

Abstract
We describe a label-free ribobase identification method, which uses ionic current measurement to resolve ribonucleoside monophosphates or diphosphates in α-hemolysin protein nanopores containing amino-cyclodextrin adapters. The accuracy of base identification is further investigated through the use of a guanidino-modified adapter. Based on these findings, an exosequencing approach is envisioned in which a processive exoribonuclease (polynucleotide phosphorylase) presents sequentially cleaved ribonucleoside diphosphates to a nanopore.
Keywords: α-hemolysin, nanopore, RNA sequencing, cyclodextrin, base identification, protein engineering
Single-molecule nanopore technology is being investigated as an ultra-rapid, low-cost platform for sequencing DNA and RNA molecules1, 2. Single-stranded DNA (ssDNA) is translocated through protein pores3, such as the heptameric staphylococcal α-hemolysin (αHL) pore4, and it was suggested that DNA sequence might be obtained from base-dependent transitions in the ionic current flowing through a pore5. Indeed, individual bases can be recognized by protein nanopores6, 7. Recently, advances in base recognition8-11 and the use of enzymes that slowly ratchet DNA strands through pores to provide improved signal-to-noise12-15 have culminated in the realization of nanopore sequencing in both the commercial arena16-18 and in academia19, 20. In a second approach, exosequencing, bases are cleaved from a DNA strand by a processive exonuclease and identified as individual nucleotides by the nanopore1, 7. Excellent identification of nucleoside monophosphates has been obtained with engineered αHL pores carrying cyclodextrin adapters, which can be non-covalently bound within the pore7 or covalently attached for continuous base identification21.
However, for both strand sequencing and exosequencing the focus has remained primarily on DNA, with RNA sequencing receiving less attention. The ability to obtain ultra-rapid RNA sequence information with nanopores would be of considerable significance. For example, it would allow the estimation of mRNA levels in cells and reveal splice patterns and other post-transcriptional modifications22-24, including potential covalent modifications that may have regulatory consequence. Such measurements will be invaluable as a tool for discovery and in medical diagnostics23, 25-27. Nanopore RNA sequencing might also be used to identify and estimate the abundance of small regulatory RNAs, such as bacterial sRNA and eukaryotic miRNA28-30.
Short ssRNA homopolymer molecules have been identified based on differences in residual current (IRES) recorded while the RNAs are translocating through the αHL pore5, 31. The transition between two homopolymer sequences poly(rA) and poly(rC) within a single translocating RNA molecule have also been observed based upon small differences in the polymer's helical structures31. We have demonstrated that individual bases in immobilized ssRNA can be distinguished based on ionic current flow32. By using the αHL NNY pore, which has superior nucleobase discrimination properties, we could distinguish between the standard bases (rG, rA, rC, and rU) and the modified bases rI, m6A and m5C. We have also shown that long ssRNAs of up to 6 kb can be translocated through an αHL pore in an applied potential33.
In the present paper, we lay the groundwork for RNA exosequencing by showing that ribonucleoside diphosphates (rNDPs) can be detected and distinguished by using engineered αHL pores containing cyclodextrin adapters within the transmembrane β barrel (Figure 1). We propose to use polynucleotide phosphorylase (PNPase), which processively cleaves ssRNA in the 3′-to-5′ direction using inorganic phosphate (Pi) to attack the phosphoester linkage and liberate rNDPs. Sequencing approaches, such as this, which distinguish natural nucleobases are advantageous1, 2 and the additional charge on rNDPs (over NMPs)7, 21 is likely to result in more efficient capture by the nanopore.
Figure 1.
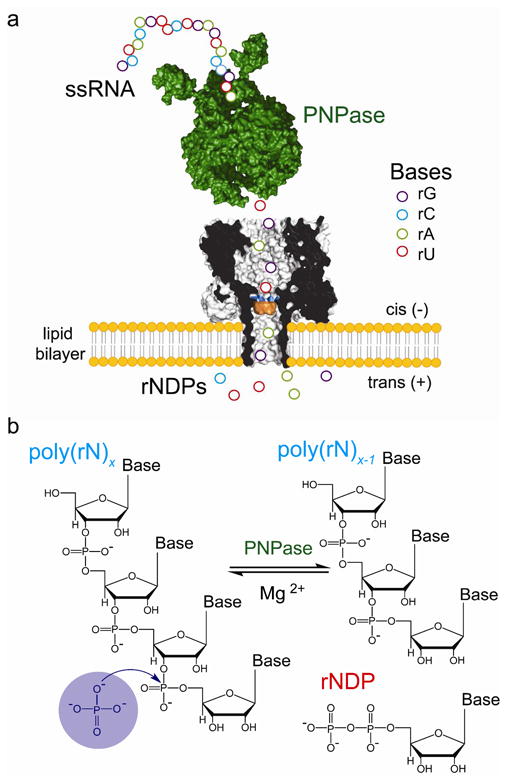
Detection of nucleotides cleaved from ssRNA by polynucleotide phosphorylase. (a) Schematic representation of a ssRNA oligonucleotide (circles) digested by polynucleotide phosphorylase (PNPase, green), one base at a time. The liberated nucleotides (rNDPs) are detected by the heptameric αHL pore (7AHL) equipped with a cyclodextrin adapter (am7βCD, orange). The mutant M113R (mutation highlighted in blue) was used in most of the experiments reported in this paper. In a functioning nanopore sequencer, the PNPase would be covalently attached to the αHL pore. (b) PNPase processively cleaves ssRNA substrates in the 3′-to-5′ direction by using inorganic phosphate (Pi) to attack the phosphodiester linkage nearest the 3′ terminus to release rNDPs.
