

Abstract
Background
Luba is one of the four historical foci of Human African Trypanosomiasis (HAT) on Bioko Island, in Equatorial Guinea. Although no human cases have been detected since 1995, T. b. gambiense was recently observed in the vector Glossina palpalis palpalis. The existence of cryptic species within this vector taxon has been previously suggested, although no data are available regarding the evolutionary history of tsetse flies populations in Bioko.
Methods
A phylogenetic analysis of 60 G. p. palpalis from Luba was performed sequencing three mitochondrial (COI, ND2 and 16S) and one nuclear (rDNA-ITS1) DNA markers. Phylogeny reconstruction was performed by Distance Based, Maximum Likelihood and Bayesian Inference methods.
Results
The COI and ND2 mitochondrial genes were concatenated and revealed 10 closely related haplotypes with a dominant one found in 61.1% of the flies. The sequence homology of the other 9 haplotypes compared to the former ranged from 99.6 to 99.9%. Phylogenetic analysis clearly clustered all island samples with flies coming from the Western African Clade (WAC), and separated from the flies belonging to the Central Africa Clade (CAC), including samples from Mbini and Kogo, two foci of mainland Equatorial Guinea. Consistent with mitochondrial data, analysis of the microsatellite motif present in the ITS1 sequence exhibited two closely related genotypes, clearly divergent from the genotypes previously identified in Mbini and Kogo.
Conclusions
We report herein that tsetse flies populations circulating in Equatorial Guinea are composed of two allopatric subspecies, one insular and the other continental. The presence of these two G. p. palpalis cryptic taxa in Equatorial Guinea should be taken into account to accurately manage vector control strategy, in a country where trypanosomiasis transmission is controlled but not definitively eliminated yet.
Keywords: Glossina palpalis palpalis, Luba, Equatorial Guinea, Tsetse, Trypanosomiasis, Phylogeny, Bioko
Background
Following the London Declaration on Neglected Tropical Diseases, Human African Trypanosomiasis (HAT) has been targeted for elimination by 2020 [1]. As Trypanosoma brucei gambiense infection actually causes 98% of the total HAT cases (the remaining are due to T. b. rhodesiense), attention must be focused on this subspecies. Among the elimination strategies, vector control can play an important role [2-4], especially in isolated populations, which can be targeted for direct intervention avoiding the reinvasion from neighbouring zones. Islands represent an ideal setting for such strategies as demonstrated by the eradication of Glossina sp. in Unguja [5] and in Principe Islands [2] after a few years of sustained control.
Tsetse flies of the palpalis group (Nemorhina subgenus) are major vectors of T. b. gambiense in West Africa [2]. This group comprises two allopatric subspecies: G. p. palpalis and G. p. gambiensis, which probably derived from an ancestral palpalis species which was isolated in several geographic points when its riverine habitat declined during the last glacial maximum [6,7]. Cumulative evidences support the recognition of G. p. gambiensis and G. p. palpalis as valid specific taxa. For example, using data from the mitochondrial gene cytochrome oxidase 1 (COI), the average genetic distance observed between G. p. palpalis and G. p. gambiensis sequences was 6.6%, which is well above the threshold of 2% divergence for inter-species comparisons [8-11]. Moreover, experimental crosses between these subspecies yielded sterile males in the offspring [12]. The phylogenetic situation is more complex since recent genetic analyses suggested the existence of at least two distinct cryptic species within G. p. palpalis[10,11]. One circulates in the Western part of Africa (named as Western African Clade or WAC), and the other in the continental part of Equatorial Guinea and the Democratic Republic of Congo (DRC) (Central African Clade or CAC). According to the available data, both types are sympatric in the Fontem focus of Cameroon [11].
In Equatorial Guinea, four historical HAT foci are classically defined: one insular (Luba, on Bioko Island) and three on the mainland (Rio Campo in the north; Mbini in the centre and Kogo in the south) [13]. Due to sustained control measures, less than 10 HAT cases are being detected every year in the three continental foci since 2009 and no cases have been recorded since 1995 in Luba. Vector control activities were never implemented in Bioko Island, and parasite elimination in humans relied on active screening of the population at risk and systematic treatment [14]. Therefore, high densities of G. p. palpalis have been observed in some localities at the south of Luba district and moderate densities in others of the epicentre of the focus [15]. Moreover the presence of T. b. gambiense has also been reported in tsetse flies of Luba despite the absence of human infections, which could be attributed to the existence of reservoirs in the wild fauna, cryptic human infections and/or low sensitivity of available diagnostic tools [15-17]. Because vector control is a key parameter to completely eradicate the parasite [2,4,5] a deep knowledge of the biology of the tsetse fly is a crucial prerequisite. In such a context, the genetic characterization of the Glossina palpalis palpalis, never performed so far in Luba, has become indispensable.
In this study we combine information from both mitochondrial DNA (mtDNA- COI, ND2 and 16S- genes) and nuclear ribosomal (rDNA-ITS1) markers to investigate the phylogeographic origin of G. p. palpalis in the focus of Luba, Bioko Island, using tsetse flies samples captured in a previous epidemiological study [15]. MtDNA has been extensively used in population and evolutionary biology of insects [18-20] and metazoa in general [21] due to their particular features: relative ease isolation, simple sequence organization, maternal inheritance, absence of recombination and rapid rate of sequence divergence allowing discrimination of recently diverged lineages [22]. On the other hand, the rDNA internal transcribed spacer 1 (ITS1) is a useful marker for both closely related species and also intraspecific populations of insects [23-25].
Methods
Sample collection
Fly sampling was carried out in September/October 2007 from five areas known to harbour G. p. palpalis (Avendaño, Drumen, Fortuny, Boloco and Las Palmas). We employed monopyramidal traps [26], which have been successfully applied for vector control and entomological surveys in Equatorial Guinea [27-29]. Details about trap distribution are provided elsewhere [15]. Tsetse flies collected were individually stored in absolute ethanol in the field until processed in the laboratory. Species identification was undertaken using the key of Brunhes et al. [30]. Tsetse flies were sent to the National Centre of Tropical Medicine, Institute of Health Carlos III (Madrid, Spain) for subsequent molecular analysis.
Molecular analysis
DNA was extracted from whole flies with SpeedTools Tissue DNA Kit (Biotools, B & M Labs, S.A) following the manufacturer instructions. We analysed three mtDNA (ND2, COI and 16S) and one nuclear (ITS1) markers in our study. COI, 16S and ITS1 sequences were amplified with primers described previously [11,31] whereas new specific primers for ND2 gene from G. p. palpalis were designed (Additional file 1: Table S1).
