

Abstract
Within T-cell-dependent germinal centers, p53 gene transcription is repressed by Bcl-6 and is thus less vulnerable to mutation. Malignant lymphomas within inflamed extranodal sites exhibit a relatively high incidence of p53 mutations. The latter might originate from normal B-cell clones manifesting activation-induced cytosine deaminase (AID) and up-regulated p53 following T-cell-independent (TI) stimulation. We here examine p53 gene transcription in such TI clones, with a focus on modulatory effects of prostaglandin E2 (PGE2), and evaluate progeny for p53 mutations. Resting IgM+IgD+CD27− B cells from human tonsils were labeled with CFSE and stimulated in vitro with complement-coated antigen surrogate, IL-4, and BAFF ± exogenous PGE2 (50 nM) or an analog specific for the EP2 PGE2 receptor. We use flow cytometry to measure p53 and AID protein within variably divided blasts, qRT-PCR of p53 mRNA from cultures with or without actinomycin D to monitor mRNA transcription/stability, and single-cell p53 RT-PCR/sequencing to assess progeny for p53 mutations. We report that EP2 signaling triggers increased p53 gene transcriptional activity in AID+ cycling blasts (P<0.01). Progeny exhibit p53 mutations at a frequency (8.5×10−4) greater than the baseline error rate (<0.8×10−4). We conclude that, devoid of the repressive influences of Bcl-6, dividing B lymphoblasts in inflamed tissues should display heightened p53 transcription and increased risk of p53 mutagenesis.—Haque, S., Yan, X. J., Rosen, L., McCormick, S., Chiorazzi, N., Mongini, P. K. A. Effects of prostaglandin E2 on p53 mRNA transcription and p53 mutagenesis during T-cell-independent human B-cell clonal expansion.
Keywords: B lymphocyte, TP53, eicosanoid, inflammation, lymphoma
B-lymphocyte immune responses are characterized by clonal proliferation and accompanying DNA damage, due to rising reactive oxygen species (ROS) and activation-induced cytosine deaminase (AID). The above stresses are expected to alert p53, the “guardian of the genome,” through p53 post-translational modifications (1). While a p53 response is activated during formation of T-cell-dependent (TD) germinal centers (GCs), the response is muted relative to the extent of DNA damage. This, in part, reflects strong repression of the p53 gene by Bcl-6, the GC master regulator (2, 3). Notably, because transcriptionally active genes are favored targets for mutagenic agents (4–7), repression likely thwarts p53 mutagenesis during formation of most B-cell memory, potentially explaining why p53 mutations are rare within TD mouse Peyer's patch B cells (8) and relatively infrequent in human GC-derived B-cell malignancies (2, 9).
Proliferative responses of shorter duration with little up-regulation of Bcl-6 are induced by T-cell-independent (TI) stimuli. TI B-cell clonal expansion occurs in extrafollicular sites (10–13) and is elicited within the splenic marginal zone (MZ) and inflamed peripheral tissues expressing antigen (e.g., microbes and apoptotic cells), costimuli from the innate immune system [including complement (C3), IL-4, IL-13, BAFF, and APRIL], and TLR ligands (14–21). The lesser clonal expansion seen in TI responses, in part, reflects p53 activity. B cells from p53-deficient mice cycle more rapidly than wild-type B cells on stimulation with B-cell antigen receptor (BCR) ligand and IL-4 (22, 23). In addition, this laboratory demonstrated that p53 protein is up-regulated and functional in promoting the death of B-cell progeny generated in response to C3d-coated multivalent Ag, IL-4, and BAFF (24). Splenic MZ hypertrophy in p53-deficient mice that resist early malignancies (25) is consistent with this p53 function in vivo.
Accruing evidence suggests that compromised p53 axis function promotes development of non-GC B-cell malignancies. MZ lymphoma is the major B-cell malignancy to emerge from p53-deficient mice without additional genetic alterations (25). Furthermore, mice with B-cell-selective p53 deletion show frequent pre-GC B-cell lymphomas, including those of MZ derivation (26, 27). In addition, in humans, B-cell p53 mutations are relatively common in malignancies of non-GC origin, e.g., MZ lymphoma, mucosa-associated lymphoid tissue lymphoma, and B-cell chronic lymphocytic leukemia (B-CLL) (9, 28–33). These observations suggest that during TI immune responses, the p53 gene is more vulnerable to mutation, and furthermore, cells with certain p53 mutations undergo positive selection.
This laboratory's extensive study of the cellular changes accompanying a dynamic clonal expansion/contraction of normal human B cells as they respond to C3d antigen, IL-4, and BAFF (24, 34–36) suggests that the latter clones are particularly prone to p53 mutation. DNA-modifying AID is highly expressed during division of these TI-activated B-cell clones (35); the robust, albeit short-lived, proliferative burst likely generates mutagenic ROS (24), and the B cells lack direct T-cell contacts needed for up-regulation of Bcl-6 (2) and high-fidelity DNA repair enzymes (37). Furthermore, replicating blasts express elevated levels of cyclooxygenase (COX-2), the downstream PGE2 synthase, mPGES-1, and the adenylate cyclase-activating PGE2 receptor EP2 (34, 35). It is important to note that COX-2 inhibitor treatment reduces AID expression, and, furthermore, exogenous PGE2 augments AID levels (35), suggesting a mechanism for AID amplification in inflamed tissues.
The present study examines whether the p53 gene is transcriptionally active within replicating lymphoblasts of this TI response and whether PGE2 signals influence p53 gene transcription and mutagenesis. This was deemed important because inflamed tissues are sites where p53-mutated B cell malignancies are often detected (30, 31, 33, 38–41).
MATERIALS AND METHODS
Resting follicular (FO) human B cells
Deidentified tonsils from elective tonsillectomy were obtained according to Institutional Review Board guidelines (with cooperation of the Departments of Pathology at New York Eye and Ear Infirmary, New York, NY, USA, and North Shore University Hospital, Manhasset, NY, USA). Methods detailed elsewhere (24, 35, 36) were used to purify a minority of resting FO B cells (high density CD27−, CD43−, CD19+, IgM+, IgD+) from tonsils for subsequent in vitro activation.
Reagents for cell culture
B cells were activated by culture with polyclonally activating surrogate for C3dg-coated, moderately multivalent antigen [dextran with covalently conjugated anti-human IgM and anti-human CD21 (BCR:CD21-L)], and human rIL-4 and rBAFF, as described previously (34, 36). These conditions induce virtually all resting B cells to express CD27 and CD86, while typically only a fraction undergo division (36).
B-cell activation
Resting B cells were cultured in an enriched RPMI 1640-based medium, optimized for growth (36). To monitor divisions, purified B cells were prelabeled with carboxyfluorescein succinimidyl ester (CFSE) as described previously (36) and cultured at 105 cells/200 μl in 96-well plates (for staining of activated blasts) or 106 cells/2 ml in 24-well plates for experiments involving lysates, RNA extraction, and sorting of single cells for p53-specific RT-PCR. PGE2/butaprost was pulsed on d 2 and 4 or d 4 only, as indicated. Stocks of PGE2 and butaprost (Cayman Chemical, Ann Arbor, MI, USA) in ethanol were stored at −80°C and diluted before use. Culture harvest was generally on d 5. In some experiments, pan-caspase inhibitor, Z-VAD-FMK (Sigma-Aldrich, St. Louis, MO, USA; 40 μM final) was added at d 3 to reduce activation-induced cell death (24). Alternatively, actinomycin D (Sigma-Aldrich; 5 μM final) was added to inhibit RNA polymerase (42).
Intracellular staining
Previously described methods for staining of p53, AID, and pH2AX (24, 35) within CFSE-lymphoblasts were employed and involved PE-anti-p53 (DO-7; BD Pharmingen, San Diego, CA, USA), PE-IgG2b mAb control (27–35; BD Pharmingen), mouse anti-AID mAb (ZA-001; Invitrogen, Carlsbad, CA, USA), and anti-pH2AX-Ser-139/140 (3F2; Thermo Scientific, Waltham, MA, USA) or IgG isotype controls, followed by PE-conjugated anti-IgG Ab.
Semiquantitative analysis of p53 mRNA
Total RNA was extracted from 1–5 × 106 cells using the Qiagen Miniprep kit (Qiagen, Gaithersburg, MD, USA) with cDNA synthesis performed using Oligo-dT primers (Invitrogen kit). PCR amplification involved the following primers: p53(A), forward 5-cagccagactgccttccg-3 and reverse 5-gcaagtcacagacttggctg-3, yielding 400-bp amplicon (exons 2a, 3, and 4); and β-actin, forward 5′-gtcctctcccaagtccacaca-3′ and reverse 5′-ctggtctcaagtcagtgtacaggtaa-3′. PCR parameters were 95°C for 30 s, 58°C for 60 s, and 72°C for 30 s for 35 cycles in a total volume of 30 µl. PCR products were resolved as described previously (24, 35).
Quantitative PCR (qPCR)
qPCR (TaqMan; Applied Biosystems, Foster City, CA, USA) of cDNA was performed (24) using the following primers and probes: p53, forward 5-aggccttggaactcaaggat-3 and reverse 5-ccctttttggacttcaggtg-3, UPL probe 12 (accession no. NM_001126114.1); and β-actin, forward 5′-ccaaccgcgagaagatga-3′ and reverse 5′-ccagaggcgtacagggatag-3′, UPL probe 64 (accession no. NM_001101.3). Probes were obtained from human universal probe library of Roche Applied Science (Indianapolis, IN, USA). qPCR involved triplicates or quadruplets of cDNA template and Eurogentec master mixes (AnaSpec, Fremont, CA, USA) with parameters as reported previously (24). Amplification was extended to 45 cycles to reveal the plateau of maximal substrate use; threshold cycle (Ct) values for data analysis represented thresholds within the linear region (routinely between 20 and 35). Fold change was calculated by comparing treated vs. untreated groups; analysis by RQ Manager 1.2 (Applied Biosystems). The reference gene β-actin was used as endogenous control, except in the supplemental experiments.
Single-cell p53-specific RT-PCR
On d 5–6, activated cultures were harvested, washed with cold PBS, and resuspended in PBS + 3% FCS. Single cells were sorted (FACS-Aria; BD Biosciences, San Jose, CA, USA) and individually deposited in each well of a 96-well PCR plate into 4.0 μl cell lysis buffer (0.5× PBS and 0.01 M DTT, Invitrogen; 2.0 U/μl RNAsin (verde) inhibitor, Promega, Madison, WI, USA; 0.25 U/μl PrimRNAse Inhibitor, Qiagen; and molecular grade water). Single lysed cells were rapidly frozen on dry ice. At the time of cDNA preparation, lysates were defrosted on ice, and 3.5 μl buffer (42.8 ng/μl random hexamer primers, Invitrogen; 1.43% Nonidet P-40, Sigma-Aldrich; 0.25 U/μl PrimRNAse Inhibitor, Qiagen; and molecular grade water) was added to each PCR-plate well and incubated at 65°C for 1 min. Plates were spun quickly at 4°C and placed on ice. To each well. 7.0 μl buffer (2.1× FS buffer from cDNA kit, Invitrogen; 0.014 M DTT, Invitrogen; 1.79 mM dNTPs, Invitrogen; 0.6 U/μl RNAsin (verde) inhibitor, Promega; 0.14 U/μl PrimRNAse Inhibitor, Qiagen; 7.1 U/μl RT-Superscript III, Invitrogen; and molecular grade water) was added. The PCR plate was tapped gently, centrifuged, and placed on a PCR thermal cycler (MJ Research, St. Bruno, QC, Canada; DNA Engine Dyad, Bio-Rad, Richmond, CA, USA) at 42°C for 5 min, 25°C for 10 min, 40°C for 60 min, 95°C for 5 min, and 4°C thereafter.
