

Abstract
An enormous amount of research has been aimed at identifying biological and environmental factors that are contributing to the current global obesity pandemic. The present paper reviews recent findings which suggest that obesity is attributable, at least in part, to a disruption of the Pavlovian control of energy regulation. Within our framework, this disruption occurs when (a) consumption of sweet-tasting, but low calorie or noncaloric, foods and beverages reduces the ability of sweet tastes to predict the postingestive caloric consequences of intake and (b) consuming diets high in saturated fat and sugar (a.k.a., Western diet) impairs hippocampal-dependent learning and memory processes that are involved with the use of interoceptive “satiety” signals to anticipate when food and eating are not followed by appetitive postingestive outcomes. The paper concludes with discussion of a “vicious-cycle’ model which links obesity to cognitive decline.
Keywords: ingestive behavior, learning, energy regulation, Western diet, hippocampus, dementia
1. Introduction
Most people in Western cultures live in places where highly palatable, energy dense foods and beverages are abundant and where advertising and other marketing ploys are aimed at keeping thoughts of these foods and the pleasures of eating them almost constantly in mind (Goris, Petersen, Stamatakis, and Veerman, 2010; Johnson, 2013). Over the past 30 years, most of us who live in these places have become overweight or obese (Ford and Mokdad, 2008). Furthermore, as our Western diet with high levels of saturated fat and processed sugars has become more popular in other societies, the obesity pandemic that seems to have started in the U.S. has become a global concern (e.g., Popkin, Adair, and Ng, 2012).
For these very good reasons, current Western and Westernized food environments are often considered to be “obesogenic” (e.g., Ard, 2007; Berthoud, 2012). However, a useful scientific explanation of why we overeat and become obese will require us to do more than merely describe and label the food environment. Specifically, even if one accepts that the abundant food-related environmental cues and the presence of energy dense food act as intended, to evoke eating when energy requirements have not been met, precisely how these stimuli continue to evoke intake when our needs for energy have been satisfied or even surpassed remains an unanswered question. The purpose of the present paper is to show how this question could be addressed using principles that are derived from a largely associative analysis of learning and memory.
To achieve this goal, we outline a model that attempts to (a) identify important stimuli and events that are critically involved with the learned control of energy intake and body weight; (b) describe the set of relationships in which these events are embedded; (c) show how this set of relationships generates a mechanism for the inhibitory control of feeding behavior; (d) discuss how aspects of the current food environment may promote overeating and obesity by interfering with specific components of this mechanism; and (e) consider how this type of interference could also result in a progressive decline in cognitive health.
2. A model of associative relations in energy regulation
The first step toward determining the role of learning and memory in any behavioral or physiological function is to identify the relevant stimuli and to describe the important relationships among them. Figure 1 outlines a model of how orosensory cues (e.g., tastes), postingestive cues produced by the arrival of calories in the gut, and energy state cues like those arising from satiety are integrated as part of the learned control of energy and body weight regulation. According to this model, tastes and other orosensory cues become associated with postingestive gastrointestinal (GI) outcomes of intake, such as those produced by the absorption of nutrients. As a consequence, orosensory food cues can come to excite memories of the appetitive postingestive stimulation produced by eating. Excitement of these memories promotes the performance of appetitive (i.e., food seeking and approach) and consummatory (e.g., eating) behaviors, as well as physiological responses that anticipate and help prepare for the arrival and absorption of nutrients in the GI tract (Buss, Kraemer-Aguiar, Maranhao, Marinho, de Souza, Wiernsperger, and Bouskela, 2012; Power and Schulkin, 2008; Smeets, Erkner, and de Graaf, 2010).
Figure 1.
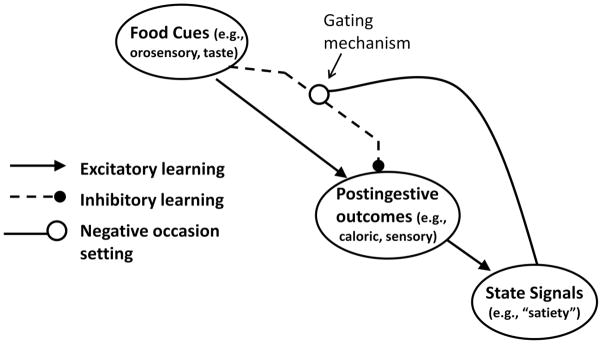
Associative model of energy and body weight regulation.
However, GI stimulation produced by intake does not always promote food approach. While the postingestive GI stimuli that are present at the outset of a meal can promote appetitive and consummatory behavior, those same types of GI stimuli are usually absent at the end of the meal and for some time thereafter. For example, after a period without eating, the first bites of a meal are often accompanied by postingestive consequences that may be evaluated as especially pleasant. By the end of a meal, the postingestive stimuli that are produced by continued eating are typically much less rewarding and may even become aversive. Therefore, orosensory food stimuli are embedded not only in an association that excites the memory of the appetitive postingestive consequences of eating, but also in associations that serve to inhibit the memory of those consequences. As a result, the relationship between food-related orosensory cues and postingestive GI stimulation is ambiguous, and animals must depend on the presence of other cues to resolve this ambiguity.
According to the model shown in Figure 1, animals can learn to use the presence of interoceptive “satiety” cues to predict that intake will not be followed by appetitive postingestive stimulation. Based on this learning, activation of the inhibitory association results in a diminished ability of orosensory cues to retrieve the memory of the appetitive postingestive stimulus consequences of intake via their excitatory association with that US representation. In the absence of satiety cues, the inhibitory association is inactive, making it more likely that conditioned food-related cues will retrieve the memory of the appetitive postingestive US representation, thereby promoting the evocation of food seeking and food intake.
The main components of this model are already well established. For example, the evidence showing that animals can associate tastes with either aversive (e.g., gastric malaise) or appetitive (e.g., nutritive or caloric) postingestive stimuli is unequivocal (Sclafani, 1997; Welzl, D’Adamo, and Lipp, 2001). The evidence is also clear that animals can learn about interoceptive “hunger” and “satiety” state stimuli (Davidson, 1993), with a number of studies showing that rats can learn to use stimuli arising from different levels of food deprivation as discriminative cues to signal that an appetitive (e.g., Davidson, Kanoski, Tracy, Walls, Clegg, and Benoit, 2005a) or an aversive US (e.g., Davidson, Flynn, and Jarrard, 1992) will be presented. Moreover, several studies show that discriminative control by such deprivation state stimuli generalizes to cues produced by hormonal or other manipulations implicated in interoceptive signals of satiety or hunger (e.g., Benoit and Davidson, 1996; Kanoski, Walls, and Davidson, 2007). Furthermore, gating mechanisms like that depicted in Figure 1 have been used to describe how animals can use conventional (e.g., auditory, visual, contextual) stimuli to disambiguate when other conditioned cues will or will not be followed by appetitive or aversive outcomes (e.g., Bouton, 2004). For example, Bouton (1994) proposed that a history of reinforced and nonreinforced training can enable a single CS to signal both the delivery and nondelivery of a US and that animals reduce ambiguity about which outcome will occur by relying on discrete, contextual, or interoceptive stimuli to gate activation of the inhibitory association (Bouton, 2004; Todd, Winterbauer, and Bouton, 2012).
Our use of the term “satiety signals” deserves additional comment. Numerous physiological events have been identified as candidate neural and hormonal satiety cues based on their sites of origin (mainly in the periphery), their sites of detection (mainly in the brain), their modes of induction (e.g., they occur as a consequence of energy intake), and their suppressive effects on appetitive and consummatory behavior (for reviews see Hellstrom, 2013; Woods, 2004). For example, much evidence implicates the hormone cholecystokin (CCK) as one satiety signal involved with meal termination (e.g., Dockray, 2012), whereas the hormone leptin is thought to inhibit intake based on information it provides about the availability of energy from longer-term bodily fat reserves (e.g., Moran and Ladenheim, 2011). These, and other hormones, are described as producing satiety based largely, but not exclusively, on findings that their endogenous release or exogenous administration is reliably accompanied by a suppression of intake. Similarly, overeating is often attributed to reductions in the release of or sensitivity to these types of satiety cues. One goal of this paper is to go beyond the identification of physiological signals that suppress intake by offering an explanation about how those signals are actually able to exert their suppressive effects on behavior. In current accounts, the links between the production or detection of satiety cues and the inhibition of behavior are often defined by little more than an arrow or a minus sign in a diagram. This paper describes a mechanism that could be used to explain those arrows and signs.
3. Implications of the model for understanding overeating and obesity
Central to the model outlined in Figure 1 are the ideas that: (a) animals rely on tastes and other food-related stimuli to activate representations of USs produced by nutritive postingestive outcomes; and (b) the ability of tastes and food-related stimuli to activate those USs is modulated by interoceptive satiety cues. Our model implies that interference with either of these two components could promote excess energy intake and body weight gain by reducing the ability to inhibit appetitive and consummatory responses to food-related cues. To evaluate the effects of interference with the first component, we assessed whether explicitly reducing the validity of sweet tastes to serve as a signal for postingestive energetic outcomes would affect energy intake and body weight regulation. To evaluate the second component, we assessed whether or not overeating and body weight gain are related to deficits in the ability to use one cue to signal that another stimulus will not be followed by an appetitive US. This is the modulatory or gating function performed by satiety signals within our model. As will be elaborated later, this function is analogous to the function of feature cues in a conditional discrimination problem known as the serial feature negative discrimination problem.
3.1. Reducing the validity of sweet taste as a signal for energetic postingestive outcomes
A longstanding principle in Pavlovian conditioning is that cues are valid signals for other events to the extent that they reliably predict the occurrence of those events (e.g., Escobar and Miller, 2004). Logically, if a cue occurs in the absence of its predicted event or if the event occurs when the cue is absent, the predictive validity of that cue for the event will be diminished (e.g., Dwyer, Haselgrove, and Jones, 2011; Rescorla, 1968; Urushihara and Miller, 2009; Wagner, Logan, Haberlandt, and Price, 1968). The model outlined in Figure 1 indicates that tastes, for example sweet, should be valid signals for caloric or nutritive postingestive outcomes. The experience of sweet taste followed by the absorption of calories is likely to be not only highly salient, but also to be encountered very early in life and remain ubiquitous throughout the lifespan.
An idea prominent since at least the time of Pavlov (Pavlov, 1910) is that orosensory stimuli such as sweet taste evoke a variety of learned physiological (cephalic phase) responses that, by anticipating the arrival and subsequent absorption of nutrients in the GI tract, help to maintain energy homeostasis (Power and Schulkin, 2008; Smeets et al., 2010). One of our initial questions was “What happens to the ability to maintain energy homeostasis when the validity of sweet taste as a signal for caloric or nutritive outcomes is reduced by the introduction of high-intensity sweeteners which provide sweet tastes, but without the delivery of energy?” Such sweeteners have been promoted as methods to curb or even reverse the ongoing obesity epidemic by satisfying the desire for sweet tastes without contributing excess energy in the form of sugars. However, when considered within the framework outlined in Figure 1, the answer to this question would be the otherwise counterintuitive suggestion that energy balance will be disrupted by such sweeteners. Within that framework, giving animals non-caloric sweeteners will promote energy dysregulation and weight gain because that experience (i.e., sweet taste without calories) will reduce the validity of sweet taste as a signal for its caloric postingestive US, thereby weakening the ability of sweet taste to evoke at least some cephalic phase responses and thus impairing the ability of animals to regulate their intake of sweet foods when they are actually high in calories.
