

Abstract
SgrAI is a sequence specific DNA endonuclease that functions through an unusual enzymatic mechanism that is allosterically activated 200-500 fold by effector DNA, with a concomitant expansion of its DNA sequence specificity. Using single-particle transmission electron microscopy to reconstruct distinct populations of SgrAI oligomers, we show that, in the presence of allosteric, activating DNA, the enzyme forms regular, repeating helical structures that are characterized by the addition of DNA-binding dimeric SgrAI subunits in a run-on manner. We also present the structure of oligomeric SgrAI at 8.6 Å resolution, demonstrating a novel conformational state of SgrAI in its activated form. Activated and oligomeric SgrAI displays key protein-protein interactions near the helix axis between its N-termini, as well as allosteric protein-DNA interactions that are required for enzymatic activation. The hybrid approach reveals an unusual mechanism of enzyme activation that explains SgrAI’s oligomerization and allosteric behavior.
INTRODUCTION
The co-evolution between parasitic phage and host bacterium represents one of nature’s most extensive struggles for survival. Competitive environmental interactions have led both parasite and host to pursue intricate and diverse strategies of adaptation and counter-adaptation in what has been metaphorically described as an evolutionary arms race (Stern and Sorek, 2011). Like higher eukaryotes, host bacteria possess an innate, non-specific immune system that accounts for the first line of defense against invading phage, which has given rise to numerous clever enzymatic mechanisms for selectively targeting and cleaving phage DNA. Restriction endonucleases (REases) are the enzymes that play a primary role in this mechanism for defense and are believed to have evolved specifically for this purpose (Pingoud and Jeltsch, 2001; Pingoud et al., 2005; Pingoud and Jeltsch, 1997).
REases recognize and cleave duplex DNA and can be categorized into one of four different types (Roberts et al., 2010). The classical Type II REase is homodimeric and cleaves DNA in a Mg2+ dependent fashion at or near its 4-6 base-pair (bp) recognition sequence (Pingoud and Jeltsch, 2001; Pingoud et al., 2005; Pingoud and Jeltsch, 1997). Of the ~4000 categorized type II REases to date, only a small fraction cleave sequences containing more than 6 bp (Roberts et al., 2010). Due to the relative rarity of such cleavage sites, these enzymes have received particular attention, both for their interesting evolutionary properties (Bilcock et al., 1999) and for their value as tools for the analysis of genomic DNA (Qiang and Schildkraut, 1987). SgrAI is one such “rare-cutting” endonuclease, which recognizes 3 degenerate primary (or canonical) octanucleotide sequences that differ in the 2nd and 7th bp – CGCCGGCG, CACCGGCG/CGCCGGTG, and CACCGGTG (Tautz et al., 1990). However, unlike any other REase, SgrAI will additionally cleave any of 14 secondary (or non-canonical) sequences (CPuCCGGPy(A/T/C) and CPuCCGGGG), but only in the presence of an activating primary site DNA containing a sufficient number of flanking bp (Bitinaite and Schildkraut, 2002; Park et al., 2010b; Wood et al., 2005). Thus, depending upon the input signal, the SgrAI REase can turn a rare DNA recognition sequence into one that is much more frequently encountered, dramatically increasing the number of DNA cleavages. How SgrAI can do this and the evolution of this mechanism is of considerable interest.
SgrAI maintains a baseline rate of cleavage for both primary and secondary sites, albeit one that is lower than its homologs (Bilcock et al., 1999; Park et al., 2010b). However, in the presence of multiple DNA cleavage sites, SgrAI acquires several properties that make the enzyme unique among known REases. First, it relaxes the requirement for highly defined nucleotide recognition sites, enabling the cleavage of a total of 17 degenerate DNA sequences through a process known as sequence-specificity expansion (Bitinaite and Schildkraut, 2002). Second, the rate of subsequent DNA cleavage for primary and secondary sites is accelerated by up to several orders of magnitude (Hingorani-Varma and Bitinaite, 2003; Park et al., 2010b). Simultaneous sequence-specificity expansion and DNA cleavage rate acceleration have been more generally known as SgrAI activation (Park et al., 2010b). In order for activation to occur, DNA containing the primary cleavage site must be present (Bitinaite and Schildkraut, 2002). The DNA must also contain a minimum number of flanking bp (Park et al., 2010b; Wood et al., 2005). This indicates that substrate DNA itself plays the role of an allosteric effector within a positively cooperative and allosteric enzymatic reaction. Thus, previous studies have hypothesized that SgrAI must exhibit at least two conformational states in order to account for its self-activating behaviour (Daniels et al., 2003; Dunten et al., 2008; Hingorani-Varma and Bitinaite, 2003; Park et al., 2010b). However, only the dimeric low-activity form has been structurally characterized to date (Dunten et al., 2008; Little et al., 2011; Park et al., 2010a).
Here we present a structural, computational, and biochemical characterization of activated and oligomeric SgrAI. This hybrid approach provides insight into the enzyme’s unusual manner of modulating activity through self-association into run-on oligomers – a phenomenon that is unique among REases. In addition, it reveals a novel, high-activity conformational state of the enzyme that suggests an evolutionary requirement for its mechanism of action within the context of innate prokaryotic immunity.
RESULTS
SgrAI forms run-on oligomers in the presence of activating DNA
Previously, it has been shown that SgrAI activation can be achieved by the presence of activating DNA of any primary cleavage site containing a sufficient number of flanking bp. These include primary-site containing plasmids (Bilcock et al., 1999; Bitinaite and Schildkraut, 2002; Wood et al., 2005), as well as uncleaved (Bitinaite and Schildkraut, 2002) and pre-cleaved (PC – containing sticky ends and resembling reaction products from DNA cleavage) (Park et al., 2010b) synthetic DNA constructs. The activated species is heterogeneous and oligomeric in form, with an average size of ~700 kDa (Daniels et al., 2003; Park et al., 2010b). To investigate the structure of oligomeric SgrAI, we used transmission electron microscopy of negatively stained complexes of purified SgrAI and PC DNA that were prepared under conditions for promoting oligomerization (Park et al., 2010b). Individual particles observed in the images were highly heterogeneous in size (Figure S1). Using an iterative workflow for aligning and reconstructing distinct populations of SgrAI oligomers (Lyumkis et al., 2013), we identified multiple classes of particles representing discrete oligomer lengths. The smallest particles were identified as an SgrAI DNA-binding dimer (DBD) (Fig. 1A), which forms the basic building block of the oligomer. Subsequently, 2D class averages and corresponding 3D reconstructions obtained using the random-conical tilt approach (Radermacher et al., 1987; 1986) were systematically arranged according to size, from a dimer of DBDs through a decamer of DBDs. Larger species, such as a tridecamer, become less well defined in the terminal regions due to flexibility along the particles (Fig. 1B). The heterogeneous distribution of oligomers in the data favors smaller sizes, with particles containing >10 DBDs becoming increasingly rare (Fig. 1C). This analysis shows that the low-activity SgrAI DBD that was previously characterized crystallographically (Dunten et al., 2008; Little et al., 2011; Park et al., 2010a) can oligomerize in a run-on fashion in the presence of activating DNA, supporting a current hypothesis from mass spectrometry data (Ma et al., 2013).
