

Abstract
Glioblastoma (GBM) is the most common brain cancer and is highly lethal in both adults and children. 2-methoxyestradiol (2ME2) is a microtubule inhibitor that potently inhibits HIF1α, GBM angiogenesis and tumor growth in preclinical models. In patients, 2ME2 exhibits low toxicity and promising but inconsistent efficacy. Given its preclinical potency and its tolerability in patients, we sought to determine whether 2ME2 therapy could be enhanced by addressing resistance via combination therapy, and with biomarkers to identify responsive glioma subgroups. We demonstrate that the PTEN-PI3K axis regulates HIF1α in glioma models. We utilized isogenic-pairs of glioma cell lines, deficient in PTEN or stably reconstituted with PTEN, to determine the role of PTEN in 2ME2 sensitivity in vitro and in vivo. Chou-Talalay synergy studies reveal significant synergy when a pan-PI3K inhibitor is combined with 2ME2. This synergistic activity was correlated with a synergistic suppression of HIF1α accumulation under hypoxic conditions in glioma models. In vivo, 2ME2 markedly inhibited tumor-induced angiogenesis and significantly reduced tumor growth only in a PTEN reconstituted GBM models in both subcutaneous and orthotopic intracranial mouse models. Collectively, these results: (1) suggest that PTEN status predicts sensitivity to 2ME2 and (2) justify exploration of 2ME2 combined with panPI3K inhibitors for the treatment of this intractable brain cancer.
Keywords: 2ME2, glioblastoma multiforme, PTEN, PI3K, angiogenesis, HIF1α
Introduction
Glioblastoma multiforme (GBM) is the most common primary malignant brain tumor in adults with an average survival of just over one year[1, 2]. Even after aggressive treatment, which includes surgery and concurrent temozolomide and radiation, survival is only extended 2.5 months[2, 3]. As standard medical practice yields little survival benefit, greater attention is being paid to personalized treatment related to the expression of specific molecular markers [4]. Phosphatase and tensin homolog (PTEN)-dependent dysregulation of signaling occurs in 40% of all GBM cases, and loss of heterozygosity (LOH) is found in 60% to 80% of cases[3]. In addition, approximately 15% of GBMs exhibit gain of function mutations of PI-3-kinase (PI3K; Class I α subunit; PIK3CA)[5, 6]. PTEN is a tumor suppressor gene involved in a number of signaling pathways, most importantly the PI3K/Akt pathway in which it serves as a phosphatase acting on PIP3, dephosphorylating PIP3 which is required for the downstream activation of PDK1 and plekstrin-homology-domain serine threonine kinase (Akt) [7]. Akt regulates a large number of downstream pathways controlling progression through the cell cycle, protein synthesis, survival, apoptosis and migration[8, 9].
The role of PTEN in controlling tumor-induced angiogenesis is well-established in brain tumors[10]. Vascular endothelial growth factor (VEGF) is a major mediator of angiogenesis that is induced under hypoxic conditions by a multistage process in which the α subunit of hypoxia-inducible factor-1(HIF-1α) plays a key role[11]. HIF-1α is rapidly degraded by the proteasome under normoxia[12, 13]. Under hypoxia, HIF-1α is translocated to the nucleus where it heterodimerizes with HIF-1β (and activates the transcription of more than 40 genes important for adaptation and survival under hypoxia[14]. The role of PTEN-PI3K signaling in the regulation of HIF1α has been implicated by several groups[15–17], however its exact role remains controversial[18, 19].
A landmark report suggested that 2-methoxyestradiol (2ME2), a natural metabolite of estradiol inhibits tumor growth and angiogenesis by dysregulating HIF[20]. This drug showed potent anti-GBM activity in preclinical models, and multiple studies highlight its potential as an anti-tumor agent[21–23]. 2ME2 has completed Phase I/II clinical trials for breast[24], renal[25] prostate[26] and GBM tumors[27]. It is currently in Phase II trials for multiple myeloma[28], and it is also in Phase II trials combined with Bevacizumab (Avastin) for carcinoid tumors[29]. So far, in patients, 2ME2 has demonstrated low toxicity with promising but mixed efficacy in several cancer types including GBM[30, 31]. The emergence of 2ME2 resistance in GBM is not understood and the mechanism of action remains unclear. While there may be any number of escape pathways leading to 2ME2 resistance, one possible candidate is loss or mutation of PTEN which maintains control over the important PI3K-Akt oncogenic pathway[32, 33]. Given the potency of 2ME2 in preclinical models, positive effects in some patients and its tolerability, we were motivated to determine whether 2ME2 for GBM treatment could potentially be significantly enhanced by (1) circumventing GBM resistance with combination therapy, and (2) the discovery of biomarkers which would identify 2ME2 responsive GBM substrata.