We reported the results of a series of experiments designed to directly assess the effects of consuming a noncaloric sweetener on the ability of sweet taste to signal calories as well as on the ability of animals to regulate their intake and body weight when they consumed sweet and high-calorie food (Davidson, Martin, Clark, and Swithers, 2011). In Experiment 1 of this series, one group of rats was pre-exposed for 14 days to a sweet, non-caloric 0.3% saccharin solution. A second, control, group received only water. Both groups were then trained with two different novel flavored solutions (cherry or grape) for 10 days with each flavor. One of these solutions contained 10% glucose and the other contained 10% polycose. While glucose and polycose are equicaloric, glucose tastes sweet to rats but polycose does not. At the conclusion of this training, intake of each flavored solution without glucose or polycose was tested. The logic of the study was that pre-exposure to non-caloric saccharin would reduce the validity of sweet taste to serve as a signal for calories so that, during training, the ability of sweet taste to compete with the novel flavor for association with a caloric postingestive US would also be reduced. As a result, learning about the novel flavor mixed with the glucose solution would be enhanced by saccharin pre-exposure compared to water pre-exposure. On the other hand, because polycose does not taste sweet to rats, weakening the validity of sweet taste to signal calories should have little impact on the ability of the “poly” taste to compete with the novel flavor for association with the same caloric outcome. Thus, pre-exposure to saccharin compared to water should have little effect on learning about the flavor presented along with the polycose solution. During testing with the flavors alone, intake was used as an index of how strongly each novel flavor had been associated with its caloric postingestive US during training.
The results of the intake test confirmed these expectations. Figure 2 shows that rats that were pre-exposed to saccharin consumed more of the flavor that had been presented in compound with glucose compared to rats that had been exposed to water. In contrast, intake of the flavored solutions that had been paired with polycose did not differ for rats that had been pre-exposed to saccharin compared to rats were given only water during the pre-exposure phase. These findings support the hypothesis that pre-exposure to saccharin reduced the validity of sweet taste as a signal for a caloric postingestive outcome. Because of this, sweet taste was less able to compete for associative strength with a novel flavor when both the sweet taste and the flavor were paired with that caloric US. The cue validity of sweet taste was not reduced for rats that received only water during the pre-exposure because these rats did not experience sweet taste without caloric consequences. The finding that intake of the flavor paired with polycose in training was not reduced by pre-exposure to saccharin was also expected. Pre-exposure to sweet taste without a caloric US should not influence the validity of poly taste as a signal for that US given that polycose does not appear to taste sweet to rodents (Bonacchi, Ackroff, and Sclafani, 2008; Treesukosol, Blonde, and Spector, 2009).
Figure 2.

Mean amount consumed (±SEM) of the glucose-paired and polycose-paired flavor solution during a 4-hr test for rats that received 0.3% saccharin solution or water during pre-training. * denotes significant difference between the saccharin and water pre-training conditions. Data are from Davidson, T. L., Martin, A. A., Clark, K., & Swithers, S. E. (2011).
An important implication of these results is that if non-caloric sweeteners impair the ability of a sweet taste to predict the delivery of calories and animals rely on this ability to maintain energy balance, then this impairment will result in the overconsumption of a maintenance diet if it also tastes sweet. Alternatively, if the maintenance diet is not sweet, then consumption of the non-caloric sweetener would be expected to have minimal effects on energy balance because disruption of the predictive relationship between sweet tastes and calories should have little effect on regulating the intake of a nonsweet diet. In Experiment 2, Davidson et al., (2011) tested this hypothesis.
Using the basic design and procedures we developed previously, we gave different groups of rats dietary supplements of 30 g of plain yogurt along with yogurt sweetened with 0.3% saccharin (~ 0.6 kcal/g) or sweetened with 20% glucose (~ 1.2 kcal/g). The rats given each type of sweetened yogurt were also assigned to one of three different maintenance diets; (1) a chow diet high in fat and carbohydrate that was sweetened with glucose (HF + glucose), (2) the same diet that had glucose replaced with an equal amount of polycose (HF + polycose), or (3) the same diet without glucose or polycose (HF plain). The energy density of the HF + glucose and HF + polycose diets was equivalent (~5.18 kcal/g), but only the HF + glucose had a distinctly sweet taste. The HF plain diet contained nominally more calories compared to the other diets (~5.48 kcal/g) but lacked the significant sweet or poly taste. Based on the rationale described above, we hypothesized that rats given the saccharin-sweetened yogurt would consume more maintenance diet and gain additional weight compared to rats given the glucose-sweetened yogurt supplement when they were maintained on the sweetened maintenance diet (HF+glucose), and that this difference would be absent or diminished for rats maintained on the chow diet without sweetener added (HF + polycose and HF plain).
Both the body weight and intake data confirmed these predictions. Figure 3 shows that the rats given yogurt with saccharin gained significantly more weight than the rats given yogurt with glucose, but only if they were maintained on the sweetened high-fat diet (HF + glucose). Consistent with these weight data, Figure 4 shows that total caloric intake (yogurt supplements plus maintenance diet) was significantly greater for rats given saccharin mixed with their yogurt compared to rats given glucose, but only when the maintenance diet was also sweetened.
Figure 3.

Mean body weight gain (±SEM) per day for rats that received yogurt sweetened with 0.3% saccharin or 20% glucose on some days and plain yogurt on other days as a function of type of high-fat (HF) maintenance diet: HF Plain (left panel), HF + 20% glucose (center panel), or HF + 20% polycose (right panel). * denotes significant difference between rats given saccharin-sweetened and glucose-sweetened yogurt. Data are from Davidson, T. L., Martin, A. A., Clark, K., & Swithers, S. E. (2011).
Figure 4.

Mean kcals consumed (±SEM) per week (yogurt + maintenance diet) for rats that received yogurt sweetened with 0.3% saccharin or 20% glucose on some days and plain yogurt on other days as a function of type of high-fat (HF) maintenance diet: HF Plain (left panel), HF + 20% glucose (center panel), or HF + 20% polycose (right panel). Analyses of variance showed that compared to exposure to glucose-sweetened yogurt, exposure to saccharin-sweetened yogurt was associated with significantly higher total intake across all four weeks of testing for rats that had been maintained on the HF + 20% glucose diet (p < .05). This pattern of differences in total intake based on sweetener was not significant for the HF Plain or HF + 20% polycose groups. Data are from Davidson, T. L., Martin, A. A., Clark, K., & Swithers, S. E. (2011).
These results are counterintuitive in that they indicate that weight gain, not weight loss, and increased, not decreased, caloric intake were promoted by consuming a noncaloric compared to a caloric sweetener. This basic finding has been replicated many times in our lab and elsewhere, and it has been obtained with noncaloric sweeteners other than saccharin (e.g., Davidson and Swithers, 2004; Feijo, Ballard, Foletto, Melo Batista, Neves, Marques Ribeiro, and Bertoluci, 2013; Pierce, Heth, Owczarczyk, Russell, and Proctor, 2007; Swithers, Baker, and Davidson, 2009; Swithers and Davidson, 2008; Swithers, Sample, and Davidson, 2013; reviewed in Swithers, Martin, and Davidson, 2011). Furthermore, similar effects have been reported following consumption of foods containing reduced-calorie fat substitutes compared to normal high-calorie fats, but only when animals are consuming a maintenance diet that is high in fat (Swithers, Ogden, and Davidson, 2011), suggesting that the principle of disrupting the relation between orosensory cues and caloric outcomes is a critical factor, rather than some specific aspect of sweetness per se.
These outcomes were anticipated by the associative model presented in Figure 1. According to the model, the ability to maintain body weight and energy balance depends, in part, on the validity of a taste to predict a caloric postingestive outcome. We showed that consuming non-caloric saccharin reduces the predictive validity of sweet taste to serve as a signal for calories—an effect that results in an impaired ability to regulate intake and body weight on a diet of sweetened, but not unsweetened, energy dense food. Moreover, the model also points to how (i.e., by what mechanism) a reduction in sweet taste cue validity may lead to positive energy balance and ensuing weight gain. Animals terminate meals well before most of the energy in the meal has been absorbed (Booth, 1977). This indicates that the production of satiety signals is also anticipatory. The model outlined in Figure 1 indicates that the postingestive stimulus consequences of intake are linked associatively to the production of satiety signals. If animals are less able to use sweet taste to predict the postingestive consequences of intake and if the production of signals themselves is dependent on the prediction of those consequences, it may be that weakening the validity of taste also reduces the strength of satiety signals.
Two types of evidence provide support for this possibility. First, Swithers, Laboy, Clark, Cooper, and Davidson (2012) reported that the release of glucagon-like peptide-1 (GLP-1) following an oral glucose load was significantly reduced for rats that had prior experience consuming saccharin-sweetened yogurt compared to rats that had experience consuming the same yogurt sweetened with glucose. The results of several earlier studies suggest that GLP-1 functions as a physiological satiety signal when its release from the intestine is stimulated by nutrient intake (e.g., Sam, Troke, Tan, and Bewick, 2012; Williams, Baskin, and Schwartz, 2009). Thus, as suggested by Swithers and her colleagues (Swithers et al., 2012), it may be that consuming non-caloric sweeteners promotes increased intake and body weight gain by suppressing the production of interoceptive satiety signals. However, as we have argued above, merely claiming that animals eat more and gain more weight because their satiety signals are weaker adds little, if anything, to the analysis of how satiety cues suppress eating behavior in the first place. The model outlined in Figure 1 addresses this question by suggesting that satiety signals suppress intake because they activate an inhibitory association between taste and other food-related cues and the memory of the postingestive consequences of eating.
Second, previous findings indicate that generalization gradients for internal satiety cues are similar to those reported for exteroceptive stimuli. Thus, reductions in the intensity of interoceptive satiety stimuli are accompanied by stimulus generalization decrements similar to those produced by reductions in the intensity of conventional types of stimuli (e.g., Davidson, 1987). Furthermore, we have shown that manipulations thought to give rise to interoceptive satiety cues, such as nutritive stomach loads and peripheral administration of CCK or leptin, also generalize to signals produced by these food deprivation intensity stimuli (Davidson, 1987; Kanoski et al., 2007). Thus within limits, less intense satiety signals should be less effective modulators of inhibitory taste-calorie associations than higher intensity satiety cues.