Detection of rNDPs with non-covalently-attached cyclodextrin adapters
We used the M113R-RL2 mutant of αHL34 (Figures S1). M113R αHL pores have previously been shown to bind cyclodextrin (CD) adapters35, which in turn bind NMPs allowing their identification by current recording7, 21. Following the earlier work, we used β-cyclodextrin with the seven primary hydroxyls replaced with amino groups (heptakis-(6-deoxy-6-amino)-β-cyclodextrin; hereafter referred to as am7βCD) (Figure S2). Because am7βCD is added to the trans compartment, the positively charged amino groups promote an extended residence time for the CD at positive applied potentials, which is long compared to the residence times of the nucleotides7, 21. We have proposed that the Arg residues at position 113 interact with nucleobase rings, while the amino groups on the cyclodextrin ring interact with phosphate groups7. By using this approach in the present study, all four standard ribonucleoside monophosphates (rGMP, rAMP, rUMP, rCMP) could be distinguished (Table S1). Here, we focus on the ribonucleoside diphosphates rGDP, rADP, rUDP and rCDP.
In the absence of am7βCD, the M113R-RL2 pore (Figure S3) remained open and passed a current IOM113R-RL2 = 123.0 ± 4.0 pA (n = 47, independent experiments) at +120 mV in 10 mM Tris-HCl, 1.2 M KCl, at pH 6.0. The addition of 80 μM am7βCD to the trans compartment produced reversible blocking events (Figure S4, Table S2) with a residual current level IRES-am7βCD = 59.0 ± 2.0 pA (n = 47) (Figure 2a, top panel). Upon the sequential addition of rNDPs (10 μM rADP; 10 μM rUDP; 10 μM rCDP; 10 μM rGDP) to the cis compartment, additional current blockades were observed originating from the CD-blocked level (Figure 2a, bottom panel). These represent the binding of rNDPs to am7βCD lodged within the M113R-RL2 pore.
Figure 2.
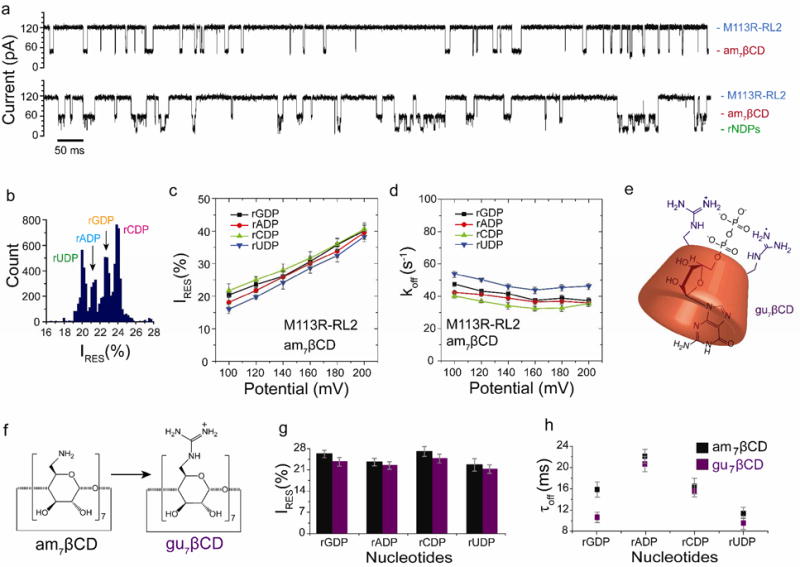
Nucleoside diphosphate discrimination with non-covalently attached cyclodextrin adapters. (a) Single-channel recordings from the homoheptameric αHL M113R-RL2 pore showing am7βCD binding (top) and rNDP detection with the am7βCD adapter (bottom). (b) Corresponding residual current (IRES%) histogram. Data were acquired in 1.2 M KCl, 25 mM Tris-HCl, pH 6.0, at +120 mV in the presence of 80 μM am7βCD (trans), 10 μM rGDP, 10 μM rADP, 10 μM rCDP and 10 μM rUDP (all cis). The results displayed are from a typical experiment. (c) Variation of residual currents (IRES%) with applied potential for each rNDP detected with the M1 13R-RL2•am7βCD pore. (d) Variation of koff with applied potential for each rNDP detected with the M1 13R-RL2•7βCD pore. Values of koff were determined by using koff = 1/τoff, where τoff is the mean dwell time for each rNDP in the pore. (e) Schematic representation of the complex of rGDP with gu7βCD, created using ChemBioDraw software (version 12.02). (f) Conversion of am7βCD to gu7βCD39. For clarity, only two of the seven guanidino groups are shown. (g) Comparison of residual currents (IRES%) for each rNDP detected with the am7βCD and gu7βCD cyclodextrins within the αHL M113R-RL2 pore (n = 22). (h) mean dwell times (τoff) for each rNDP detected with the am7βCD and gu7βCD cyclodextrins within the αHL M113R-RL2 pore (n = 22).
The blockades were plotted as IRES% histograms (Figure 2b, IRES% = (IRES/IO) × 100)). rUDP blocked the pore to the highest extent (IRES%rUDP = 20.7 ± 0.8%), followed by rADP (IRES%rADP = 21.9 ± 0.3%), rGDP (IRES%rGDP = 23.1 ± 0.7%) and rCDP (IRES%rCDP = 24.1 ± 0.7%). The difference in residual current between the two most widely dispersed current peaks in the histogram was ΔIRES%OVERALL = 3.4 ± 0.3% (n = 47) (Table S3). The products of the sequential differences (δ) between each of the four residual current levels in the histograms were also used to measure the ability of different pores to discriminate between the four nucleotide. An αHL pore that is unable to discriminate between all rNDPs has δ = 0 (i.e. the current levels of two or more rNDPs overlap). For the experiment shown in Figure 2b, δM113R-RL2= 1.4 ±0.6 at +120 mV.