PCR reactions were performed with 2 μl of each template DNA, 1X buffer (10 mM Tris–HCl, 1.5 mM MgCl2, 50 mM KCl, pH 8.3), 100 μM of each dNTP, 0.5 μM of each primer, 1 U of Fast Start Taq DNA polymerase (Roche) and double distilled water (DDW) until reaching 50 μl final volume. The thermal cycling programme started with initial denaturation step of 2 minutes at 95°C, followed by 35 cycles (30 seconds at 95°C, 30 seconds at 55–60°C and 1 minute at 72°C) and a final extension step of 5 minutes at 72°C. Results were visualized in 2% agarose gel stained with ethidium bromide under UV irradiation. After this check, we sent the amplified products to Secugen Sequencing and Molecular Diagnostics (Madrid, Spain) where they were sequenced using Sanger method.
Forward and reverse strands of all sequences were manually inspected with Sequence Scanner software v1.0 (Applied Biosystems© 2005). Sequences were trimmed and aligned using ClustalW Multiple Alignment algorithm of BioEdit Sequence Alignment Editor version 7.0.9.0 [32]. All sequences obtained in this work are available in GenBank with the following accession numbers: KF597286-91 (COI), KF597292-5 (ND2), KF597296 (16S) and KF597297-8 (ITS1).
Phylogenetic analysis
MEGA version 5 software [33] was used to calculate the pairwise and average distances expressed as number of nucleotide substitutions per site. Phylogeny reconstruction was performed with Maximum Likelihood (ML), Distance Based Neighbor-Joining (NJ) and Bayesian Inference (BI) methods. Model of evolution was inferred from data using the Find DNA/Protein Model tool of MEGA5. This software implements the Bayesian Information Criterion (BIC) [34] and corrected Akaike Information Criterion (AIC) [35] to measure the goodness-of-fit of the 24 models available. Unless otherwise specified, we applied the model selected by BIC to perform phylogenetic analysis (Table 1). For these three inference methods, all positions containing the missing data were eliminated.
Table 1.
Summary of models chosen in phylogenetic analysis
|
Marker |
COI |
ND2 |
COI + ND2 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Inference method | NJ | ML | BI | NJ | ML | BI | NJ | ML | BI |
|
Model |
T92 + Γ |
TN93 + Γ |
T92 + Γ |
HKY + Γ |
TN93 + Γ |
||||
|
BIC |
4097,84 |
4101,20 |
2186,19 |
2193,06 |
5311,13 |
||||
|
AIC |
3592,74 |
3572,45 |
2040,86 |
2034,53 |
5038,40 |
||||
|
-Lnl |
1732,16 |
1719 |
998,34 |
993,16 |
2484,13 |
||||
|
Length |
622 |
501 |
1123 |
||||||
|
Number of haplotypes |
30 |
9 |
14 |
||||||
| Overall mean distance (SE) | 0.0176 (0.0062) | 0.0093 (0.0064) | 0.0149 (0.0053) | ||||||
NJ: distance-based Neighbor-Joining, ML: Maximum Likelihood, BI: Bayesian Inference, T92: Tamura 3-parameter model [36], TN93: Tamura-Nei model [37], HKY: Hasegawa-Kishino-Yano model [38], Γ: assuming Gamma distribution (five discrete gamma categories), BIC: Bayesian Information Criterion (score), AIC: corrected Akaike Information Criterion (score), -Lnl: Maximum Likelihood value, SE: Standard Error.
MEGA5 software was also used to construct ML and distance based trees of COI, ND2 and concatenated (COI + ND2) sequences. For ML, the bootstrap method (500 replications) was chosen to test the robustness of the trees [39]. The selected substitution model was applied and rates among sites were treated as Gamma distributed with five discrete categories. We assumed no invariant sites. The ML heuristic method employed was the Nearest-Neighbor-Interchange (NNI).
Distance trees were constructed using the NJ method [40]. Gamma shape parameter was estimated from data. The bootstrap consensus tree was inferred from 2000 replicates.
BI was implemented with BEAST software [41]. This software uses Metropolis-Hasting Markov Chain Monte Carlo algorithm [42]. The default settings were generally used (1x107 generations and log parameters sampled each 1000 steps). The first 25% of trees generated was discarded as ‘burnin’. Yule process was implemented as tree prior, a simple model appropriate when studying speciation [43,44].
Assessment of genetic diversity
Under our sampling conditions, we assume that undetected haplotypes can exist. In order to assess the number of these unseen haplotypes in our studied area (Luba focus), we used two estimators that calculate haplotype richness: i) the first-order Jackknife richness estimator [45] calculated as followed: Jack1 = S0 + a1(N-1)/N, where S0 = observed number of haplotypes, a1 = number of haplotypes detected in only one fly, N = total number of flies and ii) the Bootstrap richness estimator [46] boot = S0 + Σ(1-pi)N, where S0 = observed number of haplotypes, pi = frequency of ith haplotype and N = total number of flies. Analysis was conducted in R software, with the specific ‘vegan’ package (http://cran.r-project.org, http://vegan.r-forge.r-project.org).
Phylogeographic analysis
To infer the haplotype relationships within the data sets, the median-joining network algorithm available in NETWORK v4.5.1.0 was performed [47], which combines the topology of a minimum spanning tree with a parsimonious search for the missing haplotypes.
Results
Mitochondrial markers
We amplified the mtDNA of COI (622 bp), ND2 (501 bp) and 16S (213 bp) genes for a total of sixty tsetse flies, all coming from Equatorial Guinea. Figure 1 (COI + ND2) and Table 2 show the distribution of mtDNA haplotypes regarding the sampling location. In COI gene six different haplotypes were observed with an overall genetic distance of 0.4%, ranging from 0.2% to 0.8%, in terms of number of base substitution per site. Haplotype 1 predominated over the rest (44/60), haplotype 2 was found in 9 individuals, whereas the other haplotypes were found once or twice (Table 3). For ND2, fifty-four sequences were obtained and four haplotypes were detected with an average distance of 0.4%, ranging from 0.2% to 0.5%. Haplotype 1 was the most common (45/54). Concatenated COI + ND2 yielded 10 different haplotypes and revealed that 61.1% of individuals (33/54) shared the same pattern (haplotype 11, i.e. haplotype 1 at both COI and ND2).
Figure 1.

Distribution of G. p. palpalis COI + ND2 concatenated haplotypes in Luba focus.
Table 2.