Herein, we report that the capacity of 2ME2 to inhibit GBM angiogenesis and tumor growth in vivo depends on PTEN status. Our in vitro data implicate PTEN and PI3K in the regulation of HIF1α accumulation in glioma cells. Moreover, PTEN expression correlates directly with GBM sensitivity to 2ME2. The pharmacologic inhibition of PI3K synergized with 2ME2 to suppress HIF1α accumulation and cell proliferation. In vivo PTEN status of implanted GBM cell lines influenced the anti-angiogenic and anti-tumor efficacy of 2ME2. These results support, (1) the use of PTEN status to identify 2ME2 responsive glial tumors, and (2) the potential utility of combining 2ME2 with PI3K inhibitors for GBM treatment. The data may further suggest that 2ME2 has greater activity in lower grade glial tumors (WHO grades I or II) where PTEN is rarely mutated and remains intact.
Materials and methods
Tissue culture, cells and reagents
U87MG and U373MG glioma cell lines were obtained from ATCC. Both of these human GBM cell lines lack PTEN expression but differ in terms of p53 status. The U87MG cell line retains wild type p53 whereas p53 is mutated in the U373MG cell line[34]. TheLN229vIII cells containing wild type PTEN, mutated p53 were transduced with the vIII mutated EGFR[35]. Muristerone induced U87MG GBM cell lines and their mutants G129E and G129R has been previously described [10, 36]. Generation of GFAP V12 Ras PTENfl/fl cre+, GFAP V12 Ras PTENfl/wtcre+ or GFAP V12 Ras PTENfl/fl are described in Supplementary text S1. All lines were propagated in Dulbecco’s modified Eagle’s medium (Cellgro, Invitrogen). LY294002 and 2ME2 was purchased from Calbiochem. SF1126 is a vascular targeted pan PI3K inhibitor developed by SignalRx pharmaceuticals [35]. Antibodies were purchased; Human HIF1α (Transduction Laboratories), total and phosphorylated Akt (Ser-473), phospho-ERK, ERK, PTEN and Caspase-3 (Cell Signaling Technologies).
Hypoxia and inhibitor studies
Cells were cultured in 95% O2 and 5% CO2 at 37°C and then placed in a hypoxic chamber (Billups chamber) at 1% O2, 5% CO2, and 94.9% nitrogen. Cells were pretreated with 25 μM 2ME2 alone, 25 μM LY294002, or both, followed by normoxia or hypoxia for 16 hours.
Western blots
For HIF1α Western blots, nuclear extracts were prepared using a modified Dignam protocol [37]. Extracts were electrophoresed, transferred, and immunoblotted according to standard protocols.
Proliferation and apoptosis studies
For combined dose effect and synergism analyses, the isobologram method was used [38]. Experimental details are provided in Supplementary Materials (S1). U87MG-Null and U87MG-PTEN cells were treated with 2ME2 at 25 or 50 μM concentrations for 24, 48 and 72 hours. Caspase 3 cleavage was determined using a caspase 3 specific antibody and Western blot analysis.
Animal studies
Athymic female mice (CD-1 nu/nu, 20–25 grams) were obtained from the NIH/NCI. All studies were approved by Emory University’s Institutional Animal Care and Use Committee (IACUC). 5 × 106 cells were injected subcutaneously into the right flank and treatment initiated when all tumors reached 80–100 mm3. 2ME2 (200 mg/kg) or vehicle control was injected intraperitoneally (IP) once daily until sacrifice. Terminally, tumors were excised, sectioned, and stained for CD31 (Supplementary S1). Orthotopic xenograft studies were conducted by implanting glioblastoma cells in the right frontal lobe of mice as described in Supplementary S1[10].