The rNDPs also showed variations in mean dwell time (τoff) within the CD adapter (Table S3). Longer binding events are desirable for accurate base calling as they allow a better estimate of the residual pore current. However, higher τoff values would decrease the overall rate of sequencing. For the accurate sequential reading of nucleotides, the rNDP must exit the nanopore on the trans side of the bilayer to remove the possibility of the nucleotide being re-read. The variation in the dissociation rate constant koff (1/τoff) with the applied potential can be used to determine whether a bound molecule exits a nanopore on the cis or trans side of the bilayer21, 36, 37. Therefore, the voltage dependences of the koff values for rNDPs bound to the am7βCD adapter were determined (Figure 2d, Figure S5 and Table S4). At low potentials, under which the nucleotide returns to the cis compartment, we observed high koff values for the four rNDPs (+100 mV; koff = 40 to 54 s-1) (Figure 2d). Higher potentials promoted the binding of the charged nucleotides to the cyclodextrin adapter, resulting in a decrease in koff (at +180 mV; koff= 32 to 45 s-1). At high potential, koff increases for nucleotides rCDP and rUDP suggesting their translocation through the pore. However, higher potential may be required for complete translocation of all four rNDPs.
The discrimination of rGDP rADP, rUDP and rCDP was compared over a range of physical conditions. The KCl concentration was varied from 500 mM to 1.2 M and the pH value over the range pH = 6.0 to 8.0. The applied potential was also adjusted to fine tune the separation of the peaks in the IRES% histogram. First, we found that high concentrations of KCl (1 M and 1.2 M) gave the best discrimination (1 M, ΔIRES%OVERALL = 3.1 ± 0.5% (n = 40); 1.2 M, ΔIRES%OVERALL = 3.4 ± 0.3% (n = 47) compared with 500 mM KCl (ΔIRES%OVERALL = 1.7 ± 0.4% (n = 9)). Second, the residual current levels were pH dependent. Tris-HCl buffers containing 1.2 M KCl at pH values of 6.0, 7.0, 7.5 and 8.0 were examined. Between pH 7.0 and 8.0, only three current levels were observed when the rNDPs bound to am7βCD lodged within the M113R-RL2 pore (Figure S6). At pH 6.0, all four rNDPs could be distinguished (Figure 2, Figure S6). Third, nucleotide binding was voltage dependent. At +100 mV, very few binding events were observed, suggesting that a minimum potential is required to drive the rNDPs into the CD binding site (Figure S5 and Table S4), whereas higher potentials (> +160 mV) resulted in a two-fold increase in the frequency of binding events. However, at +200 mV, the IRES% levels were too close to each other to allow the rNDPs to be distinguished (Figure 2c). ΔIRES%OVERALL = 2.3 ± 0.8%; (n = 41), hence, the optimal base discrimination was recorded within a window of +120 to +140 mV.
Two additional M113X-RL2 pores were also investigated35, 38: M113N-RL2 and M113F-RL2 (Table S2, Figure S3). Under the conditions described above (buffer: 10 mM Tris-HCl, 1.2 M KCl, at pH 6.0) M113N-RL2 bound am7βCD, but individual rNDPs could not be distinguished, because of an increase in the overlap between the blockade levels: ΔIRES%OVERALL = 1.1 ± 0.6%; (n = 6); δM113N-RL2 = 0.2 ± 0.1. M113F-RL2 gave results similar to M113R-RL2, but the peak separations between the rNDPs were smaller: ΔIRES%OVERALL = 2.4 ± 0.6%; (n = 9); δM113F-RL2 = 0.9 ± 0.4. In fact, the original mutant, αHL M113R-RL2 displayed the best discrimination with δM113R-RL2 = 1.4 ± 0.6 at +120 mV.
An alternative non-covalent adapter
In addition to the am7βCD adapter, heptakis(6-deoxy-6-guanidino)-βCD (gu7βCD)39 was tested for rNDP discrimination. The molecule comprises βCD with seven guanidinium groups in place of the primary hydroxyls (Figure 2e-f), with the guanidinium groups having a higher pKa values than the primary amines in am7βCD. We built a speculative model of the complex that is formed between rGDP and gu7βCD using ChemBioDraw software (version 12.02), compatible with Y. Astier's proposed model7. Here the rGDP molecule binds to the gu7βCD by locating its ionic phosphates close to the guanidinium residues while allowing the guanine moiety to penetrate the CD cavity (Figure 2f). At +120 mV, we observed comparable dispersion of the peaks in the IRES% histogram when gu7βCD (80 μM, trans) was used: Δ IRES%OVERALL = 4.3 ± 0.4% (n = 31), compared to ΔIRES%OVERALL = 3.4 ± 0.3% (n = 47) for am7βCD under the same optimal experimental conditions (1.2 M KCl, 10 mM Tris-HCl, at pH 6.0) Figure 2g, Table S5). The mean dwell times for rNDP (τoff) with gu7βCD (11 to 21 ms) were similar to those with am7βCD (9 to 20 ms) (Figure 2h, Table S5), with rGDP giving the largest difference: τoffrGDP (gu7βCD = 16.0 ± 1.0 ms (n = 31); τoffrGDP (gu7βCD = 16.0 ± 1.0 ms (n = 31); τoffrGDP (am7βCD = 11.0 ± 1.0 ms (n = 47) (Table 1).
Table 1.