Distribution of the mtDNA haplotypes within Luba focus
| |
COI haplotypes |
ND2 haplotypes |
COI + ND2 haplotypes |
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 1 | 2 | 3 | 4 | 11 | 21 | 12 | 13 | 31 | 41 | 14 | 51 | 54 | 61 | |
|
Las Palmas |
8 |
1 |
|
|
1 |
|
7 |
1 |
1 |
1 |
6 |
1 |
1 |
1 |
|
|
|
|
1 |
|
|
Fortuny (Boloco) |
9 |
|
|
1 |
|
|
8 |
1 |
|
|
7 |
|
1 |
|
|
1 |
|
|
|
|
|
Fortuny |
7 |
1 |
1 |
1 |
|
|
8 |
|
1 |
|
5 |
1 |
|
1 |
1 |
1 |
|
|
|
|
|
B. Drumen |
14 |
2 |
1 |
|
|
1 |
17 |
1 |
|
|
13 |
2 |
1 |
|
1 |
|
|
|
|
1 |
|
B. Avendaño |
6 |
5 |
|
|
1 |
|
5 |
2 |
|
1 |
2 |
2 |
2 |
|
|
|
1 |
1 |
|
|
| Total | 44 | 9 | 2 | 2 | 2 | 1 | 45 | 5 | 2 | 2 | 33 | 6 | 5 | 2 | 2 | 2 | 1 | 1 | 1 | 1 |
For each location is detailed the number of times a given haplotype has been obtained.
Table 3.
Distribution of ITS1 genotypes within Luba focus
| |
ITS genotypes |
|
|||
|---|---|---|---|---|---|
| Village | Genotype 1: (TA) 2 C (AT) 9 | Genotype 2: (TA) 10 | Total number of samples | ||
|
Las Palmas |
9 |
90,0% |
1 |
10,0% |
10 |
|
Fortuny/Boloco |
6 |
66,7% |
3 |
33,3% |
9 |
|
Fortuny |
7 |
70,0% |
3 |
30,0% |
10 |
|
B. Drumen |
14 |
77,8% |
4 |
22,2% |
18 |
|
B. Avendaño |
8 |
66,7% |
4 |
33,3% |
12 |
| Total | 44 | 74,6% | 15 | 25,4% | 59 |
For each location is detailed the number of times a given genotype has been obtained.
Analysis of genetic diversity seemed to be assessed with a reasonable accuracy. The number of estimated haplotypes, n = 13.93 and 11.86 calculated with Jackknife and Bootstrap estimators respectively, was close to the number of observed haplotypes (n = 10) when all the samples are considered as a unique population (Additional file 2: Table S2). This means that the haplotype structure of the population is correctly estimated even with low sample size and underestimation of the haplotype richness.
Since very limited data are available regarding the ND2 and 16S sequences in G. p. palpalis from different geographic origins, we decided to focus the phylogenetic reconstruction in COI gene. To construct more comprehensive ML, distance based NJ and BI trees we included previously published sequences of G. p. palpalis from DRC (EU591840-2), Cameroon (EU591829-31, EU591860 and EU591865), Liberia (EU591857-9), Togo (EU591838-9), Ivory Coast (EU591846-8 and EU591832-3), Burkina Faso (EU591856) and two continental foci of Equatorial Guinea, Kogo and Mbini (EU591825 and FJ767873-6). The source of these sequences is detailed elsewhere [10,11]. G. morsitans (GQ255905) was used to root the tree and G. p. gambiensis (EU591851) sequence was also placed as outgroup. Analysis was based on a total fragment of 622 bp of the COI gene.
ML, distance based NJ and BI trees showed common topologies. All mirrored two major geographic separated clusters from Central (CAC) and Western Africa (WAC) (Figure 2). NJ and BI trees showed more support in this split: 97% bootstrap and 0.9999 posterior probability in BI trees, respectively, than ML inference (88%). Within WAC, organisation was unclear and again NJ and BI methods yielded a more robust clustering. ML inference method failed to yield a clear discrimination between clades from Western Africa (50% bootstrap) whereas NJ and BI trees split samples from this area in different sister sympatric groups (with 99% bootstrap and 1.0000 posterior probability). Clusters obtained by the NJ and BI methods within the WAC are probably not artifacts since both trees separate exactly the same specimens and in spite of the lower power of the ML tree, the same trend is visible.
Figure 2.
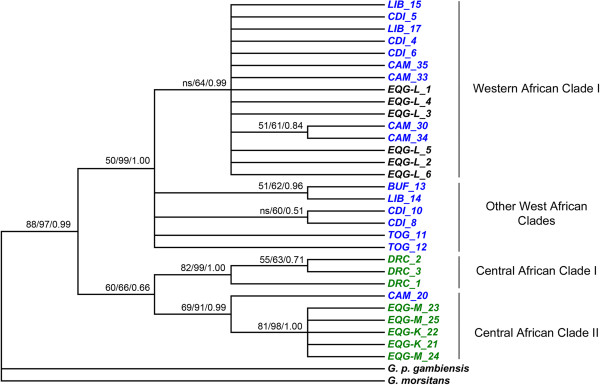
Phylogenetic reconstruction inferred for the G. p. palpalis COI dataset. Majority rule of the maximum likelihood consensus, distance consensus and Bayesian trees inferred for the COI dataset. Models of evolution used are detailed in Table 1. Numbers shown next to the nodes are the support values, i.e. the percentage of bootstrap replications (500 replicates in maximum likelihood and 2000 in distance based analysis) and posterior probability in Bayesian trees. Analysis involved 622 positions in the final dataset. EQG-L: Equatorial Guinea, Luba focus, EQG-M: Equatorial Guinea, Mbini focus, EQG-K: Equatorial Guinea, Kogo focus, DRC: Democratic Republic of Congo, CAM: Cameroon, LIB: Liberia, CDI: Cote d’Ivoire, TOG: Togo, BUF: Burkina Faso. ns: no support (<50 bootstrap or 0.5 posterior probability).
Regardless of the inference method used the COI haplotypes from the Luba focus grouped within the WAC I cluster in all trees, in clear contrast with the haplotypes from Kogo and Mbini foci (mainland Equatorial Guinea) that belong to the CAC cluster. In accordance to phylogeny trees, network analysis shows the clear separation of the two major geographic clusters (WAC and CAC) (Figure 3) together with the two subgroups corresponding to DRC and continental Equatorial Guinean haplotypes within the CAC (CAC I and II in Figure 2).
Figure 3.

Geographic representation of the COI haplotypes. A) Geographic distribution of the COI lineages; B) Haplotypes network derived from 30 haplotypes of the G. p. palpalis complex. Haplotypes are represented by circles and their frequency is proportional to the area. Network diagrams created using Phylogenetic Network software from Fluxus and using the Median Joining method as described previously [47].