PCR amplification of single-cell p53 cDNA for mutation analysis
Two rounds of PCR (at 35 cycles) amplified p53 cDNA from each cell. In the first round, 1.0 μl cDNA was used as template and Hot-Start Taq (Qiagen) was used instead of Ampli-Taq Gold (Invitrogen). In the second round, 2 μl amplicon (from first round) was used as template iwith Ampli-Taq Gold. The running procedure for semiquantitative PCR (above) was employed. Primers were p53(A) primers (above), which amplified p53 exons 2a, 3, and 4 (tonsils T602, T606, T612, and T613), and p53Ex4 primers, which amplified p53 exon 4 only (T587 and T597): forward 5′-cttgccgtcccaagcaatc-3′ and reverse 5-gcaagtcacagacttggctg-3, yielding a 270-bp amplicon.
PCR amplicon cleaning and p53 sequence analysis
Electrophoresis-tested p53+ amplicons were cleaned with the QIAquick PCR purification kit (Qiagen), and comparable amounts were sequenced by Genewiz (South Plainfield, NJ, USA). Each sample was sequenced with forward and reverse primers separately. Data were analyzed by ChromasPro and Chromas Lite software (Technylesium Pty. Ltd., South Brisbane, QLD, Australia).
Statistics
For experiments comparing p53 mRNA half-life in cultures with or without PGE2 and actinomycin D, a linear mixed model compared the mean fold change between PGE2+ and PGE2− cultures and respective controls across time (30 min and 1, 3, 8, and 20 h after actinomycin D). The interaction term between group and time assessed whether slopes in PGE2+ and PGE2− cultures were different. If the interaction term was nonsignificant, it was removed from the final model. A mixed models approach accounted for the repeated measurements over time within an experiment. For experiments comparing p53 mutation incidence in PGE2+ and PGE2− cultures, a generalized linear mixed model with a logit link was employed. (While a mixed models approach accounted for multiple measurements within an experiment with a single donor, results did not differ qualitatively, leading to the assumption of independence between observations.) Analyses were conducted in 2 ways: with the unit analysis as total nucleotides screened (mutation frequency/base pair), and with the unit analysis as total cells screened (mutation frequency/cell). Mutation frequency between variably treated cultures was compared using the χ2 test or Fisher's exact test. In the cases when the frequency of total observed mutations between 2 differing nucleotide motifs was compared, the total number of available sites with each motif within the p53 sequences analyzed was taken into account. A χ2 analysis or Fisher's exact test was used to determine whether mutations favored one motif over another. Other statistical methods are indicated in figure legends.
RESULTS
During human B-cell TI clonal expansion, maximal p53 protein expression precedes maximal AID protein
Consistent with past immunoblotting and flow cytometric studies of this laboratory (24, 35), Fig. 1A shows that both p53 and AID protein rise in a division-linked manner following the activation of resting follicular B cells from human tonsils with BCR:CD21-L, IL-4, and BAFF. The present simultaneous assessment of both proteins in the same experiments shows that p53 reaches maximal expression at earlier divisions than AID (Fig. 1B) and suggests that AID-induced DNA damage, and ensuing p53 protein stabilization, do not solely explain the elevation in p53 protein.
Figure 1.
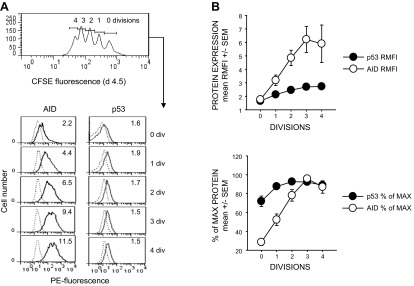
p53 protein is maximally up-regulated at earlier divisions than is AID protein. p53 and AID were simultaneously evaluated in CFSE-labeled B-cell cultures proliferating in response to BCR:CD21-L, IL-4, and BAFF. A) Representative experiment in which CFSE-labeled cells were stained intracellularly for AID or p53 on d 4.5 after activation. Top panel: gating of CFSE-labeled cells for divisions. Bottom panel: PE fluorescence within division-gated cells stained for AID or p53 (dark lines) or respective isotype control (dotted lines). B) Pooled data from 6 experiments in which p53 and AID were simultaneously evaluated. Top panel: data are expressed as ratio mean fluorescence intensity (RMFI) = ratio of the MFI of p53 or AID stained cells vs. MFI of control stained cells. Bottom panel: RMFI values from each experiment were computed as percentage of the maximal RMFI observed within all assessed divisions.
TI B lymphoblasts manifest elevated p53 mRNA
Transcription of p53 is reported to rise prior to proliferation (within 6 h) following human B-cell activation with anti-IgM and growth factors (43). We were interested in determining whether p53 transcription remains high during a TI B-cell proliferative response characterized by high AID expression (35). To do so, p53 mRNA levels in freshly isolated naive B cells were compared with that in proliferating cultures on d 4 following TI activation. Semiquantitative PCR (Fig. 2A) and qPCR (Fig. 2B) show that p53 RNA levels are notably elevated at this activation interval. Because not all CD86+, CD27+ B lymphoblasts uniformly divide in these cultures (36), we examined whether blasts representing 0 and 3–4 divisions differ in p53 mRNA expression. Viable CFSE-labeled blasts were sorted according to division status, and cDNA was assessed by qPCR (Fig. 2C). Notably, blasts representing 3–4 divisions display significantly greater p53 mRNA than undivided blasts (P=0.006). Virtually all of these divided blasts express elevated AID protein (Fig. 1A and ref. 35).
Figure 2.
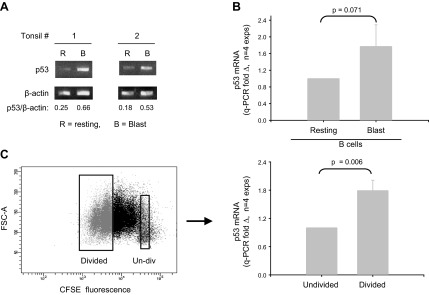
B cells replicating in response to BCR:CD21-L, IL-4, and BAFF exhibit elevated levels of p53 mRNA. A) Semiquantitative RT-PCR analysis of p53 mRNA levels in resting B cells and activated d 4 cultures stimulated with BCR:CD21-L, IL-4, and BAFF. Values represent ratios of densitometric intensity in the p53 and β-actin bands B) qPCR assessment of p53 mRNA in resting B cells vs. d 4 blasts. ΔCt values for p53 were obtained by comparisons with β-actin. Values for fold difference (Δ) were obtained by comparing ΔCt values in activated cultures with the ΔCt values of B cells prior to activation. P value shows that the differences between Δ values in resting vs. activated cells in a total of 4 replicate experiments were of borderline significance, using a 2-tailed paired Student's t test. C) Undivided blasts and divided blasts within activated cultures were sorted on the basis of CFSE fluorescence. Left panel: representative experiment. Right panel: results for p53-specific q-RT-PCR of isolated RNA. Divided blasts express significantly more p53 mRNA than do undivided blasts within the same cultures (P=0.006; 2-tailed, paired Student's t test).
Exogenous PGE2 significantly augments p53 mRNA within dividing B lymphoblasts
One notable characteristic of cycling B lymphoblasts is an increase in COX-2 and linked proteins (34, 35, 44). The elevated p53 mRNA levels in cycling blasts and evidence of substantial crosstalk between the p53 and COX-2/PGE2 axes in other cells (45–50) led us to examine whether PGE2 signaling influences p53 expression. For these experiments, exogenous PGE2 (50 nM), or vehicle alone, was pulsed into d 4 activated B cell cultures, and p53 RNA levels were subsequently assessed by qPCR. Following an early insignificant decline, PGE2-pulsed cultures showed significantly greater p53 mRNA than controls, with the peak increase at 8 h following PGE2 (P=0.0004; Fig. 3A, B). The PGE2-mediated increase was noted, regardless of whether β-actin, GAPDH, RPS20, RPS13, or RPL27 was used as housekeeping reference gene (ref. 51 and Supplemental Fig. S1). Interestingly, a comparison of undivided and divided blasts from the same cultures for sensitivity to this PGE2 effect (Fig. 3C) showed that divided blasts uniquely displayed PGE2-boosted p53 mRNA.
Figure 3.
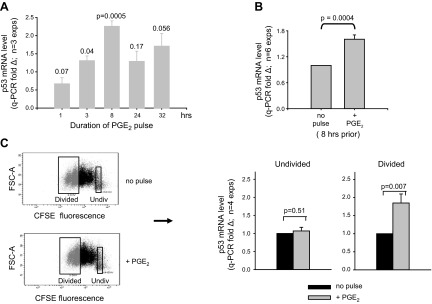
PGE2 augments p53 mRNA expression during TI clonal expansion. A) B-cell cultures activated with BCR:CD21-L, IL-4, and BAFF were pulsed with exogenous PGE2 (50 nM) or ethanol vehicle alone on d 4; mRNA was isolated at varying periods after the pulse. qPCR of cDNA was used to measure the levels of p53 mRNA in PGE2-pulsed and control cultures. Values for fold difference (Δ) were obtained by comparing ΔCt values in PGE2-pulsed cultures with the respective ΔCt values in vehicle-pulsed cultures at the same time point. Dotted line represents a value of Δ = 1 for each of the vehicle control cultures. Results are the mean ± se Δ fold difference in 3 separate experiments, where P represents significance of differences between Δ values in PGE2-pulsed vs. vehicle-pulsed cultures. B) Pooled results from a total of 6 experiments which evaluated p53 mRNA levels in cultures activated as in A with/without an 8 h pulse of PGE2 on d 4. C) Day 4 activated cultures of CFSE-labeled cells with or without pulse of exogenous PGE2 8 h earlier were harvested and sorted on the basis of their division status: undivided vs. divided (3–4 divisions). qPCR of cDNA with p53 and β-actin probes was used to monitor p53 mRNA levels, as in Fig. 2. Values for Δ in each sorted population were obtained by comparing ΔCt values in cells from PGE2-pulsed vs. unpulsed cultures. P values show that a pulse with PGE2 significantly augments p53 mRNA levels in the divided blasts (P=0.007) without affecting p53 levels in the undivided cells (P=0.51; 2-tailed, paired Student's t test).
The effects of PGE2 on p53 protein appear to be considerably more complex, as might be expected, given the many mechanisms for post-translational regulation of p53 following cell stress and DNA damage (52). Dividing d 5 lymphoblasts display significantly greater levels of p53 protein when cultures are exposed to PGE2, ranging from 10 to 1000 nM, on d 2 and 4 after TI activation (Supplemental Fig. S2). Nevertheless, if TI-activated cultures receive a similar pulse with PGE2 solely on d 4, p53 protein levels reproducibly decline by 24 h (data not shown). Additional work is needed to define the mechanisms involved. Given the rising expression of adenylate cyclase-activating PGE2 receptors in cycling B cells (35), the drop in p53 protein following an acute pulse with p53 on d 4 may involve cAMP-facilitated Mdm2 ubiquitination of p53 protein (53).
Selective EP2 agonist promotes p53 mRNA expression
We were interested in assessing whether the PGE2-driven increase in p53 mRNA involved EP2, the PGE2 receptor that increases most significantly with successive divisions (ref. 35 and Fig. 4A). This was accomplished through testing the effects of a structural analog of PGE2 that is highly selective for EP2, butaprost. Figure 4B shows that d 4 activated cultures pulsed with butaprost from 2 to 30 h earlier express significantly greater levels of p53 mRNA than vehicle-pulsed cultures. The dose response in Fig. 4C is consistent with butaprost's significantly lesser affinity for EP2, as compared to PGE2 (54).
Figure 4.