3.2 Relation to studies of the effects of high-intensiy sweeteners on energy regulation in humans
In humans, the effects of high-intensity sweeteners on learning or metabolism have not been directly explored, but some evidence is consistent with the possibility that physiological and neural responses differ in individuals who regularly consume artificial sweeteners compared to those who do not. For example, long-term epidemiological studies indicate that the risk for metabolic disorders, such as weight gain, Type 2 diabetes, metabolic syndrome, coronary heart disease, and hypertension are significantly greater in individuals who regularly consume diet sodas compared to those who do not, even when factors such as differences in dietary patterns, family history, and body mass index are accounted for (e.g., Bhupathiraju, Pan, Malik, Manson, Willett, van Dam, and Hu, 2013; Cohen, Curhan, and Forman, 2012; Duffey, Steffen, Van Horn, Jacobs, and Popkin, 2012; Fagherazzi, Vilier, Saes Sartorelli, Lajous, Balkau, and Clavel-Chapelon, 2013; Fowler, Williams, Resendez, Hunt, Hazuda, and Stern, 2008; Gardener, Rundek, Markert, Wright, Elkind, and Sacco, 2012; InterAct, 2013; Nettleton, Polak, Tracy, Burke, and Jacobs, 2009; Sakurai, Nakamura, Miura, Takamura, Yoshita, Nagasawa, Morikawa, Ishizaki, Kido, Naruse, Suwazono, Sasaki, and Nakagawa, 2013; reviewed in Swithers, 2013). In addition, imaging studies using fMRI indicate that people who are regular consumers of diet sodas have different patterns of brain responses to both caloric sweeteners and artificial sweeteners compared to those who do not consume diet sodas (Green and Murphy, 2012; Rudenga and Small, 2012). Finally, studies in humans have demonstrated that artificial sweeteners fail to elicit physiological effects of the same magnitude that would be elicited by consumption of caloric sweeteners, such as release of hormones involved in maintenance of blood glucose homeostasis like insulin, glucagon, and GLP-1 (e.g., Anton, Martin, Han, Coulon, Cefalu, Geiselman, and Williamson, 2010; Brown, Bohan Brown, Onken, and Beitz, 2011; Brown, Walter, and Rother, 2012; Ford, Peters, Martin, Sleeth, Ghatei, Frost, and Bloom, 2011; Ma, Chang, Checklin, Young, Jones, Horowitz, and Rayner, 2010; Steinert, Frey, Topfer, Drewe, and Beglinger, 2011; Wu, Zhao, Bound, Checklin, Bellon, Little, Young, Jones, Horowitz, and Rayner, 2012; see also Swithers, 2013). However, in most of those studies, the history of subjects with artificial sweeteners prior to the experiment is unknown. More recently, a small study in a cohort of obese individuals who did not regularly consume artificial sweeteners has indicated that consumption of sucralose prior to an oral glucose load resulted in higher peak plasma glucose (e.g., relative hyperglycemia), increased insulin secretion, decreased insulin clearance, and decreased insulin sensitivity (Pepino, Tiemann, Patterson, Wice, and Klein, 2013). These findings suggest that the blunted or absent responses observed in previous work could reflect an extinction resulting from subjects’ previous experience with artificial sweeteners. However, direct experimental tests of the consequences of providing experience with artificial sweeteners to naïve subjects on physiological responses to caloric sweeteners are not presently available.
3.3. Energy regulation as a solution to a serial feature negative discrimination problem
These results suggest that interfering with the first component of our model, the ability of tastes to signal postingestive outcomes, can indeed promote excess energy intake and body weight. To assess the second component, that the ability of tastes and food-related stimuli to activate USs is modulated by interoceptive satiety cues, we turn to a discussion of how satiety cues might serve a function analogous to features in serial feature negative discrimination tasks. A serial feature negative discrimination problem has the general form of A+, X→A− where target cue A (e.g., a brief tone) is followed by a US (+) on trials where A is presented alone (i.e., A+ trials), whereas no US (−) occurs on trials where feature cue X (e.g., a light or a contextual stimulus) precedes the presentation of target cue A (i.e., X→A−) trials. Within this arrangement, because stimulus A is followed by a US on some trials but not on other trials, the relationship of A to the US is ambiguous. Animals can resolve this ambiguity by learning to use stimulus X as a negative feature cue that signals that stimulus A will not be followed by the US on that trial (e.g., Morell and Davidson, 2002).
Serial feature negative training generates an inhibitory feature cue that is distinct from simple conditioned inhibitors, which are trained not serially, but in simultaneous compound with their target cues (see Chan, Morell, Jarrard, and Davidson, 2001). For example, feature cues that have been given serial feature negative training do not appear to be embedded in direct associations with the US because, unlike simple conditioned inhibitors, separate reinforcement or nonreinforcement of the serially-trained feature cues (X) alone does not abolish their capacity to signal the reinforcement or nonreinforcement of their target (A) stimuli (Holland, 1984). Furthermore, compared to simple conditioned inhibitors, there is evidence that serially-trained feature cues have a much more limited capacity to modulate responding to stimuli that do not have a history of both excitatory and inhibitory training with the same US (Morell and Davidson, 2002). Based on these and other considerations, it appears that a serially-trained negative feature cue suppresses conditioned responding by promoting the activation of an inhibitory association between its target and the US, and activation of this inhibitory association opposes or weakens the ability of the target’s excitatory association to activate that US representation (for review see Swartzentruber, 1995). This modulatory or gating function is distinct from simple conditioned inhibitors, which are instead considered to be embedded in their own direct associations with the representation of the US. As applied to the regulation of energy intake, these characteristics of serially-trained negative feature stimuli are apparent in Figure 1, in which satiety signals are depicted not as direct associates of the postingestive US, but as stimuli that gate activation of separate inhibitory taste→postingestive US associations.
The model of energy regulation depicted in Figure 1 proposes that interoceptive satiety signals inhibit appetitive and eating behavior by functioning as serial feature negative stimuli that inform when food-related cues will not be followed by appetitive postingestive consequences. Initially, the converse argument was made. Namely, that interoceptive “hunger” signals excite appetitive and eating responses by serving as serial feature positive stimuli that are informative about when food cues will be followed by those appetitive postingestive consequences (Davidson, 1993). One basis for this change in perspective is increased recognition that, in Westernized societies, humans typically do not eat in response to hunger or other internal signals of energy deficit (e.g., Woods, 2004). Rather, meals are initiated in response to environmental stimuli (e.g., time of day; advertising) that have been previously associated with food and the consequences of eating. These conditioned environmental cues evoke appetitive and eating behavior until those behaviors give rise to interoceptive meal termination or “satiety” signals. That is, in the Woods model, energy regulation depends not on “hunger” signals, but rather on the generation of physiological satiety signals that act to terminate meals and to suppress the capacity of cues in the environment to continue to stimulate additional appetitive and eating behavior.
As shown in Table 1, Woods’s model of energy regulation can be readily interpreted within the framework of the serial feature negative discrimination problem. The top portion of Table 1 depicts the structure of a conventional serial feature negative problem that employs tones and lights as stimuli along with an appetitive US (e.g., sugar pellets). As noted above, animals can solve this problem by learning that the light resolves ambiguity about the reinforcement of the tone. This solution is manifested in behavior when animals exhibit less conditioned responding (e.g., breaking a photo beam in the food magazine) on trials when the light precedes the tone compared to trials when the tone is presented alone. Table 1 also shows the key elements of Woods’s model of energy regulation arranged in a manner that corresponds to a serial FN discrimination. That is, animals learn that food cues are accompanied by an appetitive postingestive US (analogous to tone alone trials in the serial FN task) except for when satiety cues (analogous to light cues in the FN problem) are also present. Animals show that they have solved this problem by exhibiting less appetitive behavior when satiety cues and food cues occur together compared to when food cues occur alone.
Table 1.
Wood’s model interpreted as a serial feature-negative discrimination problem
| Conventional serial feature-negative discrimination problem |
| Tone+ |
| Light → Tone-(+ = appetitive reinforcement) |
| Lights inhibits the ability of the tone to retrieve the memory of appetitive reinforcement. |
| Wood’s view interpreted as a serial feature-negative problem |
| Food cues+ |
| Satiety cues → food cues-(+ = appetitive postingestive reinforcement) |
| Satiety cues inhibit the ability of environment food-related cues to retrieve the memory of appetitive postingestive reinforcement. |
Drawing analogies between an animal’s ability to solve a serial FN problem and its ability to solve the more general problem of when to feed and when to refrain from feeding will be of limited explanatory value unless common mechanisms that underlie the solution to both types of problems can be specified. For the most part, the same mechanism that describes how animals solve conventional serial feature negative discrimination problems (see above) can be used to describe how animals use their satiety signals to maintain energy balance. That is, the model diagrammed in Figure 1 proposes that food and food-related cues are embedded in an excitatory association with the appetitive postingestive US that is produced by eating. However, because eating is not always followed by an appetitive postingestive outcome (e.g., when animals are sated), food and food-related target cues are also embedded in a concurrent inhibitory association with that US. Animals can solve the problem of “when to feed” because their satiety cues serve as negative feature stimuli that selectively activate the inhibitory association, thereby reducing the ability of food cues to excite the memory of the appetitive postingestive US and evoke conditioned appetitive and consummatory responses.
The hypothesis that energy regulation depends on the ability to solve a feature negative discrimination problem suggests two possibilities: (a) impairing the ability of animals to solve these problems will produce overeating and ultimately weight gain; (b) overeating and weight gain will impair the ability to solve serial feature negative discrimination problems. Our research has evaluated both of these possibilities.
3.4. The hippocampus as a substrate for serial feature negative discrimination learning
If energy regulation depends on a learning and memory mechanism, then energy dysregulation should be one consequence of damage to the neural structures and circuits that underlie that mechanism. Holland, Lamoureux, Han, and Gallagher (1999) reported that selective neurotoxic lesions of the complete hippocampus resulted in large impairments in performance on a Pavlovian serial feature negative discrimination problem, but only a transient decline in performance on a Pavlovian serial feature positive discrimination problem. This pattern of results indicates that performance of the Pavlovian serial feature negative discrimination is dependent on the hippocampus. It should be noted that hippocampal damage has little effect on responding controlled by simple conditioned inhibitors that have been trained in simultaneous compound with their target cues (see Chan, Jarrard, and Davidson, 2003).
A variety of more recent findings also implicate the hippocampus in the regulation of energy balance and indicate that hippocampal damage has adverse effects on the regulation of energy intake and body weight. For example, several findings show that the hippocampus is involved with the ability of rats to inhibit their food-reinforced appetitive behavior (e.g., Clifton, Vickers, and Somerville, 1998; Davidson and Jarrard, 1993). In one more recent study, Chudasama, Doobay, and Liu, (2012) used a brief illumination of one of five apertures to signal that a sucrose pellet would be delivered at that location contingent upon a nose poke response. Responses to nonilluminated apertures were not reinforced. The ability to refrain from making premature responses to apertures during the intertrial interval before illumination and to refrain from making additional responses to an illuminated aperture after the sucrose pellet had already been obtained served as indices of behavioral inhibition. Rats with compete disconnection of the neural pathway from the ventral hippocampus to the prefrontal cortex exhibited increased premature responses and perseveration of post-reinforcement responding compared to rats for which ipsilateral lesions left that pathway intact in one hemisphere of the brain. Thus, eliminating direct outputs from the hippocampus to the prefrontal cortex reduced the ability of rats to inhibit their appetitive conditioned behavior. Rats with selective lesions of the hippocampus are also impaired in using their food deprivation intensity cues as discriminative stimuli to signal when either shock (Davidson and Jarrard, 1993; Hock and Bunsey, 1998) or sucrose pellets (Davidson, Kanoski, Chan, Clegg, Benoit, and Jarrard, 2010) will be presented in a common training context. This outcome is consistent with the view that the hippocampus is needed for rats to use their interoceptive energy state signals as serial feature negative stimuli to disambiguate when contextual cues will and will not be followed by a US.
Furthermore, studies using functional magnetic resonance imaging with both rats (Min, Tuor, and Chelikani, 2011) and humans (Wang, Yang, Volkow, Telang, Ma, Zhu, Wong, Tomasi, Thanos, and Fowler, 2006) have shown that gastric distention comparable to that produced by consuming a meal increases BOLD activation not only in brain areas traditionally considered to be important for energy homeostasis (e.g., hypothalamus) but also in the hippocampus and other so-called nonhomeostatic regions (e.g., amygdala, cerebellum) of the brain. Findings that a potential peripheral satiety signal triggers activity in the hippocampus are consistent with the idea that the hippocampus is a substrate for the inhibitory actions of satiety cues on behavior.
