

Abstract
Head and Neck Squamous Cell Carcinoma (HNSCC) encompasses malignancies that arise in the mucosa of the upper aerodigestive tract. Recent high throughput DNA sequencing revealed HNSCC genes mutations that contribute to several cancer cell characteristics, including dysregulation of cell proliferation and death, intracellular proinflammatory signaling, and autophagy. The PYRIN-domain containing NLR (Nucleotide-binding domain, Leucine rich Repeats – containing) proteins have recently emerged as pivotal modulators of cell death, autophagy, inflammation, and metabolism. Their close physiologic association with cancer development prompted us to determine whether mutations within the NLRP (PYRIN-containing NLR) gene family were associated with HNSCC genome instability and their clinicopathologic correlations. Catastrophic mutational events underlie cancer cell genome instability and mark a point-of-no-return in cancer cell development and generation of heterogeneity. The mutation profiles of 62 patients with primary conventional type HNSCC excluding other histologic variants were analyzed. Associations were tested using Fisher's Exact test or Mann-Whitney U test. Mutations in NLRP were associated with elevated genome instability as characterized by higher mutation rates. Clinically, NLRP mutations were more frequently found in HNSCC arising in the floor of mouth (50.0%) in comparison with HNSCC at other head and neck locations (14.8%). These mutations were clustered at the leucine rich repeats region of NLRP proteins, and affected NLRP genes were mostly localized at chromosomes 11p15.4 and 19q13.42-19q13.43. Twenty novel NLRP mutations were identified in HNSCC, and mutations in this group of genes were correlated with increased cancer cell genome mutation rates, and such features could be a potential molecular biomarker of HNSCC genome instability.
Introduction
Recent technological advances in whole exome sequencing at much greater depths provide us with an unparalleled opportunity to interrogate the human cancer genome for mutational profiles. Studies on the evolutionary history of cancer genomes reveal that catastrophic mutational events (also referred to as “kataegis”, a Greek word meaning thunderstorm or shower), which are characterized by rapid accumulation of point mutations at clustered regions, may mark a “point-of-no-return” during cancer development by giving rise to subclones of cancer cells [1], [2]. Genome instability exemplified by kataegis and chromothripsis represents a hallmark of cancer [3].
Head and Neck Squamous Cell Carcinoma (HNSCC) encompasses malignancies that arise in the mucosa of the upper aerodigestive tract, accounting for 300,000 annual deaths and ranking 6th among the most common human cancers [4]. Due to the characteristic anatomic location, the upper aerodigestive pathway, especially the oral cavity, is constantly exposed to environmental factors, many of which possess potent carcinogenic capacity. Indeed, the geographic variation of HNSCC incidence corresponds well with the exposure to risk factors including tobacco use and human papilloma virus (HPV) infection [5]. The unique environmental etiologic factors in HNSCC development suggest the functional significance of genes involved in host-environment and/or host-pathogen interactions. Novel mutations in genes regulating squamous epithelial differentiation are unveiled in recent endeavors to map HNSCC mutational landscape [6], [7]. However, the genetic signatures that may reflect the overall genome instability in HNSCC are yet to be determined. This study aims to explore the significance of mutations in a group of genes modulating host-environment interactions and their clinicopathologic correlations.
Emerging evidence place a novel gene family at the forefront of host-environment interactions. NLR (nucleotide-binding, lots of leucine-rich repeats containing) gene family (initially coined as CATERPILLER, also known as NOD and NALP) is characterized by a central nucleotide-binding domain, a C-terminal leucine rich repeats (LRR) domain, and an N-terminal effector domain. The N-terminus could be CARD, PYRIN, BIR, AD or X domain, which shows certain homology with CARD or PYRIN yet cannot be categorized into either [8]. By engaging with the formation of distinct protein complexes, NLR proteins play central roles in modulating host responses to both PAMPs (pathogen-associated molecular patterns) and DAMPs (damage-associated molecular patterns) [9], [10].
A variety of NLR-mediated signaling pathways convey meticulous regulatory mechanisms in cell death [11], inflammation [9], [10], [12], autophagy [13]–[16], and more recently carcinogenesis [17]–[24]. Among the NLR family, a subgroup of NLRs harboring an N-terminal PYRIN domain have been implicated in malignancies arising in the lower digestive tract. Indeed, genetic deficiency in Nlrp3 results in increased colitis-associated colon cancer [19], [25]. NLRP6 controls colonic microbial ecology and colorectal epithelial cells renewal; and its deficiency results in increased colitis and colorectal cancer [21]–[23]. NLRP7 variants are involved in post-molar choriocarcinoma, and a germline mutation of NLRP2 is associated with a proliferation disorder known as Beckwith-Wiedemann syndrome [26], [27].
