

Summary
Mass spectrometry analysis of intact protein complexes has emerged as an established technology for assessing the composition and connectivity within dynamic, heterogeneous multiprotein complexes at low concentrations and in the context of mixtures. As this technology continues to move forward, one of the main challenges is to integrate the information content of such intact protein complex measurements with other mass spectrometry approaches in structural biology. Methods such as H/D exchange, oxidative foot-printing, chemical cross-linking, affinity purification, and ion mobility separation add complementary information that allows access to every level of protein structure and organization. Here, we survey the structural information that can be retrieved by such experiments, demonstrate the applicability of integrative mass spectrometry approaches in structural proteomics, and look to the future to explore upcoming innovations in this rapidly-advancing area.
Keywords: Structural Genomics, Non-covalent Complexes, High-throughput, protein-ligand screening, systems biology
1 Introduction
Most critical biological processes are orchestrated by networks of macromolecular protein complexes. While such complexes are discovered continuously, our knowledge of protein-protein interactions at a global level remains limited, as demonstrated in recent genome-wide screens that revealed a surprisingly large number of biological connections previously unidentified in simple cellular systems.[1, 2] The inventory of functions performed by protein complexes can be further diversified through regulation by binding partners such as small molecule ligands,[3] metal ions,[4] and by structural rearrangements through post-translational modifications.[5] As such, characterizing multiprotein complexes that result from associations at a proteome level necessitates the analysis of mixtures of stunning complexity, heterogeneity, and dynamic range. The field of structural proteomics is primarily concerned with converting such complex cellular mixtures of interacting proteins into 3D structure information. Like other pursuits that lie under the banner of proteomics, structural proteomics is characterized by high-throughput experiments that are, by definition, less amenable to the dedicated optimization procedures that typically characterize more-traditional structural biology approaches. Therefore, it is instructive to consider the various elements of protein structure as a function of the spatial resolution required to obtain useful and biologically relevant information. Protein structures can be annotated with descriptors such as size, shape, subcomplex topology, inter-subunit distances, inter-subunit orientation, interface size, interface stability, subunit fold and, ultimately, the spatial location of individual atoms that comprise the protein complexes (Figure 1).
Figure 1.
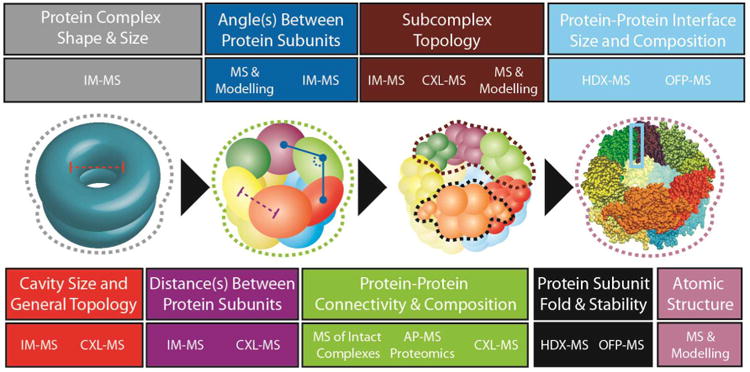
There are multiple levels of protein structure accessible through mass spectrometry-based technologies. The center section of this figure depicts the same complex at various resolution levels, progressing toward a high-resolution structure, in order to illustrate some of the structural elements accessible by mass spectrometry. The top and bottom sections of the figure are color-coded to correspond with the highlighted structural elements indicated on each structural representation shown, and they also list the specific MS technologies capable of providing the information specified. This information includes complex shape (grey), cavity size (red), distance between protein subunits (purple), inter-subunit angles (blue), protein connectivity (green) monomer topology (black), subcomplex topology (maroon), protein-protein interface structure (light blue) and atomic-level structural detail (mauve). The techniques associated with each level of structural information are indicated by abbreviations that are defined in the text.
Experimental determination of protein structures without a priori knowledge of protein function remains a challenging goal despite the rapid developments in ab-initio or bioinformatics-based prediction methods. [6] The difficulty associated with elucidating the structural features of interest for protein complexes is compounded by the highly heterogeneous, transient character of biomolecules and their low relative concentrations within physiologically-relevant samples. While analytical techniques traditionally used in structural biology, such as X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, excel at revealing structures and dynamics of biomolecules at the atomic level, the result of such experiments is often reduced to a static ‘snapshot’ of protein complex structure. Moreover, larger protein complexes and membrane proteins are less amenable to NMR or X-ray crystallography due to their common requirements for large amounts of sample and long acquisition times. Thus, characterizing and annotating the structural details of a complete set of multiprotein complexes found in cellular proteomes necessitates the development of novel structural biology tools capable of capturing the dynamic nature of heterogeneous protein complexes with high sensitivity.
A highly promising approach for addressing such challenges relies upon the integration of information acquired through multiple analytical technologies that offer complementary structural constraints. There are, however, many practical challenges in developing such an integrated approach for solving the architecture of multiprotein complexes. Mining datasets derived from several analytical tools for geometrically or topologically informative structural constraints typically involves integrating disparate expertise in data interpretation, software, and automation, in addition to finding the appropriate normalization procedures to align spatial constraints acquired by the different approaches utilized. Recently, many of these challenges were overcome to construct highly-complex structures of the nuclear pore complex, illustrating both the potential and scope of integrated approaches for applications in structural biology and structural proteomics.[7]
Recent innovations in sensitivity, speed and accuracy have established mass spectrometry (MS) as a key technology within the field of structural biology and proteomics, revealing the intricate interconnections of cellular processes.[8] MS is capable of probing the structure and dynamics of multiprotein complexes present at physiologically relevant concentrations over a wide range of solution conditions. Concurrent with developments in instrumentation, the integration of novel analytical techniques and chemical probes has strengthened the capacity of MS to characterize heterogeneous samples and retrieve structural information. Techniques like hydrogen-deuterium exchange (HDX),[9-13] chemical cross-linking (CXL),[14-16] oxidative footprinting (OFP),[17, 18], limited proteolysis,[19, 20] affinity purification (AP),[8, 21] and ion mobility separation (IMS) [22-24] have been partnered with MS as key approaches for the determination of protein structure and have established themselves as crucial tandem-technologies for revealing the structure of multiprotein complexes at various levels of structural resolution (Figure 1).
MS approaches currently being applied in structural biology and structural proteomics can be broadly categorized into those that generate spatial constraints from measurements of proteins in solution, and those that derive structural information from measurements of protein ions in the gas-phase. The latter approaches require that the structural integrity of protein complexes be maintained upon the transfer of protein to gas-phase, and MS instruments have been developed or modified with this goal in mind, specifically by increasing the ion guide pressures, incorporating low-frequency quadrupole mass analyzers, and accessing higher acceleration potentials. [25-27] Gas-phase methodologies take advantage of the desolvation process to effectively reduce sample complexity and make use of the spectrometric and spectroscopic tools available for molecular characterization in the absence of bulk solvent. MS can also be used primarily as a detector for chemical modifications designed to report on protein structure and dynamics in solution. While the integration of these two tracks of MS-based approaches is yet to be explored rigorously, the broad range of biological problems that can be investigated by each technique suggests that the combination of multiple MS approaches can provide complementary sets of information to answer previously intractable structural biology questions.
Here we review the advances in structural characterization of proteins using MS-based methods. Although relatively new compared to other technologies for analyzing protein structure, MS measurement of intact protein complexes [28-30] is now a central methodology in such experiments and we pay special attention to the role of this technology within structural proteomics. This, along with other technologies discussed herein, demonstrates that the integration of MS technologies offers a suite of tools for discovering both the composition of protein networks and their three-dimensional organization.
2 MS for Protein Primary and Secondary Protein Structure
MS approaches have played an indispensable role in identifying and characterizing the primary structure of proteins.[21] Experiments that seek to characterize protein at the individual amino acid residue level include those that involve enzymatic digestion of proteins and MS analysis of the peptide products produced (i.e., ‘bottom-up’ approaches) [31] and ‘top-down’ type experiments involving manipulation of intact, typically denatured, proteins within MS instrumentation.[32] Ongoing research in this area has been extensively reviewed, including the critical developments in tandem MS [33, 34] and automated strategies for collecting detailed information on protein sequences, composition, quantity, and dynamics. [35]
Approaches involving MS for the analysis of secondary structure, in the context of structural proteomics, are among the least developed of those discussed here. Typically, such approaches involve a focused study of small and isolated protein systems by integrating mass spectrometry with molecular dynamics simulations. For example, IM-MS has been used extensively in the analysis of peptides and small proteins and has provided, in combination with structural models generated in silico, secondary structure details for linear,[36, 37] cyclic,[38] and modified [39-41] peptide systems, as well as proteins less than ∼10 kDa.[42] Since these systems are supported by few intramolecular interactions, rearrangements of the peptide backbone can readily occur during desolvation in some cases [43, 44] and the role of solvent in protein structure can be elucidated in conjunction with gas-phase spectroscopic techniques.[45] Further, chemical labeling-MS datasets combined with molecular simulation approaches commonly employed to refine protein structures based on NMR information have been used to determine detailed structures of model protein systems over a size range similar to the IM-MS experiments discussed above.[46] Overall, while MS has often been considered a ‘low-resolution’ tool within the field of structural biology, this view is rapidly changing, especially in the context of smaller protein systems.
3 MS for Technologies for Higher-order Protein Structure
MS approaches are capable of characterizing higher-order protein structures (Figure 2), and in this section we pay special attention to those methodologies that, in our view, are most-readily integrated with the analysis of intact protein complexes by MS (see section 4). Therefore, we have structured the section below to discuss experimental results aimed at determining protein tertiary and quaternary structure according to the MS technology used to generate the structural information. In addition, we have included a section that covers recent MS results on protein-ligand interactions, focusing on those experiments that provide information on a proteome-wide scale. There are excellent reviews already available for many of the techniques discussed in this section, [8, 16, 28-30, 47, 48] some of which will appear within this special issue, and as such we will confine our comments to those experiments that are critical for integrating MS measurements of intact protein complexes with information from other MS-based technologies.
Figure 2.
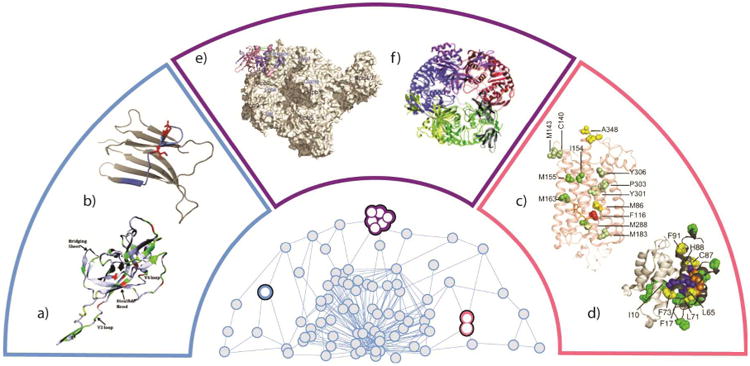
Several MS datasets from the literature are highlighted, and illustrated in context relative to how they might be used in a broader determination of a three-dimensional protein interactome model. These include studies of monomeric protein structure a) gp120-based antigen [70] and b) β-immuoglobulin [51]. Binary protein or protein-ligand systems can also be studied by MS, as in the cases of c) the GPCR-rodopsin complex [71] and d) the S100A11 dimer [69]. Finally, the structure of multiprotein systems can be studied by MS as in the cases of e) the RNA polymerase II [99] and f) the yeast exosome [182]. Each pair of data is contained within color-coded regions that correspond to the same shaded area of the 3D protein interactome cartoon at the center of the figure.