Regarding ND2 gene, five haplotypes were available in Genbank, one from Ivory Coast (EU591895), one from Liberia (EU591884), two from Cameroon (EU591897-8) and one from Equatorial Guinea (EU591905). In accordance with COI data, ML, distance based NJ and BI trees constructed with ND2 and COI + ND2 concatenated genes (Figure 4), exhibited a clear separation between the sequences of mainland Equatorial Guinean haplotypes and those from the Luba focus.
Figure 4.
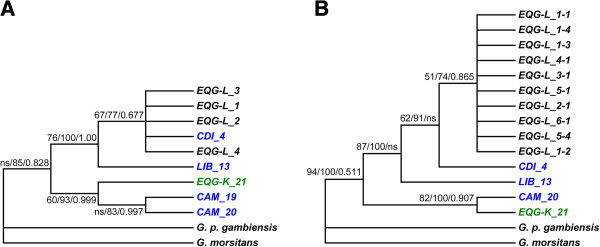
Phylogenetic reconstruction inferred for the ND2 and concatenated COI + ND2 datasets. Majority rule of the maximum likelihood consensus, distance consensus and Bayesian trees inferred for the A) ND2 and B) concatenated COI + ND2 dataset. Models of evolution used are detailed in Table 1. Numbers shown next to the nodes are the support values, i.e. the percentage of bootstrap replications (500 replicates in maximum likelihood and 2000 in distance based analysis) and posterior probability in Bayesian trees. Analysis involved 501 (ND2) or 1123 (COI + ND2) positions in the final dataset. EQG-L: Equatorial Guinea, Luba focus, EQG-K: Equatorial Guinea, Kogo focus, CAM: Cameroon, LIB: Liberia, CDI: Cote d’Ivoire. ns: no support (<50 bootstrap or 0.5 posterior probability).
The 16S sequences were 100% identical in the 34 randomly selected individuals from Luba analysed by this marker. Only one G. p. palpalis 16S sequence, from Cameroon, was available in GenBank (EU591913) before this work. Alignment of the single 16S haplotype found in Luba was performed with this Cameroonian sequence and other Glossina taxa, namely G. p. gambiensis (EU591911.1), G. fuscipes quanzensis (EU591910.1), G. fuscipes fuscipes (EU591906.1), G. tachinoides (EU591917.1), G. pallicera (EU591918.1), G. morsitans morsitans (EU591920.1) and G. pallidipes (EU591925.1) (Additional file 3: Figure S1). This alignment revealed 1.91% distance between G. p. palpalis from Luba and Cameroon, and distances ranging between 2.4% (G. p. gambiensis) and 8.97% (G. pallidipes) when comparing Luba 16S haplotype with other Glossina taxa (Maximum Likelihood Composite Model [48]).
Nuclear ITS1 marker
ITS1 size polymorphism was assessed for 59 flies. All PCR products analysed exhibited a unique size around 240 bp, similar to the size reported by Dyer et al. [11] when this fragment was amplified from flies belonging to the WAC. After sequencing, two similar genotypes defined by the (AT)n microsatellite were found. Genotype 1 was characterized by (AT)9G(TA)2 region, whereas genotype 2 exhibited (AT)10 pure repeats (Additional file 4: Figure S2). As shown in Table 3, genotype 1 was dominant in all the 5 sampled areas of the Luba focus.
The two genotypes found were aligned with ITS1 sequences of G. p. palpalis from mainland Equatorial Guinea (Kogo: J767886 and Mbini: J767887-J767888), DRC (J767892 and J767893), Guinea Conakry (EU591930), Gambia (EU591931), Burkina Faso (EU591932), Togo (EU591933), Liberia (EU591934) and Ivory Coast (EU5991935) (Additional file 4: Figure S2). The main genotype, found in 44/59 flies and defined by a (AT)9G(TA)2 domain, was not detected in the other published sequences. The second genotype, defined by a (AT)10 repeat, shared 100% homology with the sequence of a Togo sample.
Discussion
Luba focus is located at the edge between the two main clades of G. p. palpalis, one named WAC (for Western Africa Clade), including flies from Cameroon, Burkina Faso, Ivory Coast, Liberia and Togo; and the other, named CAC (for Central African Clade) represented by flies from the Democratic Republic of Congo and the mainland region of Equatorial Guinea. Phylogenetic analysis, using mtDNA markers, allowed us to cluster the G. p. palpalis population from Luba within the WAC. All the samples of this work were unambiguously included in this group and separated from the CAC regardless the phylogenetic inference method used.
Our results are consistent with the geological history of Bioko, originated by volcanic eruptions in the lower Tertiary period, around 60 million years BP. Bioko is a part of an archipelago which pertains to a large volcanic fracture originating in the South of Lake Chad and extending to Mount Cameroon in the continent [49]. Although politically belonging to Equatorial Guinea, Bioko lies closer to the Cameroon coast (ca. 30 km) rather than to the rest of Equatorial Guinea territories (more than 200 km from mainland and 700 km from Annobon island). During geoclimatic events of Quaternary period, Bioko was linked to mainland given the lower sea level, presumably to Cameroonian coast because of its geological origin. At the end of the last glaciation (around 12,000 years BP), Bioko was isolated by flooding and separated from the continent [50]. It is probable that G. p. palpalis population of Bioko was isolated from Cameroon coast after that event, as suggested for other insect vectors such as Anopheles melas and Simulium yahense[51,52].
Both mtDNA and nuclear markers show a very low genetic intra-variability. MtDNA genes data polymorphism did not exceed 0.8% and ITS1 sequences only yielded two closely related genotypes. For ITS1, the more abundant one is apparently exclusive of Luba focus, suggesting a certain degree of isolation. This is in accordance with previous studies, which found that G. p. gambiensis populations separated from the continent only by 4–5 km of sea show clear evidences of complete isolation. These results are based on wing landmarks, DNA mitochondrial markers and microsatellite dataset [53,54]. Our data should be also completed with an exhaustive coalescent population genetics analysis to support or not the hypothesis of an on-going allopatric speciation in Luba.
Because of the presence of these two sympatric ITS1 genotypes in Bioko Island one could speculate about the past occurrence of at least two separated migration events, as suggested for Anopheles gambiae in Bioko [55]. The ITS1 tandem array sequences are expected to be quickly homogenized by concerted evolution in interbreeding populations, whereas differences are usually observed between non-interbreeding ones [56,57]. However, the low difference observed between our ITS1 genotypes is based in the AT repeats, where a slight heterogeneity can be expected since microsatellite sequence could be evolving faster than the homogenization process. This phenomenon has been previously described in the tandem repeated U2 snRNA gene of primates [58,59].