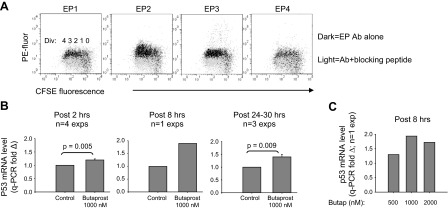
Selective agonist for EP2, a PGE2 receptor up-regulated during division, promotes heightened p53 mRNA. A) EP2 receptor expression is preferentially elevated with replicating B lymphoblasts in cultures activated by BCR:CD21-L, IL-4, and BAFF. Expression of each of 4 PGE2 receptors on B cells was assessed with receptor-specific Abs, as described elsewhere (35). EP receptor density expressed as PE fluorescence; division number revealed by CFSE fluorescence. B) p53 mRNA-up-regulating effects of an EP2 receptor-specific agonist, butaprost. Levels of p53 mRNA in activated cultures exposed on d 4 to butaprost (or ethanol vehicle) were assessed at varying intervals after the pulse. qPCR fold Δ values from experiments with a common harvest interval following the pulse were pooled and expressed as means ± se. P values indicate that the level of p53 mRNA in butaprost-pulsed cultures is significantly different from that in control-pulsed cultures. C) Dose response of the p53 mRNA-up-regulating effects of butaprost (single experiment with all doses tested).
PGE2 promotes increased p53 mRNA transcription but not greater mRNA stability
A series of experiments was performed to discern whether the PGE2-mediated boost in p53 mRNA was explained by effects on transcription and/or mechanisms that stabilize message (55–57). As a first step, we asked whether RNA polymerase activity is needed for sustained p53 mRNA levels in proliferating d 4 cultures. Figure 5 shows that this is so: p53 mRNA levels dropped significantly following 8 h exposure to actinomycin D. In addition, treatment with actinomycin D just prior to the PGE2 pulse abrogates the PGE2-induced increase in p53 mRNA.
Figure 5.

Inhibition of RNA polymerase down-regulates baseline p53 mRNA and blocks PGE2-promoted increases in p53 mRNA. B cells were activated with BCR:CD21-L, IL-4 and BAFF. After d 4 of culture, 1 set of replicate cultures received a pulse with 5 μM actinomycin D (42), while another set received vehicle alone. After a period of 15 min, a subset of each of the above were subsequently pulsed with PGE2 (50 nM) or vehicle alone and cultured for an additional 8 h. Cultures were harvested, RNA was isolated, cDNA was prepared, and levels of p53 transcript were assessed by qPCR. Mean ± se fold Δ values are shown as in Fig. 2, for a total of 4 experiments.
While the above results indicate that p53 transcription is required during the postpulse period, the PGE2-mediated boost might reflect effects on mRNA stability, rather than transcription. To address this, mRNA decay rates were compared in PGE2-pulsed and parallel nonpulsed cultures, beginning at 8 h after the pulse (Fig. 6). Any new transcription was blocked by treatment with actinomycin D. Pooled values from 4 replicate experiments show that the half-life of p53 message is ∼8 h. Notably, the decay rates (slope of plots over time) in PGE2+ and PGE2− cultures were not statistically different (Fig. 6). Thus, PGE2 does not promote greater p53 mRNA stability.
Figure 6.

PGE2-promoted rise in p53 mRNA is not explained by PGE2-induced mRNA stabilization. In order to assess whether p53 mRNA stability was affected by a pulse of exogenous PGE2, sets of replicate cultures were pulsed with PGE2 (50 nM) or vehicle alone. After 8 h of additional culture (for peak PGE2-induced elevation in p53 mRNA; Fig. 3), a series of replicates within each of the above subsets received actinomycin C (5 μM) or DMSO vehicle. RNA was isolated at varying time points after the actinomycin D or DMSO (0.5, 1, 3, 8, and 20 h). q-PCR of cDNA measured p53 transcript levels in each culture, expressed as a fraction of that present in the respective PGE2-untreated or PGE2-treated cultures with DMSO vehicle (means±se of 4 experiments). Dotted line represents the normalized level of transcript present in cultures with or without PGE2 but without actinomycin D. Data were statistically analyzed (see Materials and Methods) to compare mean fold change of p53 mRNA between PGE2-pulsed and nonpulsed cultures to their respective controls across time after actinomycin D. There was no significant interaction between time and presence of PGE2 (P<0.72), indicating that the slope of p53 mRNA expression after actinomycin D (mRNA decay) was not significantly different with or without PGE2. Nevertheless, when the interaction term was removed from the model, the main effects of cell type and time were significant. Cells without PGE2 supplement had a significantly greater p53 mRNA, relative to their controls, as compared to those pulsed with PGE2 (P<0.0001). Overall, p53 mRNA expression decreased over time for all cells relative to controls (P<0.0002). Taken together, these experiments show that the elevated p53 mRNA levels in PGE2-prepulsed cultures do not reflect diminished degradation of the p53 transcript.
Unexpectedly, p53 mRNA levels in actinomycin D-treated PGE2+ cultures were consistently lower than those in parallel PGE2− cultures. This was most evidenced early after the block in RNA synthesis (Fig. 6). One possibility is that PGE2-induced signals concomitantly enhance synthesis of negative regulators of p53 transcript stability, e.g., miR125a and miR125b (58), but further experiments are needed to resolve this.
Single-cell p53 RT-PCR shows that PGE2 signals heighten the frequency of dividing lymphoblasts with detectable p53 mRNA
Single-cell p53-specific RT-PCR analyses were initiated for the purpose of discerning whether PGE2 increases the proportion of cells with a transcriptionally active p53 gene or whether it uniformly elevates p53 mRNA transcription in all cells. Figure 7A shows representative results from the PCR amplification of p53 and β-actin cDNA from 56 sorted individual B-cell progeny from TI cultures with or without PGE2. While ∼90% of the wells containing a single B cell scored positive for β-actin, the frequency of p53+ wells was notably lower, indicating that individual progeny vary in their level of p53 transcripts. Of interest, the exogenous PGE2 pulse notably augmented the proportion of p53+ single cells (Fig. 7B). This PGE2-boosting effect was observed in each of 6 total experiments and was highly significant (P<0.001; Fig. 7C). Thus, PGE2 clearly amplifies the proportion of B lymphoblasts producing relatively high levels of p53 mRNA. Since a threshold level of p53 mRNA may be necessary for p53 amplification (detection), we cannot exclude the possibility that PGE2 positively influences p53 mRNA transcription in all progeny.
Figure 7.
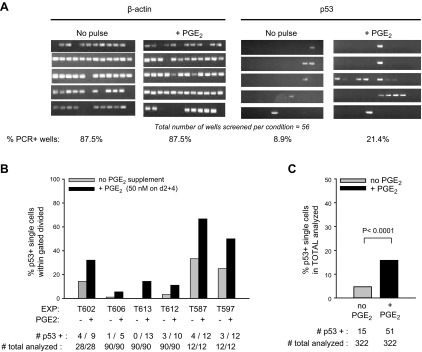
Single-cell RT-PCR shows that PGE2 augments the frequency of cells with detectable p53 mRNA. A) Single-cell RT-PCR was performed with sorted CFSE-labeled lymphoblasts (representing 3–4 divisions) from d 5 activated cultures (or d 6 for 1 experiment) with or without exogenous PGE2 (50 nM) on d 2 and 4. Shown are amplicons of β-actin and p53 from a total of 56 single cells analyzed from cultures with or without PGE2 supplement. While most wells with a single sorted cell were positive for β-actin mRNA, notably fewer were positive for p53 mRNA. B) Data from 6 separate experiments, each evaluating the frequency of p53 mRNA+ cells, as above. Below each set of bars, representing frequency of p53+ single cells within the sorted dividing population in cultures with or without PGE2, are the absolute values for number of p53 mRNA+ cells per total cells analyzed. (Note that in experiments T613, T612, T587, and T597, all cultures received a Z-VAD pulse on d 3.5 following activation; experiments T602 and T606 did not). C) Summarized data from the pooled experiments in B were subjected to statistical evaluation using a 2 × 2 contingency table and Fisher's exact test. The difference in frequency of p53 mRNA+ cells within sampled individual cells of cultures exposed/not exposed to exogenous PGE2 was highly significant (2-sided P<0.0001).
Multiple alternatively spliced transcripts of p53 are reported (59). Because identification of p53 mRNA+ cells in Fig. 7 involved primers for exons 2 and 4, we were concerned that the effects of PGE2 might be restricted to certain splice forms. Nevertheless, results from an early experiment suggest otherwise. PGE2 increased the frequency of p53 mRNA+ cells regardless of whether PCR amplification involved primers spanning p53 exons 2–4 (region A), the 3′ end of exon 4 through the 5′ end of exon 7 (region B), or exons 6–11 (region C). Within this experiment, ratios for frequency of p53+ cells with or without added PGE2 using primers for regions A, B, and C were 1.8, 4.0, and 2.0, respectively.
Single-cell RT-PCR reveals p53 mutations within occasional progeny of a TI response to BCR:CD21-L, IL-4, and BAFF
The prospect that elevated p53 transcription during a period of active replication and high AID expression might foster p53 mutations led us to sequence the purified p53 amplicons from each positive lymphoblast. We focused on upstream exons 2 through 4 because AID-induced mutations are more frequent near transcription start sites (60). This screen revealed p53 mutations, but only in the progeny of 2 of 6 experimental cultures, each involving distinct B-cell donors (Table 1). The total frequency of p53 mutations in cultures with or without exogenous PGE2 (average of 6.2×10−4 mutations/bp sequenced) was greater than an estimated background amplification error rate of <0.8 × 10−4/bp in our assays (Table 1).
Table 1.
Analysis of p53 mutation frequency in progeny of TI B-cell clones stimulated by BCR:CD21-L, IL-4, and BAFF with or without supplementary PGE2
| Experiment | Donor B cells cultureda | Total cells screened |
Total cells p53 seq+ |
Total nucleotides screenedb |
Total mutations detected |
Mutation frequency/base pairc |
Mutation frequency/celld |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No pulse | +PGE2 | No pulse | +PGE2 | No pulse | +PGE2 | No pulse | +PGE2 | No pulse | +PGE2 | No pulse | +PGE2 | ||
| 1 | T602 | 28 | 28 | 4 | 9 | 1320 | 2970 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | T606 | 90 | 90 | 1 | 5 | 330 | 1650 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | T612 | 90 | 90 | 3 | 10 | 990 | 3300 | 2 | 4 | 20 × 10−4 | 12 × 10−4 | 0.022 | 0.044 |
| 4 | T613 | 90 | 90 | 0 | 13 | 0 | 4290 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | T587 | 12 | 12 | 4 | 8 | 840 | 1680 | 0 | 0 | 0 | 0 | 0 | 0 |
| 6 | T597 | 12 | 12 | 3 | 6 | 630 | 1260 | 3 | 3 | 48 × 10−4 | 24 × 10−4 | 0.25 | 0.25 |
| Total | 322 | 322 | 15 | 51 | 4110 | 15,150 | 5 | 7 | 12 × 10−4 | 5 × 10−4e | 0.015 | 0.022e | |
Based on our prior findings that PGE2 can have both prosurvival and proapoptotic functions on B lymphoblasts, in part dependent on PGE2 dose (34), cultures were monitored for viability during sorting of PGE2-pulsed/nonpulsed single cells for p53-specific RT-PCR. Percentage viability in the d 5 cultures (excepting T612 on d 6) at time of sorting was T602 = 67/74% with/without PGE2, respectively; and similarly, T606 = 72/73%; T612 = 36/40%; T613 = 58/60%; T587 = 64/63%; T597 = 65/68% [mean of 60±5 vs. 63±5% viability in PGE2− and PGE2+ cultures, respectively; P=0.06 (nonsignificant)]. There is a high likelihood that PGE2 prosurvival effects were minimized due to the relatively high cell density of the 24-well cultures from which the sorted cells originated (106 cells/2 ml). Notably, other analyses involving cells cultured at varying densities (0.01–0.4×106 cells/0.2 ml in 96-well plates) show that supplementary PGE2 boosts lymphoblast viability only in cultures of low to moderate cell density (unpublished results). This may reflect sufficient levels of autocrine PGE2 at higher cell density. It is also possible that at the higher culture density, prosurvival effects of PGE2 on one subset of dividing blasts are masked by opposing proapoptotic effects on another subset. Consistent with the latter, viable cells with PGE2-up-regulated levels of phospho-H2AX are prominent only when cultures are pulsed with the pan-caspase inhibitor Z-VAD (see Fig. 8). In an effort to reduce cell death, cultures T612, T613, T587, and T597 received Z-VAD (40 μm) on d 3.5–4 following activation. Nevertheless, as compared to previously noted significant prosurvival effects of Z-VAD in less dense cultures (within 96-well plates; ref. 24), significant apoptosis remained. The less-than-expected Z-VAD function might reflect limited Z-VAD infiltration into the substantially larger clusters of activated cells present in the 24-well cultures seeded with 106 cells, a need for counteracting caspase function at an earlier interval in these cultures, or alternatively, activation of non-caspase-mediated death.