RESULTS
Effect of PTEN and pan PI3K inhibitors on HIF1α accumulation in glioblastoma models
Fig. 1A demonstrates that both the PTEN reconstituted U87MG-PTEN and U373MG-PTEN GBM lines expressed PTEN, while there was no PTEN expression in the parental U87MG-Null and U373MG-Null cells. The role of PTEN-PI3K pathway in HIF1α accumulation has been reported[15–17] but remains an area of intense debate in the literature[18, 19]. In order to confirm participation of PTEN in the regulation of HIF1α, we utilized the U87MG cell lines in which PTEN expression (or mutants of PTEN) was controlled by a muristerone responsive promoter [10]. We also utilized our glioma cell line derived from the GFAPV12Ras x PTENfl/fl transgenic mouse, engineered to delete one or both alleles of PTEN using a retroviral cre recombinase. PTEN blocked hypoxic accumulation of HIF1α in muristerone-induced U87PTEN and GFAPV12RasPTENfl/flcre+ cell lines, especially under serum-free conditions (Fig. 1B & C), while there was mild suppression of HIF1α in 2% serum. There was no PTEN or G129R mutant PTEN effect when cells were in 10% FBS, while under serum free conditions the G129E PTEN mutant blocked accumulation of HIF1α to some extent (Fig. 1B). The G129E mutant dephosphorylates acidic phosphopeptides (maintains protein phosphatase activity), but cannot dephosphorylate lipid substrate PtIns(3,4,5)P3, while the G129R mutant lacks protein and lipid phosphatase activity. The results suggest that PTEN regulates HIF1α accumulation under hypoxic conditions in a protein phosphatase dependent manner. To further support a role for PTEN/PI3K in control of HIF1α in glioma models, we examined the effects of a pan PI3K inhibitor, SF1126, on the accumulation of HIF1α under hypoxia. SF1126 inhibited the hypoxic accumulation of HIF1α in a dose dependent manner with complete suppression at 50 μM (Fig. 1D).
Fig. 1. PTEN-PI3K signaling is required for HIF1α accumulation in hypoxic conditions.

(A) Immunoblot confirming expression of PTEN in reterovirally reconstituted U373MG-PTEN and U87MG-PTEN cell lines while parental U373 MG and U87MG cell lines lack its expression. (B) Muristerone induced U87 PTEN, G129E and G129R cell lines induced with 0.5μmol/L of muristerone were either serum starved or kept in 2% serum or 10% serum conditions for 36 hrs and were given normoxia (21% O2) or hypoxia (1% O2) for 4 hrs followed by preparation of nuclear extracts (for HIF1α) as well as whole cell extracts. (C) GFAPV12Ras PTENfl/fl and GFAP V12 Ras PTENfl/fl cre+ cell lines were serum starved or kept in 2% serum or 10% serum conditions for 4 hrs and were kept in normoxic (21% O2) or hypoxic (1% O2) conditions for 4 hrs followed by preparation of nuclear extracts (for HIF1α) as well as whole cell extracts. (D) U87MG glioma cell line treated with 10, 25 and 50μM SF1126 inhibited HIF1α accumulation under hypoxic conditions. Cells were treated with different concentrations of inhibitor for 30 min and then placed under normoxic or hypoxic conditions (1% O2) for 4 hours followed by the preparation of cell lysates for Western blot analysis of HIF1α.
2ME2 induced AKT phosphorylation under hypoxia which was suppressed by the pan PI3K inhibitor; LY294002 and 2ME2 demonstrate synergy in blocking HIF1α accumulation in GBM cell lines
Previous reports clearly indicate that 2ME2 dysregulates HIF1α, leading to decreased angiogenesis and tumor growth [39, 40]. Fig. 1 unambiguously demonstrates that the PTEN-PI3K pathway regulates HIF1α in glioma cell lines, and taken together with reports that 2ME2 inhibits HIF1α activity[39, 40], led us to hypothesize that PTEN and/or PI3K signaling might synergistically interact with 2ME2 to control HIF1α and angiogenesis in malignant gliomas. The pan PI3K inhibitor, LY294002 potently inhibited p-Akt independent of PTEN status, while it inhibited p-ERK levels only in U87MG-PTEN reconstituted cells (Fig. 2A). In contrast, 2ME2 treatment of LN229vIII (PTEN intact, activated EGFR) cells under hypoxic conditions increased AKT activation as measured by p-AKT immunoblots. This effect was blocked by LY294002 (Fig. 2B).
Fig. 2. LY294002 and 2ME2 demonstrate synergy in blocking HIF1α accumulation in GBM cell lines.