There is also evidence from rodent models that interference with hippocampal function either as a result of permanent lesions or reversible inactivation of hippocampal neurons is associated with excess energy intake and body weight gain. Davidson, Chan, Jarrard, Kanoski, Clegg, and Benoit (2009) reported that rats with selective neurotoxic lesions of the complete hippocampus or lesions confined to the hippocampal ventral pole (i.e., the most ventral portion of the hippocampus), but not rats with lesions confined to the prefrontal cortex (PFC), ate more and gained more weight compared to unoperated and sham-operated controls. In addition, Henderson, Smith, and Parent (2013) showed that temporary inactivation of neurons in the dorsal hippocampus shortened the interval between meals for mice, providing additional evidence that hippocampus has a role in the inhibition of intake.
If the regulation of energy intake and body weight depends, at least in part, on hippocampal-dependent serial feature negative learning, then environmental factors that promote overeating and body weight gain should also be associated with impaired performance on serial feature negative discrimination problems. To test this hypothesis, Kanoski, Zhang, Zheng, and Davidson (2010) fed different groups of rats a standard (low-fat, high carbohydrate) diet of laboratory chow or a high-energy (HE) diet that contained high levels of saturated fat and processed sugar for 90 days prior to the initiation of training on serial feature negative and control serial feature positive tasks. The composition of this diet was similar to that of the human “Western diet”, so labeled because of its widespread popularity in Western and Westernized cultures. For both discrimination problems, Kanoski and his associates (2010) used procedures and parameters modeled after those employed by Holland et al., (1999), who reported that selective neurotoxic lesions of the hippocampus impaired serial feature negative learning, while leaving serial feature positive performance relatively unaffected. As expected, rats maintained on a HE diet gained more weight than chow-fed controls. Of greater interest, Figure 5 shows that rats on the HE diet were also impaired relative to the chow-fed rats on a feature negative but not on a feature positive discrimination problem, with HE diet-fed rats showing greater levels of responding on the trials in which the target cue was not reinforced. This pattern of results is consistent with the idea that the impairment reflected a relative inability to use the feature cue to signal that the target stimulus would not be followed by appetitive consequences. Thus, the adverse effects of maintenance on a HE diet on feature negative discrimination performance and the lack of effects of that diet on feature positive discrimination learning were similar to the effects of hippocampal lesions on those two types of problems reported by Holland et al (1999). An impaired ability to use feature cues to signal nonreinforcement of the target would make it difficult for rats to distinguish nonreinforced from reinforced presentations of that target. This type of “partial reinforcement” in acquisition would be expected to generate substantial net excitatory conditioning to the target cue on both L→T− and T+ trials (e.g., Rescorla and Wagner, 1972)—the pattern reported by both Holland et al., (1999) and Kanoski, et al (2010).
Figure 5.
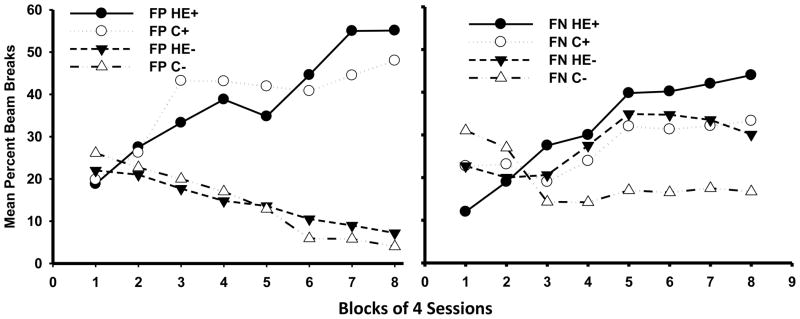
Serial feature positive (FP) and serial feature negative (FN) discrimination performance for rats fed high-energy (HE) or standard chow (C) diets ad libitum for 90 days prior to the beginning of training. The left panel shows rewarded FP+ (Light→Tone+) and nonrewarded FP− (Tone-) trials for each diet group. The right panel shows rewarded FN+ (Tone+) and nonrewarded FN − (Light→ Tone-) trials for the same groups. For the serial FP discrimination, the L→T+ vs. T− difference was significant beginning on Blocks 3 and 4 of training, and it did not vary as a function of Diet (HE vs. C). For the FN discrimination, the L→T− vs. T+ difference varied as a function of Diet. This difference was significant beginning on Blocks 5 and 6 for the rats previously fed ad libitum chow but did not achieve significance for the group previously fed ad libitum HE diet until Blocks 7–8 (all ps < .05). Data are from Kanoski, S. E., Zhang, Y., Zheng, W., & Davidson, T. L. (2010).
Kanoski et al., (2010) also identified a potential connection between intake of the “Western” HE diet and hippocampal dysfunction. The blood-brain barrier (BBB) is a specialized system comprising a microvascular endothelium that limits entry of many blood components into the brain (Persidsky, Ramirez, Haorah, and Kanmogne, 2006). Damage to the BBB has been reported to precede the development of clinical symptoms of dementia in both human patients with Alzheimer’s Disease (AD) and mouse models of AD (e.g., Ryu and McLarnon, 2009; Shimizu, Sano, Saito, Abe, Maeda, Haruki, and Kanda, 2012; van Assema, Lubberink, Bauer, van der Flier, Schuit, Windhorst, Comans, Hoetjes, Tolboom, Langer, Muller, Scheltens, Lammertsma, and van Berckel, 2012). Kanoski et al. (2010) assessed the effect of HE diet intake on the integrity of the BBB by measuring expression of proteins (e.g., occludin, claudin 5, claudin 12, Zo-1, and Zo-2) that comprise the tight junctions of the BBB.
The expression of these tight-junction proteins was reduced for rats on the HE diet. To provide more direct evidence of increased BBB permeability, they also measured the concentrations of a small molecule dye (sodium flourescein (NaFl)) that crossed the BBB. For rats that had been maintained for 90 days on a HE diet, dye concentrations in the medial prefrontal cortex (mPFC) and striatum (brain areas involved with certain types of nonhippocampal-dependent learning and memory) did not differ between the HE diet-fed rats and chow-fed controls. In contrast, following HE diet, dye concentrations in the hippocampus were nearly 100% higher than control levels. Thus, these findings suggest that the effects of HE diets on the ability to solve feature negative discrimination problems may be due, in part, to increased permeability of the BBB, which may lead to increased entry of potentially harmful substances into the brain that lead to impaired hippocampal function.
Davidson, Monnot, Neal, Martin, Horton, and Zheng (2012) extended these results to show that HE diet-induced impairments in serial feature negative discrimination performance and BBB structural integrity were both linked to the ability of that diet to promote weight gain. Rats were first reduced to 85% of their ad libitum body weight by rationing their diet of standard lab chow and then given concurrent training on a serial feature negative discrimination and on a simple discrimination problem. For the serial feature negative problem, a 5-sec tone stimulus terminated with the delivery of a sucrose pellet US on trials where the tone was presented alone, and no US was delivered on trials where the tone was preceded by the presentation of a brief light (i.e., the negative feature cue). For the simple discrimination, presentation of a 5-sec clicker stimulus was followed by the US, whereas presentation of a 5 sec white noise stimulus was not. When the rats achieved asymptotic discrimination performance on both types of problems, they were assigned to two groups matched on terminal performance on each discrimination problem. One group was then given free access to standard lab chow (CHOW) and the other was given free access to a high energy (HE) diet that contained high levels of saturated fat and sugar (dextrose) like that used by Kanoski et al., (2010).
Similar to humans, consuming a HE diet results in diet-induced obesity for some rats, whereas other rats are resistant to this effect of the HE diet (Madsen, Hansen, Paulsen, Lykkegaard, Tang-Christensen, Hansen, Levin, Larsen, Knudsen, Fosgerau, and Vrang, 2010). We measured weight gain for 28 days after introduction of the HE diet for the rats in the HE and CHOW groups. We divided the HE group into tertiles based on body weight gain 28 days after the introduction of the HE diet. The top third that gained the most body weight were classified as diet-induced obese (DIO) and the bottom third that gained the least amount of weight were classified as diet-resistant (DR). We then compared the performance of the HE group with the CHOW group on both the serial feature negative and simple discrimination problems as a function of the HE group’s DIO or DR classification. Performance on both problems was tested 7, 14, 21, and 28 days after the HE diet was introduced.
If maintaining energy balance depends on the capacity to solve a feature negative discrimination problem, then DIO rats that gain the most weight during the test phase should also be more impaired on serial feature negative performance compared to DR rats and Chow controls. If impaired discrimination performance for DIO relative to DR groups is based on nonspecific factors such as differences in level of motivation for food, level of satiety, reward, reinforcing value of the US, behavioral competence, or a global deficit in learning or memory, then such deficits in performance should be observed on both the serial feature negative and the simple discrimination problems because performance in both problems involves the same response requirement (breaking a photobeam located in the food magazine) to the same US (sucrose pellets) tested at the same time in the same rats.
Mean body weight by the end of the 28 days on the HE diet was significantly higher for rats classified as diet-induced obese (HE-DIO) compared to diet-resistant (HE-DR) rats and to CHOW controls. Body weight differences between HE-DR rats and CHOW controls did not achieve significance. The leftmost three panels of Figure 6 show that these three groups did not differ in performance on the simple discrimination problem at the end of initial training or when tested after 28 days of ad libitum access to chow or HE diet. The rightmost three panels of Figure 6 also show that serial feature negative discrimination performance did not differ substantially at the end of training. However, by the end of testing (Day 28), Group HE-DIO was impaired on the feature negative discrimination whereas Groups Chow and HE-DR were not.
Figure 6.
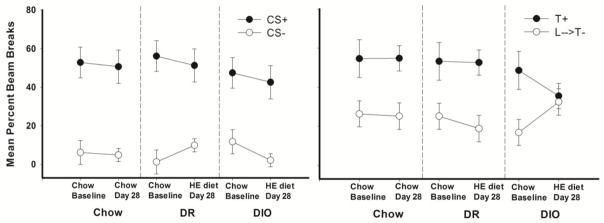
Performance on simple (CS+, CS-) and serial feature negative discriminations (T+, L→ T−) on the last day of training under food deprivation compared to a probe test 28 days after the start of ad libitum feeding for the group given standard chow (CHOW) and the diet-induced obese (DIO) and diet-resistant (DR) groups that were given high-energy diet. For each diet group, all differences shown in responding on CS+ vs. CS− trials were significant (ps < .05). All differences shown in responding on L→T− vs T+ trials were significant (ps < .05) except for responding on the Day 28 probe test for Group HE-DIO. Data from, Davidson, T. L., Monnot, A., Neal, A. U., Martin, A. A., Horton, J. J., & Zheng, W. (2012).
In this study, consuming HE diet reduced responding for DIO rats on T+ trials and increased responding somewhat on nonreinforced L→T− trials, relative to the level observed during the chow baseline period. This pattern of responding would be expected if intake of the HE diet reduced the ability to distinguish nonreinforced from reinforced presentations of the target (e.g., Rescorla and Wagner, 1972). Within our model, this nondiscriminative responding would occur to the extent that rats could no longer use the feature cue to signal nonreinforcement of the target.