In experiments 1–4, single-cell amplicons from cDNA were generated using primers for exon 2 and exon 4 (400-bp amplicon), whereas in experiments 5 and 6, amplicons were generated using primers for exon 4 alone (280-bp amplicon; 199 bp were common to both). In sequencing these amplicons, the most 5′ nucleotides were not considered due to sequence noise near the binding region for the sequencing primer.
Determining the background rate of p53 mutation in unstimulated cells is not possible due to insufficient recovery of p53 mRNA for this single-cell assay. Nevertheless, a background rate based on the frequency of introduced mutations from 2× PCR amplification of the p53 exon segment from cDNA and 1× amplification for sequencing was assessed by calculating the total nucleotides sequenced in the 4 of 6 experiments that exhibited no p53 mutations. This indicates that background error rate is <0.8 × 10−4/bp.
In an effort to avoid the bias of calculating mutation frequency on the basis of only cells with high detectable levels of p53 mRNA, values for mutation frequency per number of single cells screened were determined. While this assessment is compromised by the fact that most cells screened did not display sufficient p53 mRNA for evaluation, the calculated frequencies might be more reliable given that it is highly likely that those cells with low p53 mRNA transcription are unlikely candidates for mutation.
There was no significant association between the frequency distribution of mutations (per base pair) and culture (PGE2+ vs. PGE2−); P < 0.15 by Fisher's exact test. In addition, there was no significant association between the frequency of mutations (per cell) and culture (PGE2+ vs. PGE2−); P < 0.56 by χ2 analysis.
Somewhat surprisingly, when divided progeny of PGE2-pulsed and control cultures were compared for p53 mutations, we found no evidence that p53 mutations were more frequent in cultures pulsed with PGE2 (Table 1, columns 11–14). This was noted on the basis of mutations per total p53 mRNA+ cells sequenced (5/15 vs. 7/51 for nonpulsed vs. PGE2-pulsed cultures, respectively) as well as mutations per total cells under study (Table 1, columns 13, 14).
There is a plausible reason why PGE2-pulsed cultures did not reveal heightened p53 mutagenesis despite the concomitant elevation in AID protein and p53 transcription. Cells with PGE2-promoted DNA alterations (including p53 mutations) might have been excluded from analysis due to prior DNA damage-induced death. The data in Fig. 8 support this explanation. Notably, viable-gated blasts of PGE2-pulsed cultures expressed higher pH2AX (a measure of DNA double-strand breaks; ref. 24) than did control blasts, but only in the presence of pan-caspase inhibitor Z-VAD. This reinforces past suggestions that PGE2 promotes DNA damage in TI-activated B lymphoblasts (35) and shows that those with DNA double-strand breaks are prone to death. Thus, if p53-mutated cells were enriched in the subset with high levels of DNA damage, they would have been deleted, unless, of course, the mutation promoted survival.
Figure 8.
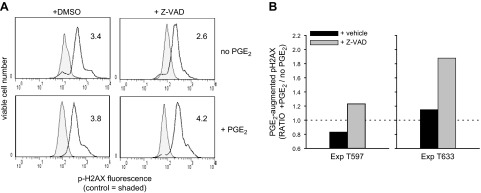
Divided progeny from PGE2-pulsed cultures manifested higher pH2AX staining (indicative of DNA double-strand breaks) when cultures were treated with pan-caspase inhibitor, Z-VAD. CFSE-labeled B cells were activated in 96-well plates (105 cells/200 μl), and activated cultures received PGE2 (50 nM), as indicated, on d 2 and 4 and 40 μM Z-VAD (or DMSO vehicle) on d 4. On d 5, cultures were harvested, fixed, and stained intracellularly with PE-anti-pH2AX (or PE-IgG control). PE MFI was determined for cells whose light scatter and CFSE fluorescence represented the viable, divided subset prior to fixation. A) Numbers represent the RMFI (ratio of pH2AX MFI/MFI IgG control MFI). B) Bars represent the ratio of the RMFI in PGE2-supplemented cultures/RMFI in nonsupplemented cultures of 2 experiments.
Next, we closely examined the type of p53 mutations in these TI cultures, both as a whole and with or without supplementary PGE2. Although numbers were limited, as seen in Table 2, a majority of the total mutations are missense, resulting in amino acid changes. These include changes in proline and cytosine that could alter protein structure. In addition, transition mutations (83% of total) significantly outnumber transversions under all conditions (Fig. 9 and Table 3; P=0.04). Both the latter finding and several characteristics concerning mutation location and type are consistent with a role for AID in p53 mutagenesis within TI-activated clones. First, the majority of p53 mutations are localized at cytosine (C) or guanine (G) residues (50% for mutated cells in experiment 3 and 83% for mutated cells in experiment 6; average=67%; Fig. 9A). Nevertheless, when corrected for total C or G sites available for mutation, this difference is not statistically significant (P=0.48). Due to a high GC/AT ratio in this region, the fraction of mutations per total C/G sites and per total A/T sites is 0.00097 and 0.00082, respectively. Second, AID-characteristic C → T and G → A transition mutations that can represent direct replication of a U:G lesion (61–63) are frequently observed: in experiment 6 (T597), 75% of total were C → T; in experiment 3 (T612), 33% of total were G → A. Third, the C → A transversion, which can occur on uracil DNA glycosylase (UNG) excision of U and replication of the resulting abasic site (62, 63) represents 17% of the total mutations. Fourth, all of the A/T mutations (2 A→G and 1 T→C in experiment 3; 1 T→C in experiment 6) are noted proximal to a C/G. These are consistent with the recognition of an AID-induced U:G lesion by an atypical mismatch repair (MMR) pathway and resulting DNA polymerase-dependent A/T mutation (63–65). Interestingly, PGE2-pulsed cultures have a statistically lower frequency of C/G-targeted mutations (P=0.018; Table 3) and favor A/T-targeted mutations (Fig. 9), albeit the latter bias is not of statistical significance (Fig. 9A and Table 3). A final attribute suggesting AID activity (66) is that several cells had >1 mutation (Fig. 9). Of the 4 mutated cells in PGE2-pulsed cultures, 50% had multiple mutations in exon 4: 1 had 3 mutations (in residues 365, 458, and 497), and the other had 2 mutations (in residues 462 and 483). Of the 4 mutated cells in nonpulsed cultures, only 1 (25%) had multiple mutations (in residues 363 and 442). Nevertheless, due to insufficient statistical power, no conclusions could be drawn concerning the effects of supplementary PGE2 on multiplicity of mutations.
Table 2.
Frequency of codon 4 missense p53 gene mutations in TI-activated human B cells
| Mutated B cell | PGE2 pulse | Total | Silent | Missense | Location |
|---|---|---|---|---|---|
| T612, experiment 3 | |||||
| Cell A | No | 2 | 0 | 2 | Codon 56 (E→K); codon 82 (P→Q) |
| Cell B | Yes | 1 | 1 | 0 | |
| Cell C | Yes | 3 | 2 | 1 | Codon 96 (S→P)a |
| T597, experiment 6 | |||||
| B3-cell A | No | 1 | 1 | 0 | |
| B4-cell B | No | 1 | 1 | 0 | |
| B5-cell C | No | 1 | 0 | 1 | Codon 72 (R→C)b |
| D2-cell D | Yes | 1 | 0 | 1 | Codon 75 (P→L) |
| D5-cell E | Yes | 2 | 0 | 2 | Codon 89 (P→T); codon 96 (S→P)a |
The codon 96 S→P missense mutation was observed twice, in PGE2-pulsed cells of 2 distinct donors, from differing experiments. This mutation occurred in a MHT motif, which can be targeted during the secondary phase of AID-driven mutagenesis (65).
This mutation occurred in exon 4 codon 72, the site of a common functionally relevant SNP in humans. This cell reverted from expression of codon 72-Arg to codon 72-Cys.
Figure 9.
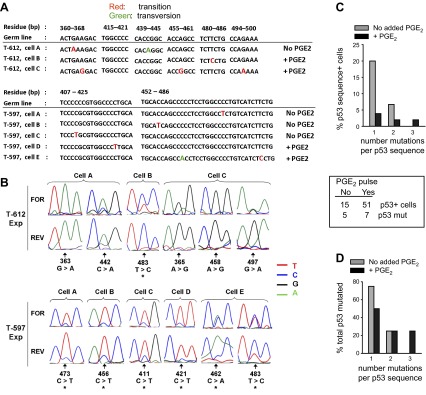
p53 mutations are present within dividing lymphoblasts of a TI response to BCR:CD21-L, IL-4, and BAFF with or withouth supplementary PGE2. A) p53 amplicons from experiments in Fig. 7 were sequenced (see Materials and Methods). Shown are sequenced segments with identified mutations from 8 distinct cells from 2 donors, as well as the corresponding germline p53 sequence (numerical positions represent residues of germline DNA beginning at the transcriptional start site). Transition mutations are in red; transversion mutations are in green. B) Segments of forward (FOR) and reverse (REV) p53 sequence chromatograms indicating the mutations detected per individual cell. Arrows indicate mutated residues. Asterisks indicate cells in which both the mutation (predominantly expressed) and the original wild-type sequence (minor) were coexpressed. Coexpression was validated by similar results on sequencing in either direction. C) Summarized data showing the percentage of total p53 sequence-positive single cells that expressed 1, 2, or 3 mutations/sequence. D) Normalized data from panel C, showing the percentage of total p53-mutated cells that expressed 1, 2, or 3 mutations/sequence. For both C and D, statistical power is too low to determine significance.
Table 3.