(A) Immunoblot showing pAKT and pERk levels in U373MG-Null and U373MG-PTEN cell lines treated with 25μM of 2ME2 either alone or in combination with 10 μM of LY294002. Cells were incubated with indicated conc. of drug for 16 hrs followed by lysate preparation and Western blot analysis using pAkt, pErk, Akt and Erk antibodies. (B) Immunoblot showing synergy between 2ME2 and LY294002 blockade in LN229 vIII cells treated with indicated amount of drug for 16 hrs.(C) U87MG -Null and U87MG-PTEN cell lines were treated with 25 μM 2ME2 (lane 4 and 8), 25 μM pan PI3K inhibitor LY294002 (lane 3 and 7), these two compounds in combination (lane 5 and 9) or no treatment (lane 2 and 6), for 16 hours at 37°C and were exposed to 1% oxygen (lanes 2–9) or 20% oxygen (lane 1). β-actin was used as loading control. Both LY294002 and 2ME2 decreased hypoxic accumulation of HIF-1α, and together they more potent than either alone. (D) Role of PTEN in 2ME2-mediated regulation of HIF1α accumulation was evaluated using U373MG-Null and U373MG-PTEN cells with mutated p53. Cells were treated with inhibitor in similar conditions as described for U87MG Null and PTEN cells. 2ME2 alone had a more pronounced effect on the PTEN-reconstituted cells than PTEN-null cells, and the combination of 2ME2 with LY294002 was more potent than either drug alone. (E). LN229VIII cells were treated with 2ME2 (10μM or 25 μM), LY294002 (5μM or 25 μM) or in combination (5μM or 12.5 μM) followed by normoxia (20% oxygen) or hypoxia (1% oxygen) for 16 hrs.
Next we explored the potential synergistic effect of these drugs on HIF1α accumulation. Figs. 2C & D show that HIF1α accumulation was suppressed in isogenic U373MG and U87MG lines treated with 2ME2 alone or in combination with LY294002. In both U87MG and U87MG-PTEN cells the combined treatment with 2ME2 and LY294002 prevented the hypoxic accumulation of HIF to an equal extent. A comparison of the effects of separate vs. combined treatment with 2ME2 and/or LY294002 in U373MG vs. U373MG-PTEN cells reveals dramatic increase in effect of combined 2ME2/LY294002 in the U373MG-PTEN reconstituted glioma cells. Therefore we conclude that for the U373MG isogenic pair, intact p53 is not required for PTEN to augment the capacity of the 2ME2/LY294002 combination to suppress HIF. Consistent with the above results, 2ME2 alone or in combination with LY294002 potently inhibited HIF1α accumulation in LN229vIII cells (Fig. 2E). These results suggest that 2ME2 can regulate HIF-1α protein expression during hypoxia more effectively when PTEN is present or PI3K is inhibited.
Synergistic activity of PI3K inhibitors and 2ME2 on proliferation
Before examining the combined effect of 2ME2 and LY294002 on proliferation, we determined the effect of 2ME2 on U87MG-Null and U87MG-PTEN cell lines (Fig. S1). Considering that a shared function of PTEN and PI3K inhibitors is to suppressPIP3 and AKT, we evaluated combined 2ME2 - LY294002 treatment in PTEN-intact LN229vIII GBM cells (wild type PTEN, mutated p53, activating vIII EGFR mutation). Cell proliferation data revealed 2ME2 and LY294002 synergism, as indicated by combination index (CI) values (Fig. 3). The CI was calculated using the Chou-Talalay equation, which takes into account both the potency (Dm or IC50, inhibitory concentration 50%) and shape of the dose curve [41]. Synergism was detected for both a 1:1 ratio and a 2:1 ratio of 2ME2 versus LY294002 for all tested doses up to the maximum 5000 nM of each drug.
Fig. 3. Synergistic activity of PI3K inhibitors and 2ME2 on proliferation.

(A) Cell proliferation data revealed synergism between 2ME2 and LY294002, as indicated by combination index (CI) values. (B) Isobologram analysis of combined IC50 doses of 2ME2 and LY294002. Line connecting single IC50 doses is the line of additivity, and the response of the two drugs used in combination at their IC50 levels are indicated on these plots. The combined effect is defined as synergistic, additive, or antagonistic when the point lies below, on, or above the line of additivity, respectively.
2ME2 induced apoptosis is not altered by PTEN status
Our next point of investigation was to determine if PTEN status influences 2ME2 mediated apoptosis induction. Interestingly, 2ME2 induced apoptosis in both U87MG-Null as well as U87MG-PTEN cell lines as revealed by a very similar kinetic pattern of caspase 3 cleavage (Fig. S2). These results suggest that apoptosis induced by 2ME2 in vitro occurred in a similar manner in PTEN positive and PTEN null GBM cells in vitro.
