

Abstract
Three new alkaloids, phomapyrrolidones A–C (1–3), bearing a cyclopenta[b]fluorene ring system were isolated from the mycelium extract of the endophytic fungal strain, Phoma sp. NRRL 46751, inhabiting Saurauia scaberrinae. Methylation of 1 afforded its N-methyl derivative 4. The planar structures and relative configurations of 1–4 were elucidated by extensive spectroscopic analysis. Phomapyrrolidones B (2) and C (3) exhibited weak antitubercular activity at subcytotoxic concentrations.
Endophytic fungi represent one of the largest and relatively underexplored resources of biologically active small-molecule natural products.1 Although endophytes were first observed with certainty over a century ago, they did not receive significant attention until the recent realization of their ecological relevance2 and the potential of yielding metabolites with diverse structures and interesting biological activities.3 It is estimated that one-third of the world's human population is infected with Mycobacterium tuberculosis4 and the problem of tuberculosis (TB) has been intensified due to an increasing number of highly susceptible individuals infected with HIV.5 Although there was a significant reduction in the number of TB cases between 1950 and 1970 as a result of the discovery of several antitubercular agents, the number of TB cases throughout the world has recently been increasing rapidly due to the emergence of multi-drug resistant Mycobacterium tuberculosis (MDRTB).4 Thus, there is an urgent need to discover and develop new, effective, and non-toxic antitubercular agents. In continuing our search for bioactive and/or novel metabolites from endosymbiotic microorganisms,6 we have investigated an EtOAc extract of the mycelium of the fungal strain Phoma sp. NRRL 46751 isolated from the lower crown of Saurauia scaberrinae (Actinidiaceae),7 a plant growing in the central highlands of Papua New Guinea. Herein we report the isolation and characterization of three alkaloids, phomapyrrolidones A–C (1–3) bearing a rare cyclopenta[b]fluorene (6/5/6/5) ring system,8 methylation of 1 to its N-methyl derivative 4, and evaluation of 1–4 for their in vitro antitubercular and cytotoxic activity. Numerous secondary metabolites are known from endophytic strains of Phoma,9 and our previous investigation of a culture broth of Phoma sp. NRRL 46751 has led to the identification of a new furandione in addition to several known fungal metabolites.7
The mycelium of the endophytic fungal strain Phoma sp. NRRL 46751 was fermented in potato dextrose broth (PDB) for 21 days, separated from the supernatent, freeze-dried, and extracted with CHCl3–MeOH (1/1). The resulting extract was suspended in H2O and extracted with EtOAc and the EtOAc fraction was subjected to solvent-solvent partitioning6b yielding hexanes and CHCl3 fractions. The hexanes fraction on gel permeation and silica gel chromatography followed by prep. TLC afforded compounds 1 and 2, whereas repeated chromatography of the CHCl3 fraction gave compound 3.
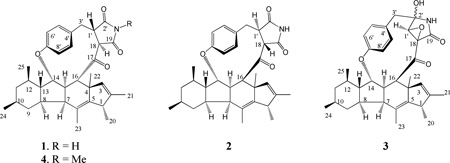