Figure 1. SgrAI forms run-on oligomers in the presence of activating DNA.

2D class averages and corresponding 3D EM density maps created by the random-conical tilt reconstruction strategy are displayed for (A) a single SgrAI DBD with the corresponding crystal structure(Dunten et al., 2008) displayed above, and subsequently for (B) elongating oligomers. Scale bars represent 150 Å. (*) The 3-DBD oligomers adopt an alternative orientation on the EM grid and, their 3D maps are therefore rotated in order to correspond to the 2D average. (C) Oligomer size distribution within the negative stain data set; the X-axis indicates the number of DBDs represented by a single oligomer identified by 2D classification; the Y-axis indicates the percentage of particles within the full data set corresponding to each DBD grouping, and in parentheses, the absolute number of particles. See also Figure S1 and Table S2.
The structure of activated and oligomeric SgrAI at 8.6 Å resolution
Individual DBD subunits within each reconstructed oligomer are organized into a left-handed helix, with ~85Å separation between adjacent subunits along the oligomeric Z axis. To improve the resolution of the EM density maps and to visualize DNA (which is not evident under most negative stain conditions, see Figure S3C-D), we prepared identical samples of SgrAI by vitrification and used cryo-electron microscopy to characterize the oligomers (Figure S2). Starting with the helical parameters measured from the 3D random-conical tilt reconstructions, we selected the largest helices identifiable within the cryo-micrographs and refined a data set of frozen hydrated oligomeric SgrAI to 8.6 Å resolution (Fig. 2A-B, Figure S2-3, and movie S1). An SgrAI DBD makes up the basic helical asymmetric unit. The final helical parameters for the assembly were determined to be 21.6 Å rise and -86.2° twist, enabling slightly more than 4 DBD subunits to be built into a single turn of the helix (Fig. 2A & movie S1). At this resolution, the major and minor grooves of the B-form DNA, as well as most alpha-helical secondary structure elements of the protein are readily visible in the EM density maps (Fig. 2C & Figure S2E). In conjunction with crystal structures of the low-activity form of SgrAI (Dunten et al., 2008; Little et al., 2011; Park et al., 2010a), the structural details of the subnanometer resolution map enabled flexible fitting of the coordinates into the EM density (Fig. 2D). The EM density can fully accommodate 12 of the 16 DNA flanking bp on either side of the helix. The outer bp likely become disordered, which can be seen by the gradual loss of density in the terminal regions (Figure 2D, arrows). Thus, the cryo-EM reconstruction together with flexible fitting provide approximate C-alpha coordinates for the activated and oligomeric form of SgrAI.
Figure 2. Structure of activated and oligomeric SgrAI.

(A) Top and side views showing the organization of helical asymmetric units (DBDs) within the SgrAI oligomers. Eight distinct DBDs have been segmented out of the cryo-EM map and are each colored differently. (B) Helical reconstruction of oligomeric SgrAI at 8.6 Å resolution, segmented into 11 individual DBDs and labeled by helical asymmetric unit. Protein components of units 1,4,6,7,9 and 2,3,5,8, 10, 11 are shaded light and dark, respectively. (C) Segmentation and different views of an individual helical asymmetric unit. Each helical asymmetric unit contains two monomeric SgrAI protein subunits (colored light and dark orange), and two copies of pre-cleaved DNA (both colored blue). (D) Flexibly fit coordinates of SgrAI (Dunten et al., 2008) into the EM density of a segmented helical asymmetric unit. Arrows mark DNA disorder in the terminal regions. Scale bar is 150 Å. See also Figure S2-S3 and Movie S1.
A conformational rearrangement establishes oligomer-specific interactions within activated SgrAI
Previous analyses of activated SgrAI have focused on kinetic and biochemical properties of the enzyme, while its high-activity structure has remained elusive (Dunten et al., 2008; Little et al., 2011; Park et al., 2010a). Here we present features of oligomeric SgrAI, characterized by single-particle electron microscopy and flexible fitting, that are specific to its high-activity form.
Flexible fitting of the atomic coordinates of SgrAI (Dunten et al., 2008) into the 8.6 Å EM density map reveals notable rearrangement within several regions of the protein (Fig. 3A). The overall RMSD for the structure is 2.1 Å. To properly position the SgrAI backbone into the EM density, internal areas of the protein require little rearrangement (e.g. 1Å RMSD for helix 247-268). However regions at, or in proximity to, critical protein-DNA interactions require greater rearrangement (e.g. 2.6 Å RMSD for loop 122-140) (Table S1). Viewed from the outside of the oligomeric assembly, each monomeric subunit undergoes a rotation and slight vertical extension about and away from its central cleavage core in a twisting fashion (Fig. 3A & movie S2). This movement slightly alters the location of C-alpha residues involved in selectivity for the outer two bp of the DNA recognition sequence, R31 and K96, although the atomic consequences of this alteration, particularly with regard to the protein-DNA interface, remain to be determined.
Figure 3. A conformational rearrangement establishes oligomer-specific interactions within activated SgrAI.
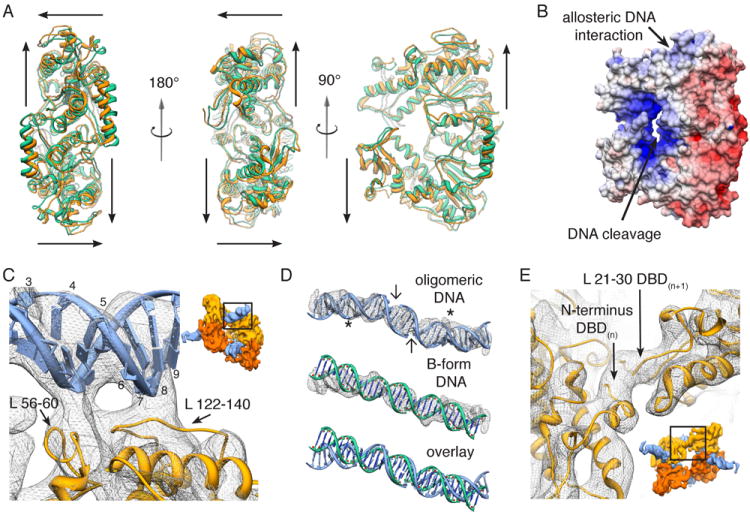
(A) Three different views of the crystal structure 3DVO(Dunten et al., 2008) (green) are shown overlaid on the flexibly fit coordinates of oligomeric SgrAI (orange). For each DBD, arrows diagram approximate movement from the starting to the ending structure (see also movie S2). (B) Surface electrostatic potential of a SgrAI DBD. (C) Close-up view of protein-DNA interactions within SgrAI’s allosteric DNA-binding site, as seen from the outside of the helix (inset). Protein loops 56-60 and 122-140, which are unique to SgrAI, are shown interacting in the vicinity of flanking bp 3-5 and 6-9, respectively, (numbered along one DNA strand) and along the minor groove of the DNA. (D) Comparison of the fit oligomeric PC-DNA with idealized B-form DNA. Oligomeric and B-form DNAs are overlaid on the EM density in mesh. Cleavage sites forming the two pre-cleaved DNAs are marked by arrows. Stars indicate the site of allosteric protein-DNA interaction. (E) Close-up view of SgrAI’s N-terminal interactions, as seen from the inside of the helix (inset). The N-terminus of DBD(n) makes apparent interactions with a portion of the N-terminal loop (aa 21-30) of DBD(n+1). See also Figure S4, Table S1 and Movie S2.