Other possible explanation for the existence of two distinct ITS1 genotypes is a more recent reintroduction of tsetse flies from continent. Bioko Island is located at 30 km West of the Cameroonian coast, the closest area of the continent. Estimated active dispersal of Glossina sp. in one day is no longer than 1.3 km (in 15–30 min/day) [60,61]. Additionally, these flies are usually unable to flight for long periods but rather in short bursts lasting between 1 and 2 minutes [62,63]. These observations make extremely unlikely a recent (posterior to the glaciations period) reproductive contact between Luba and other continental foci due to active dispersal. However, human-mediated transportation of flies may not be disregarded. This situation allowed the reinvasion of G. p. palpalis in Principe Island in 1956 despite its eradication in 1914 using mobile traps carried by workers [2]. The distance between this island (ca. 240 km) and mainland is much larger than that of Bioko. It is therefore expected that human movement between Bioko and the coast of Cameroon has been much more frequent since the island was first colonized by the Bubi ethnical group, at the end of the last glaciation [64]. Finally, although the existence of ITS1 hybrid forms was initially suggested in G. p. palpalis of Equatorial Guinea [11], this hypothesis was later rejected by the same authors using microsatellite markers [10]. A set of highly polymorphic nuclear markers should be applied in the future to definitively test this hypothesis in Luba.
The genetic markers used to assess geographical structuring of G. p. palpalis demonstrate the existence of two allopatric taxa in Equatorial Guinea, one in the insular focus, Luba, and the other in two of the mainland foci (Kogo and Mbini). G. p. palpalis populations from Rio Campo, the third mainland focus of the country, have not been studied yet. In Equatorial Guinea, vector control was implemented in Kogo and Mbini from 2002 to 2009, whereas in Luba the active detection and treatment of HAT cases was the only control method employed [14]. Vector control has proved to be efficient at controlling HAT transmission [2,5,13] since tsetse flies show particular features that make them highly susceptible to direct interventions. Firstly, tsetse flies have a very low reproductive rate, given that a female individual has probably only one reproductive mating in its life and deposits only one larva per generation (up to 12 generations with 9–10 days of interval for a lifespan of 2–3 months) [2]. As a result of this low reproductive rate, the population of the vector is usually low comparing to other diptera and small increases in mortality can lead to control [4,65] or even to population extinction [66]. Secondly, the active dispersal of tsetse flies is generally low [60,61] resulting in a more difficult re-colonization of cleared areas. Thirdly, the genetic variability observed within populations tends to be reduced as well [67,68], probably as a consequence of both low reproductive rate and limited dispersal capacity, making more difficult for the selection of new attributes such as resistance to insecticides. There are, however, other behavioural features such as feeding preferences or trap-avoidance that can vary at subspecies and even at population level. For example, within G. p. palpalis, diverse feeding preferences across foci of Cameroon have been observed [69]. Although this observation could be attributed to the opportunistic feeding behaviour of G. p. palpalis[70], it could be also associated with the existence of genetically different G. p. palpalis populations, given the probable isolation of these foci [71]. Indeed, different feeding patterns in two sympatric populations of G. p. palpalis and G. p. gambiensis have also been described in preforest areas of Cote d’Ivoire [72], demonstrating that this feature can differ between closely related subspecies. The feeding behaviour of the vector can be crucial to design an effective vector control campaign since it provides valuable information to understand the epidemiological cycle of the parasite at local level.
Conclusions
If cryptic species of G. p. palpalis are circulating as our data and previous observations suggest [11], the occurrence of both variants in Equatorial Guinea could have an important impact in the control of sleeping sickness. Are vector control methods equally effective for all foci? Do tsetse flies from Kogo and Mbini have different feeding preferences than those of Luba? Could Luba tsetse fly populations feed mainly in wild fauna allowing T. b. gambiense to persist in the focus even with absence of human infections? Are both groups equally competent for trypanosome transmission? An entomological study comparing feeding behaviour, habitat selection and parasite strain specificity of G. p. palpalis populations from the island and the continent should be conducted to ascertain if the genetic differences observed could be reflected on the vector ecology.
Competing interests
Authors declare no competing interests.
Authors’ contribution
CCO participated in the sample collection, molecular genetic studies, data analysis and wrote the manuscript. JK participated in statistical analysis. PN carried out the amplification of the genes and prepared the products for sequencing. NNB and PRNA participated in sample collection. MN, JC, PNA, JP, AB and JMB conceived the study, participated in its design and coordination, analysed the data and wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Summary of primers used.
Estimated genetic diversity, calculated in number of observed haplotypes.
Alignment of 16S found in G. p. palpalis from Luba focus. Gpp: G. p. palpalis, CAM: Cameroon, Gpg: G. p. gambiensis, Gfq: G. fuscipes quanzensis, Gff: G. fuscipes fuscipes, Gtac: G. tachinoides, Gpallic: G. pallicera, Gmm: G. morsitans morsitans, Gpallid: G. pallidipes.
Sequencing profiles and alignment of ITS1 genotypes found in Luba focus. Sequence profiles of ITS genotypes 1 and 2 (A and B, respectively). C) Alignment of ITS1 genotypes of G. p. palpalis from different origins. EQG-L: Equatorial Guinea, Luba focus, EQG-M: Equatorial Guinea, Mbini focus, EQG-K: Equatorial Guinea, Kogo focus, DRC: Democratic Republic of Congo, LIB: Liberia, CDI: Cote d’Ivoire, TOG: Togo, GAM: Gambia, BUF: Burkina Faso, GUI: Guinea.
Contributor Information
Carlos Cordon-Obras, Email: carlos.cordon@ipb.csic.es.
Jorge Cano, Email: JCano.Ortega@lshtm.ac.uk.
Jenny Knapp, Email: jknapp@univ-fcomte.fr.
Paloma Nebreda, Email: pnebreda@isciii.es.
Nicolas Ndong-Mabale, Email: nndong@crce-guinea.org.
Policarpo Ricardo Ncogo-Ada, Email: pncogo@crce-guinea.org.
Pedro Ndongo-Asumu, Email: pedrondongasumu@yahoo.es.
Miguel Navarro, Email: miguel.navarro@ipb.csic.es.
Joao Pinto, Email: JPinto@ihmt.unl.pt.
Agustin Benito, Email: abenito@isciii.es.
Jean-Mathieu Bart, Email: jmbart@isciii.es.
