

Abstract
The anaerobic sulfatase maturating enzyme from Clostridium perfringens (anSMEcpe) catalyzes the two-electron oxidation of a cysteinyl residue on a cognate protein to a formyglycyl residue (FGly) using a mechanism that involves organic radicals. The FGly residue plays a unique role as a cofactor in a class of enzymes termed arylsulfatases, which catalyze the hydrolysis of various organosulfate monoesters. anSMEcpe has been shown to be a member of the radical S-adenosylmethionine (SAM) family of enzymes, [4Fe–4S] cluster–requiring proteins that use a 5’-deoxyadenosyl 5’-radical (5’-dA•) generated from a reductive cleavage of SAM to initiate radical-based catalysis. Herein, we show that anSMEcpe contains in addition to the [4Fe–4S] cluster harbored by all radical SAM (RS) enzymes, two additional [4Fe–4S] clusters, similar to the radical SAM protein AtsB, which catalyzes the two-electron oxidation of a seryl residue to a FGly residue. We show by size-exclusion chromatography that both AtsB and anSMEcpe are monomeric proteins, and site-directed mutagenesis studies on AtsB reveal that individual Cys→Ala substitutions at seven conserved positions result in insoluble protein, consistent with those residues acting as ligands to the two additional [4Fe–4S] clusters. Ala substitutions at an additional conserved Cys residue (C291 in AtsB; C276 in anSMEcpe) afford proteins that display intermediate behavior. These proteins exhibit reduced solubility and drastically reduced activity, behavior that is conspicuously similar to that of a critical Cys residue in BtrN, another radical SAM dehydrogenase [Grove, T. L., et al (2010) Biochemistry, 49, 3783–3785]. We also show that wild-type anSMEcpe acts on peptides containing other oxidizeable amino acids at the target position. Moreover, we show that the enzyme will convert threonyl peptides to the corresponding ketone product, and also allo-threonyl peptides, but with a significantly reduced efficiency, suggesting that the proS hydrogen atom of the normal cysteinyl substrate is stereoselectively removed during turnover. Lastly, we show that the electron generated during catalysis by AtsB and anSMEcpe can utilized for multiple turnovers, albeit through a reduced flavodoxin-mediated pathway.
Radical SAM (RS)1 dehydrogenases are a burgeoning class of S-adenosylmethionine (SAM)-requiring enzymes that catalyze the two-electron oxidation of organic substrates via intermediates containing unpaired electrons (1-7). These enzymes, as do all RS proteins, contain a [4Fe–4S] cluster cofactor that is absolutely required for turnover (1-3, 8). The [4Fe–4S] cluster is coordinated by the α-amino and α-carboxylate groups of SAM, and in its reduced state, provides the essential electron for the reductive cleavage of SAM into methionine and a 5’-deoxyadenosyl 5’-radical (5’-dA•) (9, 10). The 5’-dA•, in turn, initiates turnover by abstracting a hydrogen atom (H•) from a strategic position, often cleaving unactivated or weakly activated C–H bonds (11-15). Three RS dehydrogenases spanning two distinct classes have been the subject of detailed in vitro mechanistic investigation. One, BtrN, catalyzes the third step in the biosynthetic pathway of the 2-deoxystreptamine (DOS)-containing aminoglycoside antibiotic, butirosin B, which entails the two-electron oxidation of the C3 secondary alcohol of 2-deoxy-scyllo-inosamine (DOIA) to a ketone, affording amino-2-deoxy-scyllo-inosose (amino-DOI) (Scheme 1A) (3). The remaining two, AtsB and anSMEcpe, are anaerobic sulfatase modifying enzymes (anSMEs), which catalyze the two-electron oxidation of a target seryl or cysteinyl residue on their cognate arylsulfatases to a formylglycyl (FGly) residue (Scheme 1B) (2, 4, 16, 17). The FGly residue serves as an obligate cofactor in the cleavage of a variety of sulfate monoesters by this class of enzymes (18-21). Crystallographic and mechanistic studies have shown that the FGly residue exists as a hydrate, wherein one oxygen acts as a nucleophile in the attack on the sulfur atom of the sulfate monoester. Release of sulfate is concomitant with collapse of the sulfated geminal diol to the aldehyde (22-24). This mechanism for generating the FGly cofactor is distinguished from another non-RS mechanism found in higher eukaryotes and some bacteria, which requires reducing equivalents and the intervention of dioxygen (25-28); however, in both cases the FGly cofactor is typically found in the conserved sequence motif C/S-X-P/A-S/X-R-X-X-X-L/X-T/X-G/X-R/X, with the C/S highlighted in bold type as the site of modification (16, 29).
Scheme 1.
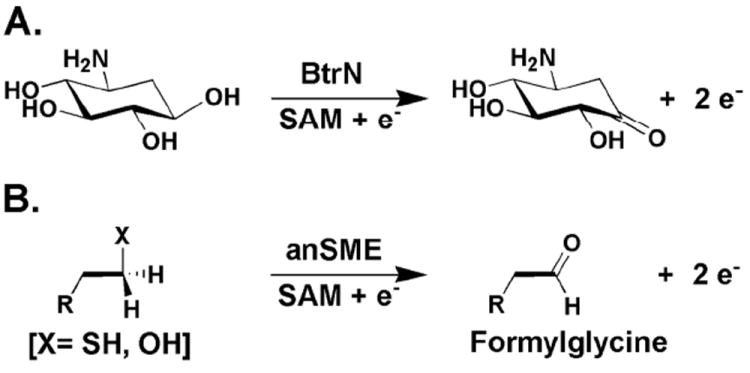
Reactions catalyzed by BtrN (A) and anSMEs (B).
Characterization of AtsB, BtrN, and anSMEcpe verified their membership in the RS superfamily of enzymes (1-3, 17, 30). In addition to their canonical CxxxCxxC motifs, which bear the Cys ligands that coordinate the iron–sulfur (Fe/S) cluster involved intimately in the cleavage of SAM, they were all shown to contain [4Fe–4S] clusters and to cleave SAM reductively to 5’-deoxyadenosine (5’-dA) and methionine during catalysis. However, the number of Fe/S clusters on these enzymes has been a subject of disagreement. In the initial characterization of BtrN, Yokoyama, et al. used quantitative analyses for iron and sulfide after reconstitution of the Fe/S cluster to demonstrate the presence of only one [4Fe–4S] cluster (presumed to be the RS Fe/S cluster) per polypeptide (8). By contrast, Grove, et al. used a combination of analytical (quantitative Fe, S2-, and protein analyses) and spectroscopic (UV-vis and Mössbauer) methods to demonstrate that BtrN harbors two [4Fe–4S] clusters (31). Using the same experimental methodology, it was also demonstrated that AtsB harbors three [4Fe–4S] clusters (2). It was suggested that one of the remaining two non-RS [4Fe–4S] clusters might coordinate to the substrate to facilitate the two-electron oxidation. For the related enzyme anSMEcpe, Benjdia, et al. reported that their reconstituted protein contained 5.7 ± 0.5 equiv of iron (sulfide not quantified). This stoichiometry in concert with characterization of the protein by UV–vis, resonance Raman, and electron paramagnetic resonance (EPR) spectroscopy led the authors to suggest that the protein most likely contained one [4Fe–4S] cluster, although they left open the possibility that it might contain two, and suggested that further studies would be required to determine this conclusively (1).
The Cys-type anSME from Clostridium perfringens (anSMEcpe) shares 48% sequence similarity with the Ser-type anSME from Klebsiella pneumoniae (AtsB). It is slightly smaller in size (370 aa vs 395 aa), but contains 18 Cys residues per polypeptide as opposed to 13 Cys residues on AtsB. Eleven Cys residues are common between the two proteins and are conserved throughout anSMEs. In light of the differences in cluster content observed between these two proteins using different strategies for protein overproduction and spectroscopic methods for Fe/S cluster characterization, we set out to characterize anSMEcpe in a quantitative manner with respect to cluster stoichiometry as well as turnover with various peptide substrates. Herein, we show that anSMEcpe harbors three [4Fe–4S]2+ clusters in its fully active form, as was found for AtsB. Thus, these results further corroborate our proposal that all natural RS-dehydrogenases require at least two [4Fe–4S] clusters for turnover (31). Moreover, we show via site-directed mutagenesis that seven Cys residues in addition to the three that coordinate the RS cluster are absolutely required, and their substitution with Ala residues affords completely insoluble proteins. Similar to findings by Grove, et al. on BtrN, one Cys residue, when substituted with Ala, affords a soluble protein that can be characterized; however, its activity is greatly diminished, supporting a key role for this residue in catalysis. Last, we show that anSMEcpe is capable of converting Cys, Ser, and SeCys residues to FGly residues, as well as threonyl residues to the corresponding keto product, while the reaction of the corresponding allo-threonyl-containing substrate does not lead to substantial formation of the keto product. Collectively these results suggest that the key step in catalysis by anSMEs is abstraction of the 3-proS H• from the substrate by the 5’-dA• intermediate. Also discussed is the fate of the second electron removed from the target Ser or Cys residue during the two-electron oxidation.
MATERIALS AND METHODS
Materials
All DNA-modifying enzymes and reagents were purchased from New England Biolabs (Ipswich, MA), as were Vent polymerase and its associated 10× buffer. Oligonucleotide primers were obtained from Integrated DNA Technologies (Coralville, IA). C. perfringens (strain NCTC 8237) genomic DNA (ATCC 13124D-5) was purchased from American Type Culture Collection (Manassas, VA). 5’-Deoxyadenosine (5’-dA), sodium sulfide (nonahydrate), sodium dithionite (DT), β-mercaptoethanol, l-tryptophan, l-(+)-arabinose, and ferric chloride were purchased from Sigma–Aldrich Chemicals (St. Louis, MO). N-(2-hydroxyethyl)piperazine-N’-(2-ethanesulfonic acid) (HEPES) was purchased from Fisher Scientific (Pittsburgh, PA), and imidazole was purchased from J. T. Baker Chemical Co (Phillipsburg, NJ). Potassium chloride and glycerol were purchased from EMD Chemicals (Gibbstown, NJ), while dithiothreitol (DTT) was purchased from Gold Biotechnology (St. Louis, MO). Coomassie blue dye-binding reagent for protein concentration determination was purchased from Pierce (Rockford, IL), as was the bovine serum albumin (BSA) standard (2 mg/mL). Talon metal affinity resin was purchased from Clontech (Mountain View, CA). Sephadex G-25 resin and NICK and NAP prepoured gel filtration columns were purchased from GE Biosciences (Piscataway, NJ). Fmoc-Thr(tBu)-OH (99%), Fmoc-allo-Thr(tBu)-OH (99%), and Fmoc-Se-4-methoxybenzyl selenocysteine (99%) were purchased from Chem-Impex International. All other chemicals were of the highest grade available.
S-Adenosyl-l-methionine (SAM) was synthesized enzymatically and purified as described previously (32). Flavodoxin (Flv) and flavodoxin reductase (Flx) were purified from E. coli BL21(DE3) containing plasmids pTYB1-Flv and pTYB1-Flx as described previously (33, 34). Fmoc-formylglycine (dimethylacetal) was kindly provided by Professor Carolyn Bertozzi and Dr. Jason Rush (UC Berkeley).
DNA sequencing was carried out at the Pennsylvania State University Nucleic Acid Facility. Analyses for iron and sulfide were performed by the procedures of Beinert (35-37). SPEX CertiPrep (Metuchen, NJ) Cläritas PPT single element Fe (1000 mg/L in 2% HNO3) was used to prepare iron standards for quantitative iron analysis. Protein concentration was measured by the procedure of Bradford using bovine serum albumin (Fraction V) as a standard (38).