Unlike its closest homologs – Cfr101, NgoMIV, and Bse634I – SgrAI contains a positively charged region in the outer periphery of the enzyme (Fig. 3B), while the rest of the protein is highly electrostatically negative (Figure S4). This region corresponds to the site of apparent contact with flanking DNA within the oligomer (Fig. 3C). Previous studies have shown that DNA flanks are required for, and play an allosteric role in activation (Bitinaite and Schildkraut, 2002; Daniels et al., 2003; Hingorani-Varma and Bitinaite, 2003; Little et al., 2011; Park et al., 2010b; Wood et al., 2005). Protein-DNA contacts appear in two positions, both contacting along the minor groove of the DNA. Protein loop 56-60 and DNA-flanks 3-5, as well as protein loop 122-140 and DNA-flanks 6-9 constitute two separate interactions evident in the EM density (Fig. 3C). Loop 56-60 contains small, uncharged residues that may interact with the backbone, sugar moieties, or DNA bases, while loop 122-140 contains three Arg residues that may interact with the DNA backbone. Both loops are specific to SgrAI and do not exist within its closest type IIF homologues, which do not form oligomers larger than tetramers. Previous crystal structures of DNA-bound SgrAI dimers show flanking DNA extending outward on both sides of the cleavage site (Dunten et al., 2008; Little et al., 2011; Park et al., 2010a). The EM density from the oligomeric form of the protein cannot accommodate a simple straight extension of idealized B-form DNA. Instead, the DNA makes an approximately 30° bend on either side of the cleavage site in order to interact with neighboring DBDs (Fig. 3D). This bend is particularly apparent along the minor groove, at and beyond the site of interaction with SgrAI. Due to apparent disorder in the terminal region, we cannot fit any of the DNA between flanking bp 12-16. DNA is known to exhibit similar local flexibility upon interaction with certain DNA-binding proteins (Peters and Maher, 2010). Thus the oligomeric form of SgrAI may facilitate stabilization of the DNA bend by slightly re-positioning the two protein subunits and providing an allosteric binding site.
SgrAI’s N-terminus represents another unique region of the protein that is unlike that of its closest homologs (Dunten et al., 2008). It has been shown to be required for activation, since mutation of Pro-27 eliminates oligomerization (Park et al., 2010a). In the oligomeric structure, the N-terminus of DBD(n) appears to interact with the N-terminal loop region of DBD(n+1) (aa 21-30) (Fig. 3E). The EM density does not directly accommodate the type of domain-swapping previously observed (Park et al., 2010a), but there is the possibility that alternative orientations may exist in the oligomeric form. The dissimilarity between SgrAI’s N-termini and those of its closest homologs, their requirement for oligomerization, and their proximity to each other within the oligomeric structure, support the conclusion that SgrAI’s N-terminal interactions facilitate the regulation of its enzymatic activity.
These data show that the oligomeric conformation of SgrAI differs from its dimeric low-activity form. The presence of neighboring DBDs and the conformational rearrangement within each individual DBD enable the formation of protein-DNA interactions between its loops and flanking DNA from the nearest neighbors, as well as protein-protein interactions within its N-termini. The rearrangement as a whole may have considerable consequences for the positioning of critical residues responsible for enzymatic activity.
SgrAI activation requires a minimum number of DNA flanking bp
Previous biochemical studies have recognized the allosteric properties of DNA and indicated that a minimal number of DNA flanking bp must be present for SgrAI activation (Park et al., 2010b; Wood et al., 2005), supporting our structural findings that protein-DNA interactions within the oligomer occur in the vicinity of the DNA minor groove and between flanking bp 3-9. To investigate the precise requirement of flanking DNA length for SgrAI activation, primary site containing DNA constructs varying in flanking DNA length and sequence (Table S3) were tested for their ability to stimulate DNA cleavage by SgrAI in single turnover cleavage assays. For both intact and pre-cleaved DNA, greater numbers of flanking bp increased SgrAI activation to a greater extent, with >8bp-flanks increasing cleavage up to a rate constant of 22/min (Table 1). All constructs with only 5 bp-flanks failed to stimulate DNA cleavage despite having low nanomolar affinity for binding (see Park et al (Park et al., 2010b) and methods). Constructs with 6-7 flanking bp provided mixed activation that was somewhat dependent upon the sequence of DNA used (and in some cases the ability of the DNA to remain annealed and/or intact), while all constructs containing 8 bp-flanks effectively stimulated cleavage. These results indicate that(under the reaction conditions and temperature at which cleavage was examined) a minimum of 6 bp-flanks is required for any SgrAI activation. This is consistent with the two allosteric protein-DNA contacts identified within the oligomeric structure that span flanking bp 3-5 and 6-9.
Table 1.
Single Turnover DNA cleavage rate constants of 32P labeled DNA 19 in the presence of added unlabeled intact and/or pre-cleaved DNA.
| Added unlabeled DNA | Type | Length of flanks (bp) | kf (min -1) (% cleaved) | ks (min -1) (% cleaved) |
|---|---|---|---|---|
| 1 (PC) | P | 16 | 22±1(Park et al., 2010b) | N/A |
| 2 (40-1) | I | 16 | 10±1.4(Park et al., 2010b) (56±12%) | 0.032±0.012(Park et al., 2010b) (44±2%) |
| 3 | P | 10 | 9.4±1.6 | N/A |
| 4 | P | 8 | 9±4 | N/A |
| 5 | P | 8 | 0.54±0.13 (63±17%) | 0.08±0.05 (37±17%) |
| 6 | I | 8 | 5.4±0.9 (63±1%) | 0.04±0.01 (37±1%) |
| 7 | P | 7 | 9±1 | N/A |
| 8 | P | 7 | 2.17±0.17 (80±4%) | 0.08±0.04 (20±4%) |
| 8 top | P | 7 | N/A | 0.19±0.02 |
| 8 bot | P | 7 | N/A | 0.15±0.03 |
| 9 | I | 7 | 1.1±0.4 | N/A |
| 10 | I | 7 | 1.4±0.6 (24±39%) | 0.04±0.04 (76±39%) |
| 11 | I | 7 | 0.8±0.3 (44±30%) | 0.02± 0.01 (56±30%) |
| 12 | I | 7 | N/A | 0.05±0.03 (100±0%) |
| 13 | I | 6 | N/A | 0.13±0.05 |
| 14 | I | 6 | 1.0±0.6 (79±5%) | 0.02±0.002 (21±5%) |
| 15 | I | 6 | N/A | 0.14±0.02 |
| 16 | P | 6 | N/A | 0.13±0.05 |
| 17 | P | 6 | N/A | 0.15±0.04 |
| 18 | I | 5 | N/A | 0.09±0.01 |
| 19 (18-1) | I | 5 | N/A | 0.16±0.01 |
| 20 | P | 5 | N/A | 0.20±0.05 |
DNA sequences are shown in Table S3. Names in parentheses are those used previously for the same DNA constructs(Park et al., 2010b). “Type” refers to either pre-cleaved DNA (“P”) or intact DNA (“I”). “Length of flanking bp” refers to the number of DNA bp outside of the octanucleotide recognition sequence. Values for kf and ks refer to rate constants for SgrAI mediated cleavage of 1 nM 32P labeled DNA 19, derived from fitting the single turnover data. If the data fits significantly better to two rate constants rather than one, the faster rate constant is assigned to kf, and the slower to ks. If only a single rate constant is used in the fit, it is assigned to kf when significantly higher than the rate constant under non-stimulating conditions, and to ks when similar to that for non-stimulating conditions. The second rate constant is then assigned an N/A, as only one rate constant can describe the data. Numbers in parentheses indicate the percentage of DNA cleaved by the fast and the slow process in the case when two are present. The values are presented as the average of three measurements ± the standard deviation. See also Table S3.