3.1 Hydrogen Deuterium Exchange MS
HDX, a technique traditionally associated with NMR, is now frequently used in conjunction with MS for monitoring protein dynamics and structure. At its core, HDX-MS enables the quantitative determination of rates at which protons in a particular region within a protein exchange with a deuterated solvent. [9, 13] Surface amide protons not participating in intermolecular interactions that comprise secondary and tertiary structural elements exchange rapidly, while those that are involved in hydrogen bonding or shielded in the protein interior exchange at a much slower rate. Thus, the technology effectively measures the solvent accessibility of a protein structure, and this information can be further used to annotate regions of a protein according to its apparent flexibility and stability. HDX-MS is amenable to a wide range of physiologically relevant solvent conditions and the exchange reaction can be efficiently quenched by rapidly lowering either the solution pH or temperature. Maintaining the conditions that prevent further HDX during proteolytic digestion and subsequent MS analysis is required to obtain accurate rates of HDX. The resolution of HDX-MS data is limited by the size of the peptide fragments generated from proteolysis or gas-phase dissociation, and also by the migration of protons in activated peptide and protein ions. Recently, significant improvements in the spatial resolution of HDX-MS information, extending to the single amino acid residue level, have been attained through both the optimization of digestion and exchange conditions [44, 45], and also by employing gas-phase fragmentation methods that suppress the scrambling of protons such as electron transfer dissociation (ETD) or electron capture dissociation (ECD).[49-51] A comparison between the deuterium levels measured by HDX-MS for β2-microglobulin and HDX-NMR shows an excellent correlation (Figure 2B), demonstrating that HDX exchange levels across a protein sequence can be annotated with resolution similar to those generated by HDX-NMR. Developments in top-down MS enable improved characterization of HDX-NMR, significantly simplifying the workflow required to analyze the architecture of protein complexes.[50]
Recently, HDX-MS has been applied to detect co-existing populations of protein conformers to exploit its ability to examine structural changes on the millisecond timescale. Such conformations include those induced upon ligand or co-factor binding [52-54], those generated during protein folding and unfolding,[55, 56] conformational mobility of nuclear receptors upon lignad binding,[57] and those related to protein stability under different solution conditions.[58] Moreover, HDX-MS shows considerable promise for studying membrane proteins, and recent results demonstrate that G-protein coupled receptors [59] and membrane-like nanodisks [60] are amenable to HDX-MS workflows. The ability of HDX-MS to measure samples over a virtually unlimited size range has also been applied to heterogeneous aggregates in an attempt to infer the degree to which intramolecular protein interactions contribute to global aggregate architecture. The progression of amyloid fibril formation was monitored by HDX-MS in order to estimate the oligomer and fibril regions inaccessible by solvent and to annotate dynamic regions of the fibril structure.[61, 62] Further HDX-MS on soluble monomers and oligomers allowed aggregate growth rates for SH3 [63] and Aβ 1-40 [64] to be determined. These studies, where measurements of protein monomers act as reporters for the structures of larger aggregates that escape detection by MS analysis represent an important bridge between HDX-MS studies and those that involve MS measurements of intact protein complexes (see Section 4.3).
3.2 Oxidative Foot-Printing MS
OFP coupled to MS probes the conformational states of protein complexes by covalently modifying surface-accessible amino acid residues through chemical oxidation. The extent of labeling depends on the protein surface area exposed to the solvent and the reactivity of exposed amino acid residues. Thus, analyzing proteolytic peptide fragments of an oxidatively-labeled protein can reveal the structures and dynamics of proteins in solution, similar to other methods such as HDX-MS and limited proteolysis. While OFP experiments can provide information similar to HDX-MS, amino acid modifications generated in OFP differ in that they are typically irreversible. Some OFP chemistries can be relatively selective for specific functional groups within proteins.[65] Thus, selecting the appropriate oxidation chemistry for the protein sequence under investigation is a crucial starting point for maximizing the information content of OFP-MS datasets.
Many OFP-MS protocols utilize oxidation chemistries designed to be both rapid and untargeted in terms of oxidation site, and most commonly involve hydroxyl radicals as the oxidative species. Activated radical precursors can rapidly generate populations of hydroxyl radicals in solution using timed laser pulses to enable the investigation of protein folding reactions with sub-millisecond time resolution.[66] In these experiments, protein folding or unfolding is triggered by a temperature or a pH jump, followed by a laser pulse for radical generation. With the incorporation of an appropriate microfluidic device that allows for rapid mixing, oxidative labeling can be precisely quenched. [67, 68] and unfolding or folding transitions can be monitored concurrently with formation of protein-protein interfaces with high temporal resolution (Figure 2D). [69] The versatility and resolution of OFP-MS experiments are best demonstrated by both the number and diversity of protein structures characterized to date. For example, studying the footprinting pattern of the loop region of HIV-1 envelope protein gp120 during its association with critical cell receptors revealed the important role of this region in the formation of higher-order protein contacts (Figure 2A). [70] Furthermore, the functional interaction between critical water molecules conserved within the trans-membrane portion of a G protein-coupled receptor during signal transmission was resolved using OFP measurements (Figure 2C). [71] In all of the applications above OFP-MS has played a crucial role in identifying and characterizing key amino acid residues involved in subunit folding and interfacial regions that result in multiprotein complex formation.
3.3 Affinity Purification Coupled to MS
The combination of AP and MS is arguably the most pervasive MS-based technology for the assessment of protein-protein interactions in vivo, and has been used to generate exhaustive interaction maps for several organisms, covering an unprecedented range of genomic sequences.[1, 72-75] The technology works by first labeling a protein of interest (the “bait”) using a molecule incorporating an affinity tag susceptible to chromatographic purification. In order to increase the selectivity of this AP step, multi-functional tags that incorporate a protease-cleavable site have been developed. The additional elution step acts to reduce the background levels of proteins bound non-specifically to both the solid-phase support and epitope tag used for separation of protein mixtures.[76] While this tandem affinity purification (TAP) strategy was developed independently from its implementation with MS, such methods have since been honed for their combination with MS detection. [77] Another critical development in the affinity purification of proteins involves the genetic modification of the bait protein to include an 8-11 residue epitope sequence for separation on a specific sepharose column.[78] The so-called ‘flag tag’ can capture protein-protein interactions weaker than TAP, however, false-positive results arising from background non-specific protein interactions needs to be carefully discriminated from biologically relevant interactions. Comparison of protein-protein interactions revealed by AP-MS against biochemical interactions determined independently verifies the high fidelity of AP-MS datasets to more-broadly informed protein-protein interaction databases.[74] AP-MS, like other technologies for screening protein-protein interactions such as yeast two hybrid and phage display, excels at revealing high-confidence binary interactions between two proteins, leading eventually to the definition of a broader protein network following the accumulation of sufficient data.
MS approaches for the analysis of affinity purified proteins can be broadly classified into ‘bottom-up’ and ‘top-down’ assessments of the protein interactions. The latter approach, which involves the analysis of intact multiprotein complexes directly by MS, is covered in detail below (Section 4). ‘Bottom-up’ MS analysis involves separation of the individual protein components and enzymatically digesting the isolated protein with its partner proteins to assess their mass and composition. Characterization of vast protein mixtures tagged across an entire proteome can be performed in a high-throughput manner by automated data acquisition and analysis. An important development in the AP-MS method involves quantitative stoichiometry determinations of isolated protein complexes. In these experiments the isolated protein is either labeled following expression,[79] separation,[80] or spiked with synthetic, labeled peptides or proteins of known concentration that are identical in sequence to those of interest,[81] in order to quantify proteins in a particular mixture and subsequently to infer the relative extent of protein expression. Comparing the relative expression levels of proteins allows an inference to be made regarding the binding stoichiometry of the isolated complex. Quantitative AP-MS is also an important tool in distinguishing interactions of biological importance from weak and non-specific interactions that result in false positives for standard TAP and flag-tag experiments. [76]
AP-MS data, acquired in a ‘bottom-up’ mode (as described above) have been utilized to assess the organization of proteins in multiple organisms, from Saccharomyces cerevisiae [72, 74, 75] to Homo sapiens. [82-85] In ground-breaking work, yeast AP-MS data have illustrated the modularity and interconnectivity of many cellular processes [72, 74]. Further, the expression levels of individual proteins were quantitatively determined to estimate the binding stoichiometries and subsequently postulate contact maps for a number of critically important protein complexes,[75] assisted by development of a scoring scheme. Such experiments have elucidated connectivity for ribosomal,[86] transcriptional,[83] prespliceosomal,[87] and kinetochore protein assemblies[88] of varying size, complexity and dynamics. Of special interest for structural proteomics are the latter two studies. In the case of the prespliceosomal A complex, EM data, along with AP-MS and CXL-MS (see Section 3.5) data are integrated to provide a more complete structural picture of a dynamic multi-MDa complex comprised of well over 100 proteins.[87] For kinetochore subcomplexes, some of which exceed 5MDa in weight, AP-MS data has been integrated with ultracentrifugation, size exclusion chromatography, fluorescence-based imaging, and other datasets to project structural models for several, previously unknown, complexes involved in the assembly of centromeric DNA, and their attachment to both chromosomes and microtubules.[88]
3.4 Chemical Cross-Linking and Chemical Labeling MS
CXL-MS strategies for obtaining higher-order structure information of protein have been under development for decades, but only now, after sufficient development of informatics tools and chemistries, has the utility of this technology matured to the point where significant endeavors in structural biology are tenable.[89] CXL-MS methods can capture interactions between flexible regions of proteins in solution by covalently linking functional groups of amino acid side chains. The covalent bonds may be formed by reaction between different components of protein complexes (intermolecular), or amino acid residues belonging to the same polypeptide (intramolecular). Identifying the cross-linked sites by MS-analysis reveals proximal amino acid residues. The length of the cross-linker serves to constrain pair-wise interaction sites in the protein sequence and imposes spatial constraints in order to eliminate candidate structural models.[90], and subsequently provides information on both the identity of the interacting partners involved in protein-protein interfaces. Such an approach has been demonstrated as an effective means of generating accurate backbone structures for small monomeric systems.[46] In combination with AP-MS, or in cases where affinity reagents are engineered into the molecules used for CXL, such data can provide high-confidence protein interaction information with limited chemical background. [91]
Like OFP, CXL agents can exploit a number of solution chemistries for targeting a specific functional group. The cross-linker additionally provides an opportunity to introduce many tunable traits in the analyte of interest, including isotopic coding, cleavability, affinity groups, linker length, and reactivity. Both formaldehyde and dithiobis-succinimidylpropionate (DSP; an amine-reactive, homobifunctional, thiolcleavable and membrane-permeable cross-linker) are used frequently in such experiments, owing to their amenability to a wide range of solution conditions. [92-99] Designing functionally tailored CXL agents that possess desirable attributes is currently an area of active research.[100, 101] Significantly, recent technical developments have produced cross-linking reagents capable of covalently modifying proteins within living cells, and allow for the assessment of protein quaternary structure under in vivo conditions. Photoactive CXL reagents have enabled the analysis of protein-protein interactions on fast timescales, and in quantitative manner for determination of the stoichiometry of proteins.[102]
MS approaches that rely upon ‘top-down’ characterization of proteins are also gaining prominence, typically involving Fourier-transform ion-cyclotron resonance (FT-ICR) and subsequent activation with one of the various fragmentation techniques such as ECD, infra-red multi-photon dissociation (IRMPD), and sustained off-resonance irradiation collision induced dissociation (SORI-CID). Such approaches can work to eliminate the need to separate proteins of interest from the cross-linking reagents prior to MS analysis.[101, 103]
Identifying cross-linked peptides within a complicated mixture of incomplete or uninformative reaction products presents significant challenges for modern bioinformatics and software development. Multiple search algorithms and new chemical linkers that yield distinct reporter ions upon CID have streamlined the CXL-MS process, and increased its sensitivity. [15] Further, validation of CXL interaction data against available high-resolution structures can minimize false-positives and allow for the discovery of novel interactions often overlooked due to protein flexibility and dynamics (Figure 2E) [99].