A significant portion of HNSCC is comprised of cancer of the oral cavity, which is not only directly exposed to a variety of PAMPs and DAMPs, but also constantly inhabited by a microbiota composed of more than 700 bacterial species [28]. Alteration of oral microbiota as seen in those with chronic adult periodontitis or poor oral hygiene has been correlated with the development of several types of cancer [29], [30]. Given the significance of NLR proteins in host inflammatory responses, autophagy, normal epithelial renewal, and emerging roles in maintaining microbiota homeostasis [13], [18], [21]–[23], their deficiency may change host responses to environmental insults and adjuvant therapeutic agents. However, their specific functions in modulating cancer cell development require further investigations. In this study, we characterize the mutational profiles of 10 NLRP genes in 62 primary conventional type HNSCC tumors, and investigate the significance of these mutations in overall cancer genome instability and their correlations with the clinicopathologic characteristics of HNSCC patients.
Results
Identification of NLRP mutations in HNSCC
A solution-phase hybrid capture and whole exome sequencing with a mean of 150-fold sequence coverage at targeted exonic regions were performed on HNSCC specimens as previously reported [6]. Among all sequenced specimens in the previous study [6], several histologic variants of primary or recurrent HNSCC existed including conventional type SCC (squamous cell carcinoma), basaloid SCC, papillary SCC, spindle cell carcinoma, adenosquamous cell carcinoma, and hybrid verrucous SCC. The latest World Health Organization classification of head and neck tumors (2005) presents a general consensus that these variants may display varied clinical courses and prognoses compared to conventional type SCC. For instance, basaloid squamous cell carcinoma and adenosquamous cell carcinoma are considered to behave more aggressively with early metastasis; papillary squamous cell carcinoma and verrucous carcinoma are slow-growing tumors and may show better prognosis than conventional HNSCC. In order to a perform analyses in a relatively more homogenous population, we restricted our study to 62 patients with primary conventional type HNSCC.
Non-silent mutations were detected in 10 of 14 human NLRP genes (NLRP1-14). The whole group of the NLRP genes was affected except for NLRP6, NLRP7, NLRP9, and NLRP13. Non-silent NLRP mutations were present in tumors from 13 patients (Table 1). Mutations in more than 1 NLRP genes were identified in 3 tumors. Despite the unknown genetic or environmental predisposition for cancer development at the floor of mouth (FOM), this site is recognized as being a high risk site for HNSCC [31]. Hence, we also reviewed the NLRP mutational profiles in 8 FOM HNSCC tumors; four tumors harbored NLRP mutations. In addition to our study of 62 conventional HNSCC, NLRP mutations have also been reported in HNSCC in an independent whole exome sequencing study of HNSCC [7] and in the Catalog of Somatic Mutations in Cancer (COSMIC) database. NLRP mutations most frequently occur at the C-terminus followed by the NBD domain (Figure 1).
Table 1. Identification of NLRP mutations in primary HNSCC.
| Chromosome Location | NLRP Genes |
| 19q13.42-19q13.43 | NLRP2, NLRP4, NLRP5, NLRP8, NLRP11, NLRP12 |
| 11p15.4 | NLRP10, NLRP14 |
| 17p13.2 | NLRP1 |
| 1q44 | NLRP3 |
Novel mutations were identified in 10 NLRP genes in HNSCC. Most mutations were involving chromosomal locations 11p15.4 and 19q13.42-19q13.43.
Figure 1. Identification of NLRP mutations in FOM HNSCC.
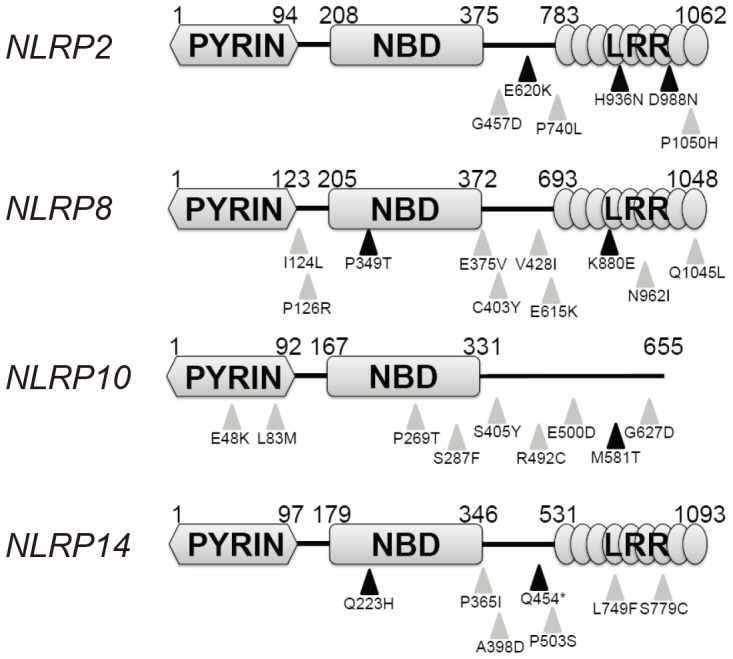
Mutations in four NLRP genes were identified in FOM HNSCC patients. Black triangles indicate novel mutations identified in this study. Gray triangles represent reported mutations in the Catalogue of Somatic Mutations in Cancer (COSMIC) database.