Phomapyrrolidone A (1) was obtained as a white solid that analyzed for C34H41NO4 by a combination of HRFABMS, 13C NMR and HSQC data, and indicated 15 degrees of unsaturation. The presence of ketone and imide carbonyl groups in 1 was inferred from its IR absorption bands at 1712 and 1778 cm−1, respectively. A D2O exchangeable singlet at δ 7.66 in its 1H NMR spectrum was suspected to be due to an NH or OH group. Methylation of 1 (CH3I/K2CO3) afforded its monomethyl derivative 4. 1H and 13C NMR chemical shifts (δH 2.92; δC 24.8) of the newly introduced methyl group in 4 suggested it to be an N–CH3. Thus, the singlet at δH 7.66 in 1 was assigned to an NH proton. The planar structure of 1 was deduced by analysis of its 1H and 13C NMR spectra (Table 1), 1H–1H COSY and HMBC data (Figure 1), and comparison of these data with those reported for related fungal alkaloids.8,10–13 In its 1H–1H COSY spectrum, 1 showed the presence of the spin systems CH3CH–, –CH2CHCH–, –CHCHCHCHCH(CH3)CH2CH(CH3)CH2CHCH–, and that due to a 1,4-disubstituted benzene ring. These data also suggested that 1 contained a succinimide moiety (δc 171.6 and 177.3) and a tetracyclic cyclopenta[b]fluor-2(3),5(6)-diene ring system bearing six methyl groups at C–1, C–2, C–4, C–6, C–10, and C–12 and that the CH of CH3CH– spin system is a part of the tetracyclic ring (Figure 1). This ring system was connected at C–16 to one of the β-carbons (C–18) of the succinimide moiety via the ketone carbonyl (C–17) as apparent from the HMBC correlations H–16/C–18, H–16/C–17, and H–18/C–17. The CHCH portion of the –CH2CHCH– spin system was shown to belong to the succinimide moiety as the NH proton of it exhibited HMBC correlations to both carbons (C–1' and C–18) of the CHCH fragment. The presence of an HMBC correlation of one of the protons of the 1,4-disubstituted benzene ring to CH2 carbon (C–3') of the –CH2CHCH– spin system suggested that this benzene ring is attached to C–3'. The HMBC correlation of H–14 (δ 4.30) to the oxygenated aromatic carbon (C–7'; δ 159.0) established the connectivity between C–14 of the tetracyclic ring system and the 1,4-disubstituted benzene ring. The relative configuration of 1 was deduced by the detailed analysis of its 1H–1H coupling constants, chemical shift and ROESY data. The vicinal coupling constant of 11.6 Hz between H–8 and H–9ax was indicative of an antiperiplanar relationship of these two protons. ROESY correlations (Figure 2) of H–9ax/Me–24, Me–24/H-11ax, H-11ax/Me–25, H–13/H–14, H–14/Me–25, H–14/H–16, H–16/H–3, H–14/H–8' and, Me–25/H–8' indicated that H–3, H–14, H–16, Me–24, Me–25, and H–8' were cofacial. Different chemical shifts observed for the chemically identical pairs of protons (H–5' and H–9'; H–6' and H–8') of the 1,4-disubstituted benzene ring suggested that it is rotation-restricted and is a part of a macrocyclic system. The cross-peaks of H–9' with one of the C–3' methylene protons (δ 3.67) and H–1' revealed that they were in the same face as above. ROESY correlations of H–3'/H–18, H–5'/H–18, H–18/Me–22, H–7/Me–22, H–18/H–15, H–15/Me–23, H–18/H–5' and H–18/H–6' suggested that they were co-facial as well. On the basis of these data and comparison of the relative stereochemistry of the related alkaloids embellicines A and B,8 the relative stereochemistry of phomapyrrolidone A (1) was deduced as depicted in Figure 2.
Table 1.
1H (400 MHz) and13C (100 MHz) NMR Data for Compounds 1–3 in CDCl3
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| Position | δC (Type) | δH, mult (J in Hz) | δC (Type) | δH, mult (J in Hz) | δC (Type) | δH, mult (J in Hz) |
| 1 | 43.2 (CH) | 2.93, q (4.8) | 43.9 (CH) | 2.97, q (7.2) | 44.2 (CH) | 2.95, q (7.2) |
| 2 | 142.5 (C) | 144.5 (C) | 141.9 (C) | |||
| 3 | 130.8(CH) | 4.40, brs | 130.3 (CH) | 4.67, t (1.4) | 133.2 (CH) | 5.28, t (1.6) |
| 4 | 54.0(C) | 56.5 (C) | 44.0 (C) | |||
| 5 | 144.4(C) | 144.4 (C) | 144.4 (C) | |||