Spectroscopic Methods
UV-visible spectra were recorded on a Cary 50 spectrometer (Varian, Walnut Creek, CA) using the associated WinUV software package for operating the instrument and manipulating the data. Mössbauer spectra were recorded on a spectrometer from WEB Research (Edina, MN), which was equipped with an SVT-400 cryostat from Janis Research Co (Wilmington, MA). Spectra were collected in constant acceleration mode in transmission geometry. Isomer shifts are quoted relative to the centroid of α-Fe at room temperature. Spectra were analyzed with the program WMOSS from WEB Research. 57Fe (97-98%) metal for Mössbauer spectroscopy was purchased from Isoflex USA (San Francisco, CA). For preparation of a 57FeSO4 solution, the solid was dissolved with heating in an anaerobic solution of 2 N H2SO4 (1.5 mol of H2SO4 per mole of 57Fe). The 57Fe solution was used as is for supplementation in E. coli culture media, or was titrated to pH 6.5 with an anaerobic solution of saturated sodium bicarbonate for in vitro reconstitution. X-band (~9.5 GHz) electron paramagnetic resonance (EPR) spectroscopy was conducted on a Bruker ESP 300 spectrometer equipped with an Oxford Instruments Model ESP 900 continuous flow cryostat. EPR parameters for various samples are provided in the appropriate figure legends.
Cloning of the cpe0635 gene from Clostridium perfringens
The gene corresponding to anSMEcpe (cpe0635) was amplified from C. perfringens genomic DNA (ATCC# 13124D-5) using the polymerase chain reaction (PCR) in combination with a forward primer containing an NdeI restriction site (underlined) (5’-CGC-GCC-CGC-ATA-TGC-CAC-CAT-TAA-GTT-TGC-TTA-TTA-AGC-3’) and a reverse primer containing a BamHI restriction site (underlined) (5’-CCG-GAT-CCG-ATT-TAA-TAT-TGT-TGG-CAA-CAT-TTA-TTA-ACC-3’). The reverse primer was designed to remove the stop codon from the C-terminus of the gene, which affords addition of a 22-amino acid C-terminal extension containing a hexahistidine tag. The PCR was conducted using a Stratagene (La Jolla, CA) Robocycler thermocyler as described previously (39), and the amplified gene was isolated and cloned into expression vector pET-26b by standard procedures. Several constructs were analyzed by DNA sequencing, which revealed that they all had identical sequences. The chosen construct was designated pCpe0635Wt.
Construction of the C15A/C19A/C22A anSMEcpe triple variant
The C15A/C19A/C22A anSMEcpe triple variant was constructed using the Stratagene QuikChange II site-directed mutagenesis kit as described previously (2). The forward primer used was 5’-CCA-TTA-AGT-TTG-CTT-ATT-AAG-CCA-GCT-TCT-AGT-GGA-GCT-AAT-TTA-AAA-GCC-ACT-TAT-GCT-3’, while the reverse primer used was 5’-CTT-AAC-ATT-TCT-ATT-ATC-ACT-TAA-AGA-ATG-ATA-AAA-AGC-ATA-AGT-GGC-TTT-TAA-ATT-AGC -3’. The underlined letters represent the altered codons.
Expression of the Cpe0635 gene and purification of anSMEcpe
Plasmid pCpe0635Wt, or constructs encoding variants of anSMEcpe, was transformed into E. coli BL21(DE3)/pDB1282 by standard methods, and the encoded Cpe0635 gene expressed as described previously for overproduction of AtsB (2). The protein was also purified as previously described. Reconstitution of the Fe/S clusters of anSMEcpe was conducted as described previously (2, 33).
Construction of Cys→Ala variants of AtsB and anSMEcpe
Single Cys→Ala substitutions in anSMEcpe (Cys276) and AtsB (Cys residues 127, 245, 270, 276, 291, 331, 334, 340, 344, and 357) were engineered using the Stratagene QuikChange II site-directed mutagenesis kit with primers listed in Table S1 as described above. Expression of the variant constructs and purification of the encoded proteins were done exactly as described previously (2).
Amino acid analysis of anSMEcpe
Amino acid analysis of anSMEcpe was carried out at the Molecular Structure Facility at the University of California–Davis (Davis, CA). The protein was exchanged by gel filtration (NICK pre-poured column) into 50 mM HEPES buffer (pH 7.5) containing 100 mM NaCl. The eluate was divided into 50 μL fractions, which were lyophilized to dryness using a Savant SpeedVac concentrator (Thermo Scientific; Waltham, MA). One fraction was used to determine the protein concentration by the procedure of Bradford before lyophilization. The remaining fractions were shipped for amino acid analysis, which was performed in quadruplicate. It was found that the concentration determined by the procedure of Bradford is an overestimate and therefore must be multiplied by 0.69 to achieve the true anSMEcpe concentration.
Synthesis and purification of substrate peptides
The following peptide substrates, each containing an N-terminal acetyl (Ac) group (except the IS peptide Kp9Ser), were synthesized at the Penn State Core Research Facilities utilizing standard Fmoc chemistry (bold and underlined type indicates target location of modification): Cp18Cys, Ac-NH2-YTAVPSCIPSRASILTGM-COOH; Kp18Cys, Ac-NH2-YYTSPMCAPARSMLLTGN-COOH; Kp18Ser, Ac-NH2-YYTSPMSAPARSMLLTGN-COOH; Kp18SeCys, Ac-NH2-YYTSPMSeCAPARSMLLTGN-COOH; Kp18Thr, Ac-NH2-YYTSPMTAPARSMLLTGN-COOH; Kp18alloThr, Ac-NH2-YYTSPMaTAPARSMLLTGN-COOH; Kp18FGly, Ac-NH2-YYTSPMfGAPARSMLLTGN-COOH; and Kp9Ser, NH2-PMSAPARSM. The first two letters of each peptide name correspond to the organism (Clostridium perfringens or Klebsiella pneumoniae) from which the peptide sequence is derived; the number corresponds to the length; and the amino acid abbreviation corresponds to the amino acid in the target position. Fmoc-S-4-methoxybenzyl selenocysteine, used in the synthesis of Kp18SeCys, was purchased from Chem-Impex International (Wood Dale, IL) and used as received. Subsequent to synthesis, the peptide (0.035 mmol, ~278 mg) was cleaved from the resin in a solution of 2% triisopropylsilane (100 μL), 100 μL water, and 2.5 % thioanisole (125 μL) in neat TFA (5 mL) containing 1.3 equiv 2,2’-dithiobis(5-nitropyridine) (14 mg) at room temperature for 2 h, after which the cleaved resin was removed by filtration. The crude peptides were then precipitated by addition of ice-cold diethyl ether (1:10 dilution). The peptide mixture was redissolved in a 50% acetonitrile solution (v/v in water) and the appropriate full-length peptide was purified by reverse-phase HPLC (Agilent 1100 System; Santa Clara, CA) using an Agilent Zorbax SBC18 (9.4 × 250 mm) semi-preparative column. A three-solvent system was employed in the separation: 0.1% trifluoroacetic acid (TFA) in water (Solvent A); 0.1% TFA in acetonitrile (Solvent B); and methanol (Solvent C). The column was equilibrated in a solution consisting of 85% Solvent A, 10% Solvent B, and 5% Solvent C. Upon injection of the crude peptide mixture, a gradient of 10-50% Solvent B was applied over 29 min, after which Solvent B was increased to 80% over 1 min. Finally, Solvent B was returned to 10% (initial conditions) over 1 min and the column was allowed to re-equilibrate for 10 min. Throughout the run Solvent C was maintained constant, the flow rate was maintained at 4 mL min-1, and detection of the peptide was monitored by UV-vis spectroscopy at 275 nm. The peak corresponding to the deprotected full-length peptide was collected and lyophilized to dryness to obtain the final product as a white solid. The peptide was then re-dissolved in water and its concentration was determined using a molar absorptivity at 274 nm of 1405 M-1 cm-1 (one Tyr residue) for Cp18Cys and 2810 M-1 cm-1 (two Tyr residues) for the remaining peptides, except for Kp9Ser. The IS peptide Kp9Ser was purified as described above with monitoring at 220 nm. Its final concentration was determined by dissolving a weighed amount in an appropriate volume of water. The purified peptides were analyzed by LC-MS using an Agilent 6410 Triple Quadrupole (QQQ) ESI-MS instrument in positive mode with an MS2 scan width of 500 – 2000 m/z to verify their masses.
Activity determination of anSMEcpe
Reactions contained in a total volume of 150 μL: 50 mM HEPES, pH 7.5, 150 mM KCl, 1 mM SAM, 3 mM DT, 1 mM peptide substrate, and either 4 μM (DT assays) or 40 μM (Flv/Flx/NADPH assays) WT anSMEcpe. Reaction mixtures lacking DT were incubated for 5 min at 37 °C, and 10 μL aliquots were removed (t=0) and added to 10 μL of a solution containing 100 mM H2SO4, 100 μM Kp9Ser (IS), and 100 μM l-tryptophan (IS) to yield final IS concentrations of 50 μM. Reactions were initiated by the addition of DT and incubated for appropriate times before being quenched as described above. The samples were subjected to centrifugation at 18,000 × g in a bench-top microcentrifuge and analyzed by LC-MS using Method 1 or Method 2 as described below. Standard curves were generated with 5’-dA or the appropriate purified peptides. All final concentrations were multiplied by a dilution factor of 2 to determine original concentrations in the assay mixtures. When the Flv/Flx/NADPH reducing system replaced DT, their concentrations were 50 μM, 15 μM, and 2 mM, respectively. When reactions were carried out with Kp18Thr or Kp18alloThr, each peptide was present at a concentration of 500 μM, and the concentrations of AtsB or anSMEcpe were adjusted to 200 μM or 100 μM, respectively. Products were analyzed as described above, as well as by MALDI MS using dinitrophenylhydrazine (DNPH) as a derivatizing agent as previously described (2).
LC-MS Method 1
HPLC with detection by mass spectrometry (LC-MS) was conducted on an Agilent Technologies (Santa Clara, CA) 1200 system, which was fitted with an autosampler for sample injection and coupled to an Agilent Technologies 6410 QQQ mass spectrometer. The system was operated with the associated MassHunter software package, which was also used for data collection and analysis. Assay mixtures were separated on an Agilent Technologies Zorbax Rapid Resolution SB-C18 column (2.4 mm × 35 mm, 3.5 μm particle size), which was equilibrated in 80% Solvent A (5 mM perfluoroheptanoic acid–6 mM ammonium formate in water, pH 3) and 20% acetonitrile at a flow rate of 0.4 mL min-1. A gradient of 20–30% acetonitrile was applied from 0 to 2 min, and then from 30 to 20% acetonitrile from 2 to 2.5 min to restore the system to initial conditions. The column was allowed to re-equilibrate for 1.5 min under initial conditions before subsequent sample injections. Detection of 5’-dA and tryptophan was performed using electrospray ionization in positive mode (ESI+) with multiple reaction monitoring. Relevant retention times and ions monitored are given in Table S2.
LC-MS Method 2
Data collection and analysis was carried out as in Method 1 with the following modifications: the column was equilibrated in 92% Solvent A (0.1% formate in water, pH 3.0) and 8% acetonitrile at a flow rate of 0.5 mL min-1. A gradient of 8–26% acetonitrile was applied from 0.5 to 2 min, and then from 26–28% acetonitrile from 2 min to 4 min. The column was restored to initial conditions from 4 min to 4.5 min and allowed to equilibrate for another 2 min before subsequent sample injections. Detection of substrates and products (Table S3) was performed using electrospray ionization in positive mode (ESI+) with MRM. Relevant retention times and ions monitored are given in Table S3.
Molecular sieve chromatography of anSMEcpe and AtsB
Molecular sieve chromatography of anSMEcpe and AtsB was performed with slight modifications of a previously described procedure (40) using an ÄKTA (GE Healthcare, Piscataway, NJ) liquid chromatography system, which was maintained inside a Coy anaerobic chamber. A HiPrep 16/60 Sephacryl S-200 HR column (GE Healthcare) column was equilibrated in a buffer composed of 10 mM HEPES pH 7.5, 500 mM KCl, 5 mM DTT, and 10% glycerol at a flow rate of 0.3 ml min-1. WT RCN anSMEcpe (100 μL of a 737 μM solution) or WT RCN AtsB (100 μL of a 568 μM solution) and standards (500 μL of 0.1 – 1 mg ml-1 solutions) were injected on the column, which was maintained at a flow rate of 0.3 mL min-1 throughout the chromatographic procedure (470 min). Adenosine (267 Da), cytochrome c (12.4 kDa), Coir albumin (75 kDa) and β-amylase (200 kDa) were used to generate a standard curve of known molecular masses, while the void volume (V0) of the column was determined using blue dextran (2,000 kDa). The elution volumes (Ve) of the standards were obtained, and the ratios of Ve V0-1 were plotted as a function of the log of their respective molecular masses. The standard curve was then used to extrapolate the apparent molecular mass of Wt RCN anSMEcpe or AtsB from their corresponding elution volumes. In some analyses, 100 nmol AtsB was combined with 125 nmol AtsA or 2 μmol Kp18Ser before injection.