DISCUSSION
In this study we show that SgrAI will assemble into a regular, repeating structure that is a run-on oligomer formed by the successive helical assembly of individual DBDs. When part of the oligomer, the structure of each of the two subunits comprising the DBD undergoes a large conformational rearrangement compared to the low-activity form characterized crystallographically (Dunten et al., 2008; Little et al., 2011; Park et al., 2010a). In this state, SgrAI maintains important interactions within its N-termini that are situated in the center of the helical SgrAI oligomer, and with flanking DNA on either side of the cleavage sites. The flanking DNA must contain a minimum of six bp to activate the enzyme under the reaction conditions described here; seven, and in particular eight flanking bp produce increased and more consistent activation. Any additional bp provide little, if any, activation increase. These biochemical results are consistent with our structural findings, which show that the protein-DNA interface occurs in two regions and involves both SgrAI-specific loops and both individual DNA strands of the minor groove (Figure 3B-C). Taken together, they suggest that this interface may contain multiple sites of contact along the minor groove, each of which adds a small degree of stability to the interaction as a whole.
SgrAI maintains an enzymatic activity that differs for primary and secondary cleavage sites. Its activity can be drastically altered by the presence of activating DNA, implying intrinsic allostery in its mechanism of action (Bitinaite and Schildkraut, 2002). Structurally, this requires the presence of at least two different conformational states of the enzyme. However, despite the biochemical differences, structures of SgrAI bound to both uncleaved (Dunten et al., 2008) and cleaved (Little et al., 2011) primary site DNA, as well as uncleaved secondary site DNA (Little et al., 2011) show few apparent differences. SgrAI bound to primary site DNA favors oligomer formation with either primary (Fig. 4A) or secondary (Fig. 4B) site DNA. However, in the presence of only secondary site DNA, oligomers do not form (Fig. 4C) (Park et al., 2010b). Therefore at least one primary site must bind to a neighboring DBD to activate the enzyme (Park et al., 2010b). Furthermore, oligomerization is temperature-dependent, since an identical DNA construct that cannot activate SgrAI at 37° is capable of low-level activation at 4° (compare construct 19 in this study to Park et al (Park et al., 2010b)). This finding implies that the limited protein-DNA interactions that occur between loop 56-60 and flanking bp 3-5 (Fig. 3C) are enough to weakly stabilize the oligomer. Presumably, the enthalpic contribution to oligomer stabilization changes little with increasing temperature, but the entropic contribution increases, and therefore at the higher temperature the limited protein-DNA interactions may be insufficient to overcome entropy leading to dissociation. Maximal stabilization is achieved only when loop 122-140 can efficiently contact bp 6-9 of flanking DNA. These data suggest that SgrAI’s two conformational states must exist in equilibrium, that the low-activity state may be inactive, and that the differences in enzymatic activity can be explained by SgrAI’s ability to access and maintain its high-activity form. The oligomer-specific structural features of the enzyme facilitate this maintenance; without sufficient DNA flanks to interact with SgrAI’s loops (Fig 3C), oligomerization will not occur (Table 1), nor will it occur without key N-terminal interactions (Park et al., 2010a). This structural and biochemical data allow us to propose that the conformationally rearranged DBD in the oligomeric form stabilizes, and thus represents, the activated enzyme. In sum, SgrAI can cleave both primary and secondary DNA in the activated conformation, albeit with different cleavage rates, the activated conformation favors oligomerization, and oligomerization in turn favors the activated conformation. The result is both sequence-specificity expansion and/or accelerated DNA cleavage (Figure 4). Future experiments will address the details of this mechanism, in particular with regard to enzyme turnover, including the balance between DBD association and dissociation from the oligomer and the kinetics of DNA release.
Figure 4. Model of SgrAI DNA-bound dimer (DBD) activation and enzymatic function.

(AC) SgrAI DBDs contain, at a minimum, two conformational states, which account for an inactive, or minimally active, (red) and an active (green) enzymatic state. The two exist in equilibrium, the extent of which is dependent upon whether (A) primary-site (blue) or (B-C) secondary-site (yellow) DNA is bound. DNA-recognition sites are displayed underneath, with the varying nucleotide in red. In the active, dimeric form, both primary- and secondary-site DNA can undergo cleavage at a slow rate that is determined by the frequency of occupation of the active state. (A) primary-site DBDs can initiate oligomerization, and they can do so with any DBD and regardless of the bound DNA sequence. (B) Secondary-site DBDs can join an oligomer containing primary-site DBDs. Oligomerization in turn stabilizes the activated conformation and facilitates attachment of additional DBDs, enabling DNA cleavage acceleration (A-B) and/or sequence-specificity expansion (B). To differentiate the distinct subunits, active & stabilized DBDs within the oligomer are shaded differently, although all are presumed to maintain equal activity. (C) Secondary-site DBDs cannot undergo oligomerization by themselves, and higher order species are not observed, although it is possible that transient oligomerzation occurs, but is undetectable under the experimental reaction conditions. The discussion section provides further mechanistic details.
Examples of enzyme regulation via run-on oligomerization are relatively uncommon. Recent screens have begun to identify other enzymes capable of self-association into filaments (An et al., 2008; Ingerson-Mahar et al., 2010; Noree et al., 2010; Werner et al., 2009), suggesting that this is a more common phenomenon that previously appreciated. However, the effect of oligomerization on enzyme activity for these enzymes is not currently known. In contrast, the more well-studied NTPases tubulin (Kueh and Mitchison, 2009), actin (Bindschadler et al., 2004), and members of the RecA family (Chen et al., 2008), form run-on oligomers with altered functional properties. Among these systems, it is typically the size, shape, binding behavior, or mechanical properties of the oligomer that is critical for function. While the enzymatic activity of these proteins may be altered with respect to the monomeric form, their substrate specificities remain unchanged. Importantly, the NTPase activity is typically altered in order to control oligomerization. The converse is true for SgrAI – its main purpose is the enzymatic function of DNA cleavage, and oligomerization appears to modulate enzymatic activity with regard to sequence-specificity expansion (and therefore alteration of substrate specificity) and cleavage rate acceleration. The mechanism by which enzymatic activity is modulated by run-on oligomerization has to our knowledge only been proposed for two other systems – acetyl-CoA carboxylase (ACC) described almost half a century ago (Vagelos et al., 1963), and the unfolded response protein, Ire1 (Korennykh et al., 2009). In the case of ACC, only very modest activation occurs with oligomerizaton (Boone et al., 2000; Brownsey et al., 2006). In the case of Ire1, an RNase activity is thought to switch on as a result of forming run-on oligomers. Neither mechanism involves the alteration of substrate specificity. Therefore SgrAI’s mechanism of activation and alteration of substrate specificity through run-on oligomerization seems to be exceptionally unusual within the enzymatic world.