Characterization of exon 4 p53 mutations in progeny of human B clones following TI activation with/without supplementary PGE2
| Mutation type | Mutations [sites (total possible sites)] |
Mutation frequency |
P | ||
|---|---|---|---|---|---|
| No pulse | +PGE2 | No pulse | +PGE2 | ||
| At G | 1 (705) | 1 (2397) | 0.00142 | 0.00042 | 0.403 |
| At C | 4 (1170) | 2 (3978) | 0.00342 | 0.00050 | 0.027* |
| At A | 0 (540) | 2 (1836) | 0 | 0.00109 | 1 |
| At T | 0 (570) | 2 (1938) | 0 | 0.00103 | 1 |
| Sum: G/C | 5 (1875) | 3 (6375) | 0.00267 | 0.00047 | 0.018* |
| Sum: A/T | 0 (1110) | 4 (3774) | 0 | 0.00106 | 0.580 |
| Total transitions | 4 (2985) | 6 (10,149) | 0.00134 | 0.00059 | 0.249 |
| Total transversions | 1 (2985) | 1 (10,149) | 0.00034 | 0.00010 | 0.403 |
| Total SYC/GRS (cold spot)a | 2 (630) | 2 (2142) | 0.00460 | 0.00135 | 0.224 |
| Total RGYW/WRCY (hot spot)a | 0 (255) | 0 (867) | 0 | 0 | NA |
| Total WA | 0 (150) | 1 (510) | 0 | 0.00196 | 1 |
| Total TW | 0 (150) | 0 (510) | 0 | 0 | NA |
| Total MHTa | 0 (255) | 2 (867) | 0 | 0.00231 | 1 |
Comparative analysis of all p53 sequences from single cells of nonpulsed and PGE2-pulsed cultures derived from 6 diverse donors was performed with the web-based SHMTool (http://scb.aecom.yu.edu/shmtool). This permits determinations of the frequency of various categories of somatic hypermutation on the basis of the base composition and total potential sites of each category available for mutation. For the above comparisons, a stretch of 199 exon 4 p53 residues that were commonly sequenced in all experiments was used. While the low frequency of mutations did not meet the minimal conditions for statistical reliability using the program's χ2 analysis, a separate statistical comparison was made with Fisher's exact test.
Letter designations for alternate nucleotides: R = A/G; S = G/C; Y = C/T; W = A/T; M = A/C; H = A/C/T (61, 65).
P < 0.05 for mutation frequency in pulsed vs. nonpulsed cohort; Fisher's exact test.
Also, some attributes incompatible with AID involvement were observed. First, the observed C/G targeted changes were not found at the C/G residues in RGYW/WRCY motifs known to be AID hot spots (refs. 8, 61, 67; Fig. 9A; and Table 3). One mutation (A→G at residue 458) was detected in the W residue of a WRCY motif, however. The latter occurred in a PGE2-pulsed B cell. Second, 3 of 4 C → T mutations found in exon 4 within experiment 6 localized at AID cold spots, SYC/GRS, in which S = G or C and Y = pyrimidine (C/T; ref. 61; Fig. 9A, B; and Table 3). Nevertheless, when the probability of targeting a cold spot over a hot spot in all sequenced cells was examined by considering the total available sites of each (42 cold spots and 17 hot spots within each commonly sequenced 199-nt span), there was no statistical difference (P=0.31). Mutations in cold spots are more consistent with the mutagenic activity of ROS, which often induces C → T transitions (68). Thus, a combination of AID-driven and non-AID (possibly ROS)-driven processes appear to be present in these TI-activated B lymphoblasts.
An intriguing finding emerged through sequencing cDNA of single cells through p53-specific RT-PCR. In several instances, both a major peak representing the p53 mutated residue and a minor peak representing the wild-type residue were observed within the same sequence chromatograms (Fig. 9B, cells designated by asterisk), and the possible implications of their presence are discussed below.
DISCUSSION
The p53 gene is a relatively rare candidate for p53 mutation during TD B-cell GC responses due in part to strong repression of p53 gene transcription by Bcl-6 (2). Nevertheless, results from the present study suggest that this is not the case for B-cell clonal expansion elicited by TI stimuli. Within dividing lymphoblasts elicited by TI stimuli typical of inflamed tissues (14–21), the p53 gene is transcriptionally active. Furthermore, sequencing of p53 RT-PCR amplicons from single blasts shows that p53 mutations arise at a relatively high overall frequency, average of ∼6.2 × 10−4 mutations per sequenced base pair. This is similar to that in human peripheral blood mononuclear cells (PBMCs) exposed to UV-irradiated hepatitis C virus, 4.6 × 10−4 mutations/bp (69), but significantly greater than in normal PBMC, 0.3 × 10−4 mutations/bp (69), and in mouse Peyer's patch GC B cells, 0.08 × 10−4 mutations/bp (8).
While the average p53 mutation rate was derived from experiments with 6 separate B-cell donors, the mutations were concentrated in the lymphoblasts of only 2 donors. Thus, the average underrepresents the mutation frequency in certain activated B-cell cultures: range of 12–48 × 10−4 mutations/bp. The reason for the variation between donors is unknown, but might involve donor epigenetic or genetic variability influencing AID activation, level of generated ROS, and/or DNA-repair enzymes. Notably, this variation does not reflect differences in the frequency of single cells scoring as p53 mRNA+. In addition, it does not reflect degree of activation, since the proliferative responses to C3d-Ag, IL-4, and BAFF were comparable in all experiments (data not shown). In addition, it seems highly unlikely that the replicating blasts with p53 mutations represented preacquired mutations in the original resting B cells. We base this on the following points: the enrichment procedure yields cells that are ≥97% IgD+ and IgM+, the strong proliferative response requires signaling via IgM (36), and no mutation was observed more than once in a given donor. Finally, it is improbable that mutations in 2/6 of the B-cell cultures represent errors during cDNA preparation or p53 amplification. The conditions for cDNA preparation and p53 RT-PCR amplification were uniform. Most notably, significant differences in mutation incidence were noted in cultures of 2 donors (T587 and T597) that were simultaneously processed for cDNA preparation, p53 RT-PCR amplification, and sequencing. Rather, our collective findings suggest that p53 mutations are more apt to develop in some backgrounds than others during this TI response.
The p53 mutations observed had characteristics consistent with either AID or ROS activity: transitions > transversions, with many transitions representing C → T. In support of AID function are the strong expression of AID in replicating blasts, seen here and previously (35); the targeting of all mutations to either C/G or to A/T proximal to C (consistent with replication of a AID-introduced U:G lesion or activity of an error-prone MMR-like pathway, respectively; refs. 62, 63, 70–72); and the discovery that several cells had 2 or 3 p53 mutations within a relatively short span of DNA, characteristic of the “processive” function of AID (66). Nevertheless, in 1 donor, C → T nucleotide changes were evidenced in AID cold spots, more compatible with ROS activity. Thus, these experiments could not attribute the expressed mutations to solely AID and suggest that multiple mechanisms are involved.
Of interest, the sequence chromatograms of several mutated cells revealed 2 mutated nucleotides per residue: the major mutated nucleotide and a more minor peak of the wild-type nucleotide. We suspect the latter may have originated from residual wild-type mRNA in the mutant cell. This explanation is consistent with recent mutation during the proliferative burst and the ∼8 h half-life of p53 mRNA in these cells. It appears less likely that the presence of both mutant and wild-type residues represents mRNA from the 2 p53 alleles of a single cell: one mutated and the other not. Of pertinence, single-cell RT-PCR of cultures from donors heterozygous for the common p53 codon 72 C → G single nucleotide polymorphism (SNP) show that allelic exclusion is prominent in these activated blasts: a single cell expressed either the p53-72C SNP or the p53-72G SNP, but not both (unpublished results). While the mutant and original p53 residue appeared to be commonly observed in cells bearing 1 mutation, in the case of 2 of 3 cells bearing multiple mutations, coexpression was suggested only for the most downstream mutation (Fig. 9B). This finding is consistent with sequential mutations being acquired during differing division cycles (66, 73). In 1 of 3 multiply mutated cells, mutant and original alleles were expressed in both mutated p53 residues, suggesting that the cell rapidly acquired these mutations within the same division cycle. It might be relevant that this cell (D5) derived from a culture receiving AID-up-regulating supplementary PGE2 (Fig. 9B and ref. 35).
A novel and potentially quite important finding of this study was that that PGE2, produced at low levels by activated B cells and in higher amounts by other inflammatory cells, significantly augments p53 mRNA transcription in activated blasts. This was demonstrated through a series of experiments involving the RNA polymerase inhibitor actinomycin D. To our knowledge, there is no prior evidence of PGE2-modulated p53 gene transcription, albeit PGE2 is reported to prolong the half-life of p53 protein through serine-15 phosphorylation (74). Mice globally overexpressing COX-2 during embryogenesis exhibit a prostaglandin-mediated, tissue-specific rise in p53 protein that occurs independently of p53 serine 15 phosphorylation (49). Whether the increased p53 protein represented elevated p53 gene transcription was not examined. Interestingly, within activated d 4–5 cultures, only the replicating blasts responded with increased p53 mRNA synthesis to PGE2. Similar augmenting effects of the selective agonist butaprost have implicated the cAMP-elevating EP2 receptor.. Our earlier findings that EP2 levels correlate directly with the frequency of divisions (35) provides a likely explanation for why the divided blasts appear to be more receptive to PGE2 signaling.
Notably, there was no statistically sound evidence that cells with PGE2-augmented p53 gene transcription had a greater frequency of p53 mutations. Nevertheless, findings of elevated DNA double-strand breaks in cultures pulsed with PGE2 (when a pan-caspase inhibitor was added to block cell death) suggest an explanation. At the time of viable lymphoblast sorting for single-cell RT-PCR and sequencing, many cells experiencing PGE2-promoted DNA changes (including p53 mutations) may have been omitted from analysis due to their recent death. In an effort to better understand whether PGE2 signaling promotes p53 mutagenesis in the absence of T-cell-dependent high-fidelity DNA repair (37), future studies will involve additional steps to more effectively block the activation-induced death characteristic of these TI clones.
The downstream mechanisms for PGE2-boosted p53 gene transcription are presently unclear, but there are several options to consider. The human p53 basal promoter upstream of exon 1 has an NF-kB motif that binds p50 and p65, as well as sites for AP-1 and Myc/Max (58, 75, 76) and is reported to be transactivated by PKCδ, in combination with Btf (77, 78). In mice, a site upstream of the basal promoter mediates c/EBPβ-regulated p53 transcription (79). Notably, there is evidence that cAMP regulates most of the above transcription factors (80–88). PGE2 might also influence p53 gene transcription in an epigenetic manner through methylation, given that cAMP-up-regulating PGE2 receptors, EP2 and EP4, regulate the expression of DNA methyl transferases (89, 90).
The above study probed for p53 gene mutations in upstream exons 2–4 on the basis of their proximity to the transcriptional start site, which should favor targeting by AID (60). However, mutations in this region are uncommon in malignancies, even within the B-cell lineage, and mutations in downstream exons encoding the DNA-binding domain are highly favored (9, 91–94). Nonetheless, exon 4 appears targeted for mutation in soft-tissue sarcomas (95), and occasional exon 4 mutations have been reported in B-CLL and high-grade non-Hodgkin's B-cell lymphomas (96, 97). While the exon 4 missense mutations revealed in this study may not be transforming, they may be functionally relevant. First, the proline-rich exon 4 domain promotes p53 interactions with other cell proteins (98–100). Second, exon 4 is important for transcription-independent p53-apoptosis, albeit not cell cycle block (101–103). Third, the common p53 exon 4 SNP in humans, which encodes Pro vs. Arg at codon 72, can significantly alter p53 function (104–108). Finally, the p53 molecule has a multitude of roles that extend beyond the regulation of cell cycle turnover and apoptosis (104, 109–114). Thus, p53 mutations that do not advance malignancy might, nevertheless, affect other B cell roles, e.g., in regulating inflammation.
In sum, when assimilated with prior observations, this study's novel findings have important implications for the process of p53 mutagenesis in vivo. They suggest that when recirculating naive B cells encounter the above synergistic TI stimuli within inflamed tissues, a short burst of proliferation will ensue. This will generate lymphoblasts expressing several COX-2 axis proteins (COX-2, mPGES-1, and EP2). Together with supplementary PGE2 from nearby inflammatory cells, this can promote expression of DNA-modifying AID (35), heighten p53 gene transcription (this study), and provide positive feedback signaling for elevated COX-2 and EP2 expression (refs. 35, 115 and unpublished results). Notably, if antigen-specific T-cell help is lacking, blasts will be deficient in p53-repressive Bcl-6 (2) and high-fidelity DNA repair enzymes (37). Together, these factors should significantly increase the likelihood that p53 mutations develop and are retained. While only a minority of the induced p53 mutations may be functionally relevant, those with a positive influence on cell survival and growth may undergo positive selection. This is particularly so given the high propensity of TI clones to succumb to p53-driven activation-induced cell death (24, 34, 36). Furthermore, the observations that p53 mutations in nontransformed synovial fibroblasts of rheumatoid arthritis patients are linked to elevated IL-6 production (116) suggest that occasional p53 mutations might promote the proinflammatory properties of B cells.