Many recent studies have described CXL reagents aimed at mitigating many of the informatics challenges inherent in the analysis of CXL-MS datasets.[16] For example, it is well known that CXL reactions can produce many potential products that encompass dead-end reactions and intra-molecular cross-links in addition to the inter-molecular linking reactions sought for protein quaternary structure analysis. [100] This situation creates an informatics bottleneck by generating a mixture of peptides many times more complex than typical ‘bottom-up’ proteomics samples.[104, 105] Recent studies have described CXL reagents that fragment to produce characteristic reporter ions as well as separated peptide ions for more-detailed MS3 experiments. [106-110] Such an approach both increases the confidence and the number of identified protein interaction sites.
Cross-linking strategies combined with MS have been applied to a number of important multiprotein systems to deduce both structure and dynamics. Several recent studies have focused on chaperone complexes, including those involved in cellular stress response and client protein binding in multiple systems. CXL-MS was critical in demonstrating that N-terminal flexibility in heat shock protein (Hsp) chaperone complexes is important for client protein recognition and binding. [111] Along with HDX-MS information, CXL-MS was used to construct a topology diagram of complexes between Hsp110 and Hsp70.[112] Further, multiple CXL-MS studies involving the protein interaction network associated with the proteosome have helped to define the interaction partners that shape the 26S protein degradation assembly, and a detailed contact map of the 19S lid complex in combination with MS measurements of the intact complex and sub-assemblies.[113]
While it is currently challenging to use CXL-MS data acquired in a high-throughput mode for proteome-wide protein topology discovery, many targeted analyses of multiprotein systems have met with a high level of success. For example, a low-resolution model of the calmodulin-melittin complex has been described using CXL-MS.[114, 115] Similar data have also been used to assess the assembly of viral coat proteins.[116] Membrane-bound complexes present distinct challenges for CXL-MS workflows, but work on rhodopsin-transducin[117, 118] as well as yeast prohibitin complexes[119] demonstrate CXL strategies capable of addressing such challenges. Perhaps the most-ambitious use of CXL has been in the construction of a model for the 50MDa nuclear pore complex, where CXL-MS data was combined with multiple other datasets to constrain a complete topological model for the 456-member assembly.[7] All of these specific examples demonstrate both the significance and enormous potential for CXL-MS in structural proteomics.
4 MS of Intact Protein Assemblies
As discussed briefly above, there are two main paradigms by which MS may interrogate the quaternary structure of multiprotein complexes: 1) by analyzing enzymatically-produced peptide fragments derived from transiently modified or co-purified proteins so as to provide information on protein network composition and topology or 2) by determining the non-covalent interaction networks of proteins intact within the mass spectrometer. The above discussion primarily centers on those approaches that conform to paradigm 1, while the following discussion is limited to those examples in the literature where paradigm 2 is employed. Throughout this review, it is has been our primary purpose to highlight those datasets where multiple types of MS measurements have been integrated to solve long-standing problems within structural proteomics (Figure 1), and have chosen to highlight those reports that utilize MS data of intact protein complexes as one of the integrated datasets used.
MS technology for the analysis of intact protein complexes is varied, but of most prevalent are quadrupole-time-of-flight (Q-ToF) platforms, utilizing nano-electrospray ionization (nESI) to generate ions. These systems are typically modified through the addition of quadrupole mass filters operating at reduced frequencies for an increased m/z range and with ion sources having tunable pressure regions for the increased collisional focusing of large ions.[26, 27] Other instrument platforms are popular, however, for the analysis of multiprotein complex systems. Fourier transform ion cyclotron resonance (FT-ICR) systems, for example, are increasing in popularity due to their ability to access tandem MS approaches that are currently more challenging to implement on Q-ToF platforms (see Section 4.1), but suffer from mass range and sensitivity issues typically absent from Q-ToF instrumentation in the analysis of large protein ions.[120, 121] Additional peripheral analytical devices can be incorporated into tandem with MS workflows aimed at the analysis of intact multiprotein complexes including size exclusion chromatography, both on-line and off-line, automated sample handling, and ion mobility (IM) separation for protein size measurements (see Section 4.4). These technologies, as well as applications of the intact MS method for multiprotein complexes are discussed in detail below.
4.1 Tandem MS Measurements
An essential tool in the analysis of multiprotein complexes by MS is tandem mass spectrometry (MS/MS), where ions are interrogated by ion selection, activation via energetic collisions with neutral gas (e.g. Ar), and dissociation (collision-induced dissociation, CID) to generate product ions that inform on the composition and stoichiometry of the ionized intact protein complex. There are several crucial mechanistic aspects of gas-phase protein complex dissociation that have been established. [122, 123] For example, the predominant fragmentation pathway for most protein complexes involves expulsion of a monomeric protein in a sequential fashion to give both highly charged subunits and lowly charged, multiply “stripped” complex ions. [120, 124, 125] Also, experimental and theoretical data indicate that a degree of protein unfolding precedes dissociation. [120, 123, 126] In general, CID has been established as an indispensible analytical tool in assigning protein stoichiometry and composition within heterogeneous samples [28].
Early experiments using blackbody infrared dissociation (BIRD) measurements on pentameric protein complexes provided the first indirect evidence that protein unfolding was intimately involved in the CID of protein complexes. [120] Following from these observations, elegant experiments utilizing protein dimers composed of monomers having disulfide bonds, and thus incapable of unfolding, further showed that protein unfolding influences the charge states acquired by product ions.[122] More recently, IM has been used to investigate the structural changes that occur to protein complexes upon collisional activation in a controlled manner. [127, 128] IM-MS experiments performed on ring-like protein complexes, and other cavity-bearing systems, indicate collision-induced compaction of the complex precedes any dramatic unfolding, through a currently unknown process. [128, 129] Detailed unfolding for the homotetrameric protein transthyretin (TTR) has further illustrated the extent and nature of unfolding of protein complexes.[127] Current understanding of the CID process for multiprotein systems is summarized in Figure 3, and most protein complex ions produced by conventional nESI or ESI methods follow the track involving the dissociation of highly-unfolded monomer ions. The charge carried by the precursor largely determines access to the other dissociation pathways shown, but the amount of charge reduction or amplification required to access these alternative pathways is not currently known universally for multiprotein complex ions.
Figure 3.

Various observed products of gas-phase protein complex dissociation are shown. This figure is color coded to correspond with techniques requiring either high (red) or low (blue) ion activation energies to access the product ions depicted. Protein complex ions activated using energetic collisions (CID, solid arrows) typically fragment via a pathway that leads to charge-reduced, monomer-stripped complexes and unfolded monomer ions (a). In references [27] and [191], exceptions to these general observations were reported (b). Electron capture dissociation (ECD, large dashed arrow) can result in the cleavage of peptide bonds within the complex and sequence-informative fragment ions as observed in reference [192] (c). Surface induced dissociation (SID, small dashed arrow) of protein complexes at higher effective energies can lead to the ejection of subcomplexes from the assembly, as observed in reference [138] (d). Charge manipulation is required to access other protein complex fragmentation pathways. Charge reduction of protein complex precursor ions, followed by collisional activation, results in either the ejection of compact protein subunits (e) or covalent fragmentation of the peptide backbone without the ejection of any protein subunits from the assembly, as observed in reference [131] (f). Charge amplification, followed by high-energy CID can lead to the ejection of large subunits from the assembly as observed in reference [193] (g), or the dissociation of protein subunits from the complex that then fragment into peptide ions as observed in reference [130] (h).
As mentioned above, the combination of charge manipulation and CID of protein complexes has provided evidence that the dissociation mechanism can be drastically influenced by precursor charge state (Figure 3), thus providing a way to significantly alter the structural information content of standard CID experiments. For example, charge amplification of the HSP 16.5 24mer (396 kDa) followed by CID has shown both increased sequential monomer losses (up to four) and fragmentation of monomers to form sequence-informative peptide ions [130]. This last point constitutes the first evidence of ‘top-down’ type sequencing of a protein that is itself stripped from an intact multiprotein complex. In addition, charge reduced complexes have been shown to produce folded protein fragment ions by IM-MS [131]. CID of further charge-reduced complexes, exceeding more than 40% charge reduction, generates fragment ions in the form of surface peptides dissociated from the intact complex [131]. Taken together, these observations suggest that CID information content can be dramatically altered to suit a wide range of experiments in structural proteomics.
While CID methodologies are among the most pervasive for the disruption and dissociation of multiprotein complexes in the gas-phase, alternative techniques are emerging that promise to provide enhanced structural information for such assemblies. Electron capture dissociation (ECD), while playing a central role in ‘top-down’ protein identification and post-translational modification assignment, is limited to only a few published examples in the context of multiprotein complex dissociation.[132, 133] In these experiments, protein complex ions are irradiated with low-energy electrons, typically on FT-ICR platforms, to produce both charge-reduced intact complexes and multiple peptide fragments resulting from backbone cleavage within the proteins that comprise the assembly. As other intact protein and peptide work in this area has shown, different bonds within the peptide backbone are preferentially broken in this experiment when compared to the CID experiments discussed above. While the ultimate analytical results of these experiments are similar to high-energy CID data for complexes, it is expected that ECD (or ETD) will be a more-effective approach to generate peptide-type fragmentation than equivalent CID experiments (Figure 3).
In addition to electron-mediated fragmentation, collisions between ions and surfaces can be used to rapidly activate protein assemblies, leading to product ions that are, in many cases, substantially different from those generated by CID. Such surface induced dissociation (SID) experiments often lead to large protein subcomplexes, presumably still in a folded configuration, to be ejected from the assembly (Figure 3). Mechanistically, such product ions are thought to arise through a shattering-type mechanism, where the large amounts of activation energy input through the SID process are not allowed to randomize throughout the complex, leading to fragments that do not inhabit the unfolded protein intermediates common in CID experiments. [134] Multiple examples of SID data for multiprotein complexes are available in the literature, encompassing many large homo-oligomers where vast numbers of subcomplexes are observed as fragment ions.[135-137] Recently, SID data for hetero-complexes have also been reported and further demonstrate the exciting potential of this technology for quickly mapping protein complex connectivity. [138]
4.2 Protein Contact Map Generation
The interactions between proteins within complexes can be represented by a reduced map of protein-protein contacts, consisting of nodes denoting individual protein subunits and connections between the nodes which indicate the non-covalent interface between a protein with another protein. MS analysis on intact protein complexes can be used to construct such contact maps allowing for inference of the topology of protein complexes. [30, 48] For example, MS data were vital in defining the organization of proteins within macromolecular assemblies such as the yeast exosome,[139] the 19S proteosome lid [113], and the eukaryotic initiation factor 3 (eIF3) [140], significantly in the absence of high-resolution structures. Comprehensive descriptions of the experimental workflows for such experiments are covered elsewhere. [139, 141] To summarize, protein assemblies are measured intact by MS, and then the assembly is partially disrupted to generate several subcomplexes. In order to reveal the entire set of protein-protein interactions, a sufficient inventory of subcomplexes is required, and accordingly both gas-phase and solution approaches have been developed with this goal in mind. Gas-phase methodologies used for the dissociation of protein complexes involving energetic activation of proteins (e.g., CID) are described in detail above (Section 4.1). Various factors contribute toward generation of sub-assemblies in solution, including solution composition (e.g. organic content), ionic strength, pH, and temperature,[139] although the mechanistic details of the protein complex disruption process under such conditions is still a subject of active research. Identifying the composition of sub-assemblies produced during the course of disruption experiment can indicate the presence and/or the absence of interactions between protein subunits. Organizing such information into a protein contact diagram, in combination with protein docking and homology modeling, can result in a pseudo-3D map of protein subunits.[141] These experiments are often performed in parallel with other MS experiments aimed at determining both the identity and the extent of post-translational modification on interacting subunits.
