthod Dentofacial Orthop 137:160.e1-160.e7. [DOI] [PubMed] [Google Scholar]
- Hopyan S, Gokgoz N, Poon R, Gensure RC, Yu C, Cole WG, et al. (2002). A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat Genet 30:306-310. [DOI] [PubMed] [Google Scholar]
- Jobert AS, Zhang P, Couvineau A, Bonaventure J, Roume J, Le Merrer M, et al. (1998). Absence of functional receptors for parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J Clin Invest 102:34-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. (2010). PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol 299:F882-F889. [DOI] [PubMed] [Google Scholar]
- Liu JG, Tabata MJ, Yamashita K, Matsumura T, Iwamoto M, Kurisu K. (1998). Developmental role of PTHrP in murine molars. Eur J Oral Sci 106(suppl 1):143-146. [DOI] [PubMed] [Google Scholar]
- Miedlich SU, Abou-Samra AB. (2008). Eliminating phosphorylation sites of the parathyroid hormone receptor type 1 differentially affects stimulation of phospholipase C and receptor internalization. Am J Physiol Endocrinol Metab 295:E665-E671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierke DF, Mao L, Pellegrini M, Piserchio A, Plati J, Tsomaia N. (2007). Structural characterization of the parathyroid hormone receptor domains determinant for ligand binding. Biochem Soc Trans 35(pt 4):721-723. [DOI] [PubMed] [Google Scholar]
- Philbrick WM, Dreyer BE, Nakchbandi IA, Karaplis AC. (1998). Parathyroid hormone-related protein is required for tooth eruption. Proc Natl Acad Sci U S A 95:11846-11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proffit WR, Vig KW. (1981). Primary failure of eruption: a possible cause of posterior open-bite. Am J Orthod 80:173-190. [DOI] [PubMed] [Google Scholar]
- Proffit WR, Frazier-Bowers SA. (2009). Mechanism and control of tooth eruption: overview and clinical implications. Orthod Craniofac Res 12:59-66. [DOI] [PubMed] [Google Scholar]
- Raj A, Rifkin SA, Andersen E, van Oudenaarden A. (2010). Variability in gene expression underlies incomplete penetrance. Nature 463:913-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipani E, Kruse K, Juppner H. (1995). A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science 268:98-100. [DOI] [PubMed] [Google Scholar]
- Schipani E, Langman CB, Parfitt AM, Jensen GS, Kikuchi S, Kooh SW, et al. (1996). Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen’s metaphyseal chondrodysplasia. N Engl J Med 335:708-714. [DOI] [PubMed] [Google Scholar]
- Schipani E, Lanske B, Hunzelman J, Luz A, Kovacs CS, Lee K, et al. (1997). Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc Natl Acad Sci USA 94:13689-13694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellzig-Eisenhauer A, Decker E, Meyer-Marcotty P, Rau C, Fiebig BS, Kress W, et al. (2010). Primary Failure of Eruption (PFE)-Clinical and Molecular Genetics Analysis. Journal of Orofacial Orthopedics/Fortschritte der Kieferorthopädie 71:6-16. [DOI] [PubMed] [Google Scholar]
- Strewler GJ. (2000). The physiology of parathyroid hormone-related protein. N Engl J Med 342:177-185. [DOI] [PubMed] [Google Scholar]
- Takasu H, Bringhurst FR. (1998). Type-1 parathyroid hormone (PTH)/PTH-related peptide (PTHrP) receptors activate phospholipase C in response to carboxyl-truncated analogs of PTH(1-34). Endocrinology 139:4293-4299. [DOI] [PubMed] [Google Scholar]
- Takasu H, Guo J, Bringhurst FR. (1999). Dual signaling and ligand selectivity of the human PTH/PTHrP receptor. J Bone Miner Res 14:11-20. [DOI] [PubMed] [Google Scholar]
- Wise GE, King GJ. (2008). Mechanisms of tooth eruption and orthodontic tooth movement. J Dent Res 87:414-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Jobert AS, Couvineau A, Silve C. (1998). A homozygous inactivating mutation in the parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand chondrodysplasia. J Clin Endocrinol Metab 83:3365-3368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.