Comparsion of kinetic parameters for rNDP identification. Comparison of the kinetic parameters of optimal nucleoside diphosphate binding to am7βCD and gu7βCD within the M113R-RL2 αHL mutant pore at pH 6.0 (+120 mV) and to the covalent α HL-(N139Q)6(N139Q/L135C)1.am6-amPDP1βCD pore at pH 7.5 (+160 mV). Values of koff were determined by using koff = 1/τoff, where τoff is the mean dwell time of each rNDP in the pore. IO and IRES% values are mean values (±S.D.) taken from Gaussian fits to event histograms. IRES% = (IRES/IO) × 100. ΔIRES%OVERALL is the difference in residual current between the two most widely separated current peaks. δ is the product of the successive differences in IRES% between the four peaks. If any two peaks overlap, then δ is zero.
| Nucleotide | M113R-RL2 (+120 mV) | αHL-(N139Q)6(N139Q/L1 35C)1.am6-amPDP1-βCD (+160 mV) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| am7βCD | gu7βCD | ||||||||
| τoff (ms) | koff (s-1) | IRES (%) | τoff (ms) | koff (s-1) | IRES (%) | τoff (ms) | koff (s-1) | IRES (%) | |
| rGDP | 11.0 ±1.0 | 91.0 ±4.0 | 23.1 ±1.4 | 16.0 ±1.2 | 43.0 ±6.0 | 25.6 ±1.2 | 26.0 ±2.0 | 39.0 ±1.0 | 28.0 ±0.2 |
| rADP | 20.0 ±1.6 | 50.0 ±6.0 | 21.9 ±0.8 | 21.0 ±1.1 | 46.0 ±6.0 | 23.0 ±1.2 | 22.0 ±2.0 | 46.0 ±1.0 | 29.4 ±0.3 |
| rCDP | 16.0 ±2.0 | 63.0 ±2.0 | 24.1 ±1.2 | 17.0 ±1.2 | 42.0 ±8.0 | 26.4 ±1.6 | 23.0 ±2.0 | 44.0 ±1.0 | 31.7 ±0.3 |
| rUDP | 9.0 ±1.0 | 111.0 ±6.0 | 20.7 ±1.0 | 11.0 ±1.0 | 49.0 ±6.0 | 22.1 ±2.0 | 18.0 ±3.0 | 56.0 ±1.0 | 32.7 ±0.3 |
| ΔIRESOVERALL(%) | 3.4 ±0.3 | 4.3 ±0.4 | 4.7 ±0.3 | ||||||
| δ | 1.4 ±0.6 | 1.9 ±0.4 | 3.2 ±1.0 | ||||||
Continuous detection of rNDPs by using a permanent cyclodextrin adapter
For sequencing applications, a permanent adapter would be desirable for continuous base detection. We have shown that CDs can be covalently attached within the αHL pore40 and used to distinguish dNMPs21. Hence, the αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD pore21 was tested for its ability to discriminate between the four rNDPs (Figure 3a, Figure S7). This αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD pore was constructed from a reactive cyclodextrin: heptakis(6-deoxy-6-amino)-6-N-mono(2-pyridyl)dithiopropanoyl-β-cyclodextrin (am6am-PDP1βCD) which contains six primary amino groups required for base detection and a reactive linker for covalent attachment to the cysteine residue (at position L135) within the barrel of the αHL (N139Q)6(N139Q/L135C)1 pore. At a potential of +160 mV, the current passed by αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD was recorded (Figure 3b, top panel), and then rGDP, rADP, rUDP and rCDP were sequentially added to the cis compartment (Figure 3b, bottom panel) and an IRES% histogram was compiled from the recording (Figure 3c). In 1 M KCl, 25 mM Tris-HCl, at pH 7.5, rGDP showed the largest block (IRES%rGDP = 27.7 ± 0.2%), followed by rADP (IRES%rADP= 29.5 ± 0.3%), rCDP (IRES%rCDP = 31.8 ± 0.3%) and rUDP (IRES%rUDP= 32.4 ± 0.3%). At lower potentials (+120 mV), rCDP and rUDP showed good separation, but the peaks for rGDP and rADP overlapped. The optimal potential was between +160 and +180 mV (Figure 3e). The outcome was comparable with the non-covalently attached am7βCD ΔIRES%OVERALL = 4.7 ± 0.3% (n = 22) with δ = 3.1 ± 1.0 (Table 1, Table S6) with the additional advantage that rNDPs are read continuously.
Figure 3.
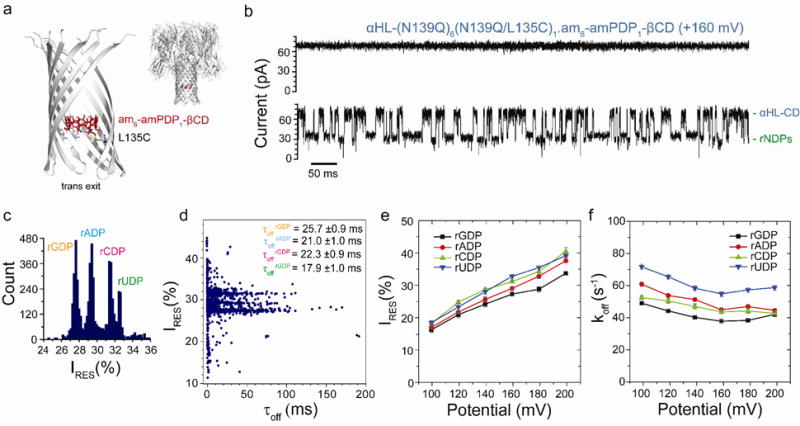
rNDP interaction with the αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD pore. (a) Structure of the αHL-(N139Q);6(N139Q/L135C)1.am6-amPDP1- βCD pore (cartoon view). The enlarged view shows a close-up of the β barrel with two subunits omitted. The cyclodextrin (am6-amPDP1-βCD) was covalently attached through a disulfide bond to position 135 in one of the seven subunits, which had been mutated to Cys (red, sticks model)21(b) Single-channel recording from the αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD pore showing continuous rNDP detection. (c) Corresponding residual current (IRES%) histogram. Data were acquired in 1 M KCl, 25 mM Tris-HCl, pH 7.5, at +160 mV in the presence of 10 μM rGDP, 10 μM rADP, 10 μM rCDP and 10 μM rUDP (all cis). The results displayed are from a typical experiment. (d) Scatter plot showing IRES% and dwell times (τoff) for rNDP binding events to the αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD pore as seen in the current trace in ‘b’. (e) Variation of residual currents (IRES%) with applied potential for each rNDP detected with the αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1βCD pore. (f) Variation of k Values of koff with applied potential for each rNDP detected with the αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD pore. Values of koff were determined by using koff = 1/τoff where τoff is the mean dwell time of the rNDP in the pore.