NLRP mutations were associated with increased cancer genome instability
Catastrophic mutational events reflect a high degree of genome instability, which is one of the hallmarks of cancer. Thus we evaluated whether mutations in NLRP genes reflected the overall cancer genome instability. Tumors without NLRP mutations harbor an average of 68 missense mutations, while those with NLRP mutations demonstrate as twice as many missense mutations across their exomes (P = 0.015) (Figure 2A). In agreement with previous findings, two recent large-scale sequencing studies also identified frequent mutations of TP53 in HNSCC [6], [7], which may drive kataegis in a fraction of the tumors. However, the missense mutation rate in tumors without TP53 mutations was comparable to that in those with TP53 mutations (Figure 2B). Despite of the similar size between the human gene family of TLR, which is comprised of 10 genes (TLR1-10), and pyrin-containing NLRs, TLR gene mutations were only identified in 7 tumors. Both selected gene members of the TLR or NLRP families, which were mutated in our cohort of 62 tumors, and total members of both families were compared in light of the length of coding regions. Our analysis showed that the coding regions of both groups of genes were similar (Figure S2). Thus, we employed the TLR gene family as another control for our gene mutation analysis. The missense mutations in tumors with TLR mutations were higher than those without TLR mutations with a marginal P value (P = 0.041) (Figure 2C). In addition, HNSCC with NLRP mutations displayed generalized elevated genome instability exemplified by general mutation rate (P = 0.0095), silent mutation rate (P = 0.016), and non-silent mutation rate (P = 0.0134) (Figure 2D). HNSCC with TP53 mutations also demonstrated higher general mutation rate and non-silent mutation rates (P = 0.041 and P = 0.038, respectively) yet similar silent mutation rates (Figure 2E). Similar to tumors with TP53 mutations, the tumors with TLR mutations more commonly demonstrated higher general mutation rates and non-silent mutation rates (P = 0.048 and P = 0.030, respectively) (Figure 2F). However the silent mutation rates between tumors with or without TLR genes mutations were comparable (Figure 2F).
Figure 2. Mutation rates comparisons.

(A) Numbers of missense and nonsense mutations were compared between tumors with or without NLRP mutations. (B) Numbers of missense and nonsense mutations were compared between tumors with or without TP53 mutations. (C) Numbers of missense and nonsense mutations were compared between tumors with or without TLR genes mutations. (D) Mutation rates [shown as mutations per million base (MB) pairs] were compared between tumors with or without NLRP mutations. (E) Mutation rates [shown as mutations per million base (MB) pairs] were compared between tumors with or without TP53 mutations. (F) Mutation rates [shown as mutations per million base (MB) pairs] were compared between tumors with or without TLR genes mutations. P value less than 0.05 was considered significant.
Demographic information of patients involved in the study
Primary conventional HNSCC has a strong predilection for males; however, this gender predilection appeared to be mitigated in patients harboring NLRP mutations, as the representation among females with HNSCC increased from 24% of all included patients in this study to almost 40% among those patients with NLRP mutations, although this difference was not statistically significant (Table 2). Neither tobacco nor alcohol use was associated with these mutations. Most examined clinical parameters, such as disease stage, histologic grade, human papilloma virus (HPV) status, 5-year survival and disease progression, did not differ between the patients with and without NLRP mutations (Table 2).
Table 2. Demographic Profile of Study Subjects with Primary Conventional HNSCC (PC-SCC).