Fate of the second reducing equivalent upon abstraction of a H• by the 5’-dA•
An anaerobic solution of DT was prepared in 1 M HEPES buffer, pH 7.5, and its concentration was determined spectrophotometrically using potassium ferricyanide (ε420 = 1020 M-1 cm-1) as a standard and assuming that 1 mol of DT reduces 2 mol of ferricyanide. Flavodoxin semiquinone (Flv•) was generated by adding 0.5 equiv of DT to 1.05 equiv of oxidized Flv (Flvox) and then incubating at 37 °C for 1 h, and its concentration was subsequently determined spectrophotometrically (ε579 = 4570 M-1 cm-1) (41). The anSMEcpe reaction was initiated by adding Flv• (204 μM final concentration) to a reaction mixture containing the following components in a final volume of 1 mL: 100 μM anSMEcpe, 50 mM HEPES, pH 7.5, 200 mM KCl, 2 mM SAM, and 2 mM Kp18Cys. The mixture was incubated at 37 °C, and at designated times, 250 μL aliquots were removed and loaded into EPR tubes, which were subsequently submerged in cryogenic isopentane (-130 °C) to rapidly freeze the solution. Monitoring of Flv• was performed by EPR at 77 K under nonsaturating conditions (see appropriate figure legends), and spin quantification was determined by comparison of the double integral of the signal to that of a 1 mM Cu(II)-EDTA standard collected under identical (nonsaturating) conditions. Low-temperature spectra were also collected at 13 K to monitor reduction of the Fe/S clusters. Product analysis was conducted in parallel by removing 10 μL aliquots of each reaction before freezing and quenching in acid as described above. The data were fitted to Equation 1, which describes a burst phase followed by a linear steady-state phase.
| Equation 1 |
RESULTS
Overproduction of anSMEcpe
The cpe0635 gene from C. perfringens was cloned into a pET-26b expression vector to yield a construct that overproduces anSMEcpe containing a C-terminal hexahistidine tag separated from the last native amino acid (aa) by a spacer of 16 aa. Sequencing of the cloned gene revealed a number of aa alterations from the sequence reported in the database (42). Subsequent recloning and resequencing of the gene indicated that these changes did not result from cloning artifacts, but were indeed authentic for this particular strain of C. perfringens. These changes include the following substitutions: D56→E, I69→T, R78→K, I177→V, R179→K, Q212→K, L224→F, S309→L, K324→R, and D341→A (Figure S1).
We employed a strategy for overproducing soluble anSMEcpe in Escherichia coli (Ec), in which the cpe0635 gene on plasmid pCpe0635Wt was coexpressed with genes from plasmid pDB1282 (33, 34, 43), which derive from an operon encoding proteins known to be involved in Fe/S cluster biosynthesis in Azotobacter vinelandii. This strategy was used successfully to overproduce sufficient amounts of soluble AtsB for biochemical and spectroscopic characterization (2). In addition, overproduction was conducted in M9 minimal medium to allow for efficient incorporation of 57Fe into the protein for analysis by Mössbauer spectroscopy. Figure 1 depicts an SDS–PAGE analysis of the purified protein, which displays migratory properties that are consistent with its molecular mass (45,740 Da) as calculated from its aa sequence. From 16 L of M9 culture, >250 mg of protein are routinely obtained. This yield is a significant improvement over that observed by Benjdia, et al. (~5 mg from 12 L of culture) (1), as well as for the previous overproduction of AtsB (2). Amino acid analysis of anSMEcpe indicates that the Bradford (38) method for protein concentration determination overestimates its concentration by a factor of 1.45 when using BSA (Fraction V) as a standard. Therefore, a correction factor of 0.69 (i.e., 1/1.45) is multiplied by the protein concentration determined by the Bradford method to yield the true protein concentration.
Figure 1.
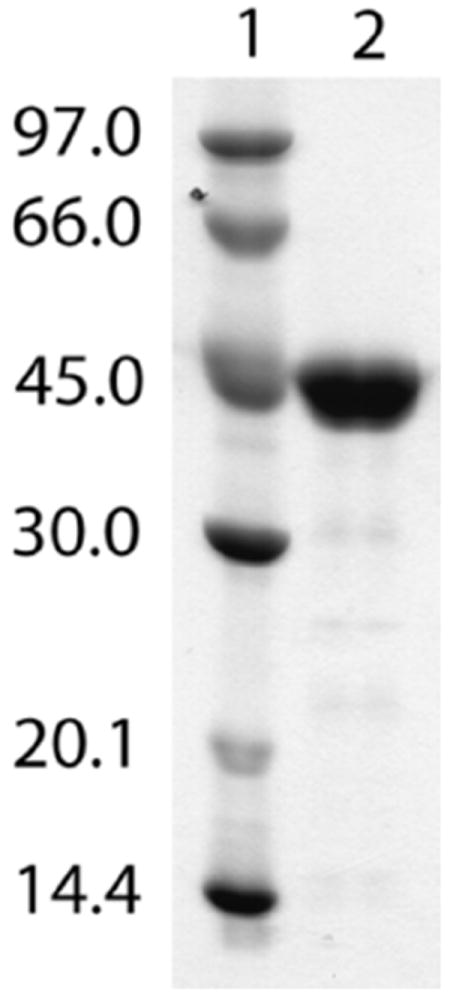
SDS–PAGE analysis of anSMEcpe. Lane 1, molecular mass markers. Lane 2, purified anSMEcpe (45,740 Da). The gel was stained with Coomassie Brilliant Blue.
Spectroscopic and analytical characterization of wild-type anSMEcpe
The as-isolated (AI) UV–vis spectrum of anSMEcpe is shown in Figure 2A (solid line). The spectrum is consistent with the presence of [4Fe–4S] clusters, showing a broad absorption that extends beyond 700 nm and a distinct feature at 397 nm. In contrast to the spectrum of the AI enzyme recorded by Benjdia, et al., there is very little evidence of [2Fe–2S] clusters (1). The ratio of the absorbance at 397 nm to that at 279 nm, which gives a qualitative assessment of cluster content, is 0.35, significantly greater than the ratio observed by Benjdia et al. (0.19), even for their reconstituted enzyme (0.29), suggesting that anSMEcpe used in this study is of significantly better quality and may be suitable for quantitative cluster analyses and rigorous biochemical characterization (34). Analytical determinations of iron and sulfide associated with AI anSMEcpe indicates 9.6 ± 0.1 of the former and 10.0 ± 0.2 of the latter, suggestive of more than one [4Fe–4S] cluster. Figure 2A also indicates that the absorbance at 397 nm is 0.207 for a 5.0 μM sample of anSMEcpe, resulting in a molar absorptivity of ~41,400 M-1 cm-1 at 397 nm. Given that average molar absorptivities in this region for inorganic model peptide-ligated [4Fe–4S] clusters in organic solvents range from 12,100 to 17,500 M-1 cm-1 (44), this analysis strongly suggests that AI anSMEcpe contains more than one [4Fe–4S] cluster, consistent with results from Fe and S2- analysis.
Figure 2.
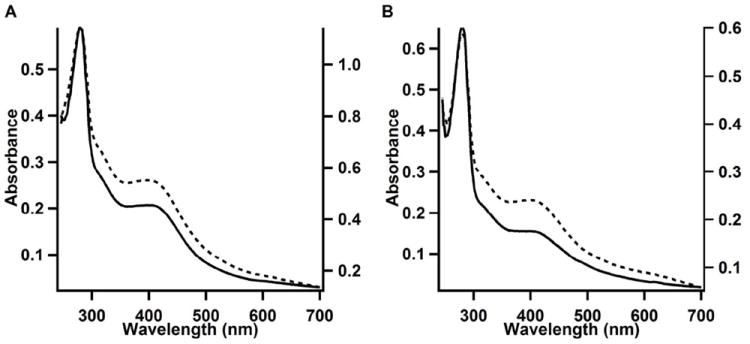
UV-visible spectra of A) AI WT anSMEcpe (5 μM, solid line, left Y-axis) and RC WT anSMEcpe (10 μM, dotted line, right Y-axis). The A279/A387 ratios of AI and RCN proteins were 2.8 and 2.1, respectively. B) UV-visible spectra of AI anSMEcpeC15A/C19A/C22A (9.4 μM, solid line, left Y-axis) and RCN anSMEcpeC15A/C19A/C22A (5.6 μM, dashed line, right Y-axis). The A279/A387 ratios of AI and RCN proteins were 4.2 and 2.4, respectively.
Reconstitution of AI anSMEcpe results in an increase in the stoichiometry of Fe (14.1 ± 0.3) and S2- (12.8 ± 0.7) associated with the protein and increased intensity in its UV-vis spectrum (Figure 2A, dashed line). Although this behavior is suggestive of increased cluster incorporation, analysis by more definitive spectroscopic techniques is required, because adventitiously bound Fe/S species derived from the reconstitution procedure can also produce similar spectra (34, 39, 45).
Mössbauer-spectroscopic characterization of wild-type anSMEcpe
To determine the type and stoichiometry of Fe/S clusters more definitively, AI and reconstituted (RCN) samples of 57Fe-enriched WT anSMEcpe were analyzed by Mössbauer spectroscopy. The 4.2-K/53-mT Mössbauer spectrum of AI anSMEcpe (523 μM; 9.6 Fe per polypeptide) is shown in Figure 3A, and is dominated by an intense quadrupole doublet. The EPR spectrum of an identical sample revealed the presence of a small amount of [3Fe–4S]+ clusters (14 μM spin, 42 μM Fe, 0.8% of total Fe) (Figure S2, red trace), corresponding to 0.8% of the total Fe (i.e. [3 Fe × 14 μM]/[5.02 mM total Fe]). Such a small amount of a paramagnetic cluster with three distinct Fe subsites is beyond the detection limit of Mössbauer spectroscopy (46). The Mössbauer spectrum can be analyzed with one broad quadrupole doublet (95% of total Fe) with parameters typical of [4Fe-4S]2+ clusters: isomer shift (δ) of 0.44 mm/s and quadrupole splitting parameter (ΔEQ) of 1.14 mm/s (solid line in Figure 3A). The weak absorption at ~0.6 mm/s (see arrow) is at a position typical of the high-energy line of spectra of [2Fe-2S]2+ clusters and is most likely associated with a small amount (~3%) of this cluster type, which is often observed as the degradation product of [4Fe-4S] clusters (46). The nature of the weak shoulder (<2% of total Fe) at ~1.7 mm/s (see arrow) is not clear. Mössbauer analysis, in addition to the stoichiometry of 9.6 Fe ions per polypeptide, therefore reveals that AI WT anSMEcpe harbors 2.3 [4Fe–4S] clusters.
Figure 3.
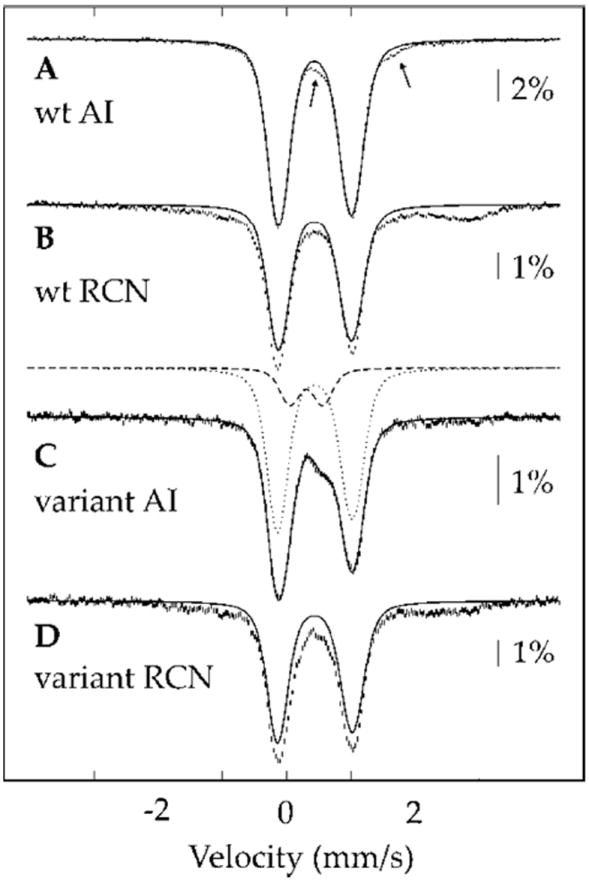
4.2-K/53-mT Mössbauer spectra of WT anSMEcpe (A and B) and anSMEcpeC15A/C19A/C22A (C and D). A and C are the AI forms, and (B) and (D) are the RCN forms. The solid lines are quadrupole doublet simulations with parameters quoted in the text.