| 6 | 128.8(C) | 130.1 (C) | 131.1 (C) | |||
| 7 | 49.3(CH) | 2.02, t | 47.5 (CH) | 1.96, t (11.9) | 46.8 (CH) | 1.93, t (11.2) |
| 8 | 41.2(CH) | 1.67, m | 43.1 (CH) | 1.51, m | 44.0 (CH) | 1.56, m |
| 9 α | 40.4(CH2) | 2.12, brd (16.0) | 40.3 (CH2) | 2.06, brd (17.6) | 40.3(CH2) | 2.09, brd (3.2) |
| 9 β | 0.68, ddd (16.0, 11.6, 4.4) | 0.68, ddd (17,6, 11.7, 5.2) | 0.73, m | |||
| 10 | 32.1(CH) | 1.56, m | 32.4 (CH) | 1.51, m | 32.5 (CH) | 1.54, m |
| 11 α | 44.6(CH2) | 0.67, ddd (16.0, 11.6, 4.4) | 44.8 (CH2) | 0.69, ddd (17.2, 11.7, 5.2) | 45.0 (CH2) | 0.74, m |
| 11 β | 1.86, m | 1.84, m | 1.81, m | |||
| 12 | 31.1(CH) | 1.92, m | 31.6 (CH) | 1.84, m | 31.8 (CH) | 1.84, m |
| 13 | 58.2(CH) | 1.05, m | 56.4 (CH) | 1.05, m | 56.1 (CH) | 1.11, m |
| 14 | 91.9(CH) | 4.30, dd (7.4, 2.4) | 88.9 (CH) | 4.33, dd (7.7, 5.1) | 88.0 (CH) | 4.26, dd (8.0, 5.6) |
| 15 | 59.3 (CH) | 1.67, t (11.6) | 51.1 (CH) | 1.51, m | 56.9 (CH) | 1.78, m |
| 16 | 60.8 (CH) | 2.97, d (11.6) | 50.8 (CH) | 1.91, d (10.1) | 52.4 (CH) | 2.04, d (10.0) |
| 17 | 201.6 (C) | 199.9 (C) | 200.9 (C) | |||
| 18 | 60.2 (CH) | 2.95, d (5.6) | 63.2 (CH) | 3.29, d (7.6) | 58.4 (C) | |
| 19 | 171.6(C) | 169.4 (C) | 167.5 (C) | |||
| 20 | 18.6 (CH3) | 0.97, d (6.8) | 18.9 (CH3) | 1.00, d (7.2) | 18.8 (CH3) | 1.02,d (7.2) |
| 21 | 14.7 (CH3) | 1.49, s | 14.9 (CH3) | 1.62, s | 14.7 (CH3) | 1.62, s |
| 22 | 28.4(CH3) | 1.18, s | 23.2 (CH3) | 0.68, s | 25.3 (CH3) | 0.74, s |
| 23 | 15.3(CH3) | 1.81, s | 15.6 (CH3) | 1.75, s | 15.5 (CH3) | 1.76,s |
| 24 | 22.5 (CH3) | 0.94, d (6.8) | 22.5 (CH3) | 0.90, d (6.6) | 22.5 (CH3) | 0.92, d (6.4) |
| 25 | 19.9 (CH3) | 1.14, d (6.8) | 20.1 (CH3) | 1.17, d (6.2) | 20.3 (CH3) | 1.13, d (6.0) |
| 1' | 43.8 (CH) | 3.83, m | 51.2 (CH) | 2.96, m | 63.9 (CH) | 3.53, m |
| 2' | 177.3(C) | 175.4 (C) | 84.2 (C) | |||
| 3' α | 34.6(CH2) | 2.43, dd (13.2, 11.2) | 36.8 (CH2) | 2.45, t (12.3) | 43.9 (CH2) | 3.15, d (14.0) |
| 3' β | 3.67, dd (13.2, 5.6) | 3.64, dd (12.3, 4.5) | 3.35, d (14.0) | |||
| 4' | 133.5 (C) | 133.7 (C) | 128.7 (C) | |||
| 5' | 133.1 (CH) | 6.97, dd (8.4, 2.0) | 129.3 (CH) | 7.05, dd (8.4, 2.4) | 129.6 (CH) | 7.19, dd (8.0, 2.0) |
| 6' | 126.3 (CH) | 6.68, dd (8.4, 2.4) | 131.9 (CH) | 6.97, dd (8.4, 2.4) | 131.1 (CH) | 7.08, dd (8.0, 2.0) |
| 7' | 159.0 (C) | 159.9 (C) | 160.3 (C) | |||
| 8' | 120.9 (CH) | 7.07, dd (8.4, 2.0) | 125.3 (CH) | 6.97, dd (8.4, 2.4) | 123.3 (CH) | 6.98, dd (8.0, 2.0) |
| 9' | 131.5 (CH) | 7.14, dd (8.4, 2.4) | 125.1 (CH) | 7.22, dd (8.4, 2.4) | 123.1 (CH) | 7.20, dd (8.0, 2.0) |
| NH | 7.66, s | 8.29, s | 6.60, brs | |||
Figure 1.

Selected COSY and HMBC correlations for 1 and HMBC correlations for 3.
Figure 2.

Selected ROESY correlations for 1–3.
The molecular formula of phomapyrrolidone B (2) was determined as C34H41NO4 by a combination of HRFABMS, 13C NMR, and HSQC data and indicated 15 degrees of unsaturation. Detailed analyses of its 1D and 2D NMR spectroscopic data (Table 1) indicated 2 to be a stereoisomer of 1 and ROESY data suggested that the relative stereochemistry at C–16, C–18, and C–1' were opposite to that of 1. ROESY correlations (Figure 2) of one of the benzylic protons (H–3'; δ 2.45) with H–9' and H–18 (δ 3.29) suggested that they are β–oriented. ROESY correlations of the other H–3' (δ 3.64) with H–1' and H–5', H–1'/H–16 and H–16/Me–22 indicated that they are α–oriented. The foregoing suggested that relative stereochemistry of phomapyrrolidone B (2) is as depicted in Figure 2.