MS data of intact complexes and subcomplexes have been used to generate such contact diagrams for large hetero-protein complexes. For example, intact MS data on the ribonuclear protein complex Cascade defined a contact map for the assembly and, along with EM datasets was used to define the overall topology of subunits within the complex, in this case a unique ‘seahorse’-type structure.[142] MS data of intact assemblies and subcomplexes was also used to define contact maps for the Trax-translin endonuclease complex,[143] RNA polymerase complexes,[144] and the DNA sliding clamp assembly from E. coli.[145] MS data contributed to the interpretation of X-ray and EM data for the 1 MDa CCT mammalian chaperone complex, leading to a comprehensive structural assignment and functional details regarding the mechanism of how the complex aides in the folding of tubulin.[146]
4.3 Protein-Ligand, Small Molecule and Drug Interactions
The analysis of protein-small molecule complexes by MS has a long history, from the earliest demonstrations of protein-ligand measurements by MS, in which the intensity of ions was often used to correlate binding affinity,[147-149] through to more recent applications to multiprotein-ligand complexes.[150, 151] In the case of an intact assembly composed of a multiprotein complex and a small-molecule ligand, minimal activation in the gas-phase is often sufficient to dissociate the ligand from the assembly. This dissociation is generally favored in protein complexes where the subunits and ligands are maintained by hydrophobic interactions [152, 153], although data indicate that even this basic rule has exceptions [154]. The past several years, however, have seen such intact protein-ligand measurements develop into a robust approach for the measurement of protein-ligand binding constants.[155-157] Furthermore, the incorporation of IM information (see Section 4.4) into such experiments has enabled collision induced unfolding (CIU) measurements as a means of assessing protein-ligand stability in ways orthogonal to MS-only probes.[158]
Chemical labeling techniques utilizing HDX and OFP-MS have also emerged as sensitive and universal approaches for quantitatively characterizing protein-ligand complexes within complex mixtures. Technologies such as SUPREX, [159] PLIMSTEX,[160] and SPROX[161] can address a wide range of protein-protein and protein-ligand complexes. Although all workflows within this category differ slightly, the general approach is that proteins, or protein mixtures, are exposed to a chemical label (e.g. a deuterating agent) and the level of labeling achieved is measured by MS to assess protein stability. Upon the addition of ligands of interest, this experiment is repeated, so the labeling patterns and extents of label incorporation can be compared. These types of technologies have been utilized to assess protein-ligand interactions on a proteome-wide scale.[162] Such data has direct relevance to the goals of structural proteomics, and points to promising future experiments where ligand binding, complex stability, and structure may be assessed on a proteome scale.
4.4 IM-MS of Intact Protein Complexes
Recently, the utility of coupling MS to IM separation has generated considerable excitement, because multiple sets of preliminary data have indicated that the molecular architecture of large protein complexes can be retained in the absence of bulk solvent [128, 163-165]. Originally applied to problems in chemical physics [166-168], trace detection [169, 170], and used for the analysis of small biomolecules for over a decade [23, 24, 171-174], IM separates ions based on their ability to traverse a chamber filled with inert neutrals under the influence of a relatively weak electric field. Ion size in the form of an orientiationally-averaged collision cross-section (CCS) is the primary information content of IM separation and established computational approaches can be used in conjunction with this information to assign the structure of small biomolecules to a high degree of precision. [175]
The past several years have witnessed numerous applications of IM-MS to multi-protein complexes in an effort to determine their structures. Early work on the Tryptophan RNA binding attenuation protein (TRAP) [128] and Aβ amyloid aggregates [165] illustrated the power of the IM-MS approach. Stand-alone IM results, principally utilizing differential mobility analyzer (DMA)-type technology were also used to analyze multiprotein complexes and demonstrate the ability of ion size measurements to inform on assembly topology and structure.[164] For example, IM results for the 20S proteosome catalytic particle (28-mer), along with half-proteosome measurements (14-mer) illustrate elegantly how size measurements of such subcomplexes can be used to refine a model of the intact assembly which they comprise. In all of the examples above, pioneering measurements were followed by a series of improvements in IM-MS technology and data analysis capabilities that now frame a field where multiprotein complexes over a large mass and size range can be routinely interrogated.
Recent examples of IM-MS technology used in the structural determination of multiprotein complex architecture typically fall into three main categories. First, IM-MS has been used extensively to refine protein contact maps derived from MS measurements. Recent experiments of the DNA clamp loader assembly (Figure 4),[176] the eukaryotic translation factor eIF3,[140] and RNA polymerase I and II complexes[144] have all utilized topologically informative IM data in addition to MS resulting in well-defined topological models. Secondly, IM-MS has been used to monitor the assembly of viral capsid proteins and assess the structure of assembly intermediates.[177] A final area of much research centers on the analysis of small oligomers involved in multiple amyloid-type diseases. Several IM-MS studies have determined topologies and stoichiometries for peptides and protein oligomer populations involved in Alzheimer's disease,[178] Type II diabetes,[179] and dialysis-related amyloidosis.[180] Recently, this work has been extended to cover several model systems for amyloid formation, and revealed critical changes in the structures of small oligomers that are potentially crucial for understanding both fibril formation and mechanisms of cytotoxicity.[181]
Figure 4.
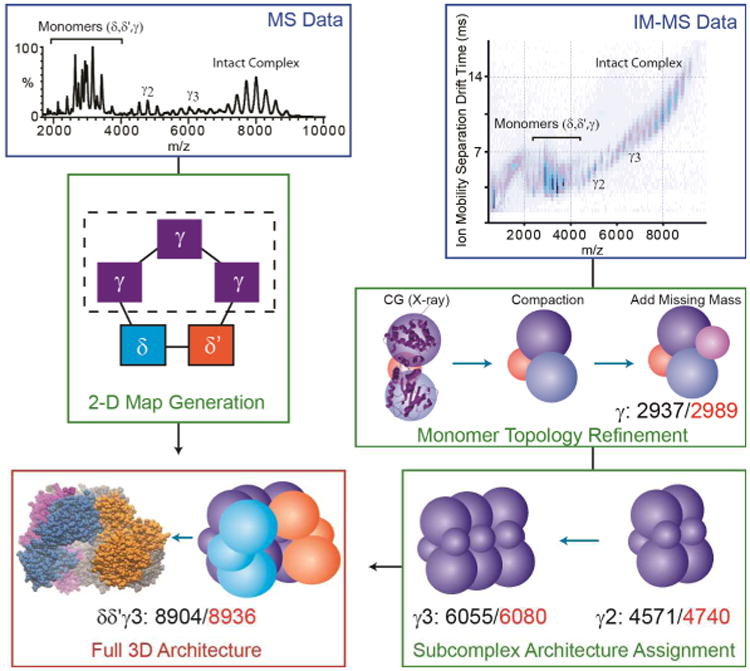
Adapted from data shown in reference [176], IM and MS data are integrated to generate a topological model of the DNA clamp loader pentamer (δδ′γγγ). First MS data on intact protein complexes and subcomplexes are recorded (blue box, upper left) and computationally analyzed to produce a contact diagram for the complex (green box, center left). The dashed box surrounding the three equivalent γ proteins within the complex indicates that the detailed connectivity between these isomass proteins cannot be determined. IM-MS data is acquired, in parallel with MS information, and size information on proteins and subcomplexes recorded (blue box, upper right). Models of all protein subunits are constructed by integrating partial X-ray data with IM distance constraints. For the γ protein, where 18% of the protein sequence is absent in the X-ray crystal structure, models must be constructed of the full length protein (shown here as a coarse-grained approximation) and the overall structure optimized to conform to IM data for the intact protein, which indicates a compact topology. Higher order and complete complexes are constructed, at each point refining the complexes based on IM measurements (green box, lower right). The resulting topology for the complex is shown in the bottom left of the diagram in the red box (right), and the X-ray structure for the intact complex is also shown (left) for comparison. Agreement between the experimental and computational models is shown in each box as a comparison between experimental (red) and theoretical (black) collision cross-section values.
4.5 Combining MS with Molecular Modeling
Computational modeling is often coupled with MS datasets as a means to predict, or verify the three-dimensional structure of biomolecules. This modeling procedure can exploit spatial constraints derived from the multiple MS-based analytical techniques described herein, although a lack of a universal software solution for managing information from different technology remains a critical bottleneck in structural proteomics. Monomeric proteins are most readily amenable to such a strategy, where distance constraints and solvent accessibility determined from CXL-MS and HDX-MS can be utilized to deduce structure (see Sections 3.1 and 3.4 above). The topology of protein complexes can be projected from connectivity information derived from methods such as CXL-MS and intact MS of protein complexes. For example, a complete model of the human exosome (Figure 2F) was constructed using this approach in advance of X-ray data, [182] and was later verified by a crystallographic structure of the same complex. [183] The construction is a combined effort of integrating intact mass spectrometry data with homology modeling and sophisticated protein-protein docking algorithms.
IM-MS experiments on protein complexes also make extensive use of computational modeling in order to develop refined structures of multiprotein assemblies. Computational analysis is required to both generate protein model structures and estimate the size of these models for comparison with the IM experiment. While complete molecular dynamics simulations, as are commonly incorporated into the analysis of IM data on smaller biomolecules, have yet to emerge for the analysis of multiprotein complex systems, molecular modeling-based topology searching plays a large role in assembling the structure of such complexes. Typically, structures of smaller units within the complex are refined against both IM-MS and other available data (Figure 5). Higher-order assemblies are then built based on distance constraints derived from IM datasets, the connectivity information obtained by MS, and data from other sources available in the literature. One active area of integrating IM-MS data with computational modeling involves determining the overall topology of small homo-oligomers important within the etiology of many neurodegenerative and amyloid-related diseases. In these studies, a set of trends which describes a correlation between the aggregate size and the number of aggregating units that comprise each oligomer is developed for different archetypal models. Aligning the experimental data determined for a range of protein aggregates with such trends for archetypal structures allows estimation of the topology of aggregates observed experimentally. [178, 180] Recent work has shown that IM can be an effective method when combined with partial X-ray data derived for the constituent proteins that comprise a complex (Figure 4), and that homology models, protein domain analysis, and elastic network modeling can be used to refine subunit models prior to complex construction. [176]
Figure 5.

Three levels of computational modeling are shown, detailing how that modeling might interface with various MS datasets. The first level (blue) shows a number of monomer or protein subunit models, ranging from simple coarse-grained representations of fixed protein density (left) to complete atomic structures of the intact protein (right). Higher-order protein models can be built using distance constraints derived from chemical cross linking or IM datasets using coarse-grained, hybrid, or atomic models in conjunction with protein-protein docking algorithms (purple). Complete models of the complex can also take the form of coarse-grained, hybrid, or complete atomic models of the intact complex.