REases are by definition non-specific enzymes in that they will cleave DNA of both phage and host. Therefore, in order to perform a protective role against invaders, they must maintain a mechanism through which they can discriminate self from non-self. One hypothesis for the biological function and evolution of SgrAI’s protective behavior is related to the unusually large genome of Streptomyces griseus, from which the enzyme originates. REases are postulated to protect their bacterial hosts from invading phage DNA, and are prevented from cleaving their host DNA by the action of a cognate methyltransferase that methylates, and thereby protects, the endonuclease target sequence in the host genome. A delicate balance must occur between the activities of the endonuclease and methyltransferase of any such restriction-modification system, such that methylation is conferred to the host, while cleavage to the invader. The large genome of S. griseus results in a greater number of potential cleavage sites, and consequently more opportunities for the endonuclease to cause damaging double stranded DNA cleavage in the host genome. To protect these sites, the cognate methyltransferase would be under selective pressure to methylate at the larger number of sites. To relieve this pressure, the longer recognition sequence of SgrAI may have evolved to reduce the total number of potential cleavage sites within the host genome. Similarly, the relatively slow basal DNA cleavage rate of SgrAI in comparison to other restriction endonucleases (Sam and Perona, 1999) also reduces the potential for DNA cleavage in the competition between methylation and DNA cleavage by the two restriction-modification enzymes. Inevitably, both of these qualities – the longer recognition sequence and the slow DNA cleavage rate – would be expected to reduce the efficacy of the SgrAI endonuclease in protecting against invading phage DNA. Activation by the spatial proximity of two unmethylated recognition sequences, which is expected to be rare in the host genome but very likely in phage DNA, combined with sequence-specificity expansion from three (primary only) to a total of seventeen (primary and secondary) different octanucleotide target sequences, would both function to increase the efficacy of SgrAI in protecting against invading phage DNA. However, such functions, particularly the expansion of sequence specificity to sites not potentially methylated by the cognate methyltransferase, could also elicit damaging DNA cleavage to the host. Oligomer formation may thus occur in order to sequester the activated SgrAI endonucleases on phage DNA, and away from the host, representing a clever defensive strategy in the phage-host competition.
EXPERIMENTAL PROCEDURES
Protein and DNA preparation
Wild type SgrAI (EC 3.1.21.4) was prepared as described (Dunten et al., 2008), and estimated at 99% purity by Coomassie stained SDS-PAGE. Purified SgrAI enzyme was dialyzed into storage buffer (20 mM Tris-OAc, (pH 8.0), 50 mM KOAc, 0.1 mM EDTA, 1 mM DTT, 50% glycerol), aliquoted into single use aliquots, flash frozen in liquid nitrogen, and stored at -80°C. The unmodified oligonucleotides (Table S3) used in this study were purchased from a commercial synthetic source, which utilized either gel purification or purification via C18 reverse phase HPLC (Aggarwal, 1990). The concentration of the DNA was measured spectrophotometrically, with an extinction coefficient calculated from standard values for the nucleotides (Fasman, 1975). The self-complementary DNA, or equimolar quantities of complementary DNA, were annealed by heating to 90°C for 10 minutes at a concentration of 1 mM, followed by slow-cooling to 4°C over 4-5 hours in a thermocycler. The concentration of the DNA was remeasured after annealing and presented in terms of duplex DNA. Because multiple freeze-thawing altered the concentration of double stranded DNA used in the assays by inducing separation of the two strands, DNA constructs used to test for SgrAI activation were aliquoted into single use amounts that were then flash frozen in liquid nitrogen and stored in - 20° C until needed. DNA was 5’ end labeled with 32P using T4 polynucleotide kinase and [γ-32P]-ATP, and excess ATP removed using G-30 spin columns.
Sample preparation for electron microscopy
Specimens were prepared for negative stain by applying 3 μ L of sample (3 μM SgrAI, 3 μM DNA 1 (PC) (Table S3) in cleavage buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM Mg(OAc)2, 0.5 mM DTT) or binding buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM Ca(OAc)2, 0.5 mM DTT) to a freshly plasma cleaned, continuous carbon grid. Sample was allowed to absorb to the carbon for 30 to 60 seconds. Excess sample was blotted from the side of the grid and replaced with 2% uranyl formate solution. Specimens were prepared for cryo-EM by applying 3 μL of sample in binding buffer to a holey carbon C-flat grid (CF-2/2-400) (Protochips, inc.) that had been plasma cleaned (Gatan, Solarus) for 5 sec. The sample was allowed to adsorb to the grid for 30 sec., then plunge-frozen into liquid ethane using a manual cryo-plunger at 4°C.
Electron microscopy data collection of negatively stained SgrAI
For the negative stain RCT dataset, data was acquired using a Tecnai F20 Twin transmission electron microscope operating at 120 keV, using a dose of 12 e-/Å2 and a nominal underfocus range of 1 to 3 μ m. Images were automatically collected at a nominal magnification of 62,000x, corresponding to a pixel size at the specimen level of 1.76 Å, at 0° and 50° tilt. Images were recorded using a Gatan 4 × 4 K pixel CCD camera utilizing the Leginon data collection software (Suloway et al., 2005).
Random-conical tilt reconstructions of separate populations of SgrAI
Experimental data were processed by the Appion software package (Lander et al., 2009), which interfaces with the Leginon database infrastructure. The contrast transfer function (CTF) for each micrograph was estimated using the ACE2 package, a variation of ACE1 (Mallick et al., 2005). CTF correction of the untilted particles was carried out by ace2image during creation of the particle stack, applying a wiener filter with a constant of 0.1. Initially, a small subset of particles were selected using the Difference of Gaussians particle picker (Voss et al., 2009). This provided a preliminary stack, which was aligned and classified in a reference-free manner using the CL2D algorithm (Sorzano et al., 2010). A template was extracted from the class averages and used to automatically select 35,581 tilted and untilted particles from the micrographs using a template-based particle picker (Roseman, 2004). After tilt-pair alignment of the tilted and untilted particle picks with TiltPicker (Voss et al., 2009), 5,442 particle tilt-pairs remained, which were binned by 2 and extracted using a boxsize of 112 pixels, corresponding to a pixel size of 3.52 Å at the specimen level. The stack was aligned and classified using ISAC (Yang et al., 2012), while maintaining approximately 100 particles per class. Classes representing each distinct SgrAI multimer were aligned to each other to create upright SgrAI references, and these were subsequently used for a reference-based alignment using SPIDER (Frank et al., 1996), followed by multivariate statistical analysis and hierarchical ascendance classification using IMAGIC (van Heel et al., 1996). This step enabled the use of the random-conical tilt pipeline implemented inside Appion (Voss et al., 2010). Particles belonging to each unique class average were reconstructed using the RCT pipeline, and the final volumes were assessed by visual evaluation and from resolution based on the FSC 0.5 criterion. We retained all unique volumes for which, after aligning the central scaffold of the SgrAI oligomer to all distinct RCT volumes, an addition of an SgrAI dimer was observed in either of the two termini at a threshold level that corresponded to the exact size of the SgrAI oligomer. Volumes were discarded based on two criteria: (i) if the volume resulted in a lower resolution than a comparable conformer and if combining their particles did not result in an improvement in resolution; (ii) if the volume lacked sufficient particle numbers to average out noise contributions. Therefore class averages that were separated at the level of classification but produced density maps with less distinguishable SgrAI dimer additions were grouped into a single reconstruction, but only if the result produced an improvement in resolution. From the 5,442 untilted particles, a total of 2,435 were included in the RCT reconstructions. All parameters for the final volumes are summarized in Table S2. Based on these parameters, the average rise and twist were approximated as 21Å and -90°, respectively. These were used to initiate the refinement of helical filaments. Alignment of the volumes to their central scaffold was performed manually in Chimera (Pettersen et al., 2004).