| All PC-SCC Cases | PC-SCC Cases with NLRP mutation | PC-SCC Cases without NLRP mutation | |||||
| n = 62 | n = 13 | n = 49 | P* | ||||
| Gender | |||||||
| Male | 47 | 75.8% | 8 | 61.5% | 39 | 79.6% | 0.27∥ |
| Female | 15 | 24.2% | 5 | 38.5% | 10 | 20.4% | |
| Age, years | |||||||
| Average (Range) | 57 (33–76) | 59 (39–75) | 56 (33–76) | 0.30§ | |||
| Cigarette Pack-Years | |||||||
| Never smoker | 10 | 16.1% | 3 | 23.1% | 7 | 14.3% | 0.62∥ |
| 1–49 py | 38 | 61.3% | 8 | 61.5% | 30 | 61.2% | |
| ≥50 py | 14 | 22.6% | 2 | 15.4% | 12 | 24.5% | |
| Alcohol Quantity† | |||||||
| Never drinker | 12 | 19.4% | 4 | 30.8% | 8 | 16.3% | 0.13∥ |
| 1–4 | 7 | 11.3% | 3 | 23.1% | 4 | 8.2% | |
| ≥5 | 41 | 66.1% | 6 | 46.2% | 35 | 71.4% | |
| Unknown | 2 | 3.2% | 0 | 0.0% | 2 | 4.1% | |
| Tumor Site | |||||||
| Oral Cavity | 31 | 50.0% | 8 | 61.5% | 23 | 46.9% | 0.98∥ |
| Oropharynx | 10 | 16.1% | 2 | 15.4% | 8 | 16.3% | |
| Hypopharynx | 8 | 12.9% | 1 | 7.7% | 7 | 14.3% | |
| Larynx | 11 | 17.7% | 2 | 15.4% | 9 | 18.4% | |
| Sinonasal | 2 | 3.2% | 0 | 0.0% | 2 | 4.1% | |
| Disease Stage | |||||||
| I | 0 | 0.0% | 0 | 0.0% | 0 | 0.0% | 0.87∥ |
| II | 5 | 1.4% | 1 | 7.7% | 4 | 8.2% | |
| III | 12 | 3.4% | 3 | 23.1% | 9 | 18.4% | |
| IV | 45 | 12.7% | 9 | 69.2% | 36 | 73.5% | |
| Histologic Grade | |||||||
| Well Differentiated | 1 | 1.6% | 0 | 0.0% | 1 | 2.0% | 0.80∥ |
| Moderately Differentiated | 39 | 62.9% | 9 | 69.2% | 30 | 61.2% | |
| Poorly Differentiated | 22 | 35.5% | 4 | 30.8% | 18 | 36.7% | |
| HPV Status | |||||||
| Positive | 10 | 16.1% | 1 | 7.7% | 9 | 18.4% | 0.67∥ |
| Negative | 52 | 83.9% | 12 | 92.3% | 40 | 81.6% | |
| Treatment | |||||||
| RT Only | 11 | 17.7% | 4 | 30.8% | 7 | 14.3% | 0.29∥ |
| CRT | 38 | 61.3% | 6 | 46.2% | 32 | 65.3% | |
| No CRT | 13 | 21.0% | 3 | 23.1% | 10 | 20.4% | |
| Unknown | 0 | 0.0% | 0 | 0.0% | 0 | 0.0% | |
| Vital Status | |||||||
| Alive | 29 | 46.8% | 6 | 46.2% | 23 | 46.9% | 1.00∥ |
| Dead | 33 | 53.2% | 7 | 53.8% | 26 | 53.1% | |
| 5-year overall survival, months | |||||||
| Median (Range) | 34.3 (2.0–60.0) | 22.1 (2.4–58.0) | 37.9 (4.6–60.0) | 0.58¶ | |||
| Disease Progression | |||||||
| No disease progression | 22 | 35.5% | 6 | 46.2% | 16 | 32.7% | 0.27∥ |
| Local/Regional Recurrence | 12 | 19.4% | 0 | 0.0% | 12 | 24.5% | |
| Second Primary | 2 | 3.2% | 0 | 0.0% | 2 | 4.1% | |
| Distant Metastasis | 8 | 12.9% | 2 | 15.4% | 6 | 12.2% | |
| Death without Recurrence | 18 | 29.0% | 5 | 38.5% | 13 | 26.5% | |
| 5-year Progression Free Survival, months | |||||||
| Median (Range) | 20.3 (2.0–60.0) | 19.1 (2.4–58.0) | 21.5 (4.6–60.0) | 0.78¶ | |||
Contingency tables were analyzed by Fisher's exact test. Survival distributions were analyzed by Log Rank test. Notes:
Typical number of alcohol drinks in 2 week period, §Mann-Whitney U test,
Fisher's exact test,
Log rank test,
Comparing patients with or without mutations in NLRP genes.