Chromatographic separation of the CHCl3 fraction of the EtOAc extract of the mycelium gave phomapyrrolidone C (3) and an additional quantity of 1. Molecular formula of 3 was determined as C34H41NO5 by a combination of HRFABMS, 13C NMR, and HSQC data and indicated 15 degrees of unsaturation. Its 1H and 13C NMR spectroscopic data were found to be similar to those of 1 except for the presence of a γ–lactam moiety in 3 in place of the succinimide moiety in 1. Differences seen included the absence of 13C NMR signals for an imide carbonyl carbon (C–2') and a methine carbon (C–18) in 3; instead it showed signals for a hemi-aminal carbon (δ 84.2, C-2') and an oxygenated quaternary carbon (δ 58.4). Compound 3 also showed an oxymethine carbon (δ 63.9, C–1') instead of a methine carbon (δ 43.4) in 1. In the HMBC spectrum of 3 (Figure 2), correlations from H–1' (δ 3.53) to carbons C–18, C–19, C–2' and C–3' were observed. Since both adjacent carbons (C–18 and C–1') were found to be oxygenated, these two carbons should belong to an oxirane ring. The relative stereochemistry of 3 was determined by the analysis of its ROESY data (Figure 2). The relative configurations of the strereogenic centers C–1, C–4, C–7, C–8, C–10, C–12, C–14, C–15 and C–16, and the orientation of aromatic ring of phomapyrrolidone C (3) were found to be similar to those of phomapyrrolidone B (2). ROESY correlations of H–5'/H–1' suggested that H–1' is α–oriented. On the basis of these data the relative stereochemistry of phomapyrrolidone C (3) was deduced to be as that shown in Figure 2.
Phomapyrrolidones B (2) and C (3) exhibited weak in vitro antitubercular activity when tested using the microplate alamar blue assay (MABA)15 for replicating cultures and the low oxygen recovery assay (LORA)16 for non-replicating Mycobacterium tuberculosis H37Pv. As depicted in Table 2, compound 3 had the lowest MIC in both assays at sub-cytotoxic concentrations; cytotoxicity activity of 1–4 against Vero cells was determined using MTS-PMS assay.17 Interestingly, phomapyrrolidone A (1) was found to be less active, while its N-methyl analogue 4 exhibited no activity in both MABA and LORA assays. When tested for their potential anticancer activity against a panel of human cancer cell lines, MCF-7 (breast adenocarcinoma), NCI-H460 (non-small cell lung cancer), SF-268 (CNS glioma), PC-3M (metastatic prostate adenocarcinoma), and MDA-MB-231 (metastatic breast adenocarcinoma), compounds 1–4 showed no activity at the highest concentration (5.0 µM) tested using Alamar Blue assay.6a
Table 2.
Antitubercular and Cytotoxic Activities of 1–4a
| MIC | IC50 | ||
|---|---|---|---|
| compound | MABA | LORA | Vero Cell |
| 1 | 20.1 | 41.1 | 17.7 (33.6) |
| 2 | 5.9 | 15.4 | 17.1 (32.4) |
| 3 | 5.2 | 13.4 | 19.4 (35.7) |
| 4 | >100 | >100 | 78.6 (145.3) |
| RMPb | 0.07 | 0.10 | 109.0 (132.5) |
| INHb | 0.03 | >17.5 | NTc |
Activities are in µg/mL; data in parentheses (IC50s) are in µM.
RMP (rifampin) and INH (isonicotinylhydrazine) were used as positive controls for the antitubercular assays and RMP as a positive control for the cytotoxicity assay.
NT = Not tested.
Although structurally related fungal alkaloids, GKK1032 A1, A2 and B from Penicillium sp. GKK1032,10 pyrrocidines A and B from LL-Cyan 426,11 hirsutellones A–E from Hirsutella nivea BCC7579,12 hirsutellone F from Trichoderma sp. BCC 7579,13 pyrrospirones A and B from Neonectria ramulariae,14 all bearing a macrocyclic ether and succinimide-derived moieties are known, it is noteworthy that only embellicines A and B from Embellisia euerka8 have been previously reported to contain the cyclopenta[b]fluorene (6/5/6/5) ring system encountered in phomapyrrolidones A–C (1–3).
Experimental Section
General Experimental Procedures
Melting points were determined with an Electrothermal melting point apparatus and are uncorrected. Optical rotations were measured with a JASCO Dip-370 digital polarimeter using MeOH as solvent. UV spectra were recorded on a Shimadzu UV-1601 UV-VIS spectrophotometer. IR spectra for KBr discs were recorded on a Shimadzu FTIR-8300 spectrometer. 1D and 2D NMR spectra were recorded in CDCl3 with a Bruker AVANCE III instrument at 400 MHz for 1H NMR and 100 MHz for 13C NMR using residual CHCl3 as internal standard. Low resolution and high resolution MS were recorded, respectively, on Shimadzu LCMS-QP8000α and JEOL HX110A spectrometers. Column chromatography was performed on flash chromatography silica gel (40 µm: J. T. Baker) or Sephadex LH-20 (25−100 µm; GE Healthcare). Analytical and preparative thin layer chromatography (TLC) were performed on pre-coated 0.25 mm thick plates of silica gel 60 F254 (Merck). Spots on analytical TLC were directly visualized under UV light and/or by spraying a solution of anisaldehyde and conc. H2SO4 in glacial acetic acid and water followed by heating.