Oligomer size distribution
An alignment and classification of a 6505-particle stack was performed with CL2D (Sorzano et al., 2010), using a boxsize of 564Å in order to accommodate the largest oligomers, and specifying 256 classes (~25 particles per class) for finer class separation. The number of DBDs accounting for each class average was manually summed. The final distribution histogram is shown in Fig. 1C, where the data was merged into 2-dimer bins
Cryo-electron microscopy data collection, raw-frame alignment, and dose-fractionation of SgrAI
Data was acquired using the Leginon software (Suloway et al., 2005) installed on a Tecnai F20 Twin transmission electron microscope operating at 200kV, with a dose of 40 e-/Å2 and a nominal underfocus ranging from 1 to 4 μm. The dose was fractionated over 20 raw frames collected on the Direct Electron DE-12 direct detection device, with each frame receiving a dose of 2 e-/Å2. 656 “movies” were automatically collected and recorded at a nominal magnification of 29,000X, corresponding to a pixel size of 1.42 Å at the specimen level. The individual frames were aligned using a Spider script that tracks the shifts between individual frames, in a manner similar to that described in Campbell et al. (Campbell et al., 2012) but without frame-averaging. Two raw-frame stacks were created, from which particles were subsequently extracted – the first contained 8 aligned frames with a total dose of 16 e-/Å2, while the second contained 16 aligned frames, corresponding to a total dose of 32 e-/Å2.
Refinement of the SgrAI helix
The RCT reconstruction of an SgrAI nonamer was used as an initial model for helical refinement of the negative stain data set. Helical refinement were performed using the IHRSR routine implemented in the SPARX package (Behrmann et al., 2012; Hohn et al., 2007). Raw particles used in the refinement were selected based on the criterion that at least 8 SgrAI dimers are present, as judged by the corresponding class averages to which they belong. This provided 1,645 particles for the refinement, aligned along the oligomerization axis, and the refinement was carried out so that the outer radius of alignment would not exceed the radius of an SgrAI octamer. The helical refinement from the RCT initial model was subsequently used to initiate refinement of the cryo-data. For the cryo-EM data set, 691 filaments were selected from 322 raw-frame aligned cryo-micrographs of SgrAI, whose CTF parameters were determined using CTFFind. These filaments were heterogeneous in overall length. Helical segments were windowed at 23 Å intervals using a box size of 272 Å (192 pixels at a pixel size of 1.42), i.e. slightly larger than the length of a helicaly asymmetric unit (Behrmann et al., 2012), and corresponding to ~90% overlap (Sachse et al., 2007). This provided 7674 helical segments. We first conducted 25 iterations of the IHRSR routine (Behrmann et al., 2012) to refine the full cryo-data set and obtain final helical parameters: -86.2° helical twist and 21.6 Å rise. Subsequently, the model from IHRSR was used for refinement in Frealign (Grigorieff, 2007) specifying different dose-fractionated stacks for the refinement (32 e-/Å2) and reconstruction (16 e-/Å2) routines. The final reconstruction was obtained from 1,918 filament segments (25% of the data), averaged 8 times to account for the helical symmetry. To avoid over-fitting, we employed the resolution-limited method for refinement (Stewart and Grigorieff, 2004). The resolution of the refinement was limited to 15 Å until the last several iterations, at which point individual particles were allowed to refine to 12 Å. Two-fold symmetry perpendicular to the helical axis was applied after the refinement and reconstruction procedure by averaging two identical maps, where one has an equivalent rotation about the XZ plane. Incorporation of two-fold symmetry in the refinement and reconstruction process had minimal affects on the nominal resolution value and did not provide obvious improvements to the map over its application post-reconstruction. The progression from the initial to the refined model is displayed in Figure S3.
Flexible fitting of the SgrAI core into the cryo-EM reconstruction
To determine the conformation of the activated enzyme, an isolated map of the SgrAI DBD was obtained using Segger (Pintilie et al., 2010) through the Chimera interface (Pettersen et al., 2004). A model of SgrAI bound to PC DNA generated using the x-ray crystal structure of SgrAI bound to an 18 bp DNA containing a primary site (PDB ID: 3DVO) and additional modeled flanking DNA in B form was flexibly fitted using Direx (Schröder et al., 2007), a geometry-based conformational sampling approach under low-resolution restraints. The refinement procedure was run for 500 steps followed by the minimization procedure of 300 steps. The following changes in the parameter set were incorporated: the radius used to compute the gradient for each atom (map_probe_sig) was set to 0.5 Å, the number of elastic restraints (den_no) was set equal to the twice the number of atoms in the system, the strength of the elastic network (den_strength) was increased to 0.4, the residue range for DEN restraints (den_resid_range) was set to 30, and the γ parameter (den_gamma) was set to 0 to avoid any optimization for individual proteins.
Electrostatic calculations
Electrostatic calculations were performed using the APBS web server (http://www.poissonboltzmann.org) (Baker et al., 2001) using default parameters.