NLRP mutations were associated with HNSCC arising in the floor of mouth (FOM)
Floor of mouth is an unequivocal high risk site for HNSCC according to the recent WHO Classification of Head and Neck Tumours (2005). In a comprehensive study of 3,360 specimens, leukoplakia at floor of mouth has the highest incidence showing epithelial dysplasia and carcinoma compared to leukoplakic lesions at all other anatomic locations in the oral cavity [32]. However, the genetic alterations that may occur in this region leading to development of tumors at this anatomical site remain poorly understood. We assessed the correlation between NLRP mutations and tumor site in 62 patients. NLRP mutations were more common in HNSCC arising in the FOM (P = 0.079 in comparison with HNSCC in other non-FOM oral cavity locations, P = 0.034 in comparison with HNSCC in other non-FOM head and neck locations) (Table 3). To determine whether the clustering of NLRP mutations in this site was because of elevated genome instability of FOM HNSCC compared to HNSCC at other sites, we evaluated missense mutation, nonsense mutation, and mutation rates. No significant differences were identified between FOM and non-FOM groups (Figure 3A, 3B), indicating that the higher rate of NLRP mutations in FOM HNSCC was not a nonspecific reflection of an overall higher mutation rate at this anatomic site. In order to further substantiate the specificity of this association, we also explored whether mutations in TP53 or TLR genes were clustered in FOM HNSCC. Although mutations of TP53 or TLR genes were seen in a group of patients with higher general mutation rate and non-silent mutation rate, these mutations were not associated with HNSCC arising in FOM (Tables S1 and S2).
Table 3. Mutations in NLRP genes were more frequently found in FOM HNSCC.
| NLRP Mut. | w/o NLRP mut. | P value | |
| FOM | 4 | 4 | |
| Non-FOM | 4 | 19 | 0.079 |
| (all other OC locations) | |||
| FOM | 4 | 4 | |
| Non-FOM | 8 | 46 | 0.034 |
| (all other H&N locations) |
Contingency table comparisons were made by Fisher's Exact test. P value of less than 0.1 was considered significant.
Figure 3. Mutation rates of FOM HNSCC.
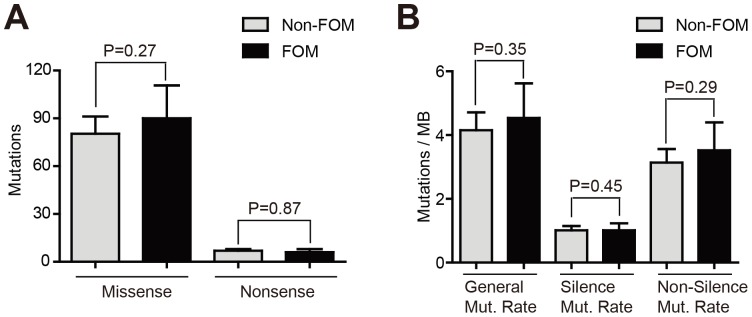
(A) Numbers of missense and nonsense mutations were compared between patients with FOM or non-FOM HNSCC. (B) Mutation rates were compared between patients with FOM or non-FOM HNSCC.
Identification of factors affecting the survival of patients with primary conventional type HNSCC
Tumors harboring NLRP mutations had significantly increased general mutation rate and missense mutations; however, the presence of these mutations was not associated with patients overall survival (Figure 4A). Although TP53 mutations were present in 67.7% of the total tumor specimens, these mutations did not affect patient survival (Figure 4B). The presence of TLR mutations had little prognostic value in patients survival (Figure 4C). Among all factors tested, only HPV infection status and tumor stage were associated with overall survival. In agreement with previous literatures [33], [34], patients with HPV-positive tumors had improved survival (P = 0.094) (Figure 4D). In fact, HPV-positive tumors demonstrated a significantly lower missense mutations and general mutation rates (Figure S1A, S1B). Despite the fact that the majority of the patients involved in this study were at advanced stage (stage III and beyond), our analysis showed low stage status positively impacted overall survival (Figure 4E). However, tumor stage was not associated with increased genome instability (Figure S1C, S1D). Neither age nor adjuvant therapies such as chemotherapy or radiotherapy were associated with patients survival (Figure 4F–G).
Figure 4. Determination of factors affecting overall survival.

Patients overall survival analyses were made between two groups: (A) patients with or without mutations in NLRP genes; (B) patients with or without mutations in TP53 genes; (C) patients with tumors that harbored TLR mutations and those without TLR mutations; (D) patients with or without HPV infections; (E) patients with low stage (stage II) or advanced stage (stage III and beyond) tumors; (F) patients of less or more than 56 years old; (G) patients who received no adjuvant therapy, radiotherapy alone or both chemotherapy and radiotherapy.