5 Emerging MS Technologies for Structural Proteomics
Several MS-based technologies currently under intense development have the potential to be valuable tools in the determination of protein structure within the context of structural proteomics. For example, ECD is used widely for obtaining sequence and identity information on proteins and peptides, but has also seen limited use as a tool to assess the structure of some monomeric proteins. [184, 185] In these experiments, backbone cleavages are taken as evidence of the level of intra-molecular interaction surrounding the cleaved region of the sequence, and cleavage frequency can be used to map relatively flexible regions of protein secondary and tertiary structures. Similarly, gas-phase IR action spectroscopy has been used extensively to assess the secondary structure of small peptides. [186] Such data are derived from photo-dissociation efficiencies observed for peptides and proteins as a function of the wavelength used to excite the ions of interest, and have recently been used to analyze structural features of gas-phase protein ions. [187] Gas-phase fluorescence measurements are also starting to provide increased numbers of datasets illuminating the structures of biomolecules in the gas-phase and relating those measurements to solution. [187, 188]
As evidenced by the many studies covered in this review, the integration of information derived from the multiple MS-based analytical tools available for protein structure is a hallmark of both the role of MS within structural biology and the future of MS within structural proteomics. Future developments are likely to find MS-based technologies integrated with many of the established structural biology tools. For example, a recent report has described preparative MS experiments used to purify protein prior to analysis by both atomic force microscopy and electron microscopy.[189] Further, multiple reports have indicated that X-ray diffraction of single protein ions will involve significant MS and IM-based filters prior to analysis to ensure protein composition and structural homogeneity. [190] Finally, the sample handing and data acquisition methods devised to conquer many of the challenges presented by protein identification experiments are readily applicable to the MS-based approaches described herein, and will undoubtedly enable high-throughput analysis of protein structures in the future. Overall, based on the breadth of literature covered here, it is easy to predict a bright future for MS-based technologies within the field of structural proteomics.
Acknowledgments
The authors thank Daniel Barsky (California State, East Bay) for his contributions to Figure 5. BTR acknowledges support from the National Institutes of Health (1-R01-GM-095832-01) and the University of Michigan.
References
- 1.Kuhner S, van Noort V, Betts MJ, Leo-Macias A, et al. Proteome organization in a genome-reduced bacterium. Science. 2009;326:1235–1240. doi: 10.1126/science.1176343. [DOI] [PubMed] [Google Scholar]
- 2.Tarassov K, Messier V, Landry CR, Radinovic S, et al. An in vivo map of the yeast protein interactome. Science. 2008;320:1465–1470. doi: 10.1126/science.1153878. [DOI] [PubMed] [Google Scholar]
- 3.Rix U, Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat Chem Biol. 2009;5:616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- 4.Shi W, Chance MR. Metalloproteomics: forward and reverse approaches in metalloprotein structural and functional characterization. Curr Opin Chem Biol. 2011;15:144–148. doi: 10.1016/j.cbpa.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young NL, Plazas-Mayorca MD, Garcia BA. Systems-wide proteomic characterization of combinatorial post-translational modification patterns. Expert Rev Proteomics. 2010;7:79–92. doi: 10.1586/epr.09.100. [DOI] [PubMed] [Google Scholar]
- 6.Yang Z. Progress and challenges in protein structure prediction. Curr Opin Struct Biol. 2008;18:342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, et al. The molecular architecture of the nuclear pore complex. Nature. 2007;450:695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- 8.Gingras AC, Gstaiger M, Raught B, Aebersold R. Analysis of protein complexes using mass spectrometry. Nat Rev Mol Cell Biol. 2007;8:645–654. doi: 10.1038/nrm2208. [DOI] [PubMed] [Google Scholar]
- 9.Englander SW. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom. 2006;17:1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engen JR. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal Chem. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaltashov IA, Bobst CE, Abzalimov RR. H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: is there a need for a top-down approach? Anal Chem. 2009;81:7892–7899. doi: 10.1021/ac901366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev Proteomics. 2011;8:43–59. doi: 10.1586/epr.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- 14.Jin Lee Y. Mass spectrometric analysis of cross-linking sites for the structure of proteins and protein complexes. Mol Biosyst. 2008;4:816–823. doi: 10.1039/b801810c. [DOI] [PubMed] [Google Scholar]
- 15.Leitner A, Walzthoeni T, Kahraman A, Herzog F, et al. Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol Cell Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petrotchenko EV, Borchers CH. Crosslinking combined with mass spectrometry for structural proteomics. Mass Spectrom Rev. 2010;29:862–876. doi: 10.1002/mas.20293. [DOI] [PubMed] [Google Scholar]
- 17.Konermann L, Stocks BB, Pan Y, Tong X. Mass spectrometry combined with oxidative labeling for exploring protein structure and folding. Mass Spectrom Rev. 2010;29:651–667. doi: 10.1002/mas.20256. [DOI] [PubMed] [Google Scholar]
- 18.Kiselar JG, Chance MR. Future directions of structural mass spectrometry using hydroxyl radical footprinting. J Mass Spectrom. 2010;45:1373–1382. doi: 10.1002/jms.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simon JH. The structural aspects of limited proteolysis of native proteins. BBA-Protein Struct M. 1998;1382:191–206. doi: 10.1016/s0167-4838(97)00175-1. [DOI] [PubMed] [Google Scholar]
- 20.Fontana A, de Laureto PP, Spolaore B, Frare E, et al. Probing protein structure by limited proteolysis. Acta Biochim Pol. 2004;51:299–321. [PubMed] [Google Scholar]
- 21.Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- 22.Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJR. Ion mobility mass spectrometry of proteins and protein assemblies. Chem Soc Rev. 2010;39:1633–1655. doi: 10.1039/b914002f. [DOI] [PubMed] [Google Scholar]
- 23.Kanu AB, Dwivedi P, Tam M, Matz L, Hill HH. Ion mobility–mass spectrometry. J Mass Spectrom. 2008;43:1–22. doi: 10.1002/jms.1383. [DOI] [PubMed] [Google Scholar]
- 24.Bohrer BC, Merenbloom SI, Koeniger SL, Hilderbrand AE, Clemmer DE. Biomolecule Analysis by Ion Mobility Spectrometry. Annu Rev Anal Chem. 2008;1:293–327. doi: 10.1146/annurev.anchem.1.031207.113001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chernushevich IV, Thomson BA. Collisional cooling of large ions in electrospray mass spectrometry. Anal Chem. 2004;76:1754–1760. doi: 10.1021/ac035406j. [DOI] [PubMed] [Google Scholar]
- 26.Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 27.van den Heuvel RH, van Duijn E, Mazon H, Synowsky SA, et al. Improving the performance of a quadrupole time-of-flight instrument for macromolecular mass spectrometry. Anal Chem. 2006;78:7473–7483. doi: 10.1021/ac061039a. [DOI] [PubMed] [Google Scholar]
- 28.Benesch JL, Ruotolo BT, Simmons DA, Robinson CV. Protein complexes in the gas phase: technology for structural genomics and proteomics. Chem Rev. 2007;107:3544–3567. doi: 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]
- 29.Heck AJR. Native mass spectrometry: a bridge between interactomics and structural biology. Nat Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 30.Zhou M, Robinson CV. When proteomics meets structural biology. Trends Biochem Sci. 2010;35:522–529. doi: 10.1016/j.tibs.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Kelleher NL. Top-down proteomics. Anal Chem. 2004;76:197A–203A. [PubMed] [Google Scholar]
- 32.Cui W, Rohrs HW, Gross ML. Top-down mass spectrometry: Recent developments, applications and perspectives. Analyst. 2011;136:3854–3864. doi: 10.1039/c1an15286f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paizs B, Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrom Rev. 2005;24:508–548. doi: 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- 34.Cooper HJ, Hakansson K, Marshall AG. The role of electron capture dissociation in biomolecular analysis. Mass Spectrom Rev. 2005;24:201–222. doi: 10.1002/mas.20014. [DOI] [PubMed] [Google Scholar]
- 35.Mann M, Hendrickson RC, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 2001;70:437–473. doi: 10.1146/annurev.biochem.70.1.437. [DOI] [PubMed] [Google Scholar]
- 36.Counterman AE, Clemmer DE. Large anhydrous polyalanine ions: Evidence for extended helices and onset of a more compact state. J Am Chem Soc. 2001;123:1490–1498. doi: 10.1021/ja9940625. [DOI] [PubMed] [Google Scholar]
- 37.Jarrold MF. Helices and sheets in vacuo. Phys Chem Chem Phys. 2007;9:1659–1671. doi: 10.1039/b612615d. [DOI] [PubMed] [Google Scholar]
- 38.Ruotolo BT, Tate CC, Russell DH. Ion mobility-mass spectrometry applied to cyclic peptide analysis: Conformational preferences of gramicidin S and linear analogs in the gas phase. J Am Soc Mass Spectrom. 2004;15:870–878. doi: 10.1016/j.jasms.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Ruotolo BT, Gillig KJ, Woods AS, Egan TF, et al. Analysis of phosphorylated peptides by ion mobility-mass spectrometry. Anal Chem. 2004;76:6727–6733. doi: 10.1021/ac0498009. [DOI] [PubMed] [Google Scholar]
- 40.Ruotolo BT, Verbeck GF, Thomson LM, Woods AS, et al. Distinguishing between phosphorylated and nonphosphorylated peptides with ion mobility-mass spectrometry. J Proteome Res. 2002;1:303–306. doi: 10.1021/pr025516r. [DOI] [PubMed] [Google Scholar]
- 41.McLean JA. The Mass-Mobility Correlation Redux: The Conformational Landscape of Anhydrous Biomolecules. J Am Soc Mass Spectrom. 2009;20:1775–1781. doi: 10.1016/j.jasms.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 42.Baumketner A, Bernstein SL, Wyttenbach T, Bitan G, et al. Amyloid beta-protein monomer structure: A computational and experimental study. Protein Sci. 2006;15:420–428. doi: 10.1110/ps.051762406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wyttenbach T, Bowers MT. Annu Rev Phys Chem, Annual Reviews, Palo Alto. 2007:511–533. doi: 10.1146/annurev.physchem.58.032806.104515. [DOI] [PubMed] [Google Scholar]
- 44.Jurneczko E, Barran PE. How useful is ion mobility mass spectrometry for structural biology? The relationship between protein crystal structures and their collision cross sections in the gas phase. Analyst. 2011;136:20–28. doi: 10.1039/c0an00373e. [DOI] [PubMed] [Google Scholar]
- 45.Rizzo TR, Stearns JA, Boyarkin OV. Spectroscopic studies of cold, gas-phase biomolecular ions. Int Rev Phys Chem. 2009;28:481–515. [Google Scholar]