Isolation of the Fungus and Culturing, Extraction, and Fractionation of EtOAc Extract of the Mycelium
Details of the isolation and identification of Phoma sp. NRRL 46751 have been presented previously.7 A seed culture of the fungus grown on PDA for two weeks was used for inoculation. Mycelia were scraped out and vortexed with sterile PDB (90 mL) and filtered through a 100 µm filter to separate spores from the mycelia. Absorbance (at 600 nm) of the spore solution was measured and adjusted to 0.6. This spore solution was used to inoculate nine 2000 mL Erlenmeyer flasks, each containing 1000 mL of the sterile medium (24 g of PDB and 6 g of sucrose in distilled H2O) and incubated at 160 rpm and 28 °C. The glucose level in the medium was monitored using glucose strips (URISCANTM), and on day 21 the glucose test indicated complete depletion of glucose in the medium. Mycelia were separated from the supernatant by filtration through Whatman No.1 filter paper, freeze-dried and extracted with CHCl3–MeOH (1/1, 4 × 700 mL). The combined extracts were evaporated under reduced pressure and the resulting residue was suspended in H2O (200 mL) and extracted with EtOAc (4 × 200 mL). The combined EtOAc extracts was washed with H2O (3 × 200 mL), dried over anhydrous Na2SO4 and evaporated under reduced pressure to give the EtOAc extract (4.374 g) as a dark brown thick liquid. A portion (4.3 g) of this was partitioned between hexane and 80% aqueous MeOH. The 80% aqueous MeOH fraction was diluted with H2O to 50% aqueous MeOH and extracted with CHCl3. Evaporation of solvents under reduced pressure yielded hexane (3.5 g) and CHCl3 (0.655 g) fractions.
Isolation of Phomopyrrolidones A and B
A portion (3.1 g) of the hexane fraction was subjected to gel permeation chromatography over a column of Sephadex LH-20 (100 g) made up in hexanes–CH2Cl2 (1/4) and eluted sequentially with hexanes–CH2Cl2 (1/4) (600 mL), CH2Cl2–acetone (3/2) (400 mL), CH2Cl2–acetone (1/4) (200 mL), CH2Cl2–MeOH (1/1) (200 mL), and MeOH (400 mL). A total of 36 fractions (50 mL each) were collected and those having similar TLC profiles were combined to give 18 fractions [F1 (1157.8 mg), F2 (984.4 mg), F3 (189.6 mg), F4 (27.6 mg), F5 (31.1 mg), F6 (52.6 mg), F7 (56.4 mg), F8 (24.4 mg), F9 (79.1 mg), F10 (49.3 mg), F11 (84.8 mg), F12 (87.2 mg), F13 (53.9 mg), F14 (18.1 mg), F15 (13.7 mg), F16 (29.9 mg), F17 (5.1 mg), F18 (1.2 mg)]. Fractions F5–F7 were combined (140.1 mg) and chromatographed over a column of silica gel (4.0 g) made up in CH2Cl2 and eluted with CH2Cl2 containing increasing amounts of MeOH affording a total of 50 fractions (6 mL each). Fractions eluted with CH2Cl2–MeOH (99.8/0.2) and early fractions eluted with CH2Cl2–MeOH (99.5/0.5) were combined to give 1 (60.4 mg). Later fractions eluted with 0.5% MeOH in CH2Cl2 were combined and separated by prep. TLC on silica gel [eluant: Hexanes–EtOAc (65/35)] to give an additional quantity of 1 (4.8 mg, Rf 0.52) and 2 (19.2 mg; Rf 0.48).
Phomapyrrolidone A (1): white solid; mp 218–220°C; [α]D + 256 (c 1.0, MeOH); UV (EtOH) λmax (log ε) 223 (3.27) nm; IR (KBr) νmax3271, 2925, 1778, 1712, 1502, 1342, 1232, 1178 cm−1; for 1H NMR and 13C NMR data, See Table 1; HRFABMS m/z 526.2966 [M–H]− (calcd for C34H40NO4 526.2963).
Phomapyrrolidone B (2): white solid; mp 186–188°C (dec); [α]D + 96 (c 1.0, MeOH); UV (EtOH) λmax (log ε) 226 (3.51) nm; IR (KBr) νmax3254, 2929, 1778, 1728, 1500, 1446, 1373, 1342, 1225, 1172 cm−1; for 1H NMR and 13C NMR data, See Table 1; HRFABMS m/z 528.3119 [M+H]+ (calcd for C34H42NO4 528.3108).
Methylation of Phomapyrrolidone A (1)
To a solution of 1 (2.3 mg) in acetone (0.5 mL) were added CH3I (0.05 mL) and K2CO3 (10 mg) and the suspension was stirred at 25°C for 14 h. After disappearance of 1 (TLC control) the reaction mixture was filtered and evaporated under reduced pressure to give N-methylphomapyrrolidone A (4, 2.3 mg).