Supplementary Material
Acknowledgments
This study was supported by funds from The Karches Foundation, The Prince Family Foundation, and The Marks Foundation; U.S. National Institutes of Health (NIH) grant R21 AR061653 (to P.K.A.M.), and NIH M01 General Clinical Research Center grant RR01853.
The authors thank Hyunjoo Lee and Jennifer Nieto for their skilled technical assistance; Herb Borrero for cell sorting; and Dr. Carlo Callissano for early help with single-cell RT-PCR. The authors also thank Dr. Betty Diamond and Dr. Michael Brunda for reviewing the manuscript and Dr. Thomas MacCarthy for helping to implement the web-based SHMTool (http://scb.aecom.yu.edu/shmtool). The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- AID
- activation-induced cytosine deaminase
- B-CLL
- B-cell chronic lymphocytic leukemia
- BCR
- B-cell antigen receptor
- CFSE
- carboxyfluorescein succinimidyl ester
- COX-2
- cyclooxygenase
- FO
- follicular
- GC
- germinal center
- MZ
- marginal zone
- PBMC
- peripheral blood mononuclear cell
- qPCR
- quantitative PCR
- ROS
- reactive oxygen species
- SNP
- single nucleotide polymorphism
- TD
- T-cell dependent
- TI
- T-cell independent
REFERENCES
- 1. Lane D. P. (1992) Cancer p53, guardian of the genome. Nature 358, 15–16 [DOI] [PubMed] [Google Scholar]
- 2. Phan R. T., Dalla-Favera R. (2004) The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 432, 635–639 [DOI] [PubMed] [Google Scholar]
- 3. Huang C., Hatzi K., Melnick A. (2013) Lineage-specific functions of Bcl-6 in immunity and inflammation are mediated by distinct biochemical mechanisms. Nat. Immunol. 14, 380–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bachl J., Carlson C., Gray-Schopfer V., Dessing M., Olsson C. (2001) Increased transcription levels induce higher mutation rates in a hypermutating cell line. J. Immunol. 166, 5051–5057 [DOI] [PubMed] [Google Scholar]
- 5. Wright B. E., Schmidt K. H., Hunt A. T., Lodmell J. S., Minnick M. F., Reschke D. K. (2011) The roles of transcription and genotoxins underlying p53 mutagenesis in vivo. Carcinogenesis 32, 1559–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fukita Y., Jacobs H., Rajewsky K. (1998) Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity 9, 105–114 [DOI] [PubMed] [Google Scholar]
- 7. Basic-Zaninovic T., Meschini R., Calcagnile A. S., Palombo F., D'Errico M., Proietti-De Sanctis L., Dogliotti D. (1995) Strand bias of ultraviolet light-induced mutations in a transcriptionally active gene in human cells. Mol. Carcinog. 14, 214–225 [DOI] [PubMed] [Google Scholar]
- 8. Liu M., Duke J. L., Richter D. J., Vinuesa C. G., Goodnow C. C., Kleinstein S. H., Schatz D. G. (2008) Two levels of protection for the B cell genome during somatic hypermutation. Nature 451, 841–845 [DOI] [PubMed] [Google Scholar]
- 9. Gaidano G., Ballerini P., Gong J. Z., Inghirami G., Neri A., Newcomb E. W., Magrath I. T., Knowles D. M., Dalla-Favera R. (1991) p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. U. S. A. 88, 5413–5417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garcia de Vinuesa C., O'Leary P., Sze D. M., Toellner K. M., MacLennan I. C. (1999) T-independent type 2 antigens induce B cell proliferation in multiple splenic sites, but exponential growth is confined to extrafollicular foci. Eur. J. Immunol. 29, 1314–1323 [DOI] [PubMed] [Google Scholar]
- 11. Hsu M. C., Toellner K. M., Vinuesa C. G., Maclennan I. C. (2006) B cell clones that sustain long-term plasmablast growth in T-independent extrafollicular antibody responses. Proc. Natl. Acad. Sci. U. S. A. 103, 5905–5910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herlands R. A., Christensen S. R., Sweet R. A., Hershberg U., Shlomchik M. J. (2008) T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity 29, 249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sweet R. A., Ols M. L., Cullen J. L., Milam A. V., Yagita H., Shlomchik M. J. (2011) Facultative role for T cells in extrafollicular Toll-like receptor-dependent autoreactive B-cell responses in vivo. Proc. Natl. Acad. Sci. U. S. A. 108, 7932–7937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Groom J., Kalled S. L., Cutler A. H., Olson C., Woodcock S. A., Schneider P., Tschopp J., Cachero T. G., Batten M., Wheway J., Mauri D., Cavill D., Gordon T. P., Mackay C. R., Mackay F. (2002) Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjögren's syndrome. J. Clin. Invest. 109, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saxena A., Alport E. C., Moshynska O., Kanthan R., Boctor M. A. (2004) Clonal B cell populations in a minority of patients with Hashimoto's thyroiditis. J. Clin. Pathol. 57, 1258–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klein R. D., Van Pelt C. S., Sabichi A. L., Dela Cerda J., Fischer S. M., Furstenberger G., Muller-Decker K. (2005) Transitional cell hyperplasia and carcinomas in urinary bladders of transgenic mice with keratin 5 promoter-driven cyclooxygenase-2 overexpression. Cancer Res. 65, 1808–1813 [DOI] [PubMed] [Google Scholar]
- 17. Aloisi F., Pujol-Borrell R. (2006) Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 6, 205–217 [DOI] [PubMed] [Google Scholar]
- 18. Bende R. J., van Maldegem F., van Noesel C. J. (2009) Chronic inflammatory disease, lymphoid tissue neogenesis and extranodal marginal zone B-cell lymphomas. Haematologica 94, 1109–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Puga I., Cols M., Barra C. M., He B., Cassis L., Gentile M., Comerma L., Chorny A., Shan M., Xu W., Magri G., Knowles D. M., Tam W., Chiu A., Bussel J. B., Serrano S., Lorente J. A., Bellosillo B., Lloreta J., Juanpere N., Alameda F., Baro T., de Heredia C. D., Toran N., Catala A., Torrebadell M., Fortuny C., Cusi V., Carreras C., Diaz G. A., Blander J. M., Farber C. M., Silvestri G., Cunningham-Rundles C., Calvillo M., Dufour C., Notarangelo L. D., Lougaris V., Plebani A., Casanova J. L., Ganal S. C., Diefenbach A., Arostegui J. I., Juan M., Yague J., Mahlaoui N., Donadieu J., Chen K., Cerutti A. (2012) B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 13, 170–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cerutti A., Puga I., Cols M. (2012) New helping friends for B cells. Eur. J. Immunol. 42, 1956–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen K., Xu W., Wilson M., He B., Miller N. W., Bengten E., Edholm E. S., Santini P. A., Rath P., Chiu A., Cattalini M., Litzman J., J B. B., Huang B., Meini A., Riesbeck K., Cunningham-Rundles C., Plebani A., Cerutti A. (2009) Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils. Nat. Immunol. 10, 889–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmidt N. W., Mayo L. D., Donner D. B., Kaplan M. H. (2006) p53 regulates Btk-dependent B cell proliferation but not differentiation. J. Leukoc. Biol. 79, 852–859 [DOI] [PubMed] [Google Scholar]
- 23. Shick L., Carman J. H., Choi J. K., Somasundaram K., Burrell M., Hill D. E., Zeng Y. X., Wang Y., Wiman K. G., Salhany K., Kadesch T. R., Monroe J. G., Donehower L. A., el-Deiry W. S. (1997) Decreased immunoglobulin deposition in tumors and increased immature B cells in p53-null mice. Cell Growth Differ. 8, 121–131 [PubMed] [Google Scholar]
- 24. Lee H., Haque S., Nieto J., Trott J., Inman J. K., McCormick S., Chiorazzi N., Mongini P. K. (2012) A p53 axis regulates B cell receptor-triggered, innate immune system-driven B cell clonal expansion. J. Immunol. 188, 6093–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ward J. M., Tadesse-Heath L., Perkins S. N., Chattopadhyay S. K., Hursting S. D., Morse H. C., 3rd (1999) Splenic marginal zone B-cell and thymic T-cell lymphomas in p53-deficient mice. Lab. Invest. 79, 3–14 [PubMed] [Google Scholar]
- 26. Gostissa M., Bianco J. M., Malkin D. J., Kutok J. L., Rodig S. J., Morse H. C., 3rd, Bassing C. H., Alt F. W. (2013) Conditional inactivation of p53 in mature B cells promotes generation of nongerminal center-derived B-cell lymphomas. Proc. Natl. Acad. Sci. U. S. A. 110, 2934–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rowh M. A., DeMicco A., Horowitz J. E., Yin B., Yang-Iott K. S., Fusello A. M., Hobeika E., Reth M., Bassing C. H. (2011) Tp53 deletion in B lineage cells predisposes mice to lymphomas with oncogenic translocations. Oncogene 30, 4757–4764 [DOI] [PubMed] [Google Scholar]
- 28. Lens D., De Schouwer P. J., Hamoudi R. A., Abdul-Rauf M., Farahat N., Matutes E., Crook T., Dyer M. J., Catovsky D. (1997) p53 abnormalities in B-cell prolymphocytic leukemia. Blood 89, 2015–2023 [PubMed] [Google Scholar]
- 29. Fabbri G., Rasi S., Rossi D., Trifonov V., Khiabanian H., Ma J., Grunn A., Fangazio M., Capello D., Monti S., Cresta S., Gargiulo E., Forconi F., Guarini A., Arcaini L., Paulli M., Laurenti L., Larocca L. M., Marasca R., Gattei V., Oscier D., Bertoni F., Mullighan C. G., Foa R., Pasqualucci L., Rabadan R., Dalla-Favera R., Gaidano G. (2011) Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J. Exp. Med. 208, 1389–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tapinos N. I., Polihronis M., Moutsopoulos H. M. (1999) Lymphoma development in Sjögren's syndrome: novel p53 mutations. Arthritis. Rheum. 42, 1466–1472 [DOI] [PubMed] [Google Scholar]
- 31. Du M., Peng H., Singh N., Isaacson P. G., Pan L. (1995) The accumulation of p53 abnormalities is associated with progression of mucosa-associated lymphoid tissue lymphoma. Blood 86, 4587–4593 [PubMed] [Google Scholar]
- 32. Baldini L., Fracchiolla N. S., Cro L. M., Trecca D., Romitti L., Polli E., Maiolo A. T., Neri A. (1994) Frequent p53 gene involvement in splenic B-cell leukemia/lymphomas of possible marginal zone origin. Blood 84, 270–278 [PubMed] [Google Scholar]
- 33. Royer B., Cazals-Hatem D., Sibilia J., Agbalika F., Cayuela J. M., Soussi T., Maloisel F., Clauvel J. P., Brouet J. C., Mariette X. (1997) Lymphomas in patients with Sjögren's syndrome are marginal zone B-cell neoplasms, arise in diverse extranodal and nodal sites, and are not associated with viruses. Blood 90, 766–775 [PubMed] [Google Scholar]
- 34. Mongini P. K., Inman J. K., Han H., Fattah R. J., Abramson S. B., Attur M. (2006) APRIL and BAFF promote increased viability of replicating human B2 cells via mechanism involving cyclooxygenase 2. J. Immunol. 176, 6736–6751 [DOI] [PubMed] [Google Scholar]