The rNDPs also showed variations in mean dwell time (τoff) within the αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD pore, with τoff values in the range 18 to 26 ms, rGDP having the longest dwell time: τoffrGDP = 26.0 ± 2.0 %, at +160 mV (Figure 3d, Table 1, Tables S6 and S7). The variations in koff (1/τoff) with the applied potential for the αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD pore were similar to the relationships obtained with non-covalently attached am7βCD (Figure 3d). An increase in koff at higher potentials (> +160 mV) was seen for all four rNDPs, indicating that these nucleotides cross into the trans compartment. The optimal base discrimination was obtained between +160 mV and +180 mV, potentials at which a high proportion of rNDPs translocate through the nanopore: at +180 mV with αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD, τoffrGDP = 25.0 ± 1.0 ms; τoffrADP = 21.0 ± 1.0 ms; τoffrCDP = 22.0 ± 1.0 ms; τoffrUDP = 17.0 ± 1.0 ms (n = 22). Future work on this can include testing the guanidinium-cyclodextrin (gu7βCD), which may also be attached using a similar strategy, to provide continuous read of nucleotides and improved accuracy of the residual current measured.
Detection of rNDP derived from ssRNA
We also used the M113R-RL2 αHL pore with am7βCD to identify rNDPs generated in situ from ssRNA by using the RNA-selective enzyme polynucleotide phosphorylase (PNPase) from Caulobacter crescentus41, 42 (Figure 4a, Figure S8). To ensure that the conditions for nucleotide discrimination were compatible with PNPase activity41, we maintained pH asymmetry: pH (cis) = 7.0 (to promote enzyme activity); pH (trans) = 6.0 (to maintain nucleotide discrimination). Two samples of RNA were used: one contained three homopolymers: oligo(rC)30, oligo(rU)30, oligo(rA)30 and the second, a heteropolymer, oligo(het)30, with all four ribobases rG, rC, rU and rA: 3′-AAAUGGACUGGCUUCGGAAGCCAAAUGGAU-5′ (Table S8). Upon the addition of PNPase and the ssRNA molecules to the cis compartment (in the absence of Pi and Mg2+ and am7βCD) a decrease in the ionic current through the nanopore from the open state level can occur when (i) ssRNA is freely translocating through the pore or (ii) the PNPase•RNA complex is captured (Figure 4a). The enzyme is too large to enter the nanopore and holds the 3′-end of the RNA substrate at the cis entrance. In this state, the ssRNA that has entered the pore causes a sequence-dependent decrease in the ionic current, but is not translocated into the trans compartment.
Figure 4.
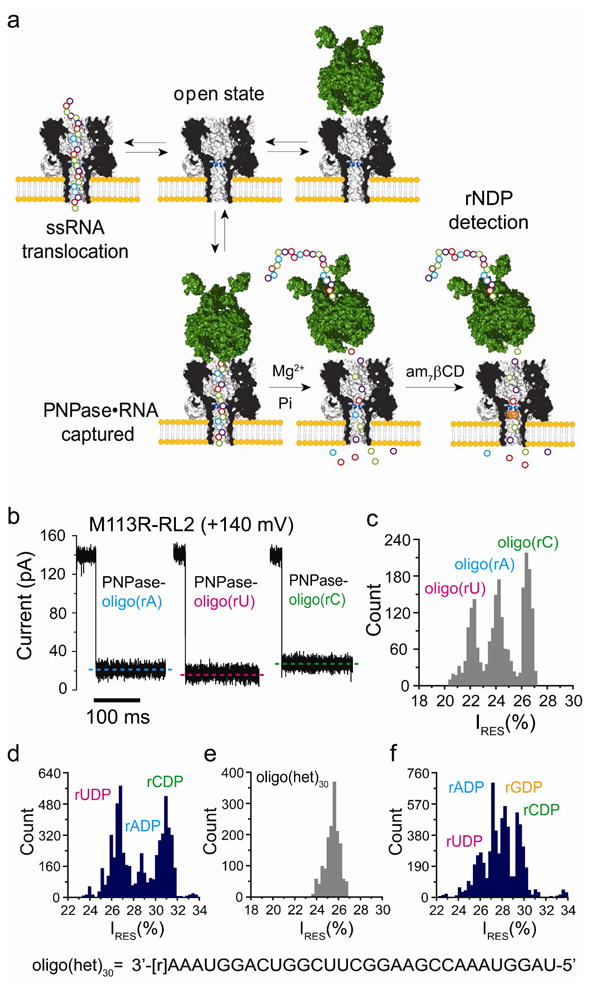
Detection of nucleosides diphosphates cleaved from ssRNA by PNPase. (a) Upon the addition of ssRNA, without the enzyme, current blockades are caused by brief ssRNA translocation events. In the presence of PNPase, additional blockades with longer residence times and/or amplitudes that differ from those of the translocation events are observed when PNPase•RNA complexes are captured. After the addition of Mg2+ and inorganic phosphate (Pi), followed by the addition of 7βCD (trans), cleaved nucleotides are detected through their interactions with the CD adapter. (b) Current traces for the capture of homopolymeric ssRNAs bound to PNPase by the αHL M113R-RL2 pore at +140 mV. (c) Histogram of the residual current levels (IRES%) after capture of homopolymers ssRNAs bound to PNPase by the αHL M113R-RL2 pore at +140 mV. (d) Histogram of the residual current levels (IRES%) for the rNDP binding events with am7βCD (trans). The rNDPs were cleaved by PNPase from the RNA homopolymers, oligo(rA)30, oligo(rC)30 and oligo(rU)30. e) Histogram of the residual current levels (IRES%) after capture of a PNPase-bound hetero-oligomeric ssRNA (oligo(het)30: 3′-[r]AAAUGGACUGGCUUCGGAAGCCAAAUGGAU-5′) inside the αHL M113R-RL2 pore at +120 mV. (f) Histogram of the residual current levels (IRES%) for rNDP binding events in the presence of am7β CD(trans). The rNDPs were cleaved by PNPase from the hetero-oligomeric ssRNA, oligo(het)30(Figure S11).