These results provide evidence that energy dysregulation leading to excessive weight gain for Group HE-DIO was accompanied by an impaired ability to solve a feature negative discrimination problem. This finding supports the hypothesis that maintaining energy balance is related to the ability to solve a serial feature negative discrimination problem in which satiety cues serve as negative feature stimuli that signal when tastes and other food-related stimuli will not be followed by an appetitive postingestive US.
Figure 7 shows that relative to DR rats and chow-fed controls, DIO rats showed increased BBB permeability as reflected by higher concentrations of NaFl in the hippocampus. This pattern of differences was not observed in the striatum or the prefrontal cortex. Thus, consuming a HE diet produced impairments in serial feature negative performance and in the regulation of body weight only in rats that also exhibited increased BBB permeability and heightened levels of NaFl in the hippocampus. If the obesity-promoting effects of HE or Western diets depend on their ability to interfere with hippocampal-dependent serial feature negative performance, this is precisely the outcome that would be expected. Figure 7 also indicates that NaFl density was higher in the prefrontal cortex for DR rats compared to DIO and chow-fed control rats. Recent, currently unpublished, results have failed to replicate this difference but did confirm the finding of higher levels of NaFl in the hippocampus of DIO rats compared to DR rats and chow controls. Previous results suggest that diet-induced obesity can alter dopamine-based reward signaling in the striatum (e.g., Sharma & Fulton, 2013). The findings of Kanoski et al., (2010) and Davidson et al., (2012) make it unlikely that this type of interference is the result of any direct effects of increased BBB permeability on the striatum.
Figure 7.

Concentration of sodium fluorescein (NaFl) in the hippocampus, prefrontal cortex, and striatum of HE diet-fed and chow-fed rats. The entire hippocampus, striatum, and prefrontal cortex were extracted bilaterally. *denotes significantly higher density of NaFl in the hippocampus for the DIO group compared to the DR group and Chow controls and a significantly higher density of NaFl in prefrontal cortex for the DR group relative to the DIO group and Chow controls (ps < .05). Data from, Davidson, T. L., Monnot, A., Neal, A. U., Martin, A. A., Horton, J. J., & Zheng, W. (2012).
3.5. Relations to findings from studies of cognitive functioning in humans
Similar to the effects of selective total and certain subtotal lesions of the hippocampus in rats, densely amnesic humans with brain damage that includes the hippocampus have been reported to exhibit hyperphagia and reduced sensitivity to interoceptive signals of hunger and satiety (Hebben, Corkin, Eichenbaum, and Shedlack, 1985; Rozin, Dow, Moscovitch, and Rajaram, 1998). The most famous of these cases is H.M., a patient who experienced near complete anterograde amnesia following a medial temporal lobe surgery aimed at reducing the frequency of his grand mal seizures that produced extensive bilateral hippocampal damage. H.M. also exhibited hyperphagia in that he would readily consume a second full meal that he received within minutes of completing a prior meal. This finding, along with H.M.’s self-reported insensitivity to changes in satiety that would have accompanied the first meal, indicates that H.M. was unable to use either the physiological satiety signals produced by the first meal or the memories associated with consuming that meal to inhibit his food intake.
A number of studies have provided evidence that the inhibition of food intake is influenced by the memory of a recent meal. Specifically, Higgs (Higgs, 2002; 2008 for review) reported that merely asking normal weight human participants to recall the lunch that they ate decreased snack consumption a few hours later compared to subjects that were asked to recall non-food items or the lunch they had consumed on the previous day. Similarly, in another study subjects that were instructed to focus on the sensory properties of the food they were eating during a fixed lunch showed greater inhibition of intake when given a snack a few hours later compared to subjects that read an article about food or were not given any specific task after eating the same lunch (Higgs and Donohoe, 2011). These data suggest that intake suppression depends on both the retrieval of the memory of the previous meal and on how well that memory was encoded originally.
More recently, Brunstrom, Burn, Sell, Collingwood, Rogers, Wilkinson, Hinton, Maynard, and Ferriday (2012) showed that the memory of a recent lunch exerts a stronger influence on subsequent intake suppression than does the actual energetic content of the meal itself. In this study, the participants were presented with either 300 ml or 500 ml of soup. However, through the use of a hidden pump that could surreptitiously add or remove soup from the bowls, half of the subjects given 300 ml of soup actually consumed 500 ml, and half given 500 ml actually consumed only 300 ml of soup. When all subjects were given a snack a few hours later, amount of intake was determined more by the amount of soup the subjects perceived and remembered that they had consumed than by the physical amount they actually consumed. Thus, in this study not only did the memory of eating soup exert a suppressive effect on subsequent intake several hours later, the suppressive effect of the memory surpassed that of the energy content of the soup. Brunstrom et al., (2012) suggested that the memory for recent eating may modulate subsequent intake by helping to interpret postingestive signals generated by physiological satiety cues, such as CCK or GLP-1, by attributing them to a recently consumed meal.
A somewhat different possibility can be derived within the framework provided by our present model. Davidson and colleagues (Davidson, 1993; Davidson et al., 1992; Davidson, Kanoski, Walls, and Jarrard, 2005b) suggested that satiety signals have no special properties relative to other types of stimuli (auditory, visual, contextual). This also applies to satiety signals that function as feature cues in serial feature negative discrimination problems. What makes an event a negative feature cue is not its origins or sensory properties, but its ability to signal when other events will not be followed by a US. Therefore, in the regulation of energy intake, it is also possible that events other than physiological satiety cues could contribute to the inhibition of energy intake by functioning as negative feature stimuli. Viewed this way, it may be that memories of recent meals act independently as satiety signals or act in concert with cues of physiological origin to form a stimulus configuration or compound that functions as a complex satiety cue. In either case, memories of recent meals may function as learned negative feature stimuli that suppress eating by inhibiting the ability of food and food-related cues to retrieve the memory of the appetitive postingestive consequences of intake.
A final point has to do with the concept of memory inhibition itself. A key component of our model is that satiety cues act to inhibit the activation of the memories of the appetitive postingestive consequences of eating. This type of memory inhibition is adaptive because when energy homeostasis has been achieved, continued retrieval of the memory of those reinforcing outcomes would be unwanted and could interfere with the performance of other adaptive functions. In recent years much data has been accumulated which support the idea that optimal memory function depends not only on the ability to retrieve information when it is needed but also on the ablity to inhibit retrieval of information when it is not needed (Healey, Campbell, Hasher, and Ossher, 2010; MacLeod and Saunders, 2008). Many of these studies also show that memory retrieval inhibition depends on brain circuits that include the hippocampus as a critical component.
For example, Depue, Burgess, Willcutt, Ruzic, and Banich (2010) compared inhibition of memory retrieval in subjects with Attention Deficit Hyperactivity Disorder (ADHD) and controls. To assess memory retrieval inhibition, the subjects were trained in a “Think/No Think” (TNT) task (Anderson and Green, 2001), which requires them to try to voluntarily suppress retrieval of a memory. In brief, the subjects were first trained with 40 different face-picture pairs. Then, in a separate phase, subjects in the Think condition were given some of the faces and told to think of the pictures that were previously associated with them. In the No Think condition they were told to actively try not to think about the pictures that had been paired with the faces that were presented. For all subjects, the TNT manipulations took place during fMRI. In a final test phase, all of the subjects were shown the faces that had been presented in the TNT phase and the faces that had not been presented (as a baseline) and were asked to report which pictures had been paired previously with each face.
The test phase results showed that while both ADHD and control groups recalled fewer pictures from the No Think than from the Think conditions, only the control group recalled fewer pictures from the No think condition than from the baseline condition in which the face-picture pairs had not been subject to either the Think or No Think manipulation. This latter finding that retrieval of pictures from the No Think condition was less than the baseline condition provides evidence for memory retrieval inhibition of the No Think pictures by control but not by ADHD subjects. However, of special interest, fMRI showed that for control, but not for ADHD subjects, successful memory retrieval inhibition was predicted by activation of the hippocampus and middle frontal gyri. Depue (2012) reviewed evidence that further substantiates this link between the hippocampus, the frontal cortex, and memory retrieval inhibition. These results indicate that at least one form of memory retrieval inhibition depends on the hippocampus. Further, they add plausibility to the idea that, similar to ADHD, impulsivity and failures of inhibitory control that are associated with overeating may also involve deficits in hippocampal-dependent memory retrieval inhibition.
A recent paper by Benoit and Anderson (2012) further elaborated on the links between memory retrieval inhibition and hippocampus. These researchers proposed that inhibition of unwanted or interfering memories could be accomplished in more than one way. In a study that used a modified TNT procedure, subjects were told to explicitly substitute and retrieve the memory of another item for an unwanted memory, or they were instructed to directly block an unwanted memory from consciousness without engaging in any distracting activity by “pushing it out of mind”. In a subsequent recall test, both the memory substitution and the direct suppression strategy produced memory inhibition retrieval below baseline level for control items that had not been subjected to either strategy. However, fMRI activity when the subjects were trying to inhibit retrieval of the unwanted memories depended on the strategy that they employed. The results indicated that successful efforts to substitute alternative for unwanted memories are accompanied by increased hippocampal activation, whereas efforts to directly suppress retrieval of unwanted memories without substitution were associated with reduced activation in the hippocampus. These results indicate that memory retrieval inhibition can be accomplished in multiple ways and that different ways may involve different types of hippocampal-dependent processes.
While there are no studies that have directly examined the hippocampal-dependence of human analogs to rodent serial feature discrimination problems, the above findings suggest that interference with the hippocampal function of humans could impair the ability of feature negative cues, including satiety signals, to inhibit memory retrieval. Furthermore, there is evidence that impaired hippocampal function in humans may also be related to the intake of Western diet and obesity. For example, Francis and Stevenson (2011) found that humans with higher self-reported HE diet intake were impaired on a neuropsychological test of hippocampal-dependent memory function, were less able to accurately recall what they consumed during a previous meal, and, consistent with the findings noted above (e.g., Brunstrom et al., 2012; Higgs, 2008), ate more during a subsequent test meal compared with subjects that reported lower HE diet intake. Another recent study reported that academic performance and inhibitory control on a “go-no go” task were negatively related to both body mass index (BMI) and body adiposity, whereas performance on a task that required less inhibitory capacity was unaffected by either of these variables (Kamijo, Khan, Pontifex, Scudder, Drollette, Raine, Evans, Castelli, and Hillman, 2012). Moreover, a number of epidemiological studies conclude that mid-life obesity and metabolic disorders, which often accompany excessive intake of Western diets, increase the risk of Alzheimer’s Disease and other cognitive dementias later in life (e.g., Gustafson, Backman, Joas, Waern, Ostling, Guo, and Skoog, 2012; Profenno, Porsteinsson, and Faraone, 2010; Whitmer, Gustafson, Barrett-Connor, Haan, Gunderson, and Yaffe, 2008). The hippocampus and hippocampal formation are sites of early pathology that is associated with these disorders (e.g., see Dhikav and Anand, 2011; Velayudhan, Proitsi, Westman, Muehlboeck, Mecocci, Vellas, Tsolaki, Kloszewska, Soininen, Spenger, Hodges, Powell, Lovestone, and Simmons, 2013). There is evidence that the hippocampal formation, which is especially vulnerable to many types of insults (e.g., Walsh and Emerich, 1988), is an early target of pathologies that later progress onto other areas of the brain that are also involved with cognition (Smith, 2002). It may be that deficits in hippocampal-dependent cognitive function are a precursor to larger, more general forms of cognitive decline. Together these types of results and other findings point to the conclusion that consumption of HE diets and the accompanying obesity may have adverse effects on cognitive functioning throughout the lifespan (Smith, Hay, Campbell, and Trollor, 2011).