Discussion
NLRP proteins are evolutionarily and functionally conserved. The NLR gene family was initially discovered through genomic database mining based on structural homology [9]. By incorporating an N-terminal effector domain, a central nucleotide-binding domain, and a variable number of LRR at the C-terminus, the 22 human NLR proteins are structurally similar to the plant R protein, which conveys resistance to pathogens [8]. The pioneering research on NLR proteins primarily focused on their pivotal roles in modulating inflammatory responses such as caspase-1 activation, MAPK, NF-κB, and mitochondria-based antiviral signaling [9], [10], [13], [35]. Several NLRs possess similar functions in facilitating the assembly of a large multimeric protein complex, coined as the inflammasome, to process pro-caspase-1 into its mature form, which induces the maturation and secretion of IL-1β and IL-18, in response to a variety of PAMPs and DAMPs [9]. Both HNSCC derived cell lines and invasive HNSCC tumor cells produce proinflammatory cytokines including IL-1β, TNF-α, and IL-6 [36], [37]. In addition, elevated salivary IL-1β has been found in oral squamous cell carcinoma patients [38]. Among the NLRP proteins, NLRP1, 2, 3, and 12 participate in the formation and activation of inflammasome [9], and mutations in these genes were identified in HNSCC.
HNSCC is notorious for its heterogeneity and frequent resistance to adjuvant therapies. In addition to the aforementioned inflammatory pathways, NLR proteins have also been implicated in the regulation of autophagy, which conveys resistance to a variety of adjuvant therapeutic agents [39]. A NLR protein NOD2 induces autophagy in dendritic cells upon engagement with muramyldipeptide [14]. NLRP4 associates with beclin1 to negatively regulate autophagy [15]. NLRX1 modulates autophagy by recruiting ATG12, ATG5, and ATG16L1 to a large mitochondrial protein complex, through an intermediary partner TUFM [13], [16]. Inhibition of autophagy is proven an effective strategy in sensitizing HNSCC cells to a number of adjuvant therapeutic agents. The majority of the mutation of NLRP genes in HNSCC are located at the C-terminal LRR domain. With the roles of NLR proteins in modulating autophagy being unveiled, it would be necessary to evaluate the function of these autophagy-related NLRs in modulating cancer cell resistance to novel adjuvant therapy.
One critical event that precedes the generation of cancer cell heterogeneity is kataegis, in which rapid mutations accumulate in “hotspots” to drive the generation of subclones of cancer cells [1], [2]. Although mutations in tumor suppressor genes such as TP53 are unequivocally involved in many HNSCC, they do not necessarily define the idiosyncratic genetic features of an individual tumor. In fact, we noted that the missense mutations were comparable between patient tumors with or without TP53 mutations. It is likely specific rarer mutational events in a subset of genes that shape the biologic features of an individual tumor, such as capability of forming subclones. These subclones may contribute to divergent responses to adjuvant treatments. It is possible that specific anatomical sites may have a propensity to develop tumors with mutations in a specific set of genes. Compared to keratinized mucosa lining the gingiva, buccal mucosa, and hard palate, mucosa lining the floor of mouth is non-keratinized, which makes this site more prone to environmental insults. Our findings that a group of genes pivotal in modulating host-environment insults interactions were frequently mutated in HNSCC arising in the floor of mouth suggest their functional significance in cancer development. Indeed, we found that mutations in NLRP genes were closely associated with higher degree of cancer genome instability. The small number of primary FOM HNSCC analyzed in this study is a limitation. However, the 62 tumors analyzed represented the common anatomic sites of primary HNSCC. In addition, we employed mutations of the TP53 gene and the TLR gene family as specificity controls. Of the genes analyzed, the association with FOM was unique to the mutations of the NLRP genes.
In agreement with previous studies [33], [34], we found HPV status and tumor stage were associated with HNSCC patients overall survival. Although genome instability exemplified by kataegis represents a defining step in driving the diversity of tumor subclones, it did not appear to be a reliable prognostic factor. For example, while HPV negative HNSCC patients had a significantly elevated level of general mutation rate, advanced stage tumors did not necessarily display worse genome instability (Figure S1). However, increased intra-tumor heterogeneity resulting from non-driver mutations in a kataegis event may substantially affect the tumor adaptation to treatment, including evolving resistance to therapy [40]. Hence, the rarer mutations especially those reflecting genome instability may not be stochastic, rather they may be the result of a collective response to PAMP/DAMP challenge and adaptive response to therapeutic inflictions, contributing to the establishment of resistance.
In Conclusion, We Further Characterized The Genetic And Clinicopathologic Profile Of The Novel Pyrin-Containing Nlr Gene Family In 62 Patients With Conventional Type Hnscc. Clinically, These Mutations Were Frequently Found In Hnscc Arising In The Floor Of Mouth. These Mutations Were Clustered At The Lrr Domain Of Nlrp Proteins; And The Affected Nlrp Genes Were Mostly Localized At Chromosomes 11p15.4 And 19q13.42-19q13.43. Mutations In The Nlrp Genes Were Associated With Enhanced Genome Instability In Hnscc.