Acknowledgments
We thank Dr Francis Raoul for his statistical assistance with R and Dr Martin Donnelly for his valuable comments. This work has been supported by ‘Fondo de Investigacion Sanitaria (FIS)’ (PI10/01128) and by VI PN de I + D + I 2008–2011, ISCIII -Subdirección General de Redes y Centros de Investigación Cooperativa RD12/0018/0001 and RD12/0018/0015 (RICET). JMB is supported by Miguel Servet Fellowship CP09/00300.
References
- Simarro P, Franco J, Diarra A, Ruiz Postigo JA, Jannin J. Diversity of human African trypanosomiasis epidemiological settings requires fine-tuning control strategies to facilitate disease elimination. Res Rep Trop Med. 2013. pp. 1–6. [DOI] [PMC free article] [PubMed]
- Kuzoe FA, Schofield CJ. Strategic review of traps and targets for tsetse and African Trypanosomiasis control. Geneva, Switzerland: World Health Organisation; 2004. (Report of Special Programme for Research and Training in Tropical Disease (TDR) TDR/IDE/TRY/051). [Google Scholar]
- Kgori PM, Modo S, Torr SJ. The use of aerial spraying to eliminate tsetse from the Okavango Delta of Botswana. Acta Trop. 2006;99(2–3):184–199. doi: 10.1016/j.actatropica.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Solano P, Torr SJ, Lehane MJ. Is vector control needed to eliminate gambiense human African trypanosomiasis? Front Cell Infect Microbiol. 2013;3:33. doi: 10.3389/fcimb.2013.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreysen MJ, Saleh KM, Ali MY, Abdulla AM, Zhu ZR, Juma KG, Dyck VA, Msangi AR, Mkonyi PA, Feldmann HU. Glossina austeni (Diptera: Glossinidae) eradicated on the island of Unguja, Zanzibar, using the sterile insect technique. J Econ Entomol. 2000;93(1):123–135. doi: 10.1603/0022-0493-93.1.123. [DOI] [PubMed] [Google Scholar]
- Challier A, Gouteaux J, Coosemans M. La limite géographique entre les sous-espèces Glossina palpalis palpalis (Rob.-Desv.) et Glossina palpalis gambiensis Vanderplanck en Afrique occidentale. Cahiers - ORSTOM Entomologie Médicale et Parasitologie. 1983;21:207–220. [Google Scholar]
- Gooding RH, Solano P, Ravel S. X-chromosome mapping experiments suggest occurrence of cryptic species in the tsetse fly Glossina palpalis palpalis. Can J Zool. 2004;82:1902–1909. doi: 10.1139/z05-002. [DOI] [Google Scholar]
- Hebert PD, Ratnasingham S, de Waard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci. 2003;270(Suppl 1):S96–S99. doi: 10.1098/rsbl.2003.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PD, Cywinska A, Ball SL, de Waard JR. Biological identifications through DNA barcodes. Proc Biol Sci. 2003;270(1512):313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer NA, Furtado A, Cano J, Ferreira F, Odete Afonso M, Ndong-Mabale N, Ndong-Asumu P, Centeno-Lima S, Benito A, Weetman D. et al. Evidence for a discrete evolutionary lineage within Equatorial Guinea suggests that the tsetse fly Glossina palpalis palpalis exists as a species complex. Mol Ecol. 2009;18(15):3268–3282. doi: 10.1111/j.1365-294X.2009.04265.x. [DOI] [PubMed] [Google Scholar]
- Dyer NA, Lawton SP, Ravel S, Choi KS, Lehane MJ, Robinson AS, Okedi LM, Hall MJ, Solano P, Donnelly MJ. Molecular phylogenetics of tsetse flies (Diptera: Glossinidae) based on mitochondrial (COI, 16S, ND2) and nuclear ribosomal DNA sequences, with an emphasis on the palpalis group. Mol Phylogenet Evol. 2008;49(1):227–239. doi: 10.1016/j.ympev.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Gooding RH. Genetic analysis of hybrid sterility in crosses of the tsetse flies Glossina palpalis palpalis and Glossina palpalis gambiensis (Diptera: Glossinidae) Can J Zool. 1997;75:1109–1117. doi: 10.1139/z97-133. [DOI] [Google Scholar]
- Simarro PP. [Current strategies in the campaign against human trypanosomiasis in the Equatorial Republic of Guinea] Rev Sanid Hig Publica (Madr) 1988;62(5–8):1483–1493. [PubMed] [Google Scholar]
- Simarro PP, Franco JR, Ndongo P, Nguema E, Louis FJ, Jannin J. The elimination of Trypanosoma brucei gambiense sleeping sickness in the focus of Luba, Bioko Island, Equatorial Guinea. Trop Med Int Health. 2006;11(5):636–646. doi: 10.1111/j.1365-3156.2006.01624.x. [DOI] [PubMed] [Google Scholar]
- Cordon-Obras C, Garcia-Estebanez C, Ndong-Mabale N, Abaga S, Ndongo-Asumu P, Benito A, Cano J. Screening of Trypanosoma brucei gambiense in domestic livestock and tsetse flies from an insular endemic focus (Luba, Equatorial Guinea) PLoS Negl Trop Dis. 2010;4(6):e704. doi: 10.1371/journal.pntd.0000704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamonneau V, Ilboudo H, Kabore J, Kaba D, Koffi M, Solano P, Garcia A, Courtin D, Laveissiere C, Lingue K. et al. Untreated human infections by Trypanosoma brucei gambiense are not 100% fatal. PLoS Negl Trop Dis. 2012;6(6):e1691. doi: 10.1371/journal.pntd.0001691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis FJ, Kohagne Tongue L, Ebo OEV, Simarro PP. [Organizing an active screening campaign for human African trypanosomiasis due to Trypanosoma brucei gambiense] Med Trop (Mars) 2008;68(1):11–16. [PubMed] [Google Scholar]
- Szymura JM, Lunt DH, Hewitt GM. The sequence and structure of the meadow grasshopper (Chorthippus parallelus) mitochondrial srRNA, ND2, COI, COII ATPase8 and 9 tRNA genes. Insect Mol Biol. 1996;5(2):127–139. doi: 10.1111/j.1365-2583.1996.tb00047.x. [DOI] [PubMed] [Google Scholar]
- Lunt DH, Zhang DX, Szymura JM, Hewitt GM. The insect cytochrome oxidase I gene: evolutionary patterns and conserved primers for phylogenetic studies. Insect Mol Biol. 1996;5(3):153–165. doi: 10.1111/j.1365-2583.1996.tb00049.x. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Szymura JM, Hewitt GM. Evolution and structural conservation of the control region of insect mitochondrial DNA. J Mol Evol. 1995;40(4):382–391. doi: 10.1007/BF00164024. [DOI] [PubMed] [Google Scholar]