The 4.2-K/53-mT Mössbauer spectrum of RCN WT anSMEcpe (173 μM; 14.2 Fe per polypeptide) (Figure 3B) is also dominated by the same intense quadrupole doublet associated with the [4Fe-4S]2+ clusters of AI WT anSMEcpe. Approximately 75% of the total Fe can be attributed to the [4Fe-4S]2+ clusters of AI anSMEcpe (Figure 3B, solid line), resulting in a stoichiometry of 2.7 [i.e. (14.2 Fe) × (0.75)/(4 Fe per cluster)] [4Fe-4S]2+ clusters per polypeptide. The remaining 25% of Fe gives rise to a broad absorption, which is attributed to unspecifically bound Fe, because the EPR spectrum of an identical sample reveals only a small amount of [3Fe-4S]+ clusters (7 μM spin, 21 μM Fe, 0.9% of total Fe) (Figure S2, black trace) and no other signals attributable to Fe/S clusters with spin state S = ½ are observed. Thus, the combination of Mössbauer spectroscopy and analytical methods strongly suggests the presence of three [4Fe-4S] clusters on anSMEcpe, as was reported for the related enzyme, AtsB, from Klebsiella pneumoniae (2).
Characterization of AI and RCN C15A/C19A/C22A triple variant anSMEcpe by Mössbauer spectroscopy
To verify the stoichiometry of three [4Fe–4S] clusters per WT anSMEcpe polypeptide, a triple variant, in which the Cys residues that ligate the RS Fe/S cluster are changed to Ala residues, was constructed (anSMEcpeC15A/C19A/C22A). This substitution of all coordinating residues to the RS Fe/S cluster with noncoordinating residues should lead to its complete elimination, resulting in a stoichiometry of two [4Fe–4S] clusters per polypeptide. anSMEcpeC15A/C19A/C22A was noticeably less stable than the WT protein, which is in contrast to that observed for AtsB, wherein the corresponding triple variant was more stable than the WT protein (2). Nonetheless, ~15 mg of 57Fe-labeled protein was isolated from 8 L of culture, considerably less than that obtained for the WT protein, but sufficient for appropriate characterization.
The UV-vis spectrum of the AI anSMEcpeC15A/C19A/C22A is still consistent with the presence of [4Fe–4S] clusters, exhibiting a pronounced feature at 397 nm and an A397/A279 ratio of 0.24 (Figure 2B, solid line), consistent with the finding of 3.2 ± 0.1 Fe and 7.5 ± 0.1 S2- per polypeptide. Reconstitution of the triple variant results in an increase in the A397/A279 ratio (0.42) (Figure 2B, dashed line) as well as iron and sulfide associated with the protein (8.8 ± 0.4 and 15.1 ± 0.9, respectively). However, the spectral features between 550 and 700 nm suggest the presence of adventitiously bound iron in this sample.
The 4.2-K/53-mT Mössbauer spectrum of AI anSMEcpeC15A/C19A/C22A (472 μM; 3.2 Fe per polypeptide) (Figure 3C) is dominated by a quadrupole doublet associated with [4Fe-4S]2+ clusters: δ = 0.44 mm/s, ΔEQ = 1.16 mm/s, 80% intensity (dotted line). In addition, the peak at 0.6 mm/s suggests the presence of [2Fe-2S]2+ clusters (δ = 0.31 mm/s, ΔEQ = 0.51 mm/s, 17% intensity, dashed line). The greater relative fraction of [2Fe-2S]2+ clusters in anSMEcpeC15A/C19A/C22A compared to that in WT anSMEcpe suggests a greater instability of the remaining [4Fe–4S] clusters in the triple variant. In addition, an identical EPR sample does not show signals of Fe/S clusters with half-integer spin ground states (Figure S2, green trace). The Mössbauer data, in concert with the observed stoichiometry of 3.2 Fe per polypeptide, indicates that AI anSMEcpeC15A/C19A/C22A contains 0.6 [4Fe-4S]2+ and 0.3 [2Fe-2S]2+ clusters per polypeptide.
Reconstitution of anSMEcpeC15A/C19A/C22A with additional Fe and sulfide leads to greater incorporation of Fe/S clusters. The 4.2-K/53-mT Mössbauer spectrum of RCN anSMEcpeC15A/C19A/C22A (281 μM; 8.8 Fe per polypeptide) (Figure 3D) is dominated by a quadrupole doublet associated with [4Fe-4S]2+ clusters (δ = 0.44 mm/s, ΔEQ = 1.16 mm/s, 70% intensity), while the remainder is associated with unspecifically bound Fe, given that an identical EPR sample does not show signals of Fe/S clusters with half-integer spin ground states (Figure S2, blue trace). Given the stoichiometry of 8.8 Fe per polypeptide, it is concluded that RCN anSMEcpeC15A/C19A/C22A harbors 1.5 [4Fe-4S] clusters. This stoichiometry clearly indicates that the triple variant harbors more than one [4Fe-4S] cluster. The fact that it does not contain a full complement of two [4Fe-4S] clusters is rationalized by the greater instability of the protein.
Gel-filtration analysis of anSMEcpe
To assess the quaternary structure of WT anSMEcpe, the RCN protein was subjected to molecular-sieve chromatography on a Sephacryl S-200 HR gel-filtration column connected to an ÄKTA preparative liquid chromatography system housed in a Coy anaerobic chamber. A series of protein standards was used to generate a plot of log molecular mass of a given standard versus Ve V0-1, wherein Ve is the elution volume of the standard and V0 is the void volume of the column. This plot was then used to extrapolate the molecular mass of anSMEcpe from its determined Ve V0-1 value. Hexahistidine-tagged anSMEcpe migrates as a symmetrical single peak of molecular mass 37,500 Da under the conditions described in Materials and Methods (Figure 4A). Its calculated molecular mass of 45,740 Da would therefore be most consistent with a monomeric quaternary structure. A similar experiment was also conducted for hexahistidine-tagged AtsB, which migrates as a symmetrical peak of molecular mass 33,500 Da (Figure 4B, blue line). Its calculated molecular mass of 46,432 Da would suggest that the protein also exhibits a monomeric quaternary structure, although the possibility of a dimeric structure exists. Interestingly, when AtsB is mixed with its peptide substrate (Kp18Ser, MW 2,001 Da) before being applied to the column, it migrates as a protein of 35,800 Da, consistent with a protein/peptide complex (Figure 4B, black line). By contrast, when it is mixed with its natural protein substrate (Kp AtsA), it migrates still as a protein of 33,500 Da (Figure 4B, red line), consistent with previous suggestions that AtsB acts on AtsA before it is folded into its native tertiary structure (17). The absence of a peak for AtsA in the chromatogram is due to monitoring at 395 nm, which allows for the selective monitoring of AtsB migration. The observation that the protein/peptide complex migrates almost exactly as the sum of the masses of the protein (33,500 Da) and peptide (2,001 Da) determined from molecular-sieve chromatography argues for a monomeric structure over a dimeric structure. Unless the protein exhibits half-of-the-sites reactivity, the protein/peptide complex for dimeric AtsB would be expected to exhibit a molecular mass of ~37,502 Da (33,500 + 4,002 Da).
Figure 4.
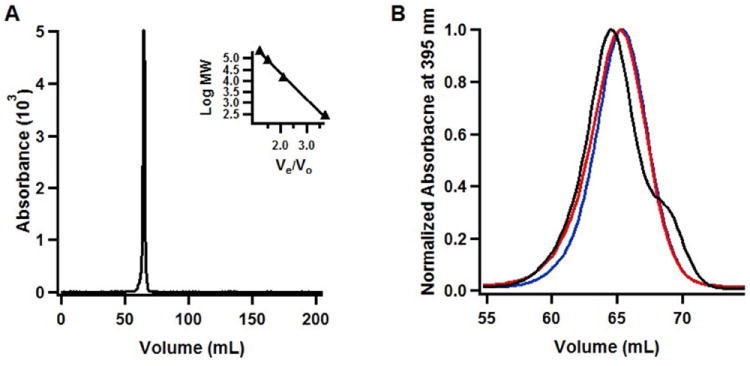
Molecular-sieve analysis of WT anSMEcpe and AtsB. A) WT RCN anSMEcpe; B) WT RCN AtsB in the absence of substrate (blue trace), in the presence of 2 mM Kp18Ser (black trace), or in the presence of WT AtsA from Kp (red trace). Molecular-sieve chromatography was conducted under anaerobic conditions as described in Materials and Methods. WT RCN anSMEcpe eluted at 64.7 mL and WT RCN AtsB eluted at 65.4 mL, yielding calculated molecular masses of 37.5 kDa and 33.5 kDa, respectively. Molecular masses were determined from a plot of known standards that were analyzed under identical conditions (Figure 4A, inset).
Activity determination of anSMEcpe
Sulfatase maturating enzymes (SMEs) act on protein substrates, installing the required FGly cofactor in arylsulfatases (18-22, 26, 47). There is a consensus sequence motif C/S-X-P-S/X-R-X-X-X-L/X-T/X-G/A-R/X found among the various protein substrates irrespective of the mechanism used to generate the FGly cofactor, in which an invariant Arg residue is separated from the Cys or Ser residue to be modified by three amino acids, the second of which is typically Pro, but which can also be Ala (16, 48). Initial activity determinations in this work were conducted with peptides used to study AtsB rather than those that mimic the natural protein substrate for anSMEcpe, given that these were on hand. The FGly modification was quantified by HPLC with detection by QQQ mass spectrometry (LC/MS) using a peptide standard of the same sequence but containing an authentic FGly residue at the target position. Figure S3 displays LC-MS data used to quantify FGly production in a typical assay, which reveals that the FGly-containing product forms at the expense of the substrate. Although the peak corresponding to the FGly product is irregular, due to the highly electrophilic nature of the aldehyde, all regions of the peak correspond to the expected m/z value for the peptide containing the FGly modification. Moreover, the FGly product migrates exactly—both with respect to retention time and shape—as a standard peptide synthesized with an FGly residue at the target position. In Figure 5a, the activity of anSMEcpe (4 μM) using Kp18Cys (500 μM) as the substrate and DT as the reductant is displayed. Formation of the FGly product (open squares) occurs with a Vmax/[ET] of 2.31 ± 0.10 min-1, while formation of 5’-dA (closed triangles) occurs with a Vmax/[ET] of 2.98 ± 0.07 min-1. In addition, ~100 turnovers take place within the 30 min span of the assay. Figure 5b depicts activity profiles of anSMEcpe (40 μM) using Kp18Cys as the substrate and the Flv/Flx/NADPH reducing system as the source of the requisite electron. Similarly to that observed for AtsB, the reaction is significantly slower under these conditions, displaying Vmax/[ET] values of 0.28 ± 0.022 min-1 and 0.26 ± 0.022 min-1 for 5’-dA (closed triangles) and FGly (open squares) formation, respectively. Importantly, for each of these assays product formation is stoichiometric with substrate consumption. In addition, these Vmax/[ET] values are significantly higher than those observed for AtsB under similar conditions (2).
Figure 5.