N-Methylphomapyrrolidone A (4): white solid; mp 178–180°C; [α]D + 248 (c 1.0, MeOH); UV (EtOH) λmax (log ε) 224 (2.78) nm; IR (KBr) νmax2953, 1703, 1502, 1434, 1379, 1282, 1232, 1126 cm−1; 1H NMR(400 MHz, CDCl3) δ 7.14 (1H, dd, J = 8.4, 2.0 Hz, H–9'), 7.04 (1H, dd, J = 8.4, 2.4 Hz, H–8'), 6.93 (1H, dd, J = 8.4, 2.4 Hz, H–6'), 6.66 (1H, dd, J = 8.4, 2.4 Hz, H–5'), 4.34, (1H, t, J = 0.8 Hz, H–3), 4.29 (1H, dd, J = 7.2, 3.6 Hz, H–14), 3.74 (1H, m, H–1'), 3.67 (1H, dd, J = 12.0, 10.4 Hz, H–3'β), 3.01 (1H, d, J = 9.2 Hz, H–16), 2.92 (3H, s, NCH3), 2.88 (1H, q, J = 4.8 Hz, H–1), 2.87 (1H, d, J = 4.8 Hz, H–18), 2.33 (1H, dd, J = 12.0, 10.4 Hz, H–3'α), 2.15 (3H, s, H3–21), 2.12 (1H, brd, J = 14.8 Hz, H–9α), 2.00 (1H, t, J = 11.6, H–7), 1.91 (1H, m, H–12), 1.80 (3H, s, H3–23), 1.77 (1H, m, H–11β), 1.64 (2H, m, H–8 and H–15), 1.53 (1H, m, H–10), 1.20 (3H, s H3–22), 1.14 (3H, d, J = 6.4 Hz, H3–25), 1.00 (1H, m, H–13), 0.95 (3H, d, J =6.4 Hz, H3–24), 0.93 (3H, d, J = 6.4 Hz, H3–20); 0.68 (1H, ddd, J = 16.0, 11.6, 4.4 Hz, H–9β); 13C NMR (100 MHz, CDCl3) δ 202.0 (C, C–17), 177.2 (C, C–2'), 171.7 (C, C–19), 159.0 (C, C–7'), 144.5 (C, C–5), 142.3 (C, C–2), 133.7 (C, C–4'), 133.2 (CH, C–5'), 131.5 (CH, C–6'), 130.8 (CH, C–3), 128.7 (C, C–6), 126.3 (CH, C–8'), 120.9 (CH, C–9'), 91.9 (CH. C–14), 60.9 (CH, C–16), 59.3 (CH, C–18), 59.3 (C, C–15), 58.3 (CH, C–13), 54.0 (C, C–4), 49.4 (C, C–7), 44.6 (CH2, C–11), 43.2 (CH, C–1), 42.8 (CH, C–1'), 41.2 (C, C–8), 40.4 (CH2, C–9), 34.7 (CH2, C–3'), 32.2 (CH, C–10), 31.2 (CH, C–12), 28.4 (CH3, CH3–22), 24.8 (CH3, N–CH3), 22.5 (CH3, CH3–24), 19.9 (CH3, C–25), 18.6 (CH3, C–20), 15.2 (CH3, C–23), 14.8 (CH3,C– 21); APCI-MS (+) mode m/z 541 [M+H]+.
Isolation of Phomapyrrolidone C (3)
A portion (0.655 g) of the CHCl3 fraction obtained above was subjected to gel permeation chromatography over a column of Sephadex LH-20 (50.0 g) made up in hexanes–CH2Cl2 (1/4) and eluted with hexanes–CH2Cl2 (1/4) (300 mL), CH2Cl2–acetone (3/2) (300 mL), CH2Cl2–acetone (1/4) (300 mL), CH2Cl2–MeOH (1/1) (200 mL) and finally with MeOH (400 mL) to yield eight fractions [F1 (210.3 mg), F2 (68.1 mg), F3 (70.8 mg), F4 (93.7 mg), F5 (54.1 mg), F6 (58.9 mg), F7 (16.4 mg) and F8 (32.9 mg)]. Fractions F1 and F2 were combined (278.4 mg) and subjected to prep. TLC (hexanes–acetone: 75/25, v/v) to give five sub-fractions [A (5.9 mg), B (18.5 mg), C (20.1 mg), D (7.2 mg), and E (63.5 mg)]. Sub-fractions C and D were combined (27.3 mg) and chromatographed over a column of silica gel (4.0 g) made up in hexanes–EtOAc (8/2) and eluted with hexane containing increasing amounts of EtOAc to give 1(5.6 mg). Fraction F4 (93.7 mg) was chromatographed over a column of silica gel (9.4 g) made up in hexanes–EtOAc (7/3) and eluted with hexane containing increasing amounts of EtOAc followed by EtOAc to give five sub-fractions [F4A (11.5 mg), F4B (5.9 mg), F4C (44.0 mg), F4D (4.3 mg), and F4E (5.7 mg)]. Fraction F5 (54.1 mg) was chromatographed over a column of silica gel (5.4 g) made up in hexanes–EtOAc (7/3) and eluted with hexane containing increasing amounts of EtOAc followed by EtOAc to give five sub-fractions [F5A (1.4 mg), F5B (2.5 mg), F5C (9.9 mg), F5D (9.3 mg), and F5E (7.0 mg)]. Sub-fractions F4C, F4D, and F5C which by TLC was found to contain a compound different from 1 and 2 were combined and chromatographed over a column of silica gel (6.0 g) made up in CH2Cl2–EtOAc (8/2) and eluted with CH2Cl2 containing increasing amounts of EtOAc to give five fractions [A (8.4 mg), B (24.2 mg), C (9.6 mg), D (6.3 mg) and E (5.8 mg)]. The fraction B (24.2 mg) containing the new compound was purified by prep. TLC (silica gel) using hexanes–acetone (65/35) as eluant to give 3 (16.9 mg, Rf 0.25).