- Caterino MS, Cho S, Sperling FA. The current state of insect molecular systematics: a thriving Tower of Babel. Annu Rev Entomol. 2000;45:1–54. doi: 10.1146/annurev.ento.45.1.1. [DOI] [PubMed] [Google Scholar]
- Harrison RG. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol Evol. 1989;4(1):6–11. doi: 10.1016/0169-5347(89)90006-2. [DOI] [PubMed] [Google Scholar]
- Vogler AP, DeSalle R. Evolution and phylogenetic information content of the ITS-1 region in the tiger beetle Cicindela dorsalis. Mol Biol Evol. 1994;11(3):393–405. doi: 10.1093/oxfordjournals.molbev.a040121. [DOI] [PubMed] [Google Scholar]
- Pereira JO, Freitas BM, Jorge DM, Torres DC, Soares CE, Grangeiro TB. Genetic variability in Melipona quinquefasciata (Hymenoptera, Apidae, Meliponini) from northeastern Brazil determined using the first internal transcribed spacer (ITS1) Genet Mol Res. 2009;8(2):641–648. doi: 10.4238/vol8-2kerr025. [DOI] [PubMed] [Google Scholar]
- Bower JE, Dowton M, Cooper RD, Beebe NW. Intraspecific concerted evolution of the rDNA ITS1 in Anopheles farauti sensu stricto (Diptera: Culicidae) reveals recent patterns of population structure. J Mol Evol. 2008;67(4):397–411. doi: 10.1007/s00239-008-9161-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouteux JP, Lancien J. [The pyramidal trap for collecting and controlling tsetse flies (Diptera: Glossinidae). Comparative trials and description of new collecting technics] Trop Med Parasitol. 1986;37(1):61–66. [PubMed] [Google Scholar]
- Simarro PP, Mas J, Lancien J, Ona Sima F, Mateo MJ, Roche J. [Epidemiology of human trypanosomiasis in the Luba focus, in Equatorial Guinea] Rev Sanid Hig Publica (Madr) 1990;64(9–10):517–534. [PubMed] [Google Scholar]
- Cano J, Descalzo MA, Ndong-Mabale N, Ndong-Asumu P, Bobuakasi L, Nzambo-Ondo S, Benito A, Roche J. Predicted distribution and movement of Glossina palpalis palpalis (Diptera: Glossinidae) in the wet and dry seasons in the Kogo trypanosomiasis focus (Equatorial Guinea) J Vector Ecol. 2007;32(2):218–225. doi: 10.3376/1081-1710(2007)32[218:PDAMOG]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Cano J, Descalzo MA, Ndong-Mabale N, Ndongo-Asumu P, Bobuakasi L, Buatiche JN, Nzambo-Ondo S, Ondo-Esono M, Benito A, Roche J. Spatial and temporal variability of the Glossina palpalis palpalis population in the Mbini focus (Equatorial Guinea) Int J Health Geogr. 2007;6:36. doi: 10.1186/1476-072X-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunhes J, Cuisance D, Geoffroy B, Hervy JP, Lebbe J. Les glossines ou mouches tsé-tsé. Logiciel d’identification. Glossina Expert. Manuel illustré d’utilisation. VI, ORSTOM, Paris: Université Paris; 1998. [Google Scholar]
- Simon C, Frati F, Bechenbach A, Crespi B, Liu H, Flook P. Evolution, weighting and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am. 1994;87:651–701. [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz GE. Estimating the dimension of a model. Ann Stat. 1978;2(6):461–464. [Google Scholar]
- Akaike H. A new look at statistical model identification. IEEE Trans Automat Control. 1974;6(19):716–723. [Google Scholar]
- Tamura K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C-content biases. Mol Biol Evol. 1992;9(4):678–687. doi: 10.1093/oxfordjournals.molbev.a040752. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22(2):160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Confidence-limits on phylogenies- an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings WK. Monte Carlo sampling methods using Markov chains and their applications. Biometrika. 1970;57(1):97–109. doi: 10.1093/biomet/57.1.97. [DOI] [Google Scholar]
- Yule GU. A mathematical theory of evolution, based on the conclusions of Dr. JC Willis, FRS. Philos Trans R Soc Lond B Biol Sci. 1925;213:21–87. doi: 10.1098/rstb.1925.0002. [DOI] [Google Scholar]
- Gernhard T. The conditioned reconstructed process. J Theor Biol. 2008;253(4):769–778. doi: 10.1016/j.jtbi.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Heltshe JF, Forrester NE. Estimating species richness using the jackknife procedure. Biometrics. 1983;39(1):1–11. doi: 10.2307/2530802. [DOI] [PubMed] [Google Scholar]
- Smith EP, van Belle G. Nonparametric estimation of species richness. Biometrics. 1984;40:119–129. doi: 10.2307/2530750. [DOI] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A. 2004;101(30):11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvar J, Mas-Coma S, Carrasco M. Modern history and physical geography of Equatorial Guinea. Rev Sanid Hig Publica (Madr) 1996;2–3:77–83. [Google Scholar]
- Worobey M, Telfer P, Souquiere S, Hunter M, Coleman CA, Metzger MJ, Reed P, Makuwa M, Hearn G, Honarvar S. et al. Island biogeography reveals the deep history of SIV. Science. 2010;329(5998):1487. doi: 10.1126/science.1193550. [DOI] [PubMed] [Google Scholar]
- Deitz KC, Athrey G, Reddy MR, Overgaard HJ, Matias A, Jawara M, Della Torre A, Petrarca V, Pinto J, Kiszewski AE. et al. Genetic isolation within the malaria mosquito Anopheles melas. Mol Ecol. 2012;21(18):4498–4513. doi: 10.1111/j.1365-294X.2012.05724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post RJ, Flook PK, Millest AL, Cheke RA, McCall PJ, Wilson MD, Mustapha M, Somiari S, Davies JB, Mank RA. et al. Cytotaxonomy, morphology and molecular systematics of the Bioko form of Simulium yahense (Diptera: Simuliidae) Bull Entomol Res. 2003;93(2):145–157. doi: 10.1079/BER2003228. [DOI] [PubMed] [Google Scholar]