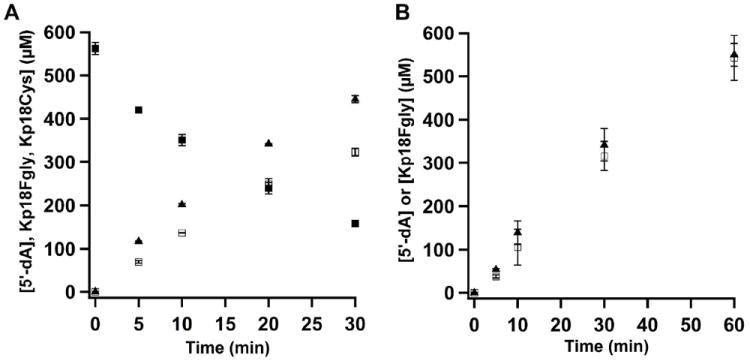
Turnover of WT RCN anSMEcpe with Kp18Cys. A) Time-dependent formation of 5′-dA (closed triangles) and Kp18FGly (open squares), and depletion of Kp18Cys (closed squares) using DT as the requisite electron donor. Reaction mixtures contained 4 μM anSMEcpe, 1 mM SAM, 0.5 mM Kp18Cys, and 3 mM dithionite. The data are the averages of three independent trials, and error bars denote one standard deviation. Vmax/[ET] values for 5’-dA and Kp18FGly formation are 2.98 ± 0.071 min-1 and 2.30 ± 0.100 min-1, respectively, while the Vmax/[ET] value for consumption of Kp18Cys is 2.37 ± 0.017 min-1. B) Time-dependent formation of 5′-dA (closed triangles) and Kp18FGly (open squares) using the Flv/Flx/NADPH reducing system. Reaction mixtures contained 40 μM anSMEcpe, 1 mM SAM, 1 mM Kp18Cys, 50 μM Flv, 15 μM Flx, and 2 mM NADPH. The data are the averages of three independent trials, and error bars denote one standard deviation. Vmax/[ET] values for 5’-dA and Kp18Cys are 0.28 ± 0.022 min-1 and 0.26 ± 0.022 min-1, respectively.
Activity determinations were also conducted with a peptide substrate that corresponds to the sequence of the natural substrate for anSMEcpe. Only substrate consumption was monitored in these assays because of lack of an FGly-containing peptide standard. However, using multiple different assays we have never observed formation of significant amounts of any intermediate species; loss of substrate peptide is always concomitant with formation of product peptide. The Vmax/[ET] for 5’-dA formation and consumption of Cp18Cys are 4.50 ± 0.052 min-1 and 1.91 ± 0.259 min-1, respectively, using DT as reductant, indicating that a significant amount of abortive cleavage of SAM occurs in the presence of this substrate (Figure S4A). In the presence of the Flv/Flx/NADPH reducing system the rates are 0.224 ± 0.003 min-1 and 0.213 ± 0.032 min-1, respectively, similar to those obtained with the Kp18Cys substrate and indicating tight coupling of SAM cleavage and FGly formation (Figure S4B).
Our previous studies indicated that AtsB can act as a Cys-type anSME, although its natural substrate bears a Ser residue at the target position. Studies by Benjdia, et al. showed that anSMEcpe can indeed oxidize Ser-containing substrates; however, the experiments were qualitative in nature and did not permit direct comparison of rates. In Figure 6, turnover of anSMEcpe with Kp18Ser is shown. As can be observed, the rates are significantly lower than that in the presence of Kp18Cys. When using DT as the reductant, Vmax/[ET] is 1.00 ± 0.029 and 0.85 ± 0.001 min-1 for formation of 5’-dA and the FGly product, respectively. When using the Flv/Flx/NADPH reducing system, Vmax/[ET] is 0.074 ± 0.009 and 0.073 ± 0.004 min-1 for formation of 5’-dA and the FGly product, respectively. These rates are approximately three-fold lower with either reductant when Kp18Ser is substituted for Kp18Cys.
Figure 6.
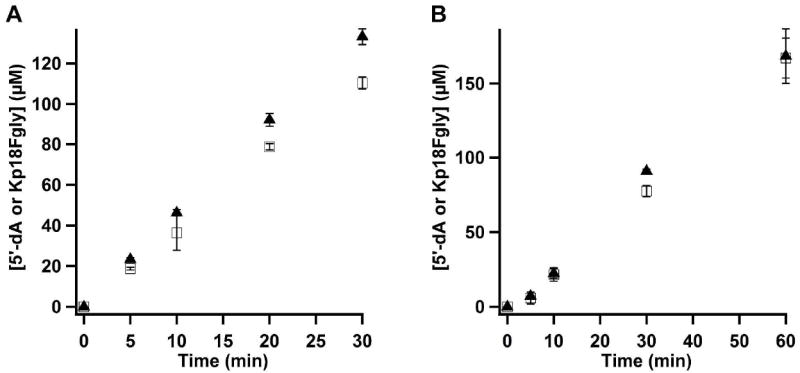
Turnover of WT RCN anSMEcpe with Kp18Ser. A) Time-dependent formation of 5′-dA (closed triangles), and Kp18FGly (open squares) in the presence of DT. Reaction mixtures contained 4 μM anSMEcpe, 1 mM SAM, 1 mM Kp18Ser, and 3 mM DT. The data are the averages of two independent trials, and error bars denote one standard deviation. Vmax/[ET] values for 5’-dA and Kp18FGly are 1.00 ± 0.029 min-1 and 0.85 ± 0.001 min-1, respectively. B) Time-dependent formation of 5′-dA (closed diamonds) and Kp18FGly (open squares) in the presence of the Flv/Flx/NADPH reducing system. Reaction mixtures contained 40 μM anSMEcpe, 1 mM SAM, 1 mM Kp18Ser, 50 μM Flv, 15 μM Flx, and 2 mM NADPH. The data are the averages of three independent trials, and error bars denote one standard deviation. Vmax/[ET] values for 5’-dA and Kp18FGly are 0.074 ± 0.009 min-1 and 0.073 ± 0.004 min-1, respectively.
The target Cys residue was also replaced with a SeCys residue, which has a number of properties that are similar to those of Cys. In addition, a substrate containing a SeCys residue would permit investigation of substrate coordination to an Fe/S cluster by selenium X-ray absorption spectroscopy (49-51). Figure S5 displays turnover of anSMEcpe in the presence of Kp18SeCys and the Flv/Flx/NADPH reducing system. The reaction is linear for the first 10 min, but becomes uncoupled at longer incubation times, which is different from that observed for substrates containing Cys or Ser at the target position. A fit to the first three time points affords Vmax/[ET] values of ~0.053 min-1 for both 5’-dA and Kp18FGly.
Fate of the second reducing equivalent upon abstraction of a H• by the 5’-dA•
All RS enzymes require the input of an electron to initiate reductive cleavage of SAM to a 5’-dA•, which is used most often to oxidize substrates by one electron via H• abstraction. In the reactions catalyzed by AtsB and anSMEcpe, the substrate is oxidized further by one electron, wherein the presumed radical intermediate transfers an electron to an undetermined acceptor. It has been suggested that the electron is returned to the RS [4Fe–4S] cluster after each turnover, implying that the introduction of one electron can prime the system for multiple turnovers as has been shown for the RS enzyme, DesII, in a reaction with a substrate analog (52). To address the fate of the remaining electron, Flv• was generated by treatment of 0.5 equiv of DT with 1.05 eq. of Flv and then added to a reaction mixture containing the following components after quantification of the Flv• concentration: anSMEcpe (100 μM), SAM (2 mM), and Kp18Cys (2 mM), and Flv• (204 μM). At designated times (1, 5, and 15 min), aliquots were removed and added to EPR tubes, which were subsequently immersed in cryogenic isopentane (~-130 °C) to quench the reaction by rapid freezing. Quantification of the change in Flv• concentration as a function of time was conducted by EPR at 77 K as described in Materials and Methods, while parallel aliquots were removed from the reaction to quantify product formation by LC/MS. As can be seen in Figure 7A, the concentration of Flv• is essentially unchanged throughout the 15 min incubation. By contrast, Figure 7B shows that greater than 200 μM product is formed (~2 turnovers) during the same time period, and that FGly formation (open squares) is tightly coupled to SAM cleavage (5’-dA, closed triangles). The open circles in Figure 7B correspond to the Flv• concentrations in Figure 7A; the slight change in concentration of Flv• during the 15 min period most likely derives from slight O2 contamination. If the sole role of Flv is to prime the reaction such that the emitted electron from the substrate radical intermediate is returned to the RS cluster to be used in a subsequent round of SAM cleavage, it would be expected that the concentration of Flv• should decrease by 50% (from 200 μM to 100 μM) within the first 3 min of the reaction, which corresponds to the time required for one full turnover. The observation that the concentration of Flv• does not change significantly over the course of multiple turnovers suggests that the ejected electron is ultimately returned to Flvox at the end of each turnover event. Consistent with this observation, parallel EPR spectra recorded at 13 K do not show evidence of a reduced [4Fe-4S] cluster (Figure S6), which would argue against recycling of the ejected electron by storing it internally on an Fe/S cluster. Whether reduction of Flvox occurs through a reduced RS [4Fe-4S] cluster intermediate or a reduced auxiliary cluster intermediate is not yet clear. Of note is the biphasic nature of the appearance of 5’-dA and Kp18FGly, indicating that a burst phase is followed by a steady-state phase. A fit of the data to an appropriate equation results in the following kinetic parameters: burst amplitude, 113 μM; kburst, 0.32 ± 0.078 min-1; kss, 0.059 ± 0.011 min-1. The burst phase, which corresponds to ~1.1 equiv of enzyme may arise from rate-limiting product release; however, we have not rigorously characterized this aspect of the reaction. A similar experiment carried out with AtsB (150 μM), SAM (1 mM), Kp18Ser (1 mM), and 75 μM Flv• showed essentially identical results, albeit with a smaller burst phase (burst amplitude, 10.6 μM; kburst, 2.0 min-1; kss, 0.015 min-1) (Figure S7).
Figure 7.
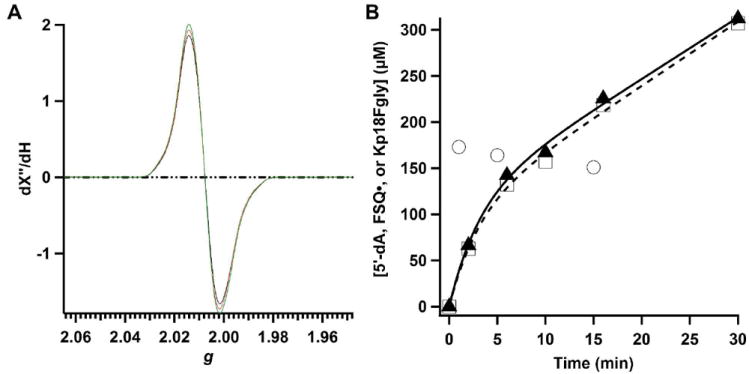
Correlation of spectral changes and product formation during anSMEcpe turnover. A) X-Band EPR (77 K) spectra of a reaction mixture containing 100 μM anSMEcpe, 2 mM SAM, 2 mM Kp18Cys and 204 μM Flv• at 1 min (red), 15 min (green), and 30 min (black). Spectra were recorded as described in Materials and Methods. B) Time-dependent quantification of Flv• (open circles), 5′-dA (closed triangles), and Kp18FGly (open squares). Lines (black line, 5’-dA; dashed line, Kp18FGly) represent fits to a burst phase followed by steady-state phase.