We recorded current blocks for the capture of PNPase-bound oligo(rA)30, oligo(rC)30 and oligo(rU)30 at +140 mV (Figure 4b, Figures S9, S10 and Table S9), and compiled the corresponding IRES% histograms (Figure 4c). PNPase-bound oligo(rU)30 blocked the pore to a greater extent (IRES%oligo(rU) = 22.2 ± 0.2%) than oligo(rA)30 (IRES%oligo(rA) = 24.3 ± 0.2%) and oligo(rC)30 (IRES%oligo(rC) = 26.6 ± 0.2%). After the addition of 5 mM Mg2+ and 10 mM Pi (10 mM NaH2PO4, pH 7.0) to the cis compartment (Figure 4a), the PNPase was activated and sequentially cleaved rNDPs from the 3′-end of the ssRNA. The liberated rNDPs were observed as binding events when am7βCD was added to the trans compartment. As expected, we observed three peaks corresponding to the three released rNDPs (Figure 4d): IRES%rUDP = 26.9 ± 0.7% < IRES%rADP=28.8 ± 0.9% < IRES%rCDP= 31.3 ± 0.6% with ΔIRES%OVERALL = 4.4 ± 0.8% and δM113R-RL2= 4.7 ± 0.8 (n = 8), (Table S9).
To test the ability of the nanopore to identify all four nucleosides diphosphates, oligo(het)30 was used. Oligo(het)30 bound to the PNPase was captured by the M113R-RL2 pore at +140 mV, giving a single peak in the residual current histogram (IRES%oligo(het)30 = 25.2 % ± 0.4%, Figure 4e, Table S10). Upon the addition of Mg2+ and Pi to cis, and subsequently am7βCD, to the trans compartment, the rNDPs were readily distinguished by their residual currents: IRES%rUDP = 25.8 ± 0.6% < IRES%rADP = 27.1 ± 0.6% < IRES%rGDP = 28.2 ± 0.9% < IRES%rCDP = 29.9 ± 0.6%; ΔIRES%OVERALL = 4.1 ± 0.8 %; δM113R-RL2= 2.4 ± 0.7 (n = 5) (Figure 4f and Figure S11, Table S10). The mean dwell times were τoffrGDP=25.0±2.0 ms, τoffrADP = 19.0 ± 1.0 ms,τoffrCDP = 21.0 ± 1.0 ms, τoffrUDP = 17.0 ± 1.0 ms (n = 5) (Table S10). These experiments support our findings with the free nucleotides and the am7βCD cyclodextrin adapter (Figure 2). The nucleotides released by PNPase give similar IRES % and τoff values (at a potential of +120 mV). Therefore, it should be possible to integrate PNPase with the αHL pore to distinguish individual nucleotides released from a ssRNA under conditions compatible with enzyme activity.
Monitoring PNPase digestion of ssRNA
The 3′-phosphorolytic activity of PNPase is dependent upon inorganic phosphate (Pi), which attacks the phosphodiester bond of ssRNA. Nanopore experiments reported this catalytic activity as an abrupt decrease in translocation-event frequency after the addition of 10 mM Pi to the cis compartment in the presence of PNPase•oligo-[P](het30) (Figure 5a,b, Table S11). In contrast, the translocation-event frequency for an oligonucleotide with a phosphorothioate backbone, oligo-[PS](het)30, which cannot be cleaved by PNPase43-45; remains unchanged after Pi addition (Figure 5b). The time course of ssRNA digestion by PNPase was determined by nanopore analysis (Figure 5c, Table S12) yielding Km = 0.4 ± 0.1 mM (derived from a Lineweaver-Burk plot) consistent with previously reported biochemical studies conducted in bulk solution41, 42, 46-50. These measurements reveal important details of PNPase kinetics which may be useful for the alignment of the ssRNA-PNPase catalytic process and the subsequent detection using the cyclodextrin adapter.
Figure 5.