4. A vicious cycle of obesity and cognitive decline
The interrelationship between energy regulation and cognitive function that is represented in our model anticipates what we (Davidson et al., 2005b; Kanoski and Davidson, 2011) have termed a “vicious-cycle of obesity and cognitive decline” (see Figure 7). That is, if eating a HE or Western diet interferes with the functioning of the hippocampus, and this interference has the effect of impairing the ability to inhibit retrieval of the memory of the appetitive postingestive consequences of energy intake by environmental food-related cues, then this impairment would increase the likelihood that those cues would evoke additional appetitive behavior and intake of the Western diet, which would give rise to further impairment in hippocampal function and memory retrieval inhibition. Unchecked, this cycle would result not only in overeating and weight gain, but also in the progressive deterioration of hippocampal-dependent cognitive functioning. Furthermore, while it may be the case that overeating produces impaired cognitive functioning, the vicious-cycle framework also allows for the possibility that cognitive impairment is a cause of overeating. Consistent with this latter interpretation, the results of a recent longitudinal study suggest that low scores on tests of cognitive abilities in young children predict subsequent excess body weight and obesity (Guxens, Mendez, Julvez, Plana, Forns, Basagana, Torrent, and Sunyer, 2009). Future research is needed to establish the direction of the relationship between obesity and cognitive performance.
5. Summary and conclusions
The analysis we have outlined in this paper proposes that efficient energy and body weight regulation depends on the operation of learning and memory mechanisms in which tastes and other environmental food cues are embedded in concurrent excitatory and inhibitory associations with the appetitive postingestive US that is produced by intake. The capacity for taste and food-related stimuli to excite the representation of that US depends on the degree to which the opposing inhibitory association is activated. Activation of that inhibitory association is gated by the presence of satiety signals of physiological or other origins, in the same way that conventional feature stimuli are thought to gate inhibitory association activation in serial feature negative discrimination problems.
We have also identified two features of the current food environment that could promote both obesity and cognitive decline by disrupting the operation of this associative mechanism. One is the introduction of dietary products that weaken the validity of sweet tastes and other orosensory stimuli as signals for nutritive or caloric postingestive outcomes. The second is the consumption of high-energy diets that weaken the ability of satiety signals to function as negative feature stimuli by compromising the structures and circuits that underlie the performance of that function. These two sources of energy dysregulation may not be completely independent in that, as our data have shown, weakening the validity of sweet taste as a signal for caloric outcomes can promote excess intake of the same high-energy or Western diets that could lead to impaired brain functioning.
There are some noteworthy things that are model does not do. This model does not rely on new learning or memory principles that were invented as a means of providing a post-hoc explanation of eating behavior. For example, the idea the predictive validity of a cue determines how well it competes with other stimuli for associative strength was not created by us to explain how the consumption of noncaloric sweeteners could disrupt energy regulation. This idea was developed and confirmed by foundational studies of the processes that underlie Pavlovian conditioning (see, Rescorla, 1968; Urushihara & Miller, 2009; Wagner et al., 1968). Similarly, the notion that one stimulus can indirectly modulate the capacity of another stimulus to activate the memorial representation of its US was not invented as a convenient device to explain how satiety cues can inhibit appetitive and consummatory responding. This idea was developed to explain how animals solve certain types of conditional discrimination problems (e.g., see Swartzentruber, 1995 for review). We have relied on the use of learning and memory constructs that have been validated previously based on empirical evidence and supporting theory, which were developed largely independently of the phenomena that we have attempted to explain.
While our analysis refers to terms such as “sweet taste”, “satiety signals”, and “appetitive” and “aversive postingestive consequences”, we used these terms descriptively—not as explanations of behavior in their own right. That is, in our model sweet taste refers to a sensory rather than a hedonic property of food; satiety signals refer to sensory events of internal origin that suppress behavior, without ascribing that suppressive effect to any motivational property of satiety itself; appetitive and aversive consequences refer to outcomes of intake that promote or suppress, respectively, behavior that is instrumental to obtaining and consuming food, without the additional claims that those outcomes incentivize or reward the behaviors that are associated with them.
Instead of appealing to hedonic, motivational, or reward processes, we conceived of sweet taste, satiety signals, and the postingestive outcomes of eating as stimuli that are components of a larger, integrative, and decidedly associative mechanism of energy and body weight regulation. We then used this mechanism to identify factors in the food environment that could cause energy dysregulation, leading to overeating and obesity, and described evidence that supported our analysis. We also described how our learning and memory model could be used as a framework for understanding recent findings that link overweight and obesity to cognitive decline.
Models and theories always provide inexact representations of the phenomena they attempt to explain. They are useful to the extent that they generate novel and testable hypotheses that can yield new information about those phenomena. We think describing energy intake and body weight regulation within a learning and memory framework yields new information about processes and mechanisms that underlie overeating and obesity and that link overeating and obesity to eventual cognitive decline. We hope that this information may provide an impetus for new ideas for effective therapies to treat both of these threats to health and well-being.
Figure 8.

Vicious-cycle model of obesity and cognitive decline (see Davidson et al. 2005; Kanoski & Davidson, 2011).
Highlights.
Pavlovian conditioning contributes to energy intake and body weight regulation.
Dietary factors can reduce the effectiveness of Pavlovian control mechanisms.
Some diets may impair hippocampal-dependent memory function
Hippocampal dysfunction may lead to a vicious-cycle of obesity and memory decline.
Acknowledgments
The preparation of this manuscript and much of the research that is described herein was supported by grants P01 HD052112 and R01 HD29792 from the National Institutes of Child Health and Development and grant R01 DK076078 from the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson MC, Green C. Suppressing unwanted memories by executive control. Nature. 2001;410:366–369. doi: 10.1038/35066572. [DOI] [PubMed] [Google Scholar]
- Anton SD, Martin CK, Han H, Coulon S, Cefalu WT, Geiselman P, Williamson DA. Effects of stevia, aspartame, and sucrose on food intake, satiety, and postprandial glucose and insulin levels. Appetite. 2010;55:37–43. doi: 10.1016/j.appet.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ard JD. Unique perspectives on the obesogenic environment. Journal of General Internal Medicine. 2007;22:1058–1060. doi: 10.1007/s11606-007-0243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit RG, Anderson MC. Opposing mechanisms support the voluntary forgetting of unwanted memories. Neuron. 2012;76:450–460. doi: 10.1016/j.neuron.2012.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit SC, Davidson TL. Interoceptive sensory signals produced by 24-hr food deprivation, pharmacological glucoprivation, and lipoprivation. Behavioral Neuroscience. 1996;110:168–180. [PubMed] [Google Scholar]
- Berthoud HR. The neurobiology of food intake in an obesogenic environment. Proceedings of the Nutrition Society. 2012;71:478–487. doi: 10.1017/S0029665112000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhupathiraju SN, Pan A, Malik VS, Manson JE, Willett WC, van Dam RM, Hu FB. Caffeinated and caffeine-free beverages and risk of type 2 diabetes. Am J Clin Nutr. 2013;97:155–166. doi: 10.3945/ajcn.112.048603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonacchi KB, Ackroff K, Sclafani A. Sucrose taste but not Polycose taste conditions flavor preferences in rats. Physiol Behav. 2008;95:235–244. doi: 10.1016/j.physbeh.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth DA. Satiety and appetite are conditioned reactions. Psychosom Med. 1977;39:76–81. doi: 10.1097/00006842-197703000-00002. [DOI] [PubMed] [Google Scholar]
- Bouton ME. Context, ambiguity, and classical conditioning. Current Directions in Psychological Science. 1994;3:49–53. [Google Scholar]
- Bouton ME. Context and behavioral processes in extinction. Learning & Memory. 2004;11:485–494. doi: 10.1101/lm.78804. [DOI] [PubMed] [Google Scholar]
- Brown AW, Bohan Brown MM, Onken KL, Beitz DC. Short-term consumption of sucralose, a nonnutritive sweetener, is similar to water with regard to select markers of hunger signaling and short-term glucose homeostasis in women. Nutr Res. 2011;31:882–888. doi: 10.1016/j.nutres.2011.10.004. [DOI] [PubMed] [Google Scholar]
- Brown RJ, Walter M, Rother KI. Effects of diet soda on gut hormones in youths with diabetes. Diabetes Care. 2012;35:959–964. doi: 10.2337/dc11-2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunstrom JM, Burn JF, Sell NR, Collingwood JM, Rogers PJ, Wilkinson LL, Hinton EC, Maynard OM, Ferriday D. Episodic Memory and Appetite Regulation in Humans. Plos One. 2012;7 doi: 10.1371/journal.pone.0050707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss C, Kraemer-Aguiar LG, Maranhao PA, Marinho C, de Souza M, Wiernsperger N, Bouskela E. Novel findings in the cephalic phase of digestion: a role for microcirculation? Physiol Behav. 2012;105:1082–1087. doi: 10.1016/j.physbeh.2011.12.004. [DOI] [PubMed] [Google Scholar]
- Chan KH, Jarrard LE, Davidson TL. The effects of selective ibotenate lesions of the hippocampus on conditioned inhibition and extinction. Cogn Affect Behav Neurosci. 2003;3:111–119. doi: 10.3758/cabn.3.2.111. [DOI] [PubMed] [Google Scholar]
- Chan KH, Morell JR, Jarrard LE, Davidson TL. Reconsideration of the role of the hippocampus in learned inhibition. Behav Brain Res. 2001;119:111–130. doi: 10.1016/s0166-4328(00)00363-6. [DOI] [PubMed] [Google Scholar]
- Chudasama Y, Doobay VM, Liu Y. Hippocampal-prefrontal cortical circuit mediates inhibitory response control in the rat. Journal of Neuroscience. 2012;32:10915–10924. doi: 10.1523/JNEUROSCI.1463-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton PG, Vickers SP, Somerville EM. Little and often: Ingestive behavior patterns following hippocampal lesions in rats. Behavioral Neuroscience. 1998;112:502–511. doi: 10.1037//0735-7044.112.3.502. [DOI] [PubMed] [Google Scholar]