Stereochemistry of AtsB and anSMEcpe
Recent studies of Benjdia, et al. verified the hypothesis that the role of the 5’-dA• in RS dehydrogenases is to abstract a hydrogen atom from the carbon undergoing oxidation, which was initially demonstrated by Yokoyama et al for BtrN (3, 53). Using a peptide containing a target Cys residue isotopically substituted at C3 with deuterium, they provided evidence via mass spectrometry and NMR for transfer of deuterium to 5’-dA. However, the C3 hydrogens of cysteine are prochiral, and it would be expected that an enzyme would act stereoselectively in the removal of an H• from this position. Given that seryl residues are oxidized to FGly both by AtsB and anSMEcpe, we assessed whether threonyl and allo-threonyl residues, which are chiral at C3, are converted into the corresponding ketone product. As shown in Figure S8, the configuration of l-threonine at its two chiral carbons is 2S,3R, while the configuration of l-allo-threonine is 2S,3S. Therefore, conversion of substrate containing a threonyl residue at the target position would require abstraction of the proS hydrogen, while conversion of a substrate containing an allo-threonyl residue at the target position would require abstraction of the proR hydrogen. Figure 8 displays the results of activity determinations with Kp18Thr and Kp18alloThr, containing l-threonyl, and l-allo-threonyl residues, respectively, at the target position. As can be observed, (Figure 8A, closed squares) ~130 μM Kp18Thr is consumed in ten min in a reaction containing 100 μM anSMEcpe and DT as the requisite reductant, and MALDI-TOF analysis of the DPNH-derivatized product (m/z = 2195.4) is consistent with its assignment as the corresponding ketone derivative (Figure S9A). By contrast, only ~20 μM Kp18alloThr is consumed under identical conditions before the reaction levels off (Figure 8B, closed squares). This amount of substrate consumption could derive from l-Thr contamination at the target position, especially given that the reaction stops abruptly. MALDI-TOF analysis of the DPNH-derivatized product (m/z = 2195.4) verifies that there is a much smaller, but observable, amount of the corresponding ketone product (Figure S9b). AtsB was also able to use Kp18Thr as a substrate, but to a lesser extent, as judged by the relative intensities of the substrates with respect to the derivatized products (Figure S10).
Figure 8.
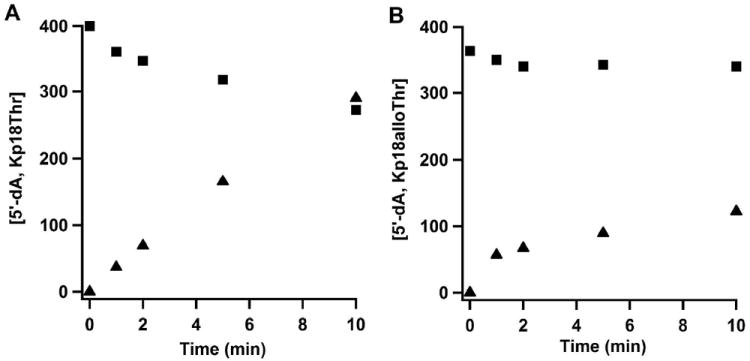
Turnover of WT RCN anSMEcpe with Kp18Thr or Kp18alloThr. A) Time-dependent formation of 5′-dA (closed triangles) and disappearance of Kp18Thr (closed squares). Reaction mixtures contained 100 μM anSMEcpe, 1 mM SAM, 0.5 mM Kp18Thr, and 3 mM DT. Vmax/[ET] values for 5’-dA formation and Kp18Thr disappearance are 0.29 min-1 and 0.11 min-1, respectively. B) Time-dependent formation of 5′-dA (closed triangles) and disappearance of Kp18alloThr (closed squares). Reaction mixtures contained 100 μM anSMEcpe, 1 mM SAM, 0.5 mM Kp18alloThr, and 3 mM DT. Vmax/[ET] values for 5’-dA formation and Kp18alloThr disappearance are 0.07 min-1 and 0.007 min-1, respectively.
Determination of cysteinyl residues that ligate the [4Fe–4S] clusters in anSMEs
AtsB contains 13 Cys residues, three of which lie in the canonical CxxxCxxC motif. Site-directed mutagenesis of the remaining ten Cys residues was conducted to determine which might coordinate the auxiliary clusters. Seven of the Cys→Ala variants (C270A, C276A, C331A, C334A, C340A, C344A, and C357A) were produced in a completely insoluble form and not studied further. Two of the variants, C127A and C245A, were freely soluble and behaved like WT AtsB in both purification and activity. The UV-vis spectra for both of these AI variants are displayed in Figure S11 (solid and dashed lines, respectively), and reveal spectral envelopes that are similar to that of the WT protein. Moreover, their A395/A280 values of 0.38 are also similar to that of the AI WT protein (0.42) (2). The AI C127A variant contained 9.8 ± 0.1 and 9.6 ± 0.5 iron and sulfide ions, respectively, per polypeptide, while the AI C245A variant contained 12.0 ± 1.1 and 15.0 ± 0.3 iron and sulfide ions, respectively, per polypeptide. Activity determinations on both of these AI proteins were conducted using the Flv/Flx/NADPH reducing system, yielding Vmax/[ET] values of 0.061 and 0.052 min-1, respectively, for the C127A and C245A variants, similar to the Vmax/[ET] value for the WT protein under similar conditions (0.040 min-1).
The C291A variant could be isolated, but was sparingly soluble and poorly behaved. The UV-vis spectrum for this variant is shown in Figure S12, and reveals a spectral envelope that is similar to that of the WT protein. Its A405/A280 value of 0.39 would indicate high cluster incorporation; however, it contained only 6.7 ± 0.1 irons and 5.6 ± 0.6 sulfides per polypeptide. Efforts to reconstitute this protein resulted in its precipitation from solution. The activity determination of this variant was not above the limit of detection of the assay when the Kp18Ser peptide was used as substrate, suggesting that this Cys residue is either structurally or functionally important in the reaction of anSMEs. When activity determinations were conducted in the presence of DT and the Kp18Cys peptide approximately 10 μM 5’-dA and 2 μM FGly product were observed after 60 min of incubation with 70 μM protein.
The equivalent Cys→Ala variant was constructed for anSMEcpe (C276A) and found, in contrast to AtsB C291A, to be stable and readily soluble. The UV-vis spectrum of this protein shows an almost identical spectral envelope as WT anSMEcpe (Figure S13). In a similar fashion as AtsB C291A, this protein was almost completely inactive in production of Kp18FGly. After a 30-min incubation in the presence of 200 μM anSMEcpe C276A, approximately 6 μM FGly was produced (Figure 9). On the other hand, SAM reductase activity was higher than that of the AtsB C291A variant, albeit less than one-half an equivalent of 5’-dA was produced after 30 min.
Figure 9.
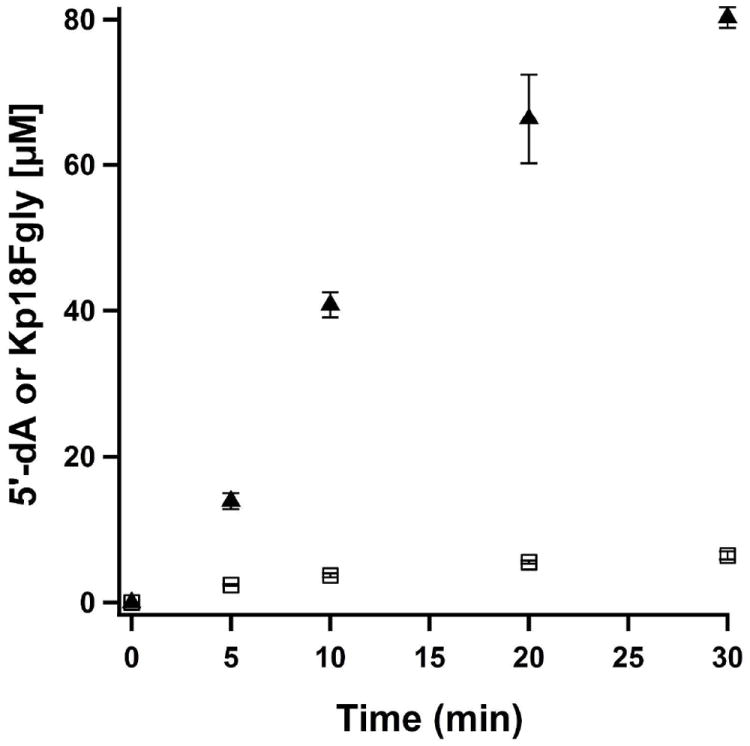
Time-dependent formation of 5′-dA (closed triangles) and Kp18FGly (open squares) with RCN anSMEcpe C276A and Kp18Cys. Reaction mixtures contained 200 μM anSMEcpe C276A, 1 mM SAM, 1 mM Kp18Cys, and 3 mM DT. The data are the averages of two independent trials, and error bars denote one standard deviation. Vmax/[ET] values for 5’-dA and Kp18FGly are 0.14 ± 0.001 min-1 and 0.001 ± 0.0001 min-1, respectively.
DISCUSSION
anSMEcpe shares 48% sequence similarity with Kp AtsB; however, it is a Cys-type anSME, and therefore its in vivo role is to catalyze the oxidation of a target Cys residue to FGly. Its mechanism of catalysis is predicted to be identical to that proposed for AtsB, except that the presumed thioaldehyde product is then hydrolyzed to the aldehyde with elimination of H2S. anSMEcpe has been characterized previously using a number of spectroscopic techniques, including UV-vis, resonance Raman, and EPR spectroscopies (1). Although the previous studies were consistent with the presence of Fe/S clusters on the enzyme, cluster content was not rigorously determined. The protein studied by Benjdia, et al. contained an N-terminal hexahistidine tag, and was overproduced largely in inclusion bodies, yielding ~5 mg of soluble protein from 12 L of growth medium. In contrast, it has been our strategy to include an accessory plasmid that harbors genes that encode proteins that are involved in Fe/S cluster biosynthesis in A. vinelandii, the homologs of which are known to encode proteins involved in Fe/S cluster biosynthesis in other organisms (34). This strategy allowed purification to near-homogeneity of >250 mg of anSMEcpe containing a C-terminal hexahistidine tag from 16 L of minimal medium. This yield is considerably higher than that reported by Benjida, et al. as well as that for AtsB (30 mg from 16 L of medium). Indeed, we find that WT anSMEcpe is a much better behaved than WT AtsB, and therefore better suited for detailed mechanistic and structural investigations.
In the work presented herein, Mössbauer spectroscopy was used in concert with analytical determinations of 57Fe content to establish not only the configuration of Fe/S clusters associated with anSMEcpe, but also the stoichiometry of each particular cluster type per anSMEcpe polypeptide. When anSMEcpe is overproduced along with proteins encoded by plasmid pDB1282, the AI enzyme contains 2.3 [4Fe–4S] clusters (95% of all 57Fe), with ~3% of all 57Fe occurring as [2Fe–2S] clusters and ~2% occurring as an undefined cluster type. Upon reconstitution of AI anSMEcpe, the protein contains ~2.7 [4Fe–4S] clusters (75% of all 57Fe), with the remaining 25% of all 57Fe existing as unspecifically bound iron. Analysis of a triple variant of anSMEcpe, in which the Cys ligands to the RS [4Fe–4S] clusters were changed to Ala residues—a state that should not allow cluster ligation—showed that the AI protein contained 0.6 [4Fe–4S] clusters and 0.3 [2Fe–2S] clusters, while the RCN triple variant contained 1.5 [4Fe–4S]2+ clusters. Our model of three [4Fe–4S] clusters per polypeptide for anSMEcpe would predict that the triple variant would harbor two [4Fe–4S] clusters. In contrast to AtsB, in which the analogous triple variant is more soluble than the WT protein, we find that the anSMEcpe triple variant is less stable and less soluble than its corresponding WT protein. We believe that the increased heterogeneity in the AI triple variant and the significantly reduced cluster content derives from the instability of this protein.