Single turnover DNA cleavage assays
Single turnover DNA cleavage measurements were performed as described previously (Park et al., 2010b) using rapid chemical quench techniques and 5’ end 32P labeled oligonucleotide substrates (typically 1.0 nM), under conditions of excess enzyme (1.0 μM), with and without the additional unlabeled DNA. All reactions were performed at 37° C in 20 mM Tris-OAc (pH 8.0), 50 mM KOAc, 10 mM Mg(OAc)2, and 1 mM DTT. Reactions were initiated by mixing 50 μl containing the DNA (1.0 nM 32P labeled and 0.5-1.0 μM unlabeled) in reaction buffer with 50 μl of enzyme (1.0 μM SgrAI) also in reaction buffer. Both solutions were preheated for 5 min at 37° C. At various times after mixing, 5 μl aliquots were withdrawn and quenched by addition to 5 μl of quench (80% formamide, 50 mM EDTA). Samples were stored at -20° C until they could be electrophoresed on 20% denaturing polyacrylamide (19:1 acrylamide:bisacrylamide, 4 M urea, 89 mM Tris, 89 mM boric acid, 2 mM EDTA) gels. Autoradiography of gels was performed without drying and a phosphor image plate exposed at 4° C for 12-17 hours. Densitometry of phosphor image plates was performed with a Typhoon Scanner (GE Healthcare Life Sciences), and integration using ImageQuant (GE Healthcare Life Sciences) or ImageJ (Abràmoff and Magalhães, 2004). The percent of product formed as a function of time was determined by integrating the density of both cleaved and uncleaved DNA bands, and normalizing to the total amount cleaved. The single turnover DNA cleavage rate constant was determined from the data using a single exponential function:
where C1 is a constant fitting the baseline, C2 is the total percent of DNA predicted to be cleaved by SgrAI, k is the rate constant, and t is the length of incubation in minutes. The data from some reactions fit poorly to a single exponential function. These were found to fit well to the sum of two exponential functions:
where C1 is a constant fitting the baseline, C2 is the total percent of DNA predicted to be cleaved by SgrAI with rate constant k1, C3 is the total percent of DNA predicted to be cleaved by SgrAI with rate constant k2, and t is the length of incubation in minutes. The rate constants are presented in Table 1 as kf and ks, where kf is the greater (faster) of the two rate constants k1 and k2, and ks is the lower (slower). For reactions fit by a single rate constant, the rate constant is listed as kf or ks in Table 1 depending on whether it was significantly greater than or similar to the unactivated rate constant (0.094±0.015 min-1) (Park et al., 2010b), respectively. Measurements were performed at least three independent times, and presented as the average±standard deviation (Table 1).
DNA binding assays
The gel shift assay (Carey, 1991) was used to measure the binding affinity of SgrAI to DNA 18 (Table 1 and Table S3) in the manner described previously (Park et al., 2010b). The equilibrium dissociation constant (KD) for DNA 18 was determined to be 0.6±0.1 nM, nearly identical to the 0.6±0.2 nM for DNA 19 determined previously (Park et al., 2010b) (Table 1 and Table S3).
Supplementary Material
HIGHLIGHTS.
Single-particle TEM identifies oligomeric SgrA/DNA as a helical run-on oligomer
The 8.6Å cryo-EM structure reveals a novel conformational state of activated SgrAI.
Within the oligomer, key interactions occur between the N-termini of SgrAI and between an allosteric site and flanking DNA.
The length of flanking DNA required for activation is determined to be 6-8 bp.
Acknowledgments
We would like to thank New England Biolabs for the kind gift of SgrAI enzyme and to Arne Moeller for help with figure graphics. This project was supported by grants from the National Science Foundation (NSF 0744732) to F.T. and N.C.H., grants from the National Center for Research Resources (RR017573) and the National Institute of General Medical Sciences (GM103310) from the National Institutes of Health to B.C. and C.S.P.
Abbreviations
- bp
base pairs
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- KOAc
potassium acetate
- MeCN
acetonitrile
- OAc
acetate
- PAGE
polyacrylamide gel electrophoresis
- SDS
sodium dodecyl sulfate
Footnotes
ACCESSION CODES
The cryo-EM reconstruction of oligomeric SgrAI and the segmented DBD subunit have been deposited in the electron-microscopy databank under accession codes ******* and *******. The flexibly fit C-alpha coordinates have been deposited in the protein databank under accession code *******.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abràmoff MD, Magalhães PJ. Image processing with ImageJ. Biophotonics Journal. 2004;11:36–42. [Google Scholar]
- Aggarwal AK. Crystallization of DNA binding proteins with oligodeoxynucleotides. Methods. 1990;1:83–90. doi: 10.1016/S1046-2023(05)80150-1. [DOI] [PubMed] [Google Scholar]
- An S, Kumar R, Sheets ED, Benkovic SJ. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008;320:103–106. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrmann E, Tao G, Stokes DL, Egelman EH, Raunser S, Penczek PA. Real-space processing of helical filaments in SPARX. J Struct Biol. 2012;177:302–313. doi: 10.1016/j.jsb.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilcock DT, Daniels LE, Bath AJ, Halford SE. Reactions of type II restriction endonucleases with 8-base pair recognition sites. J Biol Chem. 1999;274:36379–36386. doi: 10.1074/jbc.274.51.36379. [DOI] [PubMed] [Google Scholar]
- Bindschadler M, Osborn EA, Dewey CF., Jr A mechanistic model of the actin cycle. Biophysical Journal. 2004;86:2720–2739. doi: 10.1016/S0006-3495(04)74326-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitinaite J, Schildkraut I. Self-generated DNA termini relax the specificity of SgrAI restriction endonuclease. Proc Natl Acad Sci U S a. 2002;99:1164–1169. doi: 10.1073/pnas.022346799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone AN, Chan A, Kulpa JE, Brownsey RW. Bimodal activation of acetyl-CoA carboxylase by glutamate. J Biol Chem. 2000;275:10819–10825. doi: 10.1074/jbc.275.15.10819. [DOI] [PubMed] [Google Scholar]
- Brownsey RW, Boone AN, Elliott JE, Kulpa JE, Lee WM. Regulation of acetyl-CoA carboxylase. Biochem Soc Trans. 2006;34:223–227. doi: 10.1042/BST20060223. [DOI] [PubMed] [Google Scholar]
- Campbell MG, Cheng A, Brilot AF, Moeller A, Lyumkis D, Veesler D, Pan J, Harrison SC, Potter CS, Carragher B, et al. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 2012;20:1823–1828. doi: 10.1016/j.str.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey J. Gel retardation. Meth Enzymol. 1991;208:103–117. doi: 10.1016/0076-6879(91)08010-f. [DOI] [PubMed] [Google Scholar]
- Chen Z, Yang H, Pavletich NP. Mechanism of homologous recombination from the RecA–ssDNA/dsDNA structures. Nature. 2008;453:489–484. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- Daniels LE, Wood KM, Scott DJ, Halford SE. Subunit assembly for DNA cleavage by restriction endonuclease SgrAI. J Mol Biol. 2003;327:579–591. doi: 10.1016/s0022-2836(03)00143-8. [DOI] [PubMed] [Google Scholar]
- Dunten PW, Little EJ, Gregory MT, Manohar VM, Dalton M, Hough D, Bitinaite J, Horton NC. The structure of SgrAI bound to DNA; recognition of an 8 base pair target. Nucleic Acids Res. 2008;36:5405–5416. doi: 10.1093/nar/gkn510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasman GD. CRC Handbook of Biochemistry and Molecular Biology 1975 [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Grigorieff N. FREALIGN: high-resolution refinement of single particle structures. J Struct Biol. 2007;157:117–125. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Hingorani-Varma K, Bitinaite J. Kinetic analysis of the coordinated interaction of SgrAI restriction endonuclease with different DNA targets. J Biol Chem. 2003;278:40392–40399. doi: 10.1074/jbc.M304603200. [DOI] [PubMed] [Google Scholar]
- Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, Yang C, Glaeser RM, Adams PD, Ludtke SJ. SPARX, a new environment for Cryo-EM image processing. J Struct Biol. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Ingerson-Mahar M, Briegel A, Werner JN, Jensen GJ, Gitai Z. The metabolic enzyme CTP synthase forms cytoskeletal filaments. Nature. 2010;12:739–746. doi: 10.1038/ncb2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh HY, Mitchison TJ. Structural plasticity in actin and tubulin polymer dynamics. Science. 2009;325:960–963. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau P-W, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little EJ, Dunten PW, Bitinaite J, Horton NC. New clues in the allosteric activation of DNA cleavage by SgrAI: structures of SgrAI bound to cleaved primary-site DNA and uncleaved secondary-site DNA. Acta Crystallogr D Biol Crystallogr. 2011;67:67–74. doi: 10.1107/S0907444910047785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyumkis D, Doamekpor SK, Bengtson MH, Lee J-W, Toro TB, Petroski MD, Lima CD, Potter CS, Carragher B, Joazeiro CAP. Single-particle EM reveals extensive conformational variability of the Ltn1 E3 ligase. Proc Natl Acad Sci U S a. 2013;110:1702–1707. doi: 10.1073/pnas.1210041110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Shah S, Zhou M, Park CK, Wysocki VH, Horton NC. Structural Analysis of Activated SgrAI-DNA Oligomers Using Ion Mobility Mass Spectrometry. Biochemistry. 2013;52:4373–4381. doi: 10.1021/bi3013214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick SP, Carragher B, Potter CS, Kriegman DJ. ACE: Automated CTF Estimation. Ultramicroscopy. 2005;104:8–29. doi: 10.1016/j.ultramic.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Noree C, Sato BK, Broyer RM, Wilhelm JE. Identification of novel filamentforming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J Cell Biol. 2010;190:541–551. doi: 10.1083/jcb.201003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CK, Joshi HK, Agrawal A, Ghare MI, Little EJ, Dunten PW, Bitinaite J, Horton NC. Domain swapping in allosteric modulation of DNA specificity. PLoS Biol. 2010a;8:e1000554. doi: 10.1371/journal.pbio.1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CK, Stiteler AP, Shah S, Ghare MI, Bitinaite J, Horton NC. Activation of DNA cleavage by oligomerization of DNA-bound SgrAI. Biochemistry. 2010b;49:8818–8830. doi: 10.1021/bi100557v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JP, Maher LJ. DNA curvature and flexibility in vitro and in vivo. Q Rev Biophys. 2010;43:23–63. doi: 10.1017/S0033583510000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pingoud A, Jeltsch A. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001;29:3705–3727. doi: 10.1093/nar/29.18.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingoud A, Fuxreiter M, Pingoud V, Wende W. Type II restriction endonucleases: structure and mechanism. Cell Mol Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PubMed] [Google Scholar]
- Pingoud A, Jeltsch A. Recognition and Cleavage of DNA by Type-II Restriction Endonucleases. Eur J Biochem. 1997;246:1–22. doi: 10.1111/j.1432-1033.1997.t01-6-00001.x. [DOI] [PubMed] [Google Scholar]
- Pintilie GD, Zhang J, Goddard TD, Chiu W, Gossard DC. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J Struct Biol. 2010;170:427–438. doi: 10.1016/j.jsb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang BQ, Schildkraut I. Restriction endonucleases with octanucleotide recognition sequences. Meth Enzymol. 1987;155:15–21. doi: 10.1016/0076-6879(87)55005-4. [DOI] [PubMed] [Google Scholar]
- Radermacher M, Wagenknecht T, Verschoor A, Frank J. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J Microsc. 1987;146:113–136. doi: 10.1111/j.1365-2818.1987.tb01333.x. [DOI] [PubMed] [Google Scholar]
- Radermacher M, Wagenknecht T, Verschoor A, Frank J. A new 3-D reconstruction scheme applied to the 50S ribosomal subunit of E.coli. Journal of Microscopy. 1986;141:RP1–RP2. doi: 10.1111/j.1365-2818.1986.tb02693.x. [DOI] [PubMed] [Google Scholar]
- Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE--a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseman AM. FindEM--a fast, efficient program for automatic selection of particles from electron micrographs. J Struct Biol. 2004;145:91–99. doi: 10.1016/j.jsb.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Sachse C, Chen JZ, Coureux PD, Stroupe ME, Fändrich M, Grigorieff N. High-resolution electron microscopy of helical specimens: a fresh look at tobacco mosaic virus. J Mol Biol. 2007;371:812–835. doi: 10.1016/j.jmb.2007.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam MD, Perona JJ. Catalytic Roles of Divalent Metal Ions in Phosphoryl Transfer by EcoRV Endonuclease †. Biochemistry. 1999;38:6576–6586. doi: 10.1021/bi9901580. [DOI] [PubMed] [Google Scholar]
- Schröder GF, Brunger AT, Levitt M. Combining efficient conformational sampling with a deformable elastic network model facilitates structure refinement at low resolution. Structure. 2007;464:1630–1641. doi: 10.1016/j.str.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorzano CO, Bilbao-Castro JR, Shkolnisky Y, Alcorlo M, Melero R, Caffarena-Fernandez G, Li M, Xu G, Marabini R, Carazo JM. A clustering approach to multireference alignment of single-particle projections in electron microscopy. J Struct Biol. 2010;171:197–206. doi: 10.1016/j.jsb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, Sorek R. The phage-host arms race: shaping the evolution of microbes. Bioessays. 2011;33:43–51. doi: 10.1002/bies.201000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart A, Grigorieff N. Noise bias in the refinement of structures derived from single particles. Ultramicroscopy. 2004;102:67–84. doi: 10.1016/j.ultramic.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: The new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Tautz N, Kaluza K, Frey B, Jarsch M, Schmitz GG, Kessler C. SgrAI, a novel class-II restriction endonuclease from Streptomyces griseus recognizing the octanucleotide sequence 5“-CR/CCGGYG-3”. Nucleic Acids Res. 1990;18:3087. doi: 10.1093/nar/18.10.3087. [corrected] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagelos PR, Alberts AW, Martin DB. Studies on the mechanism of activation of acetyl coenzyme A carboxylase by citrate. J Biol Chem. 1963;238:533–540. [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A New Generation of the IMAGIC Image Processing System. J Struct Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: Software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss NR, Lyumkis D, Cheng A, Lau P-W, Mulder A, Lander GC, Brignole EJ, Fellmann D, Irving C, Jacovetty EL, et al. A toolbox for ab initio 3-D reconstructions in single-particle electron microscopy. J Struct Biol. 2010;169:389–398. doi: 10.1016/j.jsb.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner JN, Chen EY, Guberman JM, Zippilli AR, Irgon JJ, Gitai Z. Quantitative genome-scale analysis of protein localization in an asymmetric bacterium. Proceedings of the National Academy of Sciences. 2009;106:7858–7863. doi: 10.1073/pnas.0901781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KM, Daniels LE, Halford SE. Long-range communications between DNA sites by the dimeric restriction endonuclease SgrAI. J Mol Biol. 2005;350:240–253. doi: 10.1016/j.jmb.2005.04.053. [DOI] [PubMed] [Google Scholar]
- Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20:237–247. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.