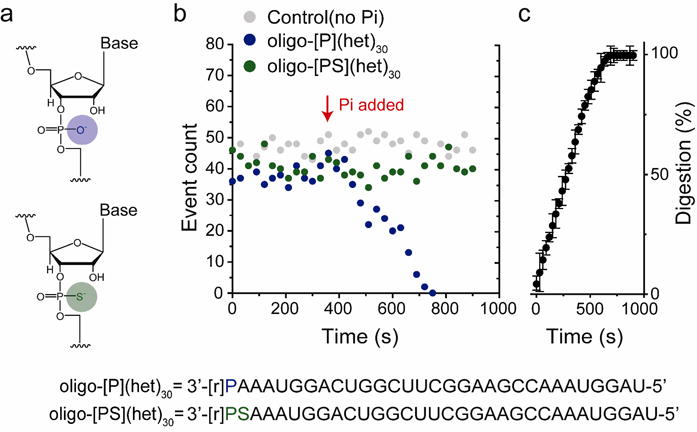
Monitoring PNPase digestion of ssRNA with a nanopore. (a) Chemical structures of the phosphodiester linkage in oligo-[P](het)30 (top, blue circle) and the phosphorothioate linkage in oligo-[PS](het)30 (bottom, green circle), which is not hydrolyzed by PNPase. (Below) Sequences of the hetero-oligomeric oligonucleotides. (b) The PNPase bound oligo-[P](het)30 ssRNA capture rate in the M113R-RL2 nanopore as a function of time. After the addition of 10 mM Pi (red arrow), the event rate sharply declines for the PNPase-oligo-[P](het)30. In contrast, the event rate is maintained for PNPase-oligo-[PS](het)30. (c) Time course for oligo-[P](het)30 ssRNA digestion by PNPase (n = 3). The plot was constructed by collecting capture events in a 30 s segment (from t= 0 s to 900 s), in the presence of Pi and Mg2+.
Conclusions
We have shown that engineered αHL nanopores with either non-covalently bound or covalently attached CD adapters accurately discriminate between the four standard rNMPs and rNDPs under optimized experimental conditions. Pores with covalently attached CD adapters are capable of continuous nucleotide detection. In addition, our platform operates under conditions compatible with PNPase activity. While many enzymes that handle nucleic acids prefer low salt conditions, PNPase is active at high KCl concentrations, allowing data acquisition under conditions suited for optimal nucleotide discrimination. The results presented here offer the groundwork to develop a platform suitable for RNA nanopore exosequencing. For high-throughput parallel analysis, thousands of pores must be integrated into arrays and feasible strategies toward this end are currently under investigation16, 51.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health, Wellcome Trust and Oxford Nanopore Technologies. BFL and SWH are supported by the Wellcome Trust. The authors thank Oxford Nanopore Technologies for the αHL-(N139Q)6(N139Q/L135C)1.am6-amPDP1-βCD protein and gu7βCD, Ellina Mikhailova for the M113X-RL2 mutants and James Clarke and Lakmal Jayasinghe for valuable discussions.
Footnotes
Supporting Information: Details of experimental procedures, nucleoside monophosphate detection, oligonucleotide sequences, and the data displayed in Figures 1-5. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions: M.A., B.L. and H.B. conceived the ideas and designed the experiments. M.A. and S.H. performed the experiments. M.A. analyzed the data. M.A. and H.B. wrote the manuscript with contributions from all the authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes: Hagan Bayley is the Founder, a Director and a share-holder of Oxford Nanopore Technologies, a company engaged in the development of nanopore sequencing technology. Work in the Bayley laboratory at the University of Oxford, including this work, is supported in part by Oxford Nanopore Technologies.
References
- 1.Bayley H. Current Opinion in Chemical Biology. 2006;10(6):628–637. doi: 10.1016/j.cbpa.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 2.Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang X, Jovanovich SB, Krstic PS, Lindsay S, Ling XS, Mastrangelo CH, Meller A, Oliver JS, Pershin YV, Ramsey JM, Riehn R, Soni GV, Tabard-Cossa V, Wanunu M, Wiggin M, Schloss JA. Nat Biotech. 2008;26(10):1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muthukumar M. Annual Review of Biophysics and Biomolecular Structure. 2007;36(1):435–450. doi: 10.1146/annurev.biophys.36.040306.132622. [DOI] [PubMed] [Google Scholar]
- 4.Song LZ, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Science. 1996;274(5294):1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 5.Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Proc Natl Acad Sci U S A. 1996;93(24):13770–3. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashkenasy N, Sánchez-Quesada J, Bayley H, Ghadiri MR. Angewandte Chemie International Edition. 2005;44(9):1401–1404. doi: 10.1002/anie.200462114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Astier Y, Braha O, Bayley H. Journal of the American Chemical Society. 2006;128(5):1705–1710. doi: 10.1021/ja057123+. [DOI] [PubMed] [Google Scholar]
- 8.Stoddart D, Heron AJ, Mikhailova E, Maglia G, Bayley H. Proc Natl Acad Sci U S A. 2009;106(19):7702–7. doi: 10.1073/pnas.0901054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purnell RF, Schmidt JJ. ACS Nano. 2009;3(9):2533–8. doi: 10.1021/nn900441x. [DOI] [PubMed] [Google Scholar]
- 10.Derrington IM, Butler TZ, Collins MD, Manrao E, Pavlenok M, Niederweis M, Gundlach JH. Proceedings of the National Academy of Sciences. 2010 doi: 10.1073/pnas.1001831107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manrao EA, Derrington IM, Pavlenok M, Niederweis M, Gundlach JH. PLoS One. 2011;6(10):e25723. doi: 10.1371/journal.pone.0025723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cockroft SL, Chu J, Amorin M, Ghadiri MR. Journal of the American Chemical Society. 2008;130(3):818–820. doi: 10.1021/ja077082c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olasagasti F, Lieberman KR, Benner S, Cherf GM, Dahl JM, Deamer DW, Akeson M. Nat Nanotechnol. 2010;5(11):798–806. doi: 10.1038/nnano.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu J, Gonzalez-Lopez M, Cockroft SL, Amorin M, Ghadiri MR. Angew Chem Int Ed Engl. 2010;49(52):10106–9. doi: 10.1002/anie.201005460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lieberman KR, Cherf GM, Doody MJ, Olasagasti F, Kolodji Y, Akeson M. J Am Chem Soc. 2010;132(50):17961–72. doi: 10.1021/ja1087612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pennisi E. Science. 2012;336(6081):534–537. doi: 10.1126/science.336.6081.534. [DOI] [PubMed] [Google Scholar]