Phomapyrrolidone C (3): white solid; mp >195°C (dec); [α]D +248 (c 1.0, MeOH); UV (EtOH) λmax (log ε) 273 (1.21), 225 (2.34) nm; IR (KBr) νmax 3413, 2954, 1733, 1504, 1450, 1404, 1373, 1227, 1137, 1105, 934 cm−1; for 1H NMR and 13C NMR data, See Table 1; HRFABMS m/z 544.3062 [M+H]+ (calcd for C34H41NO5 544.3057).
Biological Assays
Growth inhibitory activity against non-replicating Mycobacterium tuberculosis H37Pv was performed using the Microplate Alamar Blue Assay (MABA)15 and Low Oxygen Recovery Assay (LORA)16 as described previously using rifampin and isonicotinylhydrazine as positive controls. Cytotoxicity of the compounds against Vero cells and human cancer cell lines was evaluated using the MTS-PMS assay,17 and the resazurin (alamarBlue®) assay,6a as described previously using the positive controls rifampin and doxorubicin, respectively.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this work from the National Cancer Institute (RO1 CA90265) and National Institute of General Medical Sciences (P41 GM094060) are gratefully acknowledged. We thank M. X. Liu for her assistance with the preparation of cultures and some cytotoxicity assays.
Footnotes
ASSOCIATED CONTENT
Supporting Information. 1H and 13C NMR,1H –1H COSY, HSQC, HMBC and ROESY spectra of phomapyrrolidones A–C (1–3). 1H and 13C NMR spectra of N-methylphomapyrrolidone (4). These material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES AND NOTES
- 1.(a) Carroll GC. Ecology. 1988;69:2–9. [Google Scholar]; (b) Arnold AE. Fungal Biol. Rev. 2007;21:51–66. [Google Scholar]
- 2.Arnold AE, Mejia L, Kyllo D, Rojas E, Maynard Z, Herre EA. Proc. Natl. Acad. Sci. U. S. A. 2003;100:15649–15654. doi: 10.1073/pnas.2533483100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Strobel G, Daisy B, Castillo U, Harper J. J. Nat. Prod. 2004;67:257–268. doi: 10.1021/np030397v. [DOI] [PubMed] [Google Scholar]; (b) Gunatilaka AAL. J. Nat. Prod. 2006;69:509–526. doi: 10.1021/np058128n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang HW, Song YC, Tan RX. Nat. Prod. Rep. 2006;23:753–771. doi: 10.1039/b609472b. [DOI] [PubMed] [Google Scholar]; (d) Verma VC, Kharwar RN, Strobel GA. Nat. Prod. Commun. 2009;4:1511–1532. [PubMed] [Google Scholar]
- 4.Geneva, Switzerland: W.H.O. Press; 2011. Global Tuberculosis Control: W.H.O. Report; pp. 1–246. ( http://www.who.int/tb/publications/global_report/2011/gtbr11_full.pdf). [Google Scholar]
- 5.Chan ED, Iseman MD. Brit. Med. J. 2002;325:1282–1286. doi: 10.1136/bmj.325.7375.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Wang X, Bashyal BP, Wijeratne EMK, U'Ren JM, Liu MX, Gunatilaka MK, Arnold AE, Gunatilaka AAL. J. Nat. Prod. 2011;74:2052–2061. doi: 10.1021/np2000864. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wijeratne EMK, Bashyal BP, Liu MX, Rocha DD, Gunaherath GMKB, U'Ren JJ, Gunatilaka MK, Arnold AE, Whitesell L, Gunatilaka AAL. J. Nat. Prod. 2012;75:361–369. doi: 10.1021/np200769q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffman AM, Mayer SG, Strobel GA, Hess WM, Sovocool GW, Grange AH, Harper JK, Arif AM, Grant DM, Kelly-Swift EG. Phytochemistry. 2008;69:1049–1056. doi: 10.1016/j.phytochem.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 8.Ebrahim W, Aly AH, Wray V, Mándi A, Teiten M-H, Gaascht F, Orlikova B, Kassack MU, Lin W, Diederich M, Kurtán T, Debbab A, Proksch P. J. Med. Chem. 2013;56:2991–2999. doi: 10.1021/jm400034b. [DOI] [PubMed] [Google Scholar]