- 35. Lee H., Trott J. S., Haque S., McCormick S., Chiorazzi N., Mongini P. K. (2010) A cyclooxygenase-2/prostaglandin E2 pathway augments activation-induced cytosine deaminase expression within replicating human B cells. J. Immunol. 185, 5300–5314 [DOI] [PubMed] [Google Scholar]
- 36. Mongini P. K., Inman J. K., Han H., Kalled S. L., Fattah R. J., McCormick S. (2005) Innate immunity and human B cell clonal expansion: effects on the recirculating B2 subpopulation. J. Immunol. 175, 6143–6154 [DOI] [PubMed] [Google Scholar]
- 37. Wu X., Tschumper R. C., Gutierrez A., Jr., Mihalcik S. A., Nowakowski G. S., Jelinek D. F. (2010) Selective induction of DNA repair pathways in human B cells activated by CD4+ T cells. PLoS ONE 5, e15549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takakuwa T., Hongyo T., Syaifudin M., Kanno H., Matsuzuka F., Narabayashi I., Nomura T., Aozasa K. (2000) Microsatellite instability and k-ras, p53 mutations in thyroid lymphoma. Jpn. J. Cancer Res. 91, 280–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Landgren O., Rapkin J. S., Caporaso N. E., Mellemkjaer L., Gridley G., Goldin L. R., Engels E. A. (2007) Respiratory tract infections and subsequent risk of chronic lymphocytic leukemia. Blood 109, 2198–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Casabonne D., Almeida J., Nieto W. G., Romero A., Fernandez-Navarro P., Rodriguez-Caballero A., Munoz-Criado S., Diaz M. G., Benavente Y., de Sanjose S., Orfao A. (2012) Common infectious agents and monoclonal B-cell lymphocytosis: a cross-sectional epidemiological study among healthy adults. PLoS ONE 7, e52808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peng H., Chen G., Du M., Singh N., Isaacson P. G., Pan L. (1996) Replication error phenotype and p53 gene mutation in lymphomas of mucosa-associated lymphoid tissue. Am. J. Pathol. 148, 643–648 [PMC free article] [PubMed] [Google Scholar]
- 42. Abdouh M., Storring J. M., Riad M., Paquette Y., Albert P. R., Drobetsky E., Kouassi E. (2001) Transcriptional mechanisms for induction of 5-HT1A receptor mRNA and protein in activated B and T lymphocytes. J. Biol. Chem. 276, 4382–4388 [DOI] [PubMed] [Google Scholar]
- 43. Smeland E. B., Blomhoff H. K., Ohlsson R., De Lange Davies C., Funderud S., Boye E. (1988) Transcription of protooncogenes during stimulation of normal human B lymphocytes. Eur. J. Immunol. 18, 1847–1850 [DOI] [PubMed] [Google Scholar]
- 44. Fedyk E. R., Phipps R. P. (1996) Prostaglandin E2 receptors of the EP2 and EP4 subtypes regulate activation and differentiation of mouse B lymphocytes to IgE-secreting cells. Proc. Natl. Acad. Sci. U. S. A. 93, 10978–10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Subbaramaiah K., Altorki N., Chung W. J., Mestre J. R., Sampat A., Dannenberg A. J. (1999) Inhibition of cyclooxygenase-2 gene expression by p53. J. Biol. Chem. 274, 10911–10915 [DOI] [PubMed] [Google Scholar]
- 46. Choi E. M., Kim S. R., Lee E. J., Han J. A. (2009) Cyclooxygenase-2 functionally inactivates p53 through a physical interaction with p53. Biochim. Biophys. Acta 1793, 1354–1365 [DOI] [PubMed] [Google Scholar]
- 47. Liu X. H., Kirschenbaum A., Yu K., Yao S., Levine A. C. (2005) Cyclooxygenase-2 suppresses hypoxia-induced apoptosis via a combination of direct and indirect inhibition of p53 activity in a human prostate cancer cell line. J. Biol. Chem. 280, 3817–3823 [DOI] [PubMed] [Google Scholar]
- 48. Benoit V., de Moraes E., Dar N. A., Taranchon E., Bours V., Hautefeuille A., Taniere P., Chariot A., Scoazec J. Y., de Moura Gallo C. V., Merville M. P., Hainaut P. (2006) Transcriptional activation of cyclooxygenase-2 by tumor suppressor p53 requires nuclear factor-kappaB. Oncogene 25, 5708–5718 [DOI] [PubMed] [Google Scholar]
- 49. Shim M., Foley J., Anna C., Mishina Y., Eling T. (2010) Embryonic expression of cyclooxygenase-2 causes malformations in axial skeleton. J. Biol. Chem. 285, 16206–16217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. de Moraes E., Dar N. A., de Moura Gallo C. V., Hainaut P. (2007) Cross-talks between cyclooxygenase-2 and tumor suppressor protein p53: balancing life and death during inflammatory stress and carcinogenesis. Int. J. Cancer 121, 929–937 [DOI] [PubMed] [Google Scholar]
- 51. de Jonge H. J., Fehrmann R. S., de Bont E. S., Hofstra R. M., Gerbens F., Kamps W. A., de Vries E. G., van der Zee A. G., te Meerman G. J., ter Elst A. (2007) Evidence based selection of housekeeping genes. PLoS ONE 2, e898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Efeyan A., Serrano M. (2007) p53: guardian of the genome and policeman of the oncogenes. Cell Cycle 6, 1006–1010 [DOI] [PubMed] [Google Scholar]
- 53. Naderi E. H., Jochemsen A. G., Blomhoff H. K., Naderi S. Activation of cAMP signaling interferes with stress-induced p53 accumulation in ALL-derived cells by promoting the interaction between p53 and HDM2. Neoplasia 13, 653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Narumiya S., Sugimoto Y., Ushikubi F. (1999) Prostanoid receptors: structures, properties, and functions. Physiol. Rev. 79, 1193–1226 [DOI] [PubMed] [Google Scholar]
- 55. Vilborg A., Glahder J. A., Wilhelm M. T., Bersani C., Corcoran M., Mahmoudi S., Rosenstierne M., Grander D., Farnebo M., Norrild B., Wiman K. G. (2009) The p53 target Wig-1 regulates p53 mRNA stability through an AU-rich element. Proc. Natl. Acad. Sci. U. S. A. 106, 15756–15761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zou T., Mazan-Mamczarz K., Rao J. N., Liu L., Marasa B. S., Zhang A. H., Xiao L., Pullmann R., Gorospe M., Wang J. Y. (2006) Polyamine depletion increases cytoplasmic levels of RNA-binding protein HuR leading to stabilization of nucleophosmin and p53 mRNAs. J. Biol. Chem. 281, 19387–19394 [DOI] [PubMed] [Google Scholar]
- 57. Mahmoudi S., Henriksson S., Corcoran M., Mendez-Vidal C., Wiman K. G., Farnebo M. (2009) Wrap53, a natural p53 antisense transcript required for p53 induction upon DNA damage. Mol. Cell 33, 462–471 [DOI] [PubMed] [Google Scholar]
- 58. Saldana-Meyer R., Recillas-Targa F. (2011) Transcriptional and epigenetic regulation of the p53 tumor suppressor gene. Epigenetics 6, 1068–1077 [DOI] [PubMed] [Google Scholar]
- 59. Khoury M. P., Bourdon J. C. (2011) p53 Isoforms: an intracellular microprocessor? Genes Cancer 2, 453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shen H. M., Poirier M. G., Allen M. J., North J., Lal R., Widom J., Storb U. (2009) The activation-induced cytidine deaminase (AID) efficiently targets DNA in nucleosomes but only during transcription. J. Exp. Med. 206, 1057–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. MacCarthy T., Kalis S. L., Roa S., Pham P., Goodman M. F., Scharff M. D., Bergman A. (2009) V-region mutation in vitro, in vivo, and in silico reveal the importance of the enzymatic properties of AID and the sequence environment. Proc. Natl. Acad. Sci. U. S. A. 106, 8629–8634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chahwan R., Edelmann W., Scharff M. D., Roa S. (2012) AIDing antibody diversity by error-prone mismatch repair. Semin. Immunol. 24, 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li S., Zhao Y., Wang J. Y. (2013) Analysis of Ig gene hypermutation in Ung(−/−)Polh(−/−) mice suggests that UNG and A: T mutagenesis pathway target different U:G lesions. Mol. Immunol. 53, 214–217 [DOI] [PubMed] [Google Scholar]
- 64. Wilson T. M., Vaisman A., Martomo S. A., Sullivan P., Lan L., Hanaoka F., Yasui A., Woodgate R., Gearhart P. J. (2005) MSH2-MSH6 stimulates DNA polymerase eta, suggesting a role for A: T mutations in antibody genes. J. Exp. Med. 201, 637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bhattacharya P., Grigera F., Rogozin I. B., McCarty T., Morse H. C., 3rd, Kenter A. L. (2008) Identification of murine B cell lines that undergo somatic hypermutation focused to A: T and G:C residues. Eur. J. Immunol. 38, 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pham P., Bransteitter R., Petruska J., Goodman M. F. (2003) Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 424, 103–107 [DOI] [PubMed] [Google Scholar]
- 67. Dorner T., Foster S. J., Farner N. L., Lipsky P. E. (1998) Somatic hypermutation of human immunoglobulin heavy chain genes: targeting of RGYW motifs on both DNA strands. Eur. J. Immunol. 28, 3384–3396 [DOI] [PubMed] [Google Scholar]
- 68. Feig D. I., Sowers L. C., Loeb L. A. (1994) Reverse chemical mutagenesis: identification of the mutagenic lesions resulting from reactive oxygen species-mediated damage to DNA. Proc. Natl. Acad. Sci. U. S. A. 91, 6609–6613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Machida K., Cheng K. T., Sung V. M., Shimodaira S., Lindsay K. L., Levine A. M., Lai M. Y., Lai M. M. (2004) Hepatitis C virus induces a mutator phenotype: enhanced mutations of immunoglobulin and protooncogenes. Proc. Natl. Acad. Sci. U. S. A. 101, 4262–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Delbos F., Aoufouchi S., Faili A., Weill J. C., Reynaud C. A. (2007) DNA polymerase eta is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 204, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Martomo S. A., Yang W. W., Wersto R. P., Ohkumo T., Kondo Y., Yokoi M., Masutani C., Hanaoka F., Gearhart P. J. (2005) Different mutation signatures in DNA polymerase eta- and MSH6-deficient mice suggest separate roles in antibody diversification. Proc. Natl. Acad. Sci. U. S. A. 102, 8656–8661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rada C., Di Noia J. M., Neuberger M. S. (2004) Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell. 16, 163–171 [DOI] [PubMed] [Google Scholar]
- 73. Ramiro A. R., Stavropoulos P., Jankovic M., Nussenzweig M. C. (2003) Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 4, 452–456 [DOI] [PubMed] [Google Scholar]
- 74. Faour W. H., He Y., He Q. W., de Ladurantaye M., Quintero M., Mancini A., Di Battista J. A. (2001) Prostaglandin E(2) regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. J. Biol. Chem. 276, 31720–31731 [DOI] [PubMed] [Google Scholar]
- 75. Kirch H. C., Flaswinkel S., Rumpf H., Brockmann D., Esche H. (1999) Expression of human p53 requires synergistic activation of transcription from the p53 promoter by AP-1, NF-kappaB and Myc/Max. Oncogene 18, 2728–2738 [DOI] [PubMed] [Google Scholar]
- 76. Wu H., Lozano G. (1994) NF-kappa B activation of p53. A potential mechanism for suppressing cell growth in response to stress. J. Biol. Chem. 269, 20067–20074 [PubMed] [Google Scholar]