- Cohen L, Curhan G, Forman J. Association of sweetened beverage intake with incident hypertension. J Gen Intern Med. 2012;27:1127–1134. doi: 10.1007/s11606-012-2069-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TL. Learning about deprivation intensity stimuli. Behavioral Neuroscience. 1987;101:198–208. doi: 10.1037//0735-7044.101.2.198. [DOI] [PubMed] [Google Scholar]
- Davidson TL. The nature and function of interoceptive signals to feed: toward integration of physiological and learning perspectives. Psychological Review. 1993;100:640–657. doi: 10.1037/0033-295x.100.4.640. [DOI] [PubMed] [Google Scholar]
- Davidson TL, Chan K, Jarrard LE, Kanoski SE, Clegg DJ, Benoit SC. Contributions of the hippocampus and medial prefrontal cortex to energy and body weight regulation. Hippocampus. 2009;19:235–252. doi: 10.1002/hipo.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TL, Flynn FW, Jarrard LE. Potency of food deprivation intensity cues as discriminative stimuli. Journal of Experimental Psychology: Animal Behavior Processes. 1992;18:174–181. doi: 10.1037//0097-7403.18.2.174. [DOI] [PubMed] [Google Scholar]
- Davidson TL, Jarrard LE. A role for hippocampus in the utilization of hunger signals. Behavioral & Neural Biology. 1993;59:167–171. doi: 10.1016/0163-1047(93)90925-8. [DOI] [PubMed] [Google Scholar]
- Davidson TL, Kanoski SE, Chan K, Clegg DJ, Benoit SC, Jarrard LE. Hippocampal lesions impair retention of discriminative responding based on energy state cues. Behav Neurosci. 2010;124:97–105. doi: 10.1037/a0018402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TL, Kanoski SE, Tracy AL, Walls EK, Clegg D, Benoit SC. The interoceptive cue properties of ghrelin generalize to cues produced by food deprivation. Peptides. 2005a;26:1602–1610. doi: 10.1016/j.peptides.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Davidson TL, Kanoski SE, Walls EK, Jarrard LE. Memory inhibition and energy regulation. Physiol Behav. 2005b;86:731–746. doi: 10.1016/j.physbeh.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Davidson TL, Martin AA, Clark K, Swithers SE. Intake of high-intensity sweeteners alters the ability of sweet taste to signal caloric consequences: Implications for the learned control of energy and body weight regulation. Q J Exp Psychol (Colchester) 2011:1–12. doi: 10.1080/17470218.2011.552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TL, Monnot A, Neal AU, Martin AA, Horton JJ, Zheng W. The effects of a high-energy diet on hippocampal-dependent discrimination performance and blood-brain barrier integrity differ for diet-induced obese and diet-resistant rats. Physiol Behav. 2012;107:26–33. doi: 10.1016/j.physbeh.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TL, Swithers SE. A Pavlovian approach to the problem of obesity. Int J Obes Relat Metab Disord. 2004;28:933–935. doi: 10.1038/sj.ijo.0802660. [DOI] [PubMed] [Google Scholar]
- Depue BE. A neuroanatomical model of prefrontal inhibitory modulation of memory retrieval. Neurosci Biobehav Rev. 2012;36:1382–1399. doi: 10.1016/j.neubiorev.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depue BE, Burgess GC, Willcutt EG, Ruzic L, Banich MT. Inhibitory control of memory retrieval and motor processing associated with the right lateral prefrontal cortex: evidence from deficits in individuals with ADHD. Neuropsychologia. 2010;48:3909–3917. doi: 10.1016/j.neuropsychologia.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhikav V, Anand K. Potential Predictors of Hippocampal Atrophy in Alzheimer’s Disease. Drugs & Aging. 2011;28:1–11. doi: 10.2165/11586390-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Dockray GJ. Cholecystokinin. Current Opinion in Endocrinology Diabetes and Obesity. 2012;19:8–12. doi: 10.1097/MED.0b013e32834eb77d. [DOI] [PubMed] [Google Scholar]
- Duffey KJ, Steffen LM, Van Horn L, Jacobs DR, Jr, Popkin BM. Dietary patterns matter: diet beverages and cardiometabolic risks in the longitudinal Coronary Artery Risk Development in Young Adults (CARDIA) Study. Am J Clin Nutr. 2012;95:909–915. doi: 10.3945/ajcn.111.026682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer DM, Haselgrove M, Jones PM. Cue interactions in flavor preference learning: a configural analysis. J Exp Psychol Anim Behav Process. 2011;37:41–57. doi: 10.1037/a0021033. [DOI] [PubMed] [Google Scholar]
- Escobar M, Miller RR. A review of the empirical laws of basic learning in Pavlovian conditioning. International Journal of Comparative Psychology. 2004;17(2–3):279–303. [Google Scholar]
- Fagherazzi G, Vilier A, Saes Sartorelli D, Lajous M, Balkau B, Clavel-Chapelon F. Consumption of artificially and sugar-sweetened beverages and incident type 2 diabetes in the Etude Epidemiologique aupres des femmes de la Mutuelle Generale de l’Education Nationale-European Prospective Investigation into Cancer and Nutrition cohort. Am J Clin Nutr. 2013;97:517–523. doi: 10.3945/ajcn.112.050997. [DOI] [PubMed] [Google Scholar]
- Feijo FdM, Ballard CR, Foletto KC, Melo Batista BA, Neves AM, Marques Ribeiro MF, Bertoluci MC. Saccharin and aspartame, compared with sucrose, induce greater weight gain in adult Wistar rats, at similar total caloric intake levels. Appetite. 2013;60:203–207. doi: 10.1016/j.appet.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Ford ES, Mokdad AH. Epidemiology of obesity in the Western Hemisphere. J Clin Endocrinol Metab. 2008;93:S1–8. doi: 10.1210/jc.2008-1356. [DOI] [PubMed] [Google Scholar]
- Ford HE, Peters V, Martin NM, Sleeth ML, Ghatei MA, Frost GS, Bloom SR. Effects of oral ingestion of sucralose on gut hormone response and appetite in healthy normal-weight subjects. Eur J Clin Nutr. 2011;65:508–513. doi: 10.1038/ejcn.2010.291. [DOI] [PubMed] [Google Scholar]
- Fowler SP, Williams K, Resendez RG, Hunt KJ, Hazuda HP, Stern MP. Fueling the obesity epidemic? Artificially sweetened beverage use and long-term weight gain. Obesity (Silver Spring) 2008;16:1894–1900. doi: 10.1038/oby.2008.284. [DOI] [PubMed] [Google Scholar]
- Francis HM, Stevenson RJ. Higher Reported Saturated Fat and Refined Sugar Intake Is Associated With Reduced Hippocampal-Dependent Memory and Sensitivity to Interoceptive Signals. Behavioral Neuroscience. 2011;125:943–955. doi: 10.1037/a0025998. [DOI] [PubMed] [Google Scholar]
- Gardener H, Rundek T, Markert M, Wright CB, Elkind MS, Sacco RL. Diet soft drink consumption is associated with an increased risk of vascular events in the Northern Manhattan Study. J Gen Intern Med. 2012;27:1120–1126. doi: 10.1007/s11606-011-1968-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goris JM, Petersen S, Stamatakis E, Veerman JL. Television food advertising and the prevalence of childhood overweight and obesity: a multicountry comparison. Public Health Nutr. 2010;13:1003–1012. doi: 10.1017/S1368980009992850. [DOI] [PubMed] [Google Scholar]
- Green E, Murphy C. Altered processing of sweet taste in the brain of diet soda drinkers. Physiol Behav. 2012;107:560–567. doi: 10.1016/j.physbeh.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson DR, Backman K, Joas E, Waern M, Ostling S, Guo X, Skoog I. 37 years of body mass index and dementia: observations from the prospective population study of women in Gothenburg, Sweden. J Alzheimers Dis. 2012;28:163–171. doi: 10.3233/JAD-2011-110917. [DOI] [PubMed] [Google Scholar]
- Guxens M, Mendez MA, Julvez J, Plana E, Forns J, Basagana X, Torrent M, Sunyer J. Cognitive function and overweight in preschool children. Am J Epidemiol. 2009;170:438–446. doi: 10.1093/aje/kwp140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healey MK, Campbell KL, Hasher L, Ossher L. Direct evidence for the role of inhibition in resolving interference in memory. Psychol Sci. 2010;21:1464–1470. doi: 10.1177/0956797610382120. [DOI] [PubMed] [Google Scholar]
- Hebben N, Corkin S, Eichenbaum H, Shedlack K. Diminished ability to interpret and report internal states after bilateral medial temporal resection: case H.M. Behavioral Neuroscience. 1985;99:1031–1039. doi: 10.1037//0735-7044.99.6.1031. [DOI] [PubMed] [Google Scholar]
- Hellstrom PM. Satiety signals and obesity. Curr Opin Gastroenterol. 2013 doi: 10.1097/MOG.0b013e32835d9ff8. [DOI] [PubMed] [Google Scholar]
- Henderson YO, Smith GP, Parent MB. Hippocampal neurons inhibit meal onset. Hippocampus. 2013;23:100–107. doi: 10.1002/hipo.22062. [DOI] [PubMed] [Google Scholar]
- Higgs S. Memory for recent eating and its influence on subsequent food intake. Appetite. 2002;39:159–166. doi: 10.1006/appe.2002.0500. [DOI] [PubMed] [Google Scholar]
- Higgs S. Cognitive influences on food intake: the effects of manipulating memory for recent eating. Physiol Behav. 2008;94:734–739. doi: 10.1016/j.physbeh.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Higgs S, Donohoe JE. Focusing on food during lunch enhances lunch memory and decreases later snack intake. Appetite. 2011;57:202–206. doi: 10.1016/j.appet.2011.04.016. [DOI] [PubMed] [Google Scholar]
- Hock BJ, Jr, Bunsey MD. Differential effects of dorsal and ventral hippocampal lesions. Journal of Neuroscience. 1998;18:7027–7032. doi: 10.1523/JNEUROSCI.18-17-07027.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland PC. Differential renforcement of an inhibitory feature after serial and simultaneous feature negative discrimination training. Journal of Experimental Psychology-Animal Behavior Processes. 1984;10:461–475. [PubMed] [Google Scholar]
- Holland PC, Lamoureux JA, Han JS, Gallagher M. Hippocampal lesions interfere with Pavlovian negative occasion setting. Hippocampus. 1999;9:143–157. doi: 10.1002/(SICI)1098-1063(1999)9:2<143::AID-HIPO6>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- InterAct, c. Consumption of sweet beverages and type 2 diabetes incidence in European adults: results from EPIC-InterAct. Diabetologia. 2013;56:1520–1530. doi: 10.1007/s00125-013-2899-8. [DOI] [PubMed] [Google Scholar]
- Johnson AW. Eating beyond metabolic need: how environmental cues influence feeding behavior. Trends Neurosci. 2013 doi: 10.1016/j.tins.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Kamijo K, Khan NA, Pontifex MB, Scudder MR, Drollette ES, Raine LB, Evans EM, Castelli DM, Hillman CH. The relation of adiposity to cognitive control and scholastic achievement in preadolescent children. Obesity (Silver Spring) 2012;20:2406–2411. doi: 10.1038/oby.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoski SE, Davidson TL. Western diet consumption and cognitive impairment: Links to hippocampal dysfunction and obesity. Physiol Behav. 2011;103:59–68. doi: 10.1016/j.physbeh.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoski SE, Walls EK, Davidson TL. Interoceptive “satiety” signals produced by leptin and CCK. Peptides. 2007;28:988–1002. doi: 10.1016/j.peptides.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoski SE, Zhang Y, Zheng W, Davidson TL. The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. J Alzheimers Dis. 2010;21:207–219. doi: 10.3233/JAD-2010-091414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Chang J, Checklin HL, Young RL, Jones KL, Horowitz M, Rayner CK. Effect of the artificial sweetener, sucralose, on small intestinal glucose absorption in healthy human subjects. Br J Nutr. 2010;104:803–806. doi: 10.1017/S0007114510001327. [DOI] [PubMed] [Google Scholar]