- 17.Schneider GF, Dekker C. Nat Biotech. 2012;30(4):326–328. doi: 10.1038/nbt.2181. [DOI] [PubMed] [Google Scholar]
- 18.Bayley H. Physics of Life Reviews. 2012;9(2):161–163. doi: 10.1016/j.plrev.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 19.Cherf GM, Lieberman KR, Rashid H, Lam CE, Karplus K, Akeson M. Nat Biotechnol. 2012;30(4):344–8. doi: 10.1038/nbt.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manrao EA, Derrington IM, Laszlo AH, Langford KW, Hopper MK, Gillgren N, Pavlenok M, Niederweis M, Gundlach JH. Nat Biotechnol. 2012;30(4):349–53. doi: 10.1038/nbt.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. Nat Nano. 2009;4(4):265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Gerstein M, Snyder M. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marguerat S, Wilhelm BT, Bahler J. Biochem Soc T. 2008;36:1091–1096. doi: 10.1042/BST0361091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Nat Meth. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 25.Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Nature. 2009;458(7234):97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maher B. Nature. 2009;459(7244):146–7. doi: 10.1038/459146b. [DOI] [PubMed] [Google Scholar]
- 27.Wanunu M, Bhattacharya S, Xie Y, Tor Y, Aksimentiev A, Drndic M. ACS Nano. 2011;5(12):9345–9353. doi: 10.1021/nn203764j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carthew RW, Sontheimer EJ. Cell. 2009;136(4):642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wanunu M, Dadosh T, Ray V, Jin J, McReynolds L, Drndic M. Nat Nano. 2010;5(11):807–814. doi: 10.1038/nnano.2010.202. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Zheng D, Tan Q, Wang MX, Gu LQ. Nat Nano. 2011;6(10):668–674. doi: 10.1038/nnano.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akeson M, Branton D, Kasianowicz JJ, Brandin E, Deamer DW. Biophys J. 1999;77(6):3227–33. doi: 10.1016/S0006-3495(99)77153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayub M, Bayley H. Nano Lett. 2012;12(11):5637–5643. doi: 10.1021/nl3027873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cracknell JA, Japrung D, Bayley H. Nano Lett. 2013;13(6):2500–2505. doi: 10.1021/nl400560r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheley S, Gu LQ, Bayley H. Chemistry & biology. 2002;9(7):829–838. doi: 10.1016/s1074-5521(02)00172-2. [DOI] [PubMed] [Google Scholar]
- 35.Banerjee A, Mikhailova E, Cheley S, Gu LQ, Montoya M, Nagaoka Y, Gouaux E, Bayley H. Proceedings of the National Academy of Sciences. 2010;107(18):8165–8170. doi: 10.1073/pnas.0914229107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez-Quesada J, Ghadiri MR, Bayley H, Braha O. Journal of the American Chemical Society. 2000;122(48):11757–11766. [Google Scholar]
- 37.Gu LQ, Cheley S, Bayley H. Science. 2001;291(5504):636–640. doi: 10.1126/science.291.5504.636. [DOI] [PubMed] [Google Scholar]
- 38.Gu LQ, Cheley S, Bayley H. The Journal of General Physiology. 2001;118(5):481–494. doi: 10.1085/jgp.118.5.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan DQ, Izuka A, Fukudome M, Rekharsky MV, Inoue Y, Fujita K. Tetrahedron Letters. 2007;48(19):3479–3483. [Google Scholar]
- 40.Wu HC, Astier Y, Maglia G, Mikhailova E, Bayley H. Journal of the American Chemical Society. 2007;129(51):16142–16148. doi: 10.1021/ja0761840. [DOI] [PubMed] [Google Scholar]
- 41.Hardwick SW, Gubbey T, Hug I, Jenal U, Luisi BF. Open Biology. 2012;2(4) doi: 10.1098/rsob.120028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hardwick SW, Chan VSY, Broadhurst RW, Luisi BF. Nucleic Acids Research. 2011;39(4):1449–1459. doi: 10.1093/nar/gkq928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eckstein F. Annu Rev Biochem. 1985;54:367–402. doi: 10.1146/annurev.bi.54.070185.002055. [DOI] [PubMed] [Google Scholar]
- 44.Eckstein F, Gindl H. European Journal of Biochemistry. 1970;13(3):558–564. doi: 10.1111/j.1432-1033.1970.tb00961.x. [DOI] [PubMed] [Google Scholar]
- 45.Hornblower B, Coombs A, Whitaker RD, Kolomeisky A, Picone SJ, Meller A, Akeson M. Nat Meth. 2007;4(4):315–317. doi: 10.1038/nmeth1021. [DOI] [PubMed] [Google Scholar]
- 46.Nurmohamed S, Vincent HA, Titman CM, Chandran V, Pears MR, Du D, Griffin JL, Callaghan AJ, Luisi BF. J Biol Chem. 2011;286(16):14315–14323. doi: 10.1074/jbc.M110.200741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Godefroy T. European Journal of Biochemistry. 1970;14(2):222–231. doi: 10.1111/j.1432-1033.1970.tb00281.x. [DOI] [PubMed] [Google Scholar]
- 48.Chou JY, Singer MF. J Biol Chem. 1970;245(5):995-&. [PubMed] [Google Scholar]
- 49.Chou JY, Singer MF, Mcphie P. J Biol Chem. 1975;250(2):508–514. [PubMed] [Google Scholar]
- 50.Chang SA, Cozad M, Mackie GA, Jones GH. Journal of Bacteriology. 2008;190(1):98–106. doi: 10.1128/JB.00327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hall AR, Scott A, Rotem D, Mehta KK, Bayley H, Dekker C. Nat Nanotechnol. 2010;5(12):874–7. doi: 10.1038/nnano.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.