- 77. Liu H., Lu Z. G., Miki Y., Yoshida K. (2007) Protein kinase C delta induces transcription of the TP53 tumor suppressor gene by controlling death-promoting factor Btf in the apoptotic response to DNA damage. Mol. Cell. Biol. 27, 8480–8491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Abbas T., White D., Hui L., Yoshida K., Foster D. A., Bargonetti J. (2004) Inhibition of human p53 basal transcription by down-regulation of protein kinase Cdelta. J. Biol. Chem. 279, 9970–9977 [DOI] [PubMed] [Google Scholar]
- 79. Takahashi P., Polson A., Reisman D. (2011) Elevated transcription of the p53 gene in early S-phase leads to a rapid DNA-damage response during S-phase of the cell cycle. Apoptosis 16, 950–958 [DOI] [PubMed] [Google Scholar]
- 80. Zhong H., Voll R. E., Ghosh S. (1998) Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1, 661–671 [DOI] [PubMed] [Google Scholar]
- 81. Gao N., Hibi Y., Cueno M., Asamitsu K., Okamoto T. (2010) A-kinase-interacting protein 1 (AKIP1) acts as a molecular determinant of PKA in NF-kappaB signaling. J. Biol. Chem. 285, 28097–28104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang P., Zhu F., Lee N. H., Konstantopoulos K. (2010) Shear-induced interleukin-6 synthesis in chondrocytes: roles of E prostanoid (EP) 2 and EP3 in cAMP/protein kinase A- and PI3-K/Akt-dependent NF-kappaB activation. J. Biol. Chem. 285, 24793–24804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Iwahashi H., Takeshita A., Hanazawa S. (2000) Prostaglandin E2 stimulates AP-1-mediated CD14 expression in mouse macrophages via cyclic AMP-dependent protein kinase A. J. Immunol. 164, 5403–5408 [DOI] [PubMed] [Google Scholar]
- 84. Park S. W., Schonhoff C. M., Webster C. R., Anwer M. S. (2012) Protein kinase Cdelta differentially regulates cAMP-dependent translocation of NTCP and MRP2 to the plasma membrane. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G657–G665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schonhoff C. M., Gillin H., Webster C. R., Anwer M. S. (2008) Protein kinase Cdelta mediates cyclic adenosine monophosphate-stimulated translocation of sodium taurocholate cotransporting polypeptide and multidrug resistant associated protein 2 in rat hepatocytes. Hepatology 47, 1309–1316 [DOI] [PubMed] [Google Scholar]
- 86. Dumais N., Bounou S., Olivier M., Tremblay M. J. (2002) Prostaglandin E(2)-mediated activation of HIV-1 long terminal repeat transcription in human T cells necessitates CCAAT/enhancer binding protein (C/EBP) binding sites in addition to cooperative interactions between C/EBPbeta and cyclic adenosine 5′-monophosphate response element binding protein. J. Immunol. 168, 274–282 [DOI] [PubMed] [Google Scholar]
- 87. Metz R., Ziff E. (1991) cAMP stimulates the C/EBP-related transcription factor rNFIL-6 to trans-locate to the nucleus and induce c-fos transcription. Genes Dev. 5, 1754–1766 [DOI] [PubMed] [Google Scholar]
- 88. Tae H. J., Zhang S., Kim K. H. (1995) cAMP activation of CAAT enhancer-binding protein-beta gene expression and promoter I of acetyl-CoA carboxylase. J. Biol. Chem. 270, 21487–21494 [DOI] [PubMed] [Google Scholar]
- 89. Huang S. K., Scruggs A. M., Donaghy J., McEachin R. C., Fisher A. S., Richardson B. C., Peters-Golden M. (2012) Prostaglandin E(2) increases fibroblast gene-specific and global DNA methylation via increased DNA methyltransferase expression. FASEB J. 26, 3703–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xia D., Wang D., Kim S. H., Katoh H., DuBois R. N. (2012) Prostaglandin E2 promotes intestinal tumor growth via DNA methylation. Nat. Med. 18, 224–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kramata P., Lu Y. P., Lou Y. R., Singh R. N., Kwon S. M., Conney A. H. (2005) Patches of mutant p53-immunoreactive epidermal cells induced by chronic UVB Irradiation harbor the same p53 mutations as squamous cell carcinomas in the skin of hairless SKH-1 mice. Cancer Res. 65, 3577–3585 [DOI] [PubMed] [Google Scholar]
- 92. Nigro J. M., Baker S. J., Preisinger A. C., Jessup J. M., Hostetter R., Cleary K., Bigner S. H., Davidson N., Baylin S., Devilee P., Glover T., Collins F. S., Weslon A., Modali R., Harris C. C., Vogelstein B. (1989) Mutations in the p53 gene occur in diverse human tumour types. Nature 342, 705–708 [DOI] [PubMed] [Google Scholar]
- 93. Bhatia K. G., Gutierrez M. I., Huppi K., Siwarski D., Magrath I. T. (1992) The pattern of p53 mutations in Burkitt's lymphoma differs from that of solid tumors. Cancer Res. 52, 4273–4276 [PubMed] [Google Scholar]
- 94. Young K. H., Leroy K., Moller M. B., Colleoni G. W., Sanchez-Beato M., Kerbauy F. R., Haioun C., Eickhoff J. C., Young A. H., Gaulard P., Piris M. A., Oberley T. D., Rehrauer W. M., Kahl B. S., Malter J. S., Campo E., Delabie J., Gascoyne R. D., Rosenwald A., Rimsza L., Huang J., Braziel R. M., Jaffe E. S., Wilson W. H., Staudt L. M., Vose J. M., Chan W. C., Weisenburger D. D., Greiner T. C. (2008) Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood 112, 3088–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Das P., Kotilingam D., Korchin B., Liu J., Yu D., Lazar A. J., Pollock R. E., Lev D. (2007) High prevalence of p53 exon 4 mutations in soft tissue sarcoma. Cancer 109, 2323–2333 [DOI] [PubMed] [Google Scholar]
- 96. Zainuddin N., Murray F., Kanduri M., Gunnarsson R., Smedby K. E., Enblad G., Jurlander J., Juliusson G., Rosenquist R. (2011) TP53 Mutations are infrequent in newly diagnosed chronic lymphocytic leukemia. Leuk. Res. 35, 272–274 [DOI] [PubMed] [Google Scholar]
- 97. Kocialkowski S., Pezzella F., Morrison H., Jones M., Laha S., Harris A. L., Mason D. Y., Gatter K. C. (1995) Mutations in the p53 gene are not limited to classic ‘hot spots’ and are not predictive of p53 protein expression in high-grade non-Hodgkin's lymphoma. Br. J. Haematol. 89, 55–60 [DOI] [PubMed] [Google Scholar]
- 98. Zilfou J. T., Hoffman W. H., Sank M., George D. L., Murphy M. (2001) The corepressor mSin3a interacts with the proline-rich domain of p53 and protects p53 from proteasome-mediated degradation. Mol. Cell. Biol. 21, 3974–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bergamaschi D., Samuels Y., Sullivan A., Zvelebil M., Breyssens H., Bisso A., Del Sal G., Syed N., Smith P., Gasco M., Crook T., Lu X. (2006) iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat. Genet. 38, 1133–1141 [DOI] [PubMed] [Google Scholar]
- 100. Berger M., Vogt Sionov R., Levine A. J., Haupt Y. (2001) A role for the polyproline domain of p53 in its regulation by Mdm2. J. Biol. Chem. 276, 3785–3790 [DOI] [PubMed] [Google Scholar]
- 101. Sakamuro D., Sabbatini P., White E., Prendergast G. C. (1997) The polyproline region of p53 is required to activate apoptosis but not growth arrest. Oncogene 15, 887–898 [DOI] [PubMed] [Google Scholar]
- 102. Slatter T. L., Ganesan P., Holzhauer C., Mehta R., Rubio C., Williams G., Wilson M., Royds J. A., Baird M. A., Braithwaite A. W. (2010) p53-mediated apoptosis prevents the accumulation of progenitor B cells and B-cell tumors. Cell Death Differ. 17, 540–550 [DOI] [PubMed] [Google Scholar]
- 103. Chipuk J. E., Maurer U., Green D. R., Schuler M. (2003) Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 4, 371–381 [DOI] [PubMed] [Google Scholar]
- 104. Frank A. K., Leu J. I., Zhou Y., Devarajan K., Nedelko T., Klein-Szanto A., Hollstein M., Murphy M. E. (2011) The codon 72 polymorphism of p53 regulates interaction with NF-κB and transactivation of genes involved in immunity and inflammation. Mol. Cell. Biol. 31, 1201–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dumont P., Leu J. I., Della Pietra A. C., 3rd, George D. L., Murphy M. (2003) The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 33, 357–365 [DOI] [PubMed] [Google Scholar]
- 106. Zhu F., Dolle M. E., Berton T. R., Kuiper R. V., Capps C., Espejo A., McArthur M. J., Bedford M. T., van Steeg H., de Vries A., Johnson D. G. (2010) Mouse models for the p53 R72P polymorphism mimic human phenotypes. Cancer Res. 70, 5851–5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jeong B. S., Hu W., Belyi V., Rabadan R., Levine A. J. (2010) Differential levels of transcription of p53-regulated genes by the arginine/proline polymorphism: p53 with arginine at codon 72 favors apoptosis. FASEB J. 24, 1347–1353 [DOI] [PubMed] [Google Scholar]
- 108. Azzam G. A., Frank A. K., Hollstein M., Murphy M. E. (2011) Tissue-specific apoptotic effects of the p53 codon 72 polymorphism in a mouse model. Cell Cycle 10, 1352–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zheng S. J., Lamhamedi-Cherradi S. E., Wang P., Xu L., Chen Y. H. (2005) Tumor suppressor p53 inhibits autoimmune inflammation and macrophage function. Diabetes 54, 1423–1428 [DOI] [PubMed] [Google Scholar]
- 110. Zhang S., Zheng M., Kibe R., Huang Y., Marrero L., Warren S., Zieske A. W., Iwakuma T., Kolls J. K., Cui Y. (2011) Trp53 negatively regulates autoimmunity via the STAT3-Th17 axis. FASEB J. 25, 2387–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yamanishi Y., Boyle D. L., Pinkoski M. J., Mahboubi A., Lin T., Han Z., Zvaifler N. J., Green D. R., Firestein G. S. (2002) Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am. J. Pathol. 160, 123–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Murphy S. H., Suzuki K., Downes M., Welch G. L., De Jesus P., Miraglia L. J., Orth A. P., Chanda S. K., Evans R. M., Verma I. M. (2011) Tumor suppressor protein (p) 53, is a regulator of NF-kappaB repression by the glucocorticoid receptor. Proc. Natl. Acad. Sci. U. S. A. 108, 17117–17122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gudkov A. V., Gurova K. V., Komarova E. A. (2011) Inflammation and p53: a tale of two stresses. Genes Cancer 2, 503–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Carvajal L. A., Manfredi J. J. (2013) Another fork in the road-life or death decisions by the tumour suppressor p53. EMBO Rep. 14, 414–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Steinert D., Kuper C., Bartels H., Beck F. X., Neuhofer W. (2009) PGE2 potentiates tonicity-induced COX-2 expression in renal medullary cells in a positive feedback loop involving EP2-cAMP-PKA signaling. Am. J. Physiol. Cell Physiol. 296, C75–87 [DOI] [PubMed] [Google Scholar]
- 116. Yamanishi Y., Boyle D. L., Rosengren S., Green D. R., Zvaifler N. J., Firestein G. S. (2002) Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. U. S. A. 99, 10025–10030 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.