Previous site-directed mutagenesis studies on AtsB revealed, as expected, that one of the clusters is ligated by C35, C39, and C42, which are found in the canonical CxxxCxxC RS signature sequence (2). However, the large number of Cys residues (13) in the primary structure of AtsB did not readily allow determination of the ligands to the two remaining clusters, or determination of which Cys residues were partnered in the ligation of any given cluster. Given the presence of two auxiliary Fe/S clusters, our original working hypothesis was that one would be the immediate acceptor of an electron from the substrate-radical intermediate generated via H• abstraction by the 5’-dA•, and that the other cluster would act as a conduit through which the ejected electron would be transferred to an acceptor, presumed to be Flvox. This hypothesis suggested the possibility of two phenotypes for Cys→Ala variants of the cysteines coordinating the two auxiliary clusters: (1) variants that are completely inactive due to an inability to transfer an electron from the substrate radical intermediate, and (2) variants that are inactive with Flv but active with DT, presuming that oxidized DT (i.e. bisulfite) can accept an electron from the reduced auxiliary cluster (54). In an effort to determine the ligands that ligate the auxiliary clusters and perhaps provide evidence for the role(s) of these clusters, we created single Cys→Ala substitutions at the ten cysteines outside of the CxxxCxxC motif, with the intent of purifying and characterizing the corresponding proteins. We found that the behavior of the resulting variants could be grouped into three categories: those that afforded proteins that behaved essentially like WT AtsB (C127A and C245A); those that afforded completely insoluble proteins (C270A, C276A, C331A, C334A, C340A, C344A, and C357A); and one that afforded a sparingly soluble protein exhibiting measureable, but very poor, activity (C291A). Based on these observations, we feel confident that C127 and C245 play no major role in catalysis, while C270, C276, C331, C334, C340, C344, and C357 contribute ligands to the two auxiliary [4Fe-4S] clusters. The role of C291 is more difficult to assign because of its intermediate behavior. The significantly reduced activity of the C291A variant might suggest a role such as the general base to which the substrate proton is donated during the dehydrogenation reaction; however, its significantly reduced solubility might suggest that it serves as a ligand to one of the auxiliary [4Fe-4S] clusters, implying that both of these clusters are fully ligated. We note that C276 in anSMEcpe, the equivalent residue to C291 in AtsB, behaved similarly. Consistent with two fully ligated auxiliary clusters, our efforts to establish substrate ligation to an auxiliary cluster using selenium X-ray absorption spectroscopy and Kp18SeCys were unsuccessful (unpublished results). It should be mentioned that we observed a similar outcome with variants of BtrN, a RS dehydrogenase that has only one auxiliary cluster (31). This enzyme contains eight Cys residues, three of which (C16, C20, and C23) coordinate the RS cluster, and one of which behaves like the WT protein. Three additional Cys residues, which when substituted with Ala, were produced completely as insoluble aggregates, suggesting that they coordinate the auxiliary [4Fe–4S] cluster. One Cys residue, C235, behaved similarly to C291 of AtsB and C276 of anSMEcpe. Although the C235A variant of BtrN could be purified, it was poorly soluble, and exhibited a Vmax/[ET] that was less than 10% of that of the WT enzyme. If indeed both auxiliary clusters in AtsB are fully ligated by Cys residues, it is highly likely that the two auxiliary clusters in anSMEcpe and the one auxiliary cluster in BtrN are similarly ligated.
Our current studies do not allow us to deduce the role(s) of the auxiliary clusters in RS dehydrogenases. In fact, it is conceivable that they simply maintain the structural integrity of the protein. Interestingly, a subclass of the glycyl radical enzyme (GRE) activases, proteins that catalyze formation of glycyl radical cofactors on cognate enzymes, are also believed to harbor three [4Fe–4S] clusters, although the stoichiometry has not been rigorously determined (7, 55). It has been speculated that the two auxiliary clusters in the GRE activases might act as a conduit for reduction of the RS Fe/S cluster (56). This role is unlikely in AtsB and anSMEcpe, however, given that these enzymes catalyze their reactions in the presence of flavodoxin with rate constants that are equal to or better than those exhibited by many other RS enzymes that do not contain auxiliary clusters but are also activated by flavodoxin. Our studies herein, however, suggest that after each turnover, the ejected electron is returned ultimately to Flvox, given that the concentration of Flv• does not change significantly during catalysis. If the ejected electron were returned to the RS cluster as its final destination, we would expect that (i) the reaction should exhibit a lag phase (corresponding to slow reduction of the RS [4Fe–4S] cluster) followed by a faster phase (return of the ejected electron to the RS [4Fe–4S] for use in subsequent rounds of SAM cleavage) that approaches the steady-state rate of the reaction in the presence of dithionite; and (ii) the concentration of the Flv• should have been reduced by the concentration of enzyme in the assay (50%), given the burst of product corresponding to one equiv of enzyme, which suggests that all active sites are functional. Whether the electron is returned to Flvox via the auxiliary clusters or the RS cluster is currently unknown.
The RS enzyme, DesII, catalyzes a key step in the biosynthesis of d-desosamine, a deoxysugar found in a number of macrolide antibiotics. This reaction is the conversion of thymidine diphosphate (TDP)-4-amino-6-deoxy-d-glucose to TDP-3-keto-4,6-dideoxy-d-glucose, which is somewhat similar to the reaction catalyzed by the coenzyme B12-dependent enzyme, ethanolamine ammonia lyase (57). This reaction, with respect to the substrate, is redox-neutral; however, DesII catalyzes stoichiometric production of 5’-dA with respect to product rather than regeneration of SAM after each turnover, therefore requiring the input of two electrons during turnover (52). Interestingly, DesII will also catalyze a two-electron oxidation of the nonphysiological substrate, TDP-d-quinovose (4-hydroxy-6-deoxy-d-glucose), converting it to TDP-3-keto-6-deoxy-d-glucose. In this instance, although the ratio of 5’-dA to product remains 1:1, the reaction does not require external reducing equivalents once primed, suggesting that the ejected electron is returned to the RS [4Fe–4S] — the sole Fe/S cluster on the protein — after each turnover (52).
anSMEcpe and AtsB each harbor a CxxCxxxxxCxxxC motif, which our studies herein indicate contains cysteines that contribute ligands to auxiliary [4Fe–4S] clusters. Interestingly, this motif is highly conserved in a newly designated subclass of RS enzymes, TIGR04085, which are those that contain SPASM domains. The acronym SPASM derives from the finding that the founding members of this family catalyze key steps in the maturation of subtilosin, PQQ, anaerobic sulfatases, and mycofactin. In addition, the conserved cysteine-containing motif that each member shares is always C-terminal to the RS cysteine-containing motif (58, 59). Only in the anSMEs has the cluster stoichiometry been rigorously established in this subclass of RS enzymes (2), and the roles of the auxiliary cluster(s) have not been delineated in any SPASM domain-containing protein. Nevertheless, these enzymes share the characteristic of catalyzing reactions on protein or peptide substrates.
Our results with peptide substrates containing threonyl residues at the target position suggest the following working hypothesis for catalysis by AtsB and anSMEcpe. After reductive cleavage of SAM, the 5’-dA• abstracts the 3-proS H• of the substrate, yielding a substrate radical. Subsequent to electron transfer to an auxiliary cluster and loss of a substrate proton — in an order that has not been established — the ejected electron is transferred to Flvox via an auxiliary cluster. These studies herein, and future studies, will provide much needed insight into a growing class of RS enzymes — including those containing SPASM domains — that use multiple Fe/S clusters to catalyze their reactions (7).
Supplementary Material
Acknowledgments
We thank Professor Carolyn Bertozzi and Dr. Jason Rush for authentic formylglycine.
Footnotes
This work was supported by NIH Grants GM-63847 and GM-103268 (S.J.B.), the Dreyfus Foundation (Teacher Scholar Award to C.K.), and the Beckman Foundation (Young Investigator Award to C.K.). A grant from the TEAS foundation is acknowledged for support of undergraduate summer research to J.H.A.
Abbreviations: aa, amino acid; AI, as-isolated; amino-DOI, amino-2-deoxy-scyllo-inosose; anSME, anaerobic sulfatase maturating enzyme; anSMEcpe, anaerobic sulfatase maturating enzyme from Clostridium perfringens; AT, allo-threonine; AtsB, anaerobic sulfatase maturating enzyme from Klebsiella pneumoniae; BS, biotin synthase; BSA, bovine serum albumin; 5’-dA, 5’-deoxyadenosine; 5’-dA•, 5’-deoxyadenosyl 5’-radical; DOIA, 2-deoxy-scyllo-inosamine; DOS, 2-deoxystreptamine; DT, dithionite; DTT, dithiothreitol; EDTA; ethylenediaminetetraacetic acid; EPR, electron paramagnetic resonance; Fe/S, iron–sulfur; FGly, formylglycine; Flv, flavodoxin; Flx, flavodoxin reductase; HEPES, N-(2-hydroxyethyl)piperazine-N’-(2-ethanesulfonic acid); HPLC, high performance liquid chromatography; MALDI–TOF MS, matrix assisted laser desorption ionization time-of-flight mass spectrometry; IMAC, immobilized metal affinity chromatography; IPTG, isopropyl-β-D-thiogalactopyranoside; IS, internal standard; LC/MS, HPLC with detection by QQQ mass spectrometry; LS, lipoyl synthase; MRM, multiple reaction monitoring; MW, molecular weight; Ni-NTA, nickel nitrilotriacetic acid; PCR, polymerase chain reaction; PFL-AE, pyruvate formate–lyase activating enzyme; RCN, reconstituted; RS, radical SAM; SAM, S-adenosyl-l-methionine; SDS-PAGE, sodium dodecylsulfate-polyacrylamide gel electrophoresis; SeC, selenocysteine; SeCys, selenocysteine; SI, supplementary information; SME, sulfatase maturating enzyme; TFA, trifluoroacetic acid; UV-vis, UV-visible; Vo, void volume; Ve, elution volume; WT, wild-type
SUPPORTING INFORMATION AVAILABLE
Tables S1-S3, and Figures S1-S14. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Benjdia A, Subramanian S, Leprince J, Vaudry H, Johnson MK, Berteau O. Anaerobic sulfatase-maturating enzymes – first dual substrate radical S-adenosylmethionine enzymes. J Biol Chem. 2008;283:17815–17826. doi: 10.1074/jbc.M710074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grove TL, Lee KH, St Clair J, Krebs C, Booker SJ. In vitro characterization of AtsB, a radical SAM formylglycine-generating enzyme that contains three [4Fe-4S] clusters. Biochemistry. 2008;47:7523–7538. doi: 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yokoyama K, Numakura M, Kudo F, Ohmori D, Eguchi T. Characterization and mechanistic study of a radical SAM dehydrogenase in the biosynthesis of butirosin. J Am Chem Soc. 2007;129:15147–15155. doi: 10.1021/ja072481t. [DOI] [PubMed] [Google Scholar]
- 4.Benjdia A, Leprince J, Guillot A, Vaudry H, Rabot S, Berteau O. Anaerobic sulfatase-maturating enzymes: radical SAM enzymes able to catalyze in vitro sulfatase post-translational modification. J Am Chem Soc. 2007;129:3462. doi: 10.1021/ja067175e. [DOI] [PubMed] [Google Scholar]
- 5.Benjdia A, Subramanian S, Leprince J, Vaudry H, Johnson MK, Berteau O. Anaerobic sulfatase-maturating enzyme--a mechanistic link with glycyl radical-activating enzymes? FEBS J. 2010;277:1906–1920. doi: 10.1111/j.1742-4658.2010.07613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Booker SJ, Grove TL. Mechanistic and functional versatility of radical SAM enzymes. F1000 Biol Rep. 2010;2:52. doi: 10.3410/B2-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lanz ND, Booker SJ. Identification and function of auxiliary iron-sulfur clusters in radical SAM enzymes. Biochim Biophys Acta. 2012;1824:1196–1212. doi: 10.1016/j.bbapap.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Yokoyama K, Ohmori D, Kudo F, Eguchi T. Mechanistic study on the reaction of a radical SAM dehydrogenase BtrN by electron paramagnetic resonance spectroscopy. Biochemistry. 2008;47:8950–8960. doi: 10.1021/bi800509x. [DOI] [PubMed] [Google Scholar]