PTEN status influences the antitumor and anti-angiogenic activity of 2ME2
In agreement with previous reports [42], our results demonstrated increased rate of tumor growth in U373MG-Null cells implanted into nu/nu mice as compared to U87MG-PTEN cells (Fig. 4A, 4B and 4C; p < .0002). 2ME2 showed significant tumor inhibition in mice implanted with PTEN-positive LN229 vIII or PTEN-reconstituted U373-PTEN tumors (Fig. 4A, 4B and 4C). 2ME2 had no significant antitumor activity against the U373MG-Null GBM cell line in vivo (p = .877). Previous reports from our laboratory and others highlight the anti-angiogenic properties of 2ME2, PTEN and PI3K inhibitors[10, 20, 35], so we performed CD31 staining on all tumors. We found that 2ME2 clearly suppressed angiogenesis in U373MG-PTEN and LN229vIII (PTEN intact) tumors but not in U373MG-Null tumors (Fig. 4D &E). Collectively, these data imply that both the anti-angiogenic and anti-tumor effects of 2ME2 are augmented by PTEN expression within the tumor compartment.
Fig. 4. PTEN status influences the antitumor and anti-angiogenic activity of 2ME2.

Equivalent number (5 × 106 cells in 100 μl PBS) of parental U373MG PTEN-null (A), U373MGPTEN-reconstituted (B), or LN229vIII cells (C) were implanted subcutaneously in the right flank of athymic mice. Treatment with 2ME2 or vehicle control was initiated when all mice had tumors ranging in size from 80–100 mm3. The treatment was continued till tumors were harvested. 2ME2 treatment did not significantly reduce the growth rate of tumors in vivo in the PTEN-Null mice while it is effective PTEN-reconstituted U373MG tumors as well as PTEN-Wild type LN229vIII tumors (n=7). (D) CD31 staining showing reduction in microvessels in LN229vIII tumors treated with 2ME2 as compared to vehicle. (E) Bar diagrams show the quantization of CD31-positive microvessels in U373MG PTEN-Null, U373MG-PTEN and LN229vIII tumors. There was no significant difference in MVD with 2ME2 versus control U373-null, panel [i]). MVD decreased in 2ME2 treated mice versus control for PTEN-reconstituted U373MG tumors (panel [ii]) and PTEN-retaining LN229vIII cells (panel [iii]). These data suggest that 2ME2 displays greater antitumor efficacy in GBM lines that have normal PTEN function.
PTEN status determines survival with 2ME2 treatment in an orthotopic brain tumor model
To confirm that the effect of 2ME2 on tumor growth depends on PTEN status, we used a well-established orthotopic brain tumor model[10]. Median survival in mice with U373MG-Null GBM tumors was 56 days in the 2ME2 treated group vs. 45 days in vehicle controls (p = 0.87); (Fig. 5A). Median survival with U373MG-PTEN GBM tumors was 106 days in the 2ME2 treated group vs. 79 days in vehicle controls (p = 0.028; Fig. 5B. These data suggest that 2ME2 has greater activity against intracranial tumor growth when PTEN is present.
Fig. 5. PTEN status determines survival with 2ME2 treatment in an orthotopic brain tumor model.
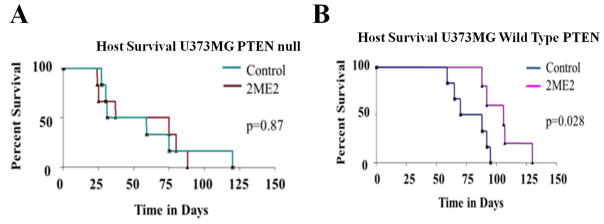
The effects of 2ME2 on survival in an orthotopic brain tumor mouse model implanted with U373MG PTEN-Null (A) and U373MG-PTEN (B) cells. Survival data represents 6–8 animals per experimental group. Beginning on post-implantation day 3, 2ME2 or a vehicle control were administered IP once daily at 200mg/kg. Among mice bearing tumors derived from U373MG PTEN-Null cells, there was no significant benefit in survival with 2ME2 versus vehicle control treatment (P=0.869). In mice bearing tumors derived from U373MGPTEN-reconstituted cells, there was a significant prolongation in survival among the 2ME2 treatment group versus vehicle controls (P= 0.028).