- 9.(a) Yang X, Strobel G, Stierle A, Hess WM, Lee J, Clardy J. Plant Sci. 1994;102:1–9. [Google Scholar]; (b) Weber RWS, Stenger E, Meffert A, Hahn M. Mycol. Res. 2004;108:662–671. doi: 10.1017/s0953756204000243. [DOI] [PubMed] [Google Scholar]; (c) Borges W, de S, Pupo MT. J. Braz. Chem. Soc. 2006;17:929–934. [Google Scholar]; (d) Krohn K, Farooq U, Flörke U, Schulz B, Draeger S, Pescitelli G, Salvadori P, Antus S, Kurtán T. Eur. J. Org. Chem. 2007;2007:3206–3211. [Google Scholar]; (e) Zhang W, Krohn K, Egold H, Draeger S, Schulz B. Eur. J. Org. Chem. 2008;2008:4320–4328. [Google Scholar]; (f) Dai J, Hussain H, Draeger S, Schulz B, Kurtan T, Pescitelli G, Flörke U, Krohn K. Nat. Prod. Comm. 2010;5:1175–1180. [PubMed] [Google Scholar]; (g) Qin S, Hussain H, Schulz B, Draeger S, Krohn K. Helv. Chim. Acta. 2010;93:169–174. [Google Scholar]; (h) Pan J-H, Deng J-J, Chen Y-G, Gao J-P, Lin Y-C, She Z-G, Gu Y-C. Helv. Chim. Acta. 2010;93:1369–1374. [Google Scholar]; (i) Strobel G, Singh SK, Riyaz-Ul-Hassan S, Mitchell AM, Geary B, Sears J. FEMS Microbiol. Lett. 2011;320:87–94. doi: 10.1111/j.1574-6968.2011.02297.x. [DOI] [PubMed] [Google Scholar]; (j) Loesgen S, Bruhn T, Meindl K, Dix I, Schulz B, Zeeck A, Bringmann G. Eur. J. Org. Chem. 2011;2011:5156–5162. [Google Scholar]; (k) Zhang L, Wang S-Q, Li X-J, Zhang A-L, Zhang Q, Gao J-M. J. Mol. Structure. 2012;1016:72–75. [Google Scholar]; (l) Kumaran RS, Choi Y-K, Lee S, Jeon HJ, Jung H, Kim HJ. Afr. J. Biotechnol. 2012;11:950–960. [Google Scholar]; (m) Wang L-W, Xu B-G, Wang J-Y, Su Z-Z, Lin F-C, Zhang C-L, Kubicek CP. Appl. Microbiol. Biot. 2012;93:1231–1239. doi: 10.1007/s00253-011-3472-3. [DOI] [PubMed] [Google Scholar]
- 10.Oikawa H. J. Org. Chem. 2003;68:3552–3557. doi: 10.1021/jo0267596. [DOI] [PubMed] [Google Scholar]
- 11.He H, Yang HY, Bigelis R, Solum EH, Greenstein M, Carter GT. Tetrahedron Lett. 2002;43:1633–1636. [Google Scholar]
- 12.Isaka M, Rugseree N, Maithip P, Kongsaeree P, Prabpai S, Thebtaranonth Y. Tetrahedron. 2005;61:5577–5583. [Google Scholar]
- 13.Isaka M, Prathumpai W, Wongsa P, Tanticharoen M. Org. Lett. 2006;8:2815–2817. doi: 10.1021/ol060926x. [DOI] [PubMed] [Google Scholar]
- 14.Shiono Y, Shimanuki K, Hiramatsu F, Koseki T, Tetsuya M, Fujisawa N, Kimura K-I. Bioorg. Med. Chem. Lett. 2008;18:6050–6053. doi: 10.1016/j.bmcl.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 15.Collins LA, Franzblau SG. Antimicrob. Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. Antimicrob. Agents Chermother. 2007;51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falzari K, Zhu Z, Pan D, Liu H, Hongmanee P, Franzblau SG. Antimicrob. Agents Chemother. 2005;49:1447–1454. doi: 10.1128/AAC.49.4.1447-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.