- Solano P, Ravel S, Bouyer J, Camara M, Kagbadouno MS, Dyer N, Gardes L, Herault D, Donnelly MJ, De Meeus T. The population structure of Glossina palpalis gambiensis from island and continental locations in Coastal Guinea. PLoS Negl Trop Dis. 2009;3(3):e392. doi: 10.1371/journal.pntd.0000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara M, Caro-Riano H, Ravel S, Dujardin JP, Hervouet JP, De Meeus T, Kagbadouno MS, Bouyer J, Solano P. Genetic and morphometric evidence for population isolation of Glossina palpalis gambiensis (Diptera: Glossinidae) on the Loos islands, Guinea. J Med Entomol. 2006;43(5):853–860. doi: 10.1603/0022-2585(2006)43[853:GAMEFP]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Moreno M, Salgueiro P, Vicente JL, Cano J, Berzosa PJ, de Lucio A, Simard F, Caccone A, Do Rosario VE, Pinto J. et al. Genetic population structure of Anopheles gambiae in Equatorial Guinea. Malar J. 2007;6:137. doi: 10.1186/1475-2875-6-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D. Concerted evolution: molecular mechanism and biological implications. Am J Hum Genet. 1999;64(1):24–30. doi: 10.1086/302221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JC, Pinto J, Charlwood JD, Gentile G, Santolamazza F, Simard F, del la Torre A, Donnelly MJ, Caccone A. Exploring the origin and degree of genetic isolation of Anopheles gambiae from the islands of Sao Tome and Prıncipe, potential sites for testing transgenic-based vector control. Evol Appl. 2008;4(1):631–644. doi: 10.1111/j.1752-4571.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Pavelitz T, Kidd JR, Kidd KK, Weiner AM. Concerted evolution of the tandemly repeated genes encoding human U2 snRNA (the RNU2 locus) involves rapid intrachromosomal homogenization and rare interchromosomal gene conversion. EMBO J. 1997;16(3):588–598. doi: 10.1093/emboj/16.3.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Weiner AM. Concerted evolution of the tandemly repeated genes encoding primate U2 small nuclear RNA (the RNU2 locus) does not prevent rapid diversification of the (CT)n.(GA)n microsatellite embedded within the U2 repeat unit. Genomics. 1995;30(3):583–593. doi: 10.1006/geno.1995.1280. [DOI] [PubMed] [Google Scholar]
- Williams B, Dransfield R, Brightwell R. The control of tsetse flies in relation to fly movement and trapping efficiency. J Appl Biol. 1992;29:163–179. [Google Scholar]
- Bursell E, Taylor P. An energy budget for Glossina (Diptera: Glossinidae) Bull Entomol Res. 1980;78:281–291. [Google Scholar]
- Bursell E. Quantitative aspects of proline utilization during flight in tsetse flies. Physiol Entomol. 1978;3:265–272. doi: 10.1111/j.1365-3032.1978.tb00158.x. [DOI] [Google Scholar]
- Brady J. The circadian organization of behaviour: time keeping in the tsetse fly, a model system. Adv Stud Behav. 1988;18:153–159. [Google Scholar]
- Mateu E, Comas D, Calafell F, Perez-Lezaun A, Abade A, Bertranpetit J. A tale of two islands: population history and mitochondrial DNA sequence variation of Bioko and Sao Tome, Gulf of Guinea. Ann Hum Genet. 1997;61(Pt 6):507–518. doi: 10.1046/j.1469-1809.1997.6160507.x. [DOI] [PubMed] [Google Scholar]
- Vale GA, Bursell E, Hargrove JW. Catching-out the tsetse fly. Parasitol Today. 1985;1(4):106–110. doi: 10.1016/0169-4758(85)90005-5. [DOI] [PubMed] [Google Scholar]
- Hargrove JW. Optimized simulation of the control of tsetse flies Glossina pallidipes and G. m. morsitans (Diptera: Glossinidae) using odour-baited targets in Zimbabwe. Bull Entomol Res. 2003;93(1):19–29. doi: 10.1079/BER2002205. [DOI] [PubMed] [Google Scholar]
- Gooding RH, Krafsur ES. Tsetse genetics: contributions to biology, systematics, and control of tsetse flies. Annu Rev Entomol. 2005;50:101–123. doi: 10.1146/annurev.ento.50.071803.130443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafsur ES. Tsetse fly population genetics: an indirect approach to dispersal. Trends Parasitol. 2003;19(4):162–166. doi: 10.1016/S1471-4922(03)00034-5. [DOI] [PubMed] [Google Scholar]
- Simo G, Njiokou F, Mbida Mbida JA, Njitchouang GR, Herder S, Asonganyi T, Cuny G. Tsetse fly host preference from sleeping sickness foci in Cameroon: epidemiological implications. Infect Genet Evol. 2008;8(1):34–39. doi: 10.1016/j.meegid.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Clausen PH, Adeyemi I, Bauer B, Breloeer M, Salchow F, Staak C. Host preferences of tsetse (Diptera: Glossinidae) based on bloodmeal identifications. Med Vet Entomol. 1998;12(2):169–180. doi: 10.1046/j.1365-2915.1998.00097.x. [DOI] [PubMed] [Google Scholar]
- Melachio TT, Simo G, Ravel S, De Meeus T, Causse S, Solano P, Lutumba P, Asonganyi T, Njiokou F. Population genetics of Glossina palpalis palpalis from central African sleeping sickness foci. Parasit Vectors. 2011;4:140. doi: 10.1186/1756-3305-4-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spath J. Feeding patterns of three sympatric tsetse species (Glossina spp.) (Diptera: Glossinidae) in the preforest zone of Cote d’Ivoire. Acta Trop. 2000;75(1):109–118. doi: 10.1016/S0001-706X(99)00096-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of primers used.
Estimated genetic diversity, calculated in number of observed haplotypes.
Alignment of 16S found in G. p. palpalis from Luba focus. Gpp: G. p. palpalis, CAM: Cameroon, Gpg: G. p. gambiensis, Gfq: G. fuscipes quanzensis, Gff: G. fuscipes fuscipes, Gtac: G. tachinoides, Gpallic: G. pallicera, Gmm: G. morsitans morsitans, Gpallid: G. pallidipes.
Sequencing profiles and alignment of ITS1 genotypes found in Luba focus. Sequence profiles of ITS genotypes 1 and 2 (A and B, respectively). C) Alignment of ITS1 genotypes of G. p. palpalis from different origins. EQG-L: Equatorial Guinea, Luba focus, EQG-M: Equatorial Guinea, Mbini focus, EQG-K: Equatorial Guinea, Kogo focus, DRC: Democratic Republic of Congo, LIB: Liberia, CDI: Cote d’Ivoire, TOG: Togo, GAM: Gambia, BUF: Burkina Faso, GUI: Guinea.