DISCUSSION
In this report, we utilized isogenic sets of glioblastoma cell lines reconstituted with PTEN or mutants of PTEN to investigate the potential role of PTEN and PI3K signaling as it relates to in vitro and in vivo sensitivity to 2ME2. PTEN participates in the control of glioma angiogenesis, and it is reported that PTEN, PI-3K and 2ME2 have anti-angiogenic properties and regulate HIF1α [10, 21, 39]. PTEN is a key tumor suppressor and dysregulation of the PTEN-PI3K-Akt axis has been implicated in the expansion and therapeutic resistance of GBM and other cancers [32, 33]. Considering the frequency of PTEN and PI3K mutations in GBM, and that the utility of 2ME2 for GBM therapy is diminished by the emergence of resistance, our studies were designed to determine specifically (1) if PTEN status could potentially inform the stratification of GBM in terms of predicted response to 2ME2, and (2) whether blockade of PI3K signaling combined with 2ME2 treatment would enhance anti-GBM efficacy. To date, no reports have emerged to examine the relationship between 2ME2 sensitivity and PTEN status in malignant glioma pathogenesis and treatment. We found that 2ME2 inhibited both hypoxic accumulation of HIF1α and GBM cell proliferation in vitro, effects that were significantly potentiated by the addition of the pan-PI3K inhibitor, LY294002 or the presence of PTEN (Fig. 3 & 4).
The role of the PTEN-PI3K pathway in stabilizing HIF1α is controversial [19]. Our present studies with muristerone induced U87MG and GFAPV12 Ras cell lines clearly demonstrate that PTEN blocks HIF1α accumulation in glioma cells (Fig. 1). On the same note, work done by Zundel et al suggests that loss of PTEN mediates HIF1α mediated gene transcription[17]. Moreover, consistent with a previous report [15], our results also showed that treatment with PI3K inhibitors SF1126 or LY294002 suppressed HIF1α accumulation in glioma cell lines (Fig. 1 & 3). Most importantly, treatment with 2ME2 and LY294002 displayed a synergistic effect in suppressing cell proliferation as well as to block HIF1α accumulation, a finding which has significant potential clinical relevance (Fig. 3 & 4).
The second potentially clinically relevant conclusion from this study is that PTEN status within the tumor compartment may serve as a biomarker for GBM sensitivity to treatment with 2ME2. Our results suggest that 2ME2 suppression of HIF1α stabilization, tumor growth and angiogenesis depends on PTEN status (Fig. 3, 6–8). Interestingly, the effect of PTEN reconstitution on these phenotypes occurred in an isogenic pair of U373MG cell lines which are mutated in p53. Hence, these data suggest that a functional p53 axis is not required for PTEN to exert these effects on 2ME2 sensitivity in vitro and in vivo. A recent clinical report in 145 patients by Said et al demonstrated that patients with p53 mutations have augmented response to anti-angiogenic therapy with Avastin[43]. A number of recent reports link p53 to the control of HIF1α and both PTEN and PI3K are known to regulate p53 via mdm2 phosphorylation, eliciting speculation that this key signaling axis (PTEN- HIF1α -p53) may be an important determinant of malignant glioma pathogenesis and therapeutics [44, 45]. Finally, it has been reported that the PTEN promoter is regulated by p53 adding additional complexity to the interpretation our model systems[46].
2ME2 reduced microvascular density and tumor growth in mice implanted with U373MG-PTEN GBM tumors, and significantly extended survival of mice bearing orthotopic wild type PTEN-intact tumors. This suggests that the antitumor efficacy of 2ME2 on GBM relates to an anti-angiogenic effect within the stromal compartment rather than solely a direct tumor cell cytotoxic effect. 2ME2 suppressed HIF1α, an inducer of angiogenesis, more potently in PTEN-intact cells. Previously, reports addressing 2ME2 effects on tubulin in both the tumor and endothelial cell compartments indicate that 2ME2 may disrupt tumor- endothelium crosstalk, suppressing angiogenesis [20].
In aggregate, the data in this study suggest that 2ME2 may be more efficacious in patients whose GBM tumors express a wild type PTEN tumor suppressor protein. It has previously been reported that PTEN status is predictive of responsiveness to EGFR inhibitor therapy in GBM[47] Therefore, it is reasonable to suggest that PTEN genetic and protein analysis in a patients tumor may serve as predictive biomarkers of response to 2ME2 therapy. Moreover, our data highlight the potential clinical utility of investigating combined pharmacologic inhibition of PI3K isoforms with 2ME2 for GBM therapy. These data may further suggest that 2ME2 may have greater activity in lower grade glial tumor (WHO grades I or II) where PTEN is rarely mutated and remains intact.
Supplementary Material
Acknowledgments
We thank Dr. H.K Shu for providing the EGFRvIII transduced LN229 cell line. This work was supported in part by a grant from EntreMed, Inc. 9640 Medical Center Drive, Rockville, MD 20850 and NIH grants CA94233 and HL091365 to Dr. Donald L. Durden, and a grant from the Musella Foundation and Heroes of Hope Coalition to Dr. Carrie Muh.