Materials and Methods
Ethics statement
All clinical data including patients' demographic information, tumor histologic type and grading, genetic mutation identity, adjuvant treatment information, and vital status were made available through the Specialized Program of Research Excellence (SPORE) in Head and Neck Cancer neoplasm of the University of Pittsburgh. Patients included in this study were enrolled into the Head and Neck tumor bank protocol. This protocol requires written consent and was approved by the Institutional Review Board of the University of Pittsburgh.
Study subjects
Whole exome sequencing was performed on 62 patients with primary or recurrent HNSCC as previously described [6]. Only patients with primary conventional type squamous cell carcinoma were included. Recurrent tumors or other histologic variants, such as basaloid squamous cell carcinoma, papillary squamous cell carcinoma, spindle cell carcinoma, adenosquamous cell carcinoma, and hybrid verrucous squamous cell carcinoma, were excluded. All participating patients were Caucasians, and other demographic information was summarized in Table 2.
Data Deposition
Identified mutations associated with our recent whole exome sequencing effort were made available in dbGaP with the accession # phs000370.v1.p1 as previously described [6]. All novel mutations were also available through the COSMIC database. In order to differentiate the novel mutations associated with tumors analyzed in this study and those that had been present in the COSMIC database, we highlighted all novel mutations with a black triangle in Figure 1.
Gene family coding region calculations
The numbers of amino acids of each member of the TLR or NLRP families were retrieved from the National Center for Biotechnology Information (NCBI) protein database, and the lengths of the coding regions were determined by the number of the amino acids multiplied by three. The comparison was analyzed by Mann-Whitney U test, and a P value of less than 0.05 was considered significant.
Statistical Analyses
Comparisons of mutation rates and average ages between the two groups were made by Mann-Whitney U test. Fisher's exact test was employed to analyze contingency tables. Survival distributions were analyzed by Log Rank test. Analyses were made using Graphpad Prism 5.0 (Graphpad Software, Inc.). P value of less than 0.1 was considered to be significant.
Supporting Information
Mutation rates comparisons. (A) Numbers of missense and nonsense mutations were compared between patients with or without HPV infection. (B) Mutation rates were compared between patients with or without HPV infection. (C) Numbers of missense and nonsense mutations were compared between patients with low stage or advanced stage SCC. (D) Mutation rates were compared between patients with low stage or advanced stage SCC. P value less than 0.05 was considered significant.
(TIF)
Coding region lengths comparisons. (A) The coding region lengths between selected members of the TLR and NLRP gene families, which were mutated in our cohort, were compared by Mann-Whitney U test. (B) The coding region lengths between the total members of the TLR and NLRP gene families were compared by Mann-Whitney U test. P value of less than 0.05 was considered significant.
(TIF)
Mutations of the TP53 gene were not enriched in FOM HNSCC. Contingency table comparisons were made by Fisher's Exact test to investigate whether mutations of the TP53 gene were more frequently seen in HNSCC arising FOM. P value of less than 0.1 was considered significant.
(DOCX)
Mutations of the TLR genes were not enriched in FOM HNSCC. Contingency table comparisons were made by Fisher's Exact test to investigate whether mutations of the TLR gene family were more frequently seen in HNSCC arising FOM. P value of less than 0.1 was considered significant.
(DOCX)
Funding Statement
This work was supported by P50CA097190 (JRG) the Specialized Program of Research Excellence (SPORE) in Head and Neck Cancer neoplasm of the University of Pittsburgh and K07CA137140 (AME). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, et al. (2012) Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, et al. (2012) The life history of 21 breast cancers. Cell 149: 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 4. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, et al. (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127: 2893–2917. [DOI] [PubMed] [Google Scholar]
- 5. van Monsjou HS, Balm AJ, van den Brekel MM, Wreesmann VB (2010) Oropharyngeal squamous cell carcinoma: a unique disease on the rise? Oral Oncol 46: 780–785. [DOI] [PubMed] [Google Scholar]
- 6. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, et al. (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333: 1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, et al. (2011) Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333: 1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, et al. (2008) The NLR gene family: a standard nomenclature. Immunity 28: 285–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davis BK, Wen H, Ting JP (2011) The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu Rev Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ting JP, Duncan JA, Lei Y (2010) How the noninflammasome NLRs function in the innate immune system. Science 327: 286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ting JP, Willingham SB, Bergstralh DT (2008) NLRs at the intersection of cell death and immunity. Nat Rev Immunol 8: 372–379. [DOI] [PubMed] [Google Scholar]
- 12. Lamkanfi M, Dixit VM (2009) Inflammasomes: guardians of cytosolic sanctity. Immunol Rev 227: 95–105. [DOI] [PubMed] [Google Scholar]