- 46.Fabris D, Yu ET. Elucidating the higher-order structure of biopolymers by structural probing and mass spectrometry: MS3D. J Mass Spectrom. 2010;45:841–860. doi: 10.1002/jms.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinz A. Investigation of protein-protein interactions in living cells by chemical crosslinking and mass spectrometry. Anal Bioanal Chem. 2010;397:3433–3440. doi: 10.1007/s00216-009-3405-5. [DOI] [PubMed] [Google Scholar]
- 48.Sharon M, Robinson CV. Annu Rev Biochem, Annual Reviews, Palo Alto. 2007:167–193. doi: 10.1146/annurev.biochem.76.061005.090816. [DOI] [PubMed] [Google Scholar]
- 49.Rand KD, Adams CM, Zubarev RA, Jorgensen TJ. Electron capture dissociation proceeds with a low degree of intramolecular migration of peptide amide hydrogens. J Am Chem Soc. 2008;130:1341–1349. doi: 10.1021/ja076448i. [DOI] [PubMed] [Google Scholar]
- 50.Pan J, Han J, Borchers CH, Konermann L. Hydrogen/deuterium exchange mass spectrometry with top-down electron capture dissociation for characterizing structural transitions of a 17 kDa protein. J Am Chem Soc. 2009;131:12801–12808. doi: 10.1021/ja904379w. [DOI] [PubMed] [Google Scholar]
- 51.Rand KD, Zehl M, Jensen ON, Jorgensen TJ. Protein hydrogen exchange measured at single-residue resolution by electron transfer dissociation mass spectrometry. Anal Chem. 2009;81:5577–5584. doi: 10.1021/ac9008447. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J, Chalmers MJ, Stayrook KR, Burris LL, et al. Hydrogen/deuterium exchange reveals distinct agonist/partial agonist receptor dynamics within vitamin D receptor/retinoid X receptor heterodimer. Structure. 2010;18:1332–1341. doi: 10.1016/j.str.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.West GM, Chien EY, Katritch V, Gatchalian J, et al. Ligand-Dependent Perturbation of the Conformational Ensemble for the GPCR beta(2) Adrenergic Receptor Revealed by HDX. Structure. 2011 doi: 10.1016/j.str.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marcsisin SR, Narute PS, Emert-Sedlak LA, Kloczewiak M, et al. On the solution conformation and dynamics of the HIV-1 viral infectivity factor. J Mol Biol. 2011;410:1008–1022. doi: 10.1016/j.jmb.2011.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morgan CR, Hebling CM, Rand KD, Stafford DW, et al. Conformational transitions in the membrane scaffold protein of phospholipid bilayer nanodiscs. Mol Cell Proteomics. 2011;10:M111 010876. doi: 10.1074/mcp.M111.010876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pan J, Han J, Borchers CH, Konermann L. Characterizing short-lived protein folding intermediates by top-down hydrogen exchange mass spectrometry. Anal Chem. 2010;82:8591–8597. doi: 10.1021/ac101679j. [DOI] [PubMed] [Google Scholar]
- 57.Choi JH, Banks AS, Kamenecka TM, Busby SA, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang L, Lane LC, Smith DL. Detecting structural changes in viral capsids by hydrogen exchange and mass spectrometry. Protein Sci. 2001;10:1234–1243. doi: 10.1110/ps.100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X, Chien EY, Chalmers MJ, Pascal BD, et al. Dynamics of the beta2-adrenergic G-protein coupled receptor revealed by hydrogen-deuterium exchange. Anal Chem. 2010;82:1100–1108. doi: 10.1021/ac902484p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hebling CM, Morgan CR, Stafford DW, Jorgenson JW, et al. Conformational analysis of membrane proteins in phospholipid bilayer nanodiscs by hydrogen exchange mass spectrometry. Anal Chem. 2010;82:5415–5419. doi: 10.1021/ac100962c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449:233–237. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 62.Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, et al. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol. 2011;18:504–506. doi: 10.1038/nsmb.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carulla N, Zhou M, Giralt E, Robinson CV, Dobson CM. Structure and intermolecular dynamics of aggregates populated during amyloid fibril formation studied by hydrogen/deuterium exchange. Acc Chem Res. 2010;43:1072–1079. doi: 10.1021/ar9002784. [DOI] [PubMed] [Google Scholar]
- 64.Williams AD, Sega M, Chen M, Kheterpal I, et al. Structural properties of Abeta protofibrils stabilized by a small molecule. Proc Natl Acad Sci U S A. 2005;102:7115–7120. doi: 10.1073/pnas.0408582102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mendoza VL, Vachet RW. Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev. 2009;28:785–815. doi: 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 67.Stocks BB, Konermann L. Structural characterization of short-lived protein unfolding intermediates by laser-induced oxidative labeling and mass spectrometry. Anal Chem. 2009;81:20–27. doi: 10.1021/ac801888h. [DOI] [PubMed] [Google Scholar]
- 68.Chen J, Rempel DL, Gross ML. Temperature jump and fast photochemical oxidation probe submillisecond protein folding. J Am Chem Soc. 2010;132:15502–15504. doi: 10.1021/ja106518d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stocks BB, Rezvanpour A, Shaw GS, Konermann L. Temporal development of protein structure during S100A11 folding and dimerization probed by oxidative labeling and mass spectrometry. J Mol Biol. 2011;409:669–679. doi: 10.1016/j.jmb.2011.04.028. [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Qin Y, Ilchenko S, Bohon J, et al. Structural analysis of a highly glycosylated and unliganded gp120-based antigen using mass spectrometry. Biochemistry. 2010;49:9032–9045. doi: 10.1021/bi1011332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Angel TE, Gupta S, Jastrzebska B, Palczewski K, Chance MR. Structural waters define a functional channel mediating activation of the GPCR, rhodopsin. Proc Natl Acad Sci U S A. 2009;106:14367–14372. doi: 10.1073/pnas.0901074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gavin AC, Bosche M, Krause R, Grandi P, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 73.Butland G, Peregrin-Alvarez JM, Li J, Yang W, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 74.Gavin AC, Aloy P, Grandi P, Krause R, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 75.Krogan NJ, Cagney G, Yu H, Zhong G, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 76.Gavin AC, Maeda K, Kuhner S. Recent advances in charting protein-protein interaction: mass spectrometry-based approaches. Curr Opin Biotechnol. 2011;22:42–49. doi: 10.1016/j.copbio.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 77.Rigaut G, Shevchenko A, Rutz B, Wilm M, et al. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 78.Einhauer A, Jungbauer A. The FLAG peptide, a versatile fusion tag for the purification of recombinant proteins. J Biochem Biophys Methods. 2001;49:455–465. doi: 10.1016/s0165-022x(01)00213-5. [DOI] [PubMed] [Google Scholar]
- 79.Blagoev B, Kratchmarova I, Ong SE, Nielsen M, et al. A proteomics strategy to elucidate functional protein-protein interactions applied to EGF signaling. Nat Biotechnol. 2003;21:315–318. doi: 10.1038/nbt790. [DOI] [PubMed] [Google Scholar]
- 80.Ross PL, Huang YN, Marchese JN, Williamson B, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 81.Gygi SP, Rist B, Gerber SA, Turecek F, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 82.Bouwmeester T, Bauch A, Ruffner H, Angrand PO, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 83.Jeronimo C, Forget D, Bouchard A, Li Q, et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell. 2007;27:262–274. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jao DL, Chen KY. Tandem affinity purification revealed the hypusine-dependent binding of eukaryotic initiation factor 5A to the translating 80S ribosomal complex. J Cell Biochem. 2006;97:583–598. doi: 10.1002/jcb.20658. [DOI] [PubMed] [Google Scholar]
- 87.Behzadnia N, Golas MM, Hartmuth K, Sander B, et al. Composition and three-dimensional EM structure of double affinity-purified, human prespliceosomal A complexes. EMBO J. 2007;26:1737–1748. doi: 10.1038/sj.emboj.7601631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Wulf P, McAinsh AD, Sorger PK. Hierarchical assembly of the budding yeast kinetochore from multiple subcomplexes. Genes Dev. 2003;17:2902–2921. doi: 10.1101/gad.1144403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rappsilber J. The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J Struct Biol. 2011;173:530–540. doi: 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee YJ, Lackner LL, Nunnari JM, Phinney BS. Shotgun cross-linking analysis for studying quaternary and tertiary protein structures. J Proteome Res. 2007;6:3908–3917. doi: 10.1021/pr070234i. [DOI] [PubMed] [Google Scholar]
- 91.Vasilescu J, Guo XC, Kast J. Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics. 2004;4:3845–3854. doi: 10.1002/pmic.200400856. [DOI] [PubMed] [Google Scholar]
- 92.Chu F, Shan SO, Moustakas DT, Alber F, et al. Unraveling the interface of signal recognition particle and its receptor by using chemical cross-linking and tandem mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:16454–16459. doi: 10.1073/pnas.0407456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chu F, Maynard JC, Chiosis G, Nicchitta CV, Burlingame AL. Identification of novel quaternary domain interactions in the Hsp90 chaperone, GRP94. Protein Sci. 2006;15:1260–1269. doi: 10.1110/ps.052065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maiolica A, Cittaro D, Borsotti D, Sennels L, et al. Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol Cell Proteomics. 2007;6:2200–2211. doi: 10.1074/mcp.M700274-MCP200. [DOI] [PubMed] [Google Scholar]
- 95.Schulz DM, Kalkhof S, Schmidt A, Ihling C, et al. Annexin A2/P11 interaction: new insights into annexin A2 tetramer structure by chemical crosslinking, high-resolution mass spectrometry, and computational modeling. Proteins. 2007;69:254–269. doi: 10.1002/prot.21445. [DOI] [PubMed] [Google Scholar]
- 96.Pimenova T, Nazabal A, Roschitzki B, Seebacher J, et al. Epitope mapping on bovine prion protein using chemical cross-linking and mass spectrometry. J Mass Spectrom. 2008;43:185–195. doi: 10.1002/jms.1280. [DOI] [PubMed] [Google Scholar]
- 97.Dimova K, Kalkhof S, Pottratz I, Ihling C, et al. Structural insights into the calmodulin-Munc13 interaction obtained by cross-linking and mass spectrometry. Biochemistry. 2009;48:5908–5921. doi: 10.1021/bi900300r. [DOI] [PubMed] [Google Scholar]
- 98.Trnka MJ, Burlingame AL. Topographic studies of the GroEL-GroES chaperonin complex by chemical cross-linking using diformyl ethynylbenzene: the power of high resolution electron transfer dissociation for determination of both peptide sequences and their attachment sites. Mol Cell Proteomics. 2010;9:2306–2317. doi: 10.1074/mcp.M110.003764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen ZA, Jawhari A, Fischer L, Buchen C, et al. Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sinz A. Chemical cross-linking and mass spectrometry for mapping three-dimensional structures of proteins and protein complexes. J Mass Spectrom. 2003;38:1225–1237. doi: 10.1002/jms.559. [DOI] [PubMed] [Google Scholar]