- 9.Walsby CJ, Ortillo D, Yang J, Nnyepi MR, Broderick WE, Hoffman BM, Broderick JB. Spectroscopic approaches to elucidating novel iron-sulfur chemistry in the “radical-SAM” protein superfamily. Inorg Chem. 2005;44:727–741. doi: 10.1021/ic0484811. [DOI] [PubMed] [Google Scholar]
- 10.Dowling DP, Vey JL, Croft AK, Drennan CL. Structural diversity in the AdoMet radical enzyme superfamily. Biochim Biophys Acta. 2012;1824:1178–1195. doi: 10.1016/j.bbapap.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Booker SJ. Anaerobic functionalization of unactivated C–H bonds. Curr Opin Chem Biol. 2009;13:58–73. doi: 10.1016/j.cbpa.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontecave M, Mulliez E, Ollagnier-de Choudens S. Adenosylmethionine as a source of 5’-deoxyadenosyl radicals. Curr Opin Chem Biol. 2001;5:506–511. doi: 10.1016/s1367-5931(00)00237-4. [DOI] [PubMed] [Google Scholar]
- 13.Frey PA, Magnusson OT. S-Adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin? Chem Rev. 2003;103:2129–2148. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 14.Challand MR, Driesener RC, Roach PL. Radical S-adenosylmethionine enzymes: mechanism, control and function. Nat Prod Rep. 2011;28:1696–1721. doi: 10.1039/c1np00036e. [DOI] [PubMed] [Google Scholar]
- 15.Hiscox MJ, Driesener RC, Roach PL. Enzyme catalyzed formation of radicals from S-adenosylmethionine and inhibition of enzyme activity by the cleavage products. Biochim Biophys Acta. 2012;1824:1165–1177. doi: 10.1016/j.bbapap.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 16.Berteau O, Guillot A, Benjdia A, Rabot S. A new type of bacterial sulfatase reveals a novel maturation pathway in prokaryotes. J Biol Chem. 2006;281:22464–22470. doi: 10.1074/jbc.M602504200. [DOI] [PubMed] [Google Scholar]
- 17.Fang Q, Peng J, Dierks T. Post-translational formylglycine modification of bacterial sulfatases by the radical S-adenosylmethionine protein AtsB. J Biol Chem. 2004;279:14570–14578. doi: 10.1074/jbc.M313855200. [DOI] [PubMed] [Google Scholar]
- 18.Hanson SR, Best MD, Wong C-H. Sulfatases: structure, mechanism biological activity, inhibition, and synthetic utility. Angew Chem Int Ed. 2004;43:5736–5763. doi: 10.1002/anie.200300632. [DOI] [PubMed] [Google Scholar]
- 19.Parenti G, Meroni G, Ballabio A. The sulfatase gene family. Curr Opin Genet Dev. 1997;7:386–391. doi: 10.1016/s0959-437x(97)80153-0. [DOI] [PubMed] [Google Scholar]
- 20.Schirmer A, Kolter R. Computational analysis of bacterial sulfatases and their modifying enzymes. Chem Biol. 1998;5:R181–R186. doi: 10.1016/s1074-5521(98)90154-5. [DOI] [PubMed] [Google Scholar]
- 21.von Figura K, Schmidt B, Selmer T, Dierks T. A novel protein modification generating an aldehyde group in sulfatases: its role in catalysis and disease. Bioessays. 1998;20:505–510. doi: 10.1002/(SICI)1521-1878(199806)20:6<505::AID-BIES9>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 22.Boltes I, Czapinska H, Kahnert A, von Bülow R, Dierks T, Schmidt B, von Figura K, Kertesz MA, Usón I. 1.3 Å structure of arylsulfatase from Pseudomonas aeruginosa establishes the catalytic mechanism of sulfate ester cleavage in the sulfatase family. Structure. 2001;9:483–491. doi: 10.1016/s0969-2126(01)00609-8. [DOI] [PubMed] [Google Scholar]
- 23.Lukatela G, Krauss N, Theis K, Selmer T, Gieselmann V, von Figura K, Saenger W. Crystal structure of human arylsulfatase A: the aldehyde function and the metal ion at the active site suggest a novel mechanism for sulfate ester hydrolysis. Biochemistry. 1998;37:3654–3664. doi: 10.1021/bi9714924. [DOI] [PubMed] [Google Scholar]
- 24.Recksiek M, Selmer T, Dierks T, Schmidt B, von Figura K. Sulfatases, trapping of the sulfated enzyme intermediate by substituting the active site formylglycine. J Biol Chem. 1998;273:6096–6103. doi: 10.1074/jbc.273.11.6096. [DOI] [PubMed] [Google Scholar]
- 25.Cosma MP, Pepe S, Annunziata I, Newbold RF, Grompe M, Parenti G, Ballabio A. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113:445–456. doi: 10.1016/s0092-8674(03)00348-9. [DOI] [PubMed] [Google Scholar]
- 26.Dierks T, Dickmanns A, Preusser-Kunze A, Schmidt B, Mariappan M, von Figura K, Ficner R, Rudolph MG. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell. 2005;121:541–552. doi: 10.1016/j.cell.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Dierks T, Schmidt B, Borissenko LV, Peng J, Preusser A, Mariappan M, von Figura K. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human Cα-formylglycine generating enzyme. Cell. 2003;113:435–444. doi: 10.1016/s0092-8674(03)00347-7. [DOI] [PubMed] [Google Scholar]
- 28.Carlson BL, Ballister ER, Skordalakes E, King DS, Breidenbach MA, Gilmore SA, Berger JM, Bertozzi CR. Function and structure of a prokaryotic formylglycine-generating enzyme. J Biol Chem. 2008;283:20117–20125. doi: 10.1074/jbc.M800217200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt B, Selmer T, Ingendoh A, von Figura K. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell. 1995;82:271–278. doi: 10.1016/0092-8674(95)90314-3. [DOI] [PubMed] [Google Scholar]
- 30.Benjdia A, Deho G, Rabot S, Berteau O. First evidences for a third sulfatase maturation system in prokaryotes from E-coli aslB and ydeM deletion mutants. FEBS Lett. 2007;581:1009–1014. doi: 10.1016/j.febslet.2007.01.076. [DOI] [PubMed] [Google Scholar]
- 31.Grove TL, Ahlum JH, Sharma P, Krebs C, Booker SJ. A consensus mechanism for radical SAM-dependent dehydrogenation? BtrN contains two [4Fe–4S] clusters. Biochemistry. 2010;49:3783–3785. doi: 10.1021/bi9022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwig DF, Booker SJ. Insight into the polar reactivity of the onium chalcogen analogues of S-adenosyl-L-methionine. Biochemistry. 2004;43:13496–13509. doi: 10.1021/bi048693+. [DOI] [PubMed] [Google Scholar]
- 33.Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu-Gogonea C, Souder MG, Tu L, Booker SJ. Lipoyl synthase requires two equivalents of S-adenosyl-L-methionine to synthesize one equivalent of lipoic acid. Biochemistry. 2004;43:6378–6386. doi: 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- 34.Lanz ND, Grove TL, Gogonea CB, Lee KH, Krebs C, Booker SJ. RlmN and AtsB as models for the overproduction and characterization of radical SAM proteins. Methods Enzymol. 2012;516:125–152. doi: 10.1016/B978-0-12-394291-3.00030-7. [DOI] [PubMed] [Google Scholar]
- 35.Beinert H. Micro methods for the quantitative determination of iron and copper in biological material. Methods Enzymol. 1978;54:435–445. doi: 10.1016/s0076-6879(78)54027-5. [DOI] [PubMed] [Google Scholar]
- 36.Beinert H. Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy MC, Kent TA, Emptage M, Merkle H, Beinert H, Münck E. Evidence for the formation of a linear [3Fe-4S] cluster in partially unfolded aconitase. J Biol Chem. 1984;259:14463–14471. [PubMed] [Google Scholar]
- 38.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye-binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 39.Cicchillo RM, Lee KH, Baleanu-Gogonea C, Nesbitt NM, Krebs C, Booker SJ. Escherichia coli lipoyl synthase binds two distinct [4Fe-4S] clusters per polypeptide. Biochemistry. 2004;43:11770–11781. doi: 10.1021/bi0488505. [DOI] [PubMed] [Google Scholar]
- 40.Cicchillo RM, Baker MA, Schnitzer EJ, Newman EB, Krebs C, Booker SJ. Escherichia coli L-serine deaminase requires a [4Fe–4S] cluster in catalysis. J Biol Chem. 2004;279:32418–32425. doi: 10.1074/jbc.M404381200. [DOI] [PubMed] [Google Scholar]
- 41.Jenkins CM, Waterman MR. NADPH–flavodoxin redutase and flavodoxin from Escherichia coli: Characteristics as a soluble microsomal P450 reductase. Biochemistry. 1998;37:6106–6113. doi: 10.1021/bi973076p. [DOI] [PubMed] [Google Scholar]
- 42.The_UniProt_Consortium. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2007;35:D193–D197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson DC, Unciuleac MC, Dean DR. Controlled expression and functional analysis of iron-sulfur cluster biosynthetic components within Azotobacter vinelandii. J Bacteriol. 2006;188:7551–7561. doi: 10.1128/JB.00596-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holm RH, Ibers JA. Synthetic analogues of the active sites of iron–sulfur proteins. In: Lovenberg W, editor. Iron-Sulfur Proteins. Academic Press; New York: 1977. [Google Scholar]
- 45.Cosper MM, Jameson GNL, Hernández HL, Krebs C, Huynh BH, Johnson MK. Characterization of the cofactor composition of Escherichia coli biotin synthase. Biochemistry. 2004;43:2007–2021. doi: 10.1021/bi0356653. [DOI] [PubMed] [Google Scholar]
- 46.Krebs C, Henshaw TF, Cheek J, Huynh BH, Broderick JB. Conversion of 3Fe–4S to 4Fe–4S clusters in native pyruvate formate-lyase activating enzyme: Mössbauer characterization and implications for mechanism. J Am Chem Soc. 2000;122:12497–12506. [Google Scholar]
- 47.Ghosh D. Human sulfatases: a structural perspective to catalysis. Cell Mol Life Sci. 2007;64:2013–2022. doi: 10.1007/s00018-007-7175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dierks T, Lecca MR, Schlotterhose P, Schmidt B, von Figura K. Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J. 1999;18:2084–2091. doi: 10.1093/emboj/18.8.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cosper NJ, Booker SJ, Ruzicka F, Frey PA, Scott RA. Direct FeS cluster involvement in generation of a radical in lysine 2,3-aminomutase. Biochemistry. 2000;39:15668–15673. doi: 10.1021/bi0022184. [DOI] [PubMed] [Google Scholar]
- 50.Eidsness MA, Scott RA, Prickril BC, DerVartanian DV, Legall J, Moura I, Moura JJG, Peck HDJ. Evidence for selenocysteine coordination to the active site nickel in the [NiFeSe]hydrogenases from Desulfovibrio baculatus. Proc Natl Acad Sci U S A. 1989;86:147–151. doi: 10.1073/pnas.86.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shokes JE, Duin EC, Bauer C, Jaun B, H R, Koch J, Scott RA. Direct interaction of coenzyme M with the active-site Fe–S cluster of heterodisulfide reductase. FEBS Lett. 2005;579:1741–1744. doi: 10.1016/j.febslet.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 52.Ruszczycky MW, Choi SH, Liu HW. Stoichiometry of the redox neutral deamination and oxidative dehydrogenation reactions catalyzed by the radical SAM enzyme DesII. J Am Chem Soc. 2010;132:2359–2369. doi: 10.1021/ja909451a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benjdia A, Leprince J, Sandström C, Vaudry H, Berteau O. Mechanistic investigations of anaerobic sulfatase-maturating enzyme: Direct Cβ H-atom abstraction catalyzed by a radical AdoMet enzyme. J Am Chem Soc. 2009;131:8348–8349. doi: 10.1021/ja901571p. [DOI] [PubMed] [Google Scholar]
- 54.Mayhew SG. The redox potential of dithionite and SO2− from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur J Biochem. 1978;85:535–547. doi: 10.1111/j.1432-1033.1978.tb12269.x. [DOI] [PubMed] [Google Scholar]
- 55.Demick JM, Lanzilotta WN. Radical SAM activation of the B12-independent glycerol dehydratase results in formation of 5’-deoxy-5’-(methylthio)adenosine and not 5’-deoxyadenosine. Biochemistry. 2011;50:440–442. doi: 10.1021/bi101255e. [DOI] [PubMed] [Google Scholar]
- 56.Yu L, Blaser M, Andrei PI, Pierik AJ, Selmer T. 4-Hydroxyphenylacetate decarboxylases: Properties of a novel subclass of glycyl radical enzymes systems. Biochemistry. 2006;45:9584–9592. doi: 10.1021/bi060840b. [DOI] [PubMed] [Google Scholar]
- 57.Banerjee R, Ragsdale SW. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 58.Haft DH, Basu MK. Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J Bacteriol. 2011;193:2745–2755. doi: 10.1128/JB.00040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haft DH, Selengut JD, Richter RA, Harkins D, Basu MK, Beck E. TIGRFAMS and genome properties in 2013. Nuc Acids Res. 2013;41:D387–D395. doi: 10.1093/nar/gks1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.