- 13. Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, et al. (2012) The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 36: 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cooney R, Baker J, Brain O, Danis B, Pichulik T, et al. (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16: 90–97. [DOI] [PubMed] [Google Scholar]
- 15. Jounai N, Kobiyama K, Shiina M, Ogata K, Ishii KJ, et al. (2011) NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol 186: 1646–1655. [DOI] [PubMed] [Google Scholar]
- 16. Lei Y, Wen H, Ting JP (2013) The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy 9: 432–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, et al. (2011) The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20: 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, et al. (2012) NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity 36: 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, et al. (2010) The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 207: 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zaki MH, Lamkanfi M, Kanneganti TD (2011) The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol 32: 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen GY, Liu M, Wang F, Bertin J, Nunez G (2011) A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol 186: 7187–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, et al. (2011) Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A 108: 9601–9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Strowig T, Henao-Mejia J, Elinav E, Flavell R (2012) Inflammasomes in health and disease. Nature 481: 278–286. [DOI] [PubMed] [Google Scholar]
- 25. Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD (2010) IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol 185: 4912–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meyer E, Lim D, Pasha S, Tee LJ, Rahman F, et al. (2009) Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome). PLoS Genet 5: e1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Slim R, Coullin P, Diatta AL, Chebaro W, Courtin D, et al. (2012) NLRP7 and the genetics of post-molar choriocarcinomas in Senegal. Mol Hum Reprod 18: 52–56. [DOI] [PubMed] [Google Scholar]
- 28. Ahn J, Chen CY, Hayes RB (2012) Oral microbiome and oral and gastrointestinal cancer risk. Cancer Causes Control 23: 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meyer MS, Joshipura K, Giovannucci E, Michaud DS (2008) A review of the relationship between tooth loss, periodontal disease, and cancer. Cancer Causes Control 19: 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meurman JH (2010) Oral microbiota and cancer. J Oral Microbiol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mashberg A, Samit A (1995) Early diagnosis of asymptomatic oral and oropharyngeal squamous cancers. CA Cancer J Clin 45: 328–351. [DOI] [PubMed] [Google Scholar]
- 32. Neville BW, Day TA (2002) Oral cancer and precancerous lesions. CA Cancer J Clin 52: 195–215. [DOI] [PubMed] [Google Scholar]
- 33. Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, et al. (2010) Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med 363: 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Rorke MA, Ellison MV, Murray LJ, Moran M, James J, et al. (2012) Human papillomavirus related head and neck cancer survival: A systematic review and meta-analysis. Oral Oncol [DOI] [PubMed] [Google Scholar]
- 35. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, et al. (2008) NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451: 573–577. [DOI] [PubMed] [Google Scholar]
- 36. Woods KV, El-Naggar A, Clayman GL, Grimm EA (1998) Variable expression of cytokines in human head and neck squamous cell carcinoma cell lines and consistent expression in surgical specimens. Cancer Res 58: 3132–3141. [PubMed] [Google Scholar]
- 37. Pries R, Wollenberg B (2006) Cytokines in head and neck cancer. Cytokine Growth Factor Rev 17: 141–146. [DOI] [PubMed] [Google Scholar]
- 38. Brailo V, Vucicevic-Boras V, Lukac J, Biocina-Lukenda D, Zilic-Alajbeg I, et al. (2011) Salivary and serum interleukin 1 beta, interleukin 6 and tumor necrosis factor alpha in patients with leukoplakia and oral cancer. Med Oral Patol Oral Cir Bucal [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Collisson EA, Cho RJ, Gray JW (2012) What are we learning from the cancer genome? Nat Rev Clin Oncol [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mutation rates comparisons. (A) Numbers of missense and nonsense mutations were compared between patients with or without HPV infection. (B) Mutation rates were compared between patients with or without HPV infection. (C) Numbers of missense and nonsense mutations were compared between patients with low stage or advanced stage SCC. (D) Mutation rates were compared between patients with low stage or advanced stage SCC. P value less than 0.05 was considered significant.
(TIF)
Coding region lengths comparisons. (A) The coding region lengths between selected members of the TLR and NLRP gene families, which were mutated in our cohort, were compared by Mann-Whitney U test. (B) The coding region lengths between the total members of the TLR and NLRP gene families were compared by Mann-Whitney U test. P value of less than 0.05 was considered significant.
(TIF)
Mutations of the TP53 gene were not enriched in FOM HNSCC. Contingency table comparisons were made by Fisher's Exact test to investigate whether mutations of the TP53 gene were more frequently seen in HNSCC arising FOM. P value of less than 0.1 was considered significant.
(DOCX)
Mutations of the TLR genes were not enriched in FOM HNSCC. Contingency table comparisons were made by Fisher's Exact test to investigate whether mutations of the TLR gene family were more frequently seen in HNSCC arising FOM. P value of less than 0.1 was considered significant.
(DOCX)