- 101.Sinz A. Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- 102.Petrotchenko EV, Xiao K, Cable J, Chen Y, et al. BiPS, a photocleavable, isotopically coded, fluorescent cross-linker for structural proteomics. Mol Cell Proteomics. 2009;8:273–286. doi: 10.1074/mcp.M800265-MCP200. [DOI] [PubMed] [Google Scholar]
- 103.Novak P, Giannakopulos AE. Chemical cross-linking and mass spectrometry as structure determination tools. Eur J Mass Spectrom (Chichester, Eng) 2007;13:105–113. doi: 10.1255/ejms.868. [DOI] [PubMed] [Google Scholar]
- 104.Panchaud A, Singh P, Shaffer SA, Goodlett DR. xComb: A Cross-Linked Peptide Database Approach to Protein-Protein Interaction Analysis. J Proteome Res. 2010;9:2508–2515. doi: 10.1021/pr9011816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gao QX, Xue S, Doneanu CE, Shaffer SA, et al. Pro-CrossLink. Software tool for protein cross-linking and mass spectrometry. Anal Chem. 2006;78:2145–2149. doi: 10.1021/ac051339c. [DOI] [PubMed] [Google Scholar]
- 106.Muller MQ, Dreiocker F, Ihling CH, Schafer M, Sinz A. Cleavable Cross-Linker for Protein Structure Analysis: Reliable Identification of Cross-Linking Products by Tandem MS. Anal Chem. 2010;82:6958–6968. doi: 10.1021/ac101241t. [DOI] [PubMed] [Google Scholar]
- 107.Dreiocker F, Muller MQ, Sinz A, Schafer M. Collision-induced dissociative chemical cross-linking reagent for protein structure characterization: applied Edman chemistry in the gas phase. J Mass Spectrom. 2010;45:178–189. doi: 10.1002/jms.1702. [DOI] [PubMed] [Google Scholar]
- 108.Lu YL, Tanasova M, Borhan B, Reid GE. Ionic Reagent for Controlling the Gas-Phase Fragmentation Reactions of Cross-Linked Peptides. Anal Chem. 2008;80:9279–9287. doi: 10.1021/ac801625e. [DOI] [PubMed] [Google Scholar]
- 109.Soderblom EJ, Bobay BG, Cavanagh J, Goshe MB. Tandem mass spectrometry acquisition approaches to enhance identification of protein-protein interactions using low-energy collision-induced dissociative chemical crosslinking reagents. Rapid Commun Mass Spectrom. 2007;21:3395–3408. doi: 10.1002/rcm.3213. [DOI] [PubMed] [Google Scholar]
- 110.Hoopmann MR, Weisbrod CR, Bruce JE. Improved Strategies for Rapid Identification of Chemically Cross-Linked Peptides Using Protein Interaction Reporter Technology. J Proteome Res. 2010;9:6323–6333. doi: 10.1021/pr100572u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jaya N, Garcia V, Vierling E. Substrate binding site flexibility of the small heat shock protein molecular chaperones. Proc Natl Acad Sci U S A. 2009;106:15604–15609. doi: 10.1073/pnas.0902177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Andreasson C, Fiaux J, Rampelt H, Druffel-Augustin S, Bukau B. Insights into the structural dynamics of the Hsp110-Hsp70 interaction reveal the mechanism for nucleotide exchange activity. Proc Natl Acad Sci U S A. 2008;105:16519–16524. doi: 10.1073/pnas.0804187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sharon M, Taverner T, Ambroggio XI, Deshaies RJ, Robinson CV. Structural organization of the 19S proteasome lid: insights from MS of intact complexes. PLoS Biol. 2006;4:e267. doi: 10.1371/journal.pbio.0040267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Scaloni A, Miraglia N, Orru S, Amodeo P, et al. Topology of the calmodulin-melittin complex. J Mol Biol. 1998;277:945–958. doi: 10.1006/jmbi.1998.1629. [DOI] [PubMed] [Google Scholar]
- 115.Schulz DM, Ihling C, Clore GM, Sinz A. Mapping the topology and determination of a low-resolution three-dimensional structure of the calmodulin-melittin complex by chemical cross-linking and high-resolution FTICRMS: direct demonstration of multiple binding modes. Biochemistry. 2004;43:4703–4715. doi: 10.1021/bi036149f. [DOI] [PubMed] [Google Scholar]
- 116.Kang S, Hawkridge AM, Johnson KL, Muddiman DC, Prevelige PE., Jr Identification of subunit-subunit interactions in bacteriophage P22 procapsids by chemical cross-linking and mass spectrometry. J Proteome Res. 2006;5:370–377. doi: 10.1021/pr050356f. [DOI] [PubMed] [Google Scholar]
- 117.Cai K, Itoh Y, Khorana HG. Mapping of contact sites in complex formation between transducin and light-activated rhodopsin by covalent crosslinking: use of a photoactivatable reagent. Proc Natl Acad Sci U S A. 2001;98:4877–4882. doi: 10.1073/pnas.051632898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Itoh Y, Cai K, Khorana HG. Mapping of contact sites in complex formation between light-activated rhodopsin and transducin by covalent crosslinking: use of a chemically preactivated reagent. Proc Natl Acad Sci U S A. 2001;98:4883–4887. doi: 10.1073/pnas.051632998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Back JW, Sanz MA, De Jong L, De Koning LJ, et al. A structure for the yeast prohibitin complex: Structure prediction and evidence from chemical crosslinking and mass spectrometry. Protein Sci. 2002;11:2471–2478. doi: 10.1110/ps.0212602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Felitsyn N, Kitova EN, Klassen JS. Thermal decomposition of a gaseous multiprotein complex studied by blackbody infrared radiative dissociation. Investigating the origin of the asymmetric dissociation behavior. Anal Chem. 2001;73:4647–4661. doi: 10.1021/ac0103975. [DOI] [PubMed] [Google Scholar]
- 121.Kitova EN, Kitov PI, Bundle DR, Klassen JS. The observation of multivalent complexes of Shiga-like toxin with globotriaoside and the determination of their stoichiometry by nanoelectrospray Fourier-transform ion cyclotron resonance mass spectrometry. Glycobiology. 2001;11:605–611. doi: 10.1093/glycob/11.7.605. [DOI] [PubMed] [Google Scholar]
- 122.Jurchen JC, Garcia DE, Williams ER. Further studies on the origins of asymmetric charge partitioning in protein homodimers. J Am Soc Mass Spectrom. 2004;15:1408–1415. doi: 10.1016/j.jasms.2004.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jurchen JC, Williams ER. Origin of asymmetric charge partitioning in the dissociation of gas-phase protein homodimers. J Am Chem Soc. 2003;125:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Versluis C, van der Staaij A, Stokvis E, Heck AJ, de Craene B. Metastable ion formation and disparate charge separation in the gas-phase dissection of protein assemblies studied by orthogonal time-of-flight mass spectrometry. J Am Soc Mass Spectrom. 2001;12:329–336. doi: 10.1016/S1044-0305(00)00227-0. [DOI] [PubMed] [Google Scholar]
- 125.Rostom AA, Sunde M, Richardson SJ, Schreiber G, et al. Dissection of multi-protein complexes using mass spectrometry: subunit interactions in transthyretin and retinol-binding protein complexes. Proteins. 1998;(2):3–11. doi: 10.1002/(sici)1097-0134(1998)33:2+<3::aid-prot2>3.3.co;2-8. [DOI] [PubMed] [Google Scholar]
- 126.Benesch JL, Aquilina JA, Ruotolo BT, Sobott F, Robinson CV. Tandem mass spectrometry reveals the quaternary organization of macromolecular assemblies. Chem Biol. 2006;13:597–605. doi: 10.1016/j.chembiol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 127.Ruotolo BT, Hyung SJ, Robinson PM, Giles K, et al. Ion mobility-mass spectrometry reveals long-lived, unfolded intermediates in the dissociation of protein complexes. Angew Chem Int Ed Engl. 2007;46:8001–8004. doi: 10.1002/anie.200702161. [DOI] [PubMed] [Google Scholar]
- 128.Ruotolo BT, Giles K, Campuzano I, Sandercock AM, et al. Evidence for macromolecular protein rings in the absence of bulk water. Science. 2005;310:1658–1661. doi: 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- 129.Duijn Ev, Barendregt A, Synowsky S, Versluis C, Heck AJR. Chaperonin Complexes Monitored by Ion Mobility Mass Spectrometry. J Am Chem Soc. 2009;131:1452–1459. doi: 10.1021/ja8055134. [DOI] [PubMed] [Google Scholar]
- 130.Benesch JL, Ruotolo BT, Sobott F, Wildgoose J, et al. Quadrupole-time-of-flight mass spectrometer modified for higher-energy dissociation reduces protein assemblies to peptide fragments. Anal Chem. 2009;81:1270–1274. doi: 10.1021/ac801950u. [DOI] [PubMed] [Google Scholar]
- 131.Pagel K, Hyung SJ, Ruotolo BT, Robinson CV. Alternate dissociation pathways identified in charge-reduced protein complex ions. Anal Chem. 2010;82:5363–5372. doi: 10.1021/ac101121r. [DOI] [PubMed] [Google Scholar]
- 132.Ge Y, Lawhorn BG, ElNaggar M, Strauss E, et al. Top down characterization of larger proteins (45 kDa) by electron capture dissociation mass spectrometry. J Am Chem Soc. 2002;124:672–678. doi: 10.1021/ja011335z. [DOI] [PubMed] [Google Scholar]
- 133.Horn DM, Zubarev RA, McLafferty FW. Automated de novo sequencing of proteins by tandem high-resolution mass spectrometry. Proc Natl Acad Sci U S A. 2000;97:10313–10317. doi: 10.1073/pnas.97.19.10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wysocki VH, Joyce KE, Jones CM, Beardsley RL. Surface-induced dissociation of small molecules, peptides, and non-covalent protein complexes. J Am Soc Mass Spectrom. 2008;19:190–208. doi: 10.1016/j.jasms.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jones CM, Beardsley RL, Galhena AS, Dagan S, et al. Symmetrical gas-phase dissociation of noncovalent protein complexes via surface collisions. J Am Chem Soc. 2006;128:15044–15045. doi: 10.1021/ja064586m. [DOI] [PubMed] [Google Scholar]
- 136.Galhena AS, Dagan S, Jones CM, Beardsley RL, Wysocki VH. Surface-induced dissociation of peptides and protein complexes in a quadrupole/time-of-flight mass spectrometer. Anal Chem. 2008;80:1425–1436. doi: 10.1021/ac701782q. [DOI] [PubMed] [Google Scholar]
- 137.Beardsley RL, Jones CM, Galhena AS, Wysocki VH. Noncovalent protein tetramers and pentamers with “n” charges yield monomers with n/4 and n/5 charges. Anal Chem. 2009;81:1347–1356. doi: 10.1021/ac801883k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Blackwell AE, Dodds ED, Bandarian V, Wysocki VH. Revealing the quaternary structure of a heterogeneous noncovalent protein complex through surface-induced dissociation. Anal Chem. 2011;83:2862–2865. doi: 10.1021/ac200452b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hernandez H, Robinson CV. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- 140.Zhou M, Sandercock AM, Fraser CS, Ridlova G, et al. Mass spectrometry reveals modularity and a complete subunit interaction map of the eukaryotic translation factor eIF3. Proc Natl Acad Sci U S A. 2008;105:18139–18144. doi: 10.1073/pnas.0801313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Taverner T, Hernandez H, Sharon M, Ruotolo BT, et al. Subunit architecture of intact protein complexes from mass spectrometry and homology modeling. Acc Chem Res. 2008;41:617–627. doi: 10.1021/ar700218q. [DOI] [PubMed] [Google Scholar]
- 142.Jore MM, Lundgren M, van Duijn E, Bultema JB, et al. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat Struct Mol Biol. 2011;18:529–536. doi: 10.1038/nsmb.2019. [DOI] [PubMed] [Google Scholar]
- 143.Tian Y, Simanshu DK, Ascano M, Diaz-Avalos R, et al. Multimeric assembly and biochemical characterization of the Trax-translin endonuclease complex. Nat Struct Mol Biol. 2011;18:658–664. doi: 10.1038/nsmb.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lane LA, Fernandez-Tornero C, Zhou M, Morgner N, et al. Mass spectrometry reveals stable modules in holo and apo RNA polymerases I and III. Structure. 2011;19:90–100. doi: 10.1016/j.str.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 145.Park AY, Jergic S, Politis A, Ruotolo BT, et al. A single subunit directs the assembly of the Escherichia coli DNA sliding clamp loader. Structure. 2010;18:285–292. doi: 10.1016/j.str.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Munoz IG, Yebenes H, Zhou M, Mesa P, et al. Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin. Nat Struct Mol Biol. 2011;18:14–19. doi: 10.1038/nsmb.1971. [DOI] [PubMed] [Google Scholar]