- MacLeod MD, Saunders J. Retrieval inhibition and memory distortion - Negative consequences of an adaptive process. Current Directions in Psychological Science. 2008;17:26–30. [Google Scholar]
- Madsen AN, Hansen G, Paulsen SJ, Lykkegaard K, Tang-Christensen M, Hansen HS, Levin BE, Larsen PJ, Knudsen LB, Fosgerau K, Vrang N. Long-term characterization of the diet-induced obese and diet-resistant rat model: a polygenetic rat model mimicking the human obesity syndrome. Journal of Endocrinology. 2010;206:287–296. doi: 10.1677/JOE-10-0004. [DOI] [PubMed] [Google Scholar]
- Min DK, Tuor UI, Chelikani PK. Gastric distention induced functional magnetic resonance signal changes in the rodent brain. Neuroscience. 2011;179:151–158. doi: 10.1016/j.neuroscience.2011.01.051. [DOI] [PubMed] [Google Scholar]
- Moran TH, Ladenheim EE. Adiposity signaling and meal size control. Physiol Behav. 2011;103:21–24. doi: 10.1016/j.physbeh.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morell JR, Davidson TL. Transfer across unconditioned stimuli in serial feature discrimination training. Journal of Experimental Psychology: Animal Behavior Processes. 2002;28:83–96. [PubMed] [Google Scholar]
- Nettleton JA, Polak JF, Tracy R, Burke GL, Jacobs DR., Jr Dietary patterns and incident cardiovascular disease in the Multi-Ethnic Study of Atherosclerosis. Am J Clin Nutr. 2009;90:647–654. doi: 10.3945/ajcn.2009.27597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov IP. The work of the digestive glands. London: C. Griffin, Co. Ltd; 1910. [Google Scholar]
- Pepino MY, Tiemann CD, Patterson BW, Wice BM, Klein S. Sucralose Affects Glycemic and Hormonal Responses to an Oral Glucose Load. Diabetes Care. 2013 doi: 10.2337/dc12-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1:223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- Pierce WD, Heth CD, Owczarczyk JC, Russell JC, Proctor SD. Overeating by young obesity-prone and lean rats caused by tastes associated with low energy foods. Obesity (Silver Spring) 2007;15:1969–1979. doi: 10.1038/oby.2007.235. [DOI] [PubMed] [Google Scholar]
- Popkin BM, Adair LS, Ng SW. Global nutrition transition and the pandemic of obesity in developing countries. Nutr Rev. 2012;70:3–21. doi: 10.1111/j.1753-4887.2011.00456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power ML, Schulkin J. Anticipatory physiological regulation in feeding biology: cephalic phase responses. Appetite. 2008;50:194–206. doi: 10.1016/j.appet.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profenno LA, Porsteinsson AP, Faraone SV. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol Psychiatry. 2010;67:505–512. doi: 10.1016/j.biopsych.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Rescorla RA. Probability of shock in the presence and absence of CS in fear conditioning. J Comp Physiol Psychol. 1968;66:1–5. doi: 10.1037/h0025984. [DOI] [PubMed] [Google Scholar]
- Rescorla RA, Wagner AR, editors. A theory of Pavlovian conditioning: Variations in the effectiveness of reinforcement and nonreinforcement. New York: Appleton-Century-Crofts; 1972. [Google Scholar]
- Rozin P, Dow S, Moscovitch M, Rajaram S. What causes humans to begin and end a meal? A role for memory for what has been eaten, as evidenced by a study of multiple meal eating in amnesic patients. Psychological Science. 1998;9:392–396. [Google Scholar]
- Rudenga KJ, Small DM. Amygdala response to sucrose consumption is inversely related to artificial sweetener use. Appetite. 2012;58:504–507. doi: 10.1016/j.appet.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu JK, McLarnon JG. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J Cell Mol Med. 2009;13:2911–2925. doi: 10.1111/j.1582-4934.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M, Nakamura K, Miura K, Takamura T, Yoshita K, Nagasawa SY, Morikawa Y, Ishizaki M, Kido T, Naruse Y, Suwazono Y, Sasaki S, Nakagawa H. Sugar-sweetened beverage and diet soda consumption and the 7-year risk for type 2 diabetes mellitus in middle-aged Japanese men. Eur J Nutr. 2013 doi: 10.1007/s00394-013-0523-9. [DOI] [PubMed] [Google Scholar]
- Sam AH, Troke RC, Tan TM, Bewick GA. The role of the gut/brain axis in modulating food intake. Neuropharmacology. 2012;63:46–56. doi: 10.1016/j.neuropharm.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Sclafani A. Learned controls of ingestive behavior. Appetite. 1997;29:153–158. doi: 10.1006/appe.1997.0120. [DOI] [PubMed] [Google Scholar]
- Sharma S, Fulton S. Diet-induced obesity promotes depressive-like behaviour that is associated with neural adaptations in brain reward circuitry. International Journal of Obesity. 2013;37:382–389. doi: 10.1038/ijo.2012.48. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sano Y, Saito K, Abe MA, Maeda T, Haruki H, Kanda T. Pericyte-derived Glial Cell Line-derived Neurotrophic Factor Increase the Expression of Claudin-5 in the Blood-brain Barrier and the Blood-nerve Barrier. Neurochemical Research. 2012;37:401–409. doi: 10.1007/s11064-011-0626-8. [DOI] [PubMed] [Google Scholar]
- Smeets PA, Erkner A, de Graaf C. Cephalic phase responses and appetite. Nutr Rev. 2010;68:643–655. doi: 10.1111/j.1753-4887.2010.00334.x. [DOI] [PubMed] [Google Scholar]
- Smith AD. Imaging the progression of Alzheimer pathology through the brain. Proc Natl Acad Sci U S A. 2002;99:4135–4137. doi: 10.1073/pnas.082107399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Hay P, Campbell L, Trollor JN. A review of the association between obesity and cognitive function across the lifespan: implications for novel approaches to prevention and treatment. Obesity Reviews. 2011;12:740–755. doi: 10.1111/j.1467-789X.2011.00920.x. [DOI] [PubMed] [Google Scholar]
- Steinert RE, Frey F, Topfer A, Drewe J, Beglinger C. Effects of carbohydrate sugars and artificial sweeteners on appetite and the secretion of gastrointestinal satiety peptides. Br J Nutr. 2011;105:1320–1328. doi: 10.1017/S000711451000512X. [DOI] [PubMed] [Google Scholar]
- Swartzentruber D. Modulatory mechanisms in Pavlovian conditioninig. Animal Learning & Behavior. 1995;23:123–143. [Google Scholar]
- Swithers SE. Artificial sweeteners produce the counterintuitive effect of inducing metabolic derangements. Trends in Endocrinology and Metabolism. 2013 doi: 10.1016/j.tem.2013.05.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swithers SE, Baker CR, Davidson TL. General and persistent effects of high-intensity sweeteners on body weight gain and caloric compensation in rats. Behav Neurosci. 2009;123:772–780. doi: 10.1037/a0016139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swithers SE, Davidson TL. A role for sweet taste: calorie predictive relations in energy regulation by rats. Behav Neurosci. 2008;122:161–173. doi: 10.1037/0735-7044.122.1.161. [DOI] [PubMed] [Google Scholar]
- Swithers SE, Laboy AF, Clark K, Cooper S, Davidson TL. Experience with the high-intensity sweetener saccharin impairs glucose homeostasis and GLP-1 release in rats. Behav Brain Res. 2012;233:1–14. doi: 10.1016/j.bbr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swithers SE, Martin AA, Davidson TL. High-intensity sweeteners and energy balance. Physiol Behav. 2011;100:55–62. doi: 10.1016/j.physbeh.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swithers SE, Ogden SB, Davidson TL. Fat substitutes promote weight gain in rats consuming high-fat diets. Behav Neurosci. 2011;125:512–518. doi: 10.1037/a0024404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swithers SE, Sample CH, Davidson TL. Adverse effects of high-intensity sweeteners on energy intake and weight control in male and obesity-prone female rats. Behav Neurosci. 2013;127:262–274. doi: 10.1037/a0031717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd TP, Winterbauer NE, Bouton ME. Contextual control of appetite. Renewal of inhibited food-seeking behavior in sated rats after extinction. Appetite. 2012;58:484–489. doi: 10.1016/j.appet.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treesukosol Y, Blonde GD, Spector AC. T1R2 and T1R3 subunits are individually unnecessary for normal affective licking responses to Polycose: implications for saccharide taste receptors in mice. Am J Physiol Regul Integr Comp Physiol. 2009;296:R855–865. doi: 10.1152/ajpregu.90869.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urushihara K, Miller RR. Stimulus Competition Between a Discrete Cue and a Training Context: Cue Competition Does Not Result From the Division of a Limited Resource. Journal of Experimental Psychology-Animal Behavior Processes. 2009;35:197–211. doi: 10.1037/a0013763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Assema DME, Lubberink M, Bauer M, van der Flier WM, Schuit RC, Windhorst AD, Comans EFI, Hoetjes NJ, Tolboom N, Langer O, Muller M, Scheltens P, Lammertsma AA, van Berckel BNM. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain. 2012;135:181–189. doi: 10.1093/brain/awr298. [DOI] [PubMed] [Google Scholar]
- Velayudhan L, Proitsi P, Westman E, Muehlboeck JS, Mecocci P, Vellas B, Tsolaki M, Kloszewska I, Soininen H, Spenger C, Hodges A, Powell J, Lovestone S, Simmons A. Entorhinal cortex thickness predicts cognitive decline in Alzheimer’s disease. J Alzheimers Dis. 2013;33:755–766. doi: 10.3233/JAD-2012-121408. [DOI] [PubMed] [Google Scholar]
- Wagner AR, Logan FA, Haberlandt K, Price T. Stimulus selection in animal discrimination learning. J Exp Psychol. 1968;76:171–180. doi: 10.1037/h0025414. [DOI] [PubMed] [Google Scholar]
- Walsh TJ, Emerich DF. THE HIPPOCAMPUS AS A COMMON TARGET OF NEUROTOXIC AGENTS. Toxicology. 1988;49:137–140. doi: 10.1016/0300-483x(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Yang J, Volkow ND, Telang F, Ma Y, Zhu W, Wong CT, Tomasi D, Thanos PK, Fowler JS. Gastric stimulation in obese subjects activates the hippocampus and other regions involved in brain reward circuitry. Proc Natl Acad Sci U S A. 2006;103:15641–15645. doi: 10.1073/pnas.0601977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welzl H, D’Adamo P, Lipp HP. Conditioned taste aversion as a learning and memory paradigm. Behavioural Brain Research. 2001;125:205–213. doi: 10.1016/s0166-4328(01)00302-3. [DOI] [PubMed] [Google Scholar]
- Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008;71:1057–1064. doi: 10.1212/01.wnl.0000306313.89165.ef. [DOI] [PubMed] [Google Scholar]
- Williams DL, Baskin DG, Schwartz MW. Evidence that Intestinal Glucagon-Like Peptide-1 Plays a Physiological Role in Satiety. Endocrinology. 2009;150:1680–1687. doi: 10.1210/en.2008-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SC. Gastrointestinal satiety signals I. An overview of gastrointestinal signals that influence food intake. Am J Physiol Gastrointest Liver Physiol. 2004;286:G7–13. doi: 10.1152/ajpgi.00448.2003. [DOI] [PubMed] [Google Scholar]
- Wu T, Zhao BR, Bound MJ, Checklin HL, Bellon M, Little TJ, Young RL, Jones KL, Horowitz M, Rayner CK. Effects of different sweet preloads on incretin hormone secretion, gastric emptying, and postprandial glycemia in healthy humans. Am J Clin Nutr. 2012;95:78–83. doi: 10.3945/ajcn.111.021543. [DOI] [PubMed] [Google Scholar]