Footnotes
COI statement
D. Durden discloses financial conflict of interest related to the development of SF1126. This aspect has been reviewed by the UCSD committee on conflict of interest.
References
- 1.Buckner JC. Factors influencing survival in high-grade gliomas. Semin Oncol. 2003;30(6 Suppl 19):10–4. doi: 10.1053/j.seminoncol.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 2.DeAngelis LM. Brain tumors. N Engl J Med. 2001;344(2):114–23. doi: 10.1056/NEJM200101113440207. [DOI] [PubMed] [Google Scholar]
- 3.Srividya MR, et al. Homozygous 10q23/PTEN deletion and its impact on outcome in glioblastoma: a prospective translational study on a uniformly treated cohort of adult patients. Neuropathology. 2011;31(4):376–83. doi: 10.1111/j.1440-1789.2010.01178.x. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 5.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallia GL, et al. PIK3CA gene mutations in pediatric and adult glioblastoma multiforme. Mol Cancer Res. 2006;4(10):709–14. doi: 10.1158/1541-7786.MCR-06-0172. [DOI] [PubMed] [Google Scholar]
- 7.Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003;546(1):108–12. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- 8.Castellino RC, Muh CR, Durden DL. PI-3 kinase-PTEN signaling node: an intercept point for the control of angiogenesis. Curr Pharm Des. 2009;15(4):380–8. doi: 10.2174/138161209787315873. [DOI] [PubMed] [Google Scholar]
- 9.Sun H, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96(11):6199–204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen S, et al. PTEN controls tumor-induced angiogenesis. PNAS. 2001;98(8):4622–4627. doi: 10.1073/pnas.081063798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forsythe JA, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16(9):4604–13. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivan M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 13.Jaakkola P, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 14.Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64(5–6):993–8. doi: 10.1016/s0006-2952(02)01168-1. [DOI] [PubMed] [Google Scholar]
- 15.Jiang BH, et al. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 2001;12(7):363–9. [PubMed] [Google Scholar]
- 16.Zhong H, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60(6):1541–5. [PubMed] [Google Scholar]
- 17.Zundel W, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14(4):391–6. [PMC free article] [PubMed] [Google Scholar]
- 18.Alvarez-Tejado M, et al. Lack of evidence for the involvement of the phosphoinositide 3-kinase/Akt pathway in the activation of hypoxia-inducible factors by low oxygen tension. J Biol Chem. 2002;277(16):13508–17. doi: 10.1074/jbc.M200017200. [DOI] [PubMed] [Google Scholar]
- 19.Arsham AM, et al. Phosphatidylinositol 3-kinase/Akt signaling is neither required for hypoxic stabilization of HIF-1 alpha nor sufficient for HIF-1-dependent target gene transcription. J Biol Chem. 2002;277(17):15162–70. doi: 10.1074/jbc.M111162200. [DOI] [PubMed] [Google Scholar]
- 20.Mabjeesh NJ, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3(4):363–75. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 21.Kirches E, Warich-Kirches M. 2-methoxyestradiol as a potential cytostatic drug in gliomas? Anticancer Agents Med Chem. 2009;9(1):55–65. doi: 10.2174/187152009787047725. [DOI] [PubMed] [Google Scholar]
- 22.Kang SH, et al. Antitumor effect of 2-methoxyestradiol in a rat orthotopic brain tumor model. Cancer Res. 2006;66(24):11991–7. doi: 10.1158/0008-5472.CAN-06-1320. [DOI] [PubMed] [Google Scholar]
- 23.Lis A, et al. 2-Methoxyestradiol inhibits proliferation of normal and neoplastic glial cells, and induces cell death, in vitro. Cancer Lett. 2004;213:57–65. doi: 10.1016/j.canlet.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 24.James J, et al. Phase I safety, pharmacokinetic and pharmacodynamic studies of 2-methoxyestradiol alone or in combination with docetaxel in patients with locally recurrent or metastatic breast cancer. Invest New Drugs. 2007;25(1):41–8. doi: 10.1007/s10637-006-9008-5. [DOI] [PubMed] [Google Scholar]
- 25.Bruce JY, et al. A phase II study of 2-methoxyestradiol nanocrystal colloidal dispersion alone and in combination with sunitinib malate in patients with metastatic renal cell carcinoma progressing on sunitinib malate. Invest New Drugs. 2012;30(2):794–802. doi: 10.1007/s10637-010-9618-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sweeney C, et al. A phase II multicenter, randomized, double-blind, safety trial assessing the pharmacokinetics, pharmacodynamics, and efficacy of oral 2-methoxyestradiol capsules in hormone-refractory prostate cancer. Clin Cancer Res. 2005;11(18):6625–33. doi: 10.1158/1078-0432.CCR-05-0440. [DOI] [PubMed] [Google Scholar]
- 27.ClinicalTrials.gov. Phase 2 Study of Panzem Nanocrystal Colloidal Dispersion (NCD) in Combination With Fixed-Dose Temozolomide to Patients With Recurrent Glioblastoma Multiforme (GBM) Available from: http://clinicaltrials.gov/ct2/show/NCT00481455.