- 147.Ganem B, Li YT, Henion JD. Observation of noncovalent enzyme substrate and enzyme product complexes by ion-spray mass-spectrometry. J Am Chem Soc. 1991;113:7818–7819. [Google Scholar]
- 148.Katta V, Chait BT. Observation of the heme globin complex in native myoglobin by electrospray-ionization mass-spectrometry. J Am Chem Soc. 1991;113:8534–8535. [Google Scholar]
- 149.Hu PF, Loo JA. Determining calcium-binding stoichiometry and cooperativity of parvalbumin and calmodulin by mass-spectrometry. J Mass Spectrom. 1995;30:1076–1082. [Google Scholar]
- 150.McCammon MG, Scott DJ, Keetch CA, Greene LH, et al. Screening transthyretin amyloid fibril inhibitors: Characterization of novel multiprotein, multiligand complexes by mass spectrometry. Structure. 2002;10:851–863. doi: 10.1016/s0969-2126(02)00771-2. [DOI] [PubMed] [Google Scholar]
- 151.Kitova EN, Seo M, Roy PN, Klassen JS. Elucidating the intermolecular interactions within a desolvated protein-ligand complex. An experimental and computational study. J Am Chem Soc. 2008;130:1214–1226. doi: 10.1021/ja075333b. [DOI] [PubMed] [Google Scholar]
- 152.Robinson CV, Chung EW, Kragelund BB, Knudsen J, et al. Probing the Nature of Noncovalent Interactions by Mass Spectrometry. A Study of Protein–CoA Ligand Binding and Assembly. J Am Chem Soc. 1996;118:8646–8653. [Google Scholar]
- 153.Loo JA. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom Rev. 1997;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 154.Bovet C, Wortmann A, Eiler S, Granger F, et al. Estrogen receptor-ligand complexes measured by chip-based nanoelectrospray mass spectrometry: an approach for the screening of endocrine disruptors. Protein Sci. 2007;16:938–946. doi: 10.1110/ps.062664107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wortmann A, Jecklin MC, Touboul D, Badertscher M, Zenobi R. Binding constant determination of high-affinity protein-ligand complexes by electrospray ionization mass spectrometry and ligand competition. J Mass Spectrom. 2008;43:600–608. doi: 10.1002/jms.1355. [DOI] [PubMed] [Google Scholar]
- 156.Liu J, Konermann L. Protein-protein binding affinities in solution determined by electrospray mass spectrometry. J Am Soc Mass Spectrom. 2011;22:408–417. doi: 10.1007/s13361-010-0052-1. [DOI] [PubMed] [Google Scholar]
- 157.Kitova EN, Soya N, Klassen JS. Identifying Specific Small-Molecule Interactions Using Electrospray Ionization Mass Spectrometry. Anal Chem. 2011;83:5160–5167. doi: 10.1021/ac200244u. [DOI] [PubMed] [Google Scholar]
- 158.Hyung SJ, Robinson CV, Ruotolo BT. Gas-phase unfolding and disassembly reveals stability differences in ligand-bound multiprotein complexes. Chem Biol. 2009;16:382–390. doi: 10.1016/j.chembiol.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 159.Ghaemmaghami S, Fitzgerald MC, Oas TG. A quantitative, high-throughput screen for protein stability. Proc Natl Acad Sci U S A. 2000;97:8296–8301. doi: 10.1073/pnas.140111397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhu MM, Rempel DL, Du Z, Gross ML. Quantification of protein-ligand interactions by mass spectrometry, titration, and H/D exchange: PLIMSTEX. J Am Chem Soc. 2003;125:5252–5253. doi: 10.1021/ja029460d. [DOI] [PubMed] [Google Scholar]
- 161.West GM, Tang L, Fitzgerald MC. Thermodynamic analysis of protein stability and ligand binding using a chemical modification- and mass spectrometry-based strategy. Anal Chem. 2008;80:4175–4185. doi: 10.1021/ac702610a. [DOI] [PubMed] [Google Scholar]
- 162.Dearmond PD, Xu Y, Strickland EC, Daniels KG, Fitzgerald MC. Thermodynamic Analysis of Protein-Ligand Interactions in Complex Biological Mixtures using a Shotgun Proteomics Approach. J Proteome Res. 2011 doi: 10.1021/pr200403c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ruotolo BT, Robinson CV. Aspects of native proteins are retained in vacuum. Curr Opin Chem Biol. 2006;10:402–408. doi: 10.1016/j.cbpa.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 164.Loo JA, Berhane B, Kaddis CS, Wooding KM, et al. Electrospray ionization mass spectrometry and ion mobility analysis of the 20S proteasome complex. J Am Soc Mass Spectrom. 2005;16:998–1008. doi: 10.1016/j.jasms.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 165.Bernstein SL, Wyttenbach T, Baumketner A, Shea JE, et al. Amyloid beta-protein: monomer structure and early aggregation states of Abeta42 and its Pro19 alloform. J Am Chem Soc. 2005;127:2075–2084. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]
- 166.Bowers MT, Kemper PR, von Helden G, van Koppen PA. Gas-phase ion chromatography: transition metal state selection and carbon cluster formation. Science. 1993;260:1446–1451. doi: 10.1126/science.260.5113.1446. [DOI] [PubMed] [Google Scholar]
- 167.Kemper PR, Bowers MT. Electronic-state chromatography: application to first-row transition-metal ions. J Phys Chem. 1991;95:5134–5146. [Google Scholar]
- 168.Jarrold MF. Drift Tube Studies of Atomic Clusters. J Phys Chem. 1995;99:11–21. [Google Scholar]
- 169.Eiceman GA. Advances in Ion Mobility Spectrometry: 1980-1990. Cr Rev Anal Chem. 1991;22:471–490. [Google Scholar]
- 170.Baumbach JI, Eiceman GA. Ion Mobility Spectrometry: Arriving On Site and Moving Beyond a Low Profile. Appl Spectrosc. 1999;53:338A–355A. doi: 10.1366/0003702991947847. [DOI] [PubMed] [Google Scholar]
- 171.von Helden G, Wyttenbach T, Bowers MT. Conformation of Macromolecules in the Gas Phase: Use of Matrix-Assisted Laser Desorption Methods in Ion Chromatography. Science. 1995;267:1483–1485. doi: 10.1126/science.267.5203.1483. [DOI] [PubMed] [Google Scholar]
- 172.Clemmer DE, Jarrold MF. Ion Mobility Measurements and their Applications to Clusters and Biomolecules. J Mass Spectrom. 1997;32:577–592. [Google Scholar]
- 173.Valentine SJ, Liu X, Plasencia MD, Hilderbrand AE, et al. Developing liquid chromatography ion mobility mass spectometry techniques. Expert Rev Proteomic. 2005;2:553–565. doi: 10.1586/14789450.2.4.553. [DOI] [PubMed] [Google Scholar]
- 174.McLean JA, Ruotolo BT, Gillig KJ, Russell DH. Ion mobility–mass spectrometry: a new paradigm for proteomics. Int J Mass Spectrom. 2005;240:301–315. [Google Scholar]
- 175.Tao L, Dahl D, Pérez L, Russell D. The contributions of molecular framework to IMS collision cross-sections of gas-phase peptide ions. J Am Soc Mass Spectrom. 2009;20:1593–1602. doi: 10.1016/j.jasms.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 176.Politis A, Park AY, Hyung SJ, Barsky D, et al. Integrating ion mobility mass spectrometry with molecular modelling to determine the architecture of multiprotein complexes. PLoS One. 2010;5:e12080. doi: 10.1371/journal.pone.0012080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Uetrecht C, Barbu IM, Shoemaker GK, van Duijn E, Heck AJ. Interrogating viral capsid assembly with ion mobility-mass spectrometry. Nat Chem. 2011;3:126–132. doi: 10.1038/nchem.947. [DOI] [PubMed] [Google Scholar]
- 178.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, et al. Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dupuis NF, Wu C, Shea JE, Bowers MT. The amyloid formation mechanism in human IAPP: dimers have beta-strand monomer-monomer interfaces. J Am Chem Soc. 2011;133:7240–7243. doi: 10.1021/ja1081537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Smith DP, Radford SE, Ashcroft AE. Elongated oligomers in beta2-microglobulin amyloid assembly revealed by ion mobility spectrometry-mass spectrometry. Proc Natl Acad Sci U S A. 2010;107:6794–6798. doi: 10.1073/pnas.0913046107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Smith DP, Woods LA, Radford SE, Ashcroft AE. Structure and Dynamics of Oligomeric Intermediates in beta(2)-Microglobulin Self-Assembly. Biophys J. 2011;101:1238–1247. doi: 10.1016/j.bpj.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hernandez H, Dziembowski A, Taverner T, Seraphin B, Robinson CV. Subunit architecture of multimeric complexes isolated directly from cells. EMBO Rep. 2006;7:605–610. doi: 10.1038/sj.embor.7400702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Liu Q, Greimann JC, Lima CD. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell. 2006;127:1223–1237. doi: 10.1016/j.cell.2006.10.037. [DOI] [PubMed] [Google Scholar]
- 184.Breuker K, McLafferty FW. Native electron capture dissociation for the structural characterization of noncovalent interactions in native cytochrome c. Angew Chem Int Edit. 2003;42:4900–4904. doi: 10.1002/anie.200351705. [DOI] [PubMed] [Google Scholar]
- 185.Breuker K, McLafferty FW. The thermal unfolding of native cytochrome c in the transition from solution to gas phase probed by native electron capture dissociation. Angew Chem Int Edit. 2005;44:4911–4914. doi: 10.1002/anie.200500668. [DOI] [PubMed] [Google Scholar]
- 186.Polfer NC, Oomens J. Vibrational spectroscopy of bare and solvated ionic complexes of biological relevance. Mass Spectrom Rev. 2009;28:468–494. doi: 10.1002/mas.20215. [DOI] [PubMed] [Google Scholar]
- 187.Oomens J, Polfer N, Moore DT, van der Meer L, et al. Charge-state resolved mid-infrared spectroscopy of a gas-phase protein. Phys Chem Chem Phys. 2005;7:1345–1348. doi: 10.1039/b502322j. [DOI] [PubMed] [Google Scholar]
- 188.Bian QZ, Forbes MW, Talbot FO, Jockusch RA. Gas-phase fluorescence excitation and emission spectroscopy of mass-selected trapped molecular ions. Phys Chem Chem Phys. 2010;12:2590–2598. doi: 10.1039/b921076h. [DOI] [PubMed] [Google Scholar]
- 189.Benesch JLP, Ruotolo BT, Simmons DA, Barrera NP, et al. Separating and visualisiing protein assemblies by means of preparative mass spectrometry and microscopy. J Struct Biol. 2010;172:161–168. doi: 10.1016/j.jsb.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 190.Kim J, Kim KH, Lee JH, Ihee H. Ultrafast X-ray diffraction in liquid, solution and gas: present status and future prospects. Acta Crystallogr A. 2010;66:270–280. doi: 10.1107/S0108767309052052. [DOI] [PubMed] [Google Scholar]
- 191.Aquilina JA. The major toxin from the Australian Common Brown Snake is a hexamer with unusual gas-phase dissociation properties. Proteins. 2009;75:478–485. doi: 10.1002/prot.22259. [DOI] [PubMed] [Google Scholar]
- 192.Zhang H, Cui WD, Wen JZ, Blankenship RE, Gross ML. Native Electrospray and Electron-Capture Dissociation in FTICR Mass Spectrometry Provide Top-Down Sequencing of a Protein Component in an Intact Protein Assembly. J Am Soc Mass Spectrom. 2010;21:1966–1968. doi: 10.1016/j.jasms.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Boeri Erba E, Ruotolo BT, Barsky D, Robinson CV. Ion mobility-mass spectrometry reveals the influence of subunit packing and charge on the dissociation of multiprotein complexes. Anal Chem. 2010;82:9702–9710. doi: 10.1021/ac101778e. [DOI] [PubMed] [Google Scholar]