- 28.ClinicalTrials.gov. A Phase 2 Study With Panzem in Patients With Relapsed or Plateau Phase Multiple Myeloma. Available from: http://clinicaltrials.gov/ct2/show/NCT00592579.
- 29.ClinicalTrials.gov. A Combination Study to Determine the Safety and Efficacy of Panzem NCD With Avastin in Metastatic Carcinoid Tumors. Available from: http://clinicaltrials.gov/ct2/show/NCT00328497.
- 30.Dahut WL, et al. Phase I clinical trial of oral 2-methoxyestradiol, an antiangiogenic and apoptotic agent, in patients with solid tumors. Cancer Biol Ther. 2006;5(1):22–7. doi: 10.4161/cbt.5.1.2349. [DOI] [PubMed] [Google Scholar]
- 31.Mooberry SL. New insights into 2-methoxyestradiol, a promising antiangiogenic and antitumor agent. Curr Opin Oncol. 2003;15(6):425–30. doi: 10.1097/00001622-200311000-00004. [DOI] [PubMed] [Google Scholar]
- 32.Duman BB, et al. PTEN, Akt, MAPK, p53 and p95 expression to predict trastuzumab resistance in HER2 positive breast cancer. J buon. 2013;18(1):44–50. [PubMed] [Google Scholar]
- 33.Sami A, Karsy M. Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: novel therapeutic agents and advances in understanding. Tumour Biol. 2013 doi: 10.1007/s13277-013-0800-5. [DOI] [PubMed] [Google Scholar]
- 34.Wischhusen J, et al. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene. 2003;22(51):8233–45. doi: 10.1038/sj.onc.1207198. [DOI] [PubMed] [Google Scholar]
- 35.Garlich JR, et al. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 2008;68(1):206–15. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 36.Rong Y, et al. PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res. 2005;65(4):1406–13. doi: 10.1158/0008-5472.CAN-04-3376. [DOI] [PubMed] [Google Scholar]
- 37.Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19(9):2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peirce SK, et al. The PI-3 kinase-Akt-MDM2-survivin signaling axis in high-risk neuroblastoma: a target for PI-3 kinase inhibitor intervention. Cancer Chemother Pharmacol. 2011;68(2):325–35. doi: 10.1007/s00280-010-1486-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mabjeesh NJ, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–375. doi: 10.1016/s1535-6108(03)00077-1. [DOI] [PubMed] [Google Scholar]
- 40.Ricker JL, et al. 2-Methoxyestradiol inhibits hypoxia-inducible factor 1α, tumor growth, and angiogenesis and augments paclitaxel efficacy in head and neck squamous cell carcinoma. Clin Cancer Res. 2004;10:8665–8673. doi: 10.1158/1078-0432.CCR-04-1393. [DOI] [PubMed] [Google Scholar]
- 41.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 42.Su JD, et al. PTEN and phosphatidylinositol 3′-kinase inhibitors up-regulate p53 and block tumor-induced angiogenesis: evidence for an effect on the tumor and endothelial compartment. Cancer Res. 2003;63(13):3585–92. [PubMed] [Google Scholar]
- 43.Said R, et al. P53 Mutations in Advanced Cancers: Clinical Characteristics, Outcomes, and Correlation between Progression-Free Survival and Bevacizumab-Containing Therapy. Oncotarget. 2013;4(5):705–14. doi: 10.18632/oncotarget.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011;2:e164. doi: 10.1038/cddis.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen D, et al. Direct interactions between HIF-1 alpha and Mdm2 modulate p53 function. J Biol Chem. 2003;278(16):13595–8. doi: 10.1074/jbc.C200694200. [DOI] [PubMed] [Google Scholar]
- 46.Stambolic V, et al. Regulation of PTEN transcription by p53. Mol Cell. 2001;8(2):317–25. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- 47.Mellinghoff IK, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.