

Abstract
BACKGROUND
Genome-wide association studies (GWAS) have discovered loci that predispose to asthma. To integrate these new discoveries with emerging models of asthma pathobiology, research is needed to test how genetic discoveries relate to developmental and biological characteristics of asthma.
METHODS
We derived a multi-locus profile of genetic risk from published GWAS of asthma case status. We then tested associations between this “genetic risk score” and developmental and biological characteristics of asthma in a population-based long-running birth cohort, the Dunedin Longitudinal Study (n=1,037). We evaluated asthma onset, persistence, atopy, airway hyperresponsiveness, incompletely reversible airflow obstruction, and asthma-related school and work absenteeism and hospitalization during 9 prospective assessments spanning ages 9–38 years, when 95% of surviving cohort members were seen.
INTERPRETATION
Cohort members at higher genetic risk experienced asthma onset earlier in life (HR=1.12 [1.01–1.26]). Childhood-onset asthma cases at higher genetic risk were more likely to become life-course-persistent asthma cases (RR=1.36 [1.14–1.63]). Asthma cases at higher genetic risk more often manifested atopy (RR=1.07 [1.01–1.14]), airway hyperresponsiveness (RR=1.16 [1.03–1.32]), and incompletely reversible airflow obstruction (RR=1.28 [1.04–1.57]). They were also more likely to miss school or work due to asthma (IRR=1.38 [1.02–1.86]) and to be hospitalized with breathing problems (HR=1.38 [1.07–1.79]). Genotypic information about asthma risk was independent of and additive to information derived from cohort members’ family histories of asthma.
CONCLUSIONS
Findings from this population study confirm that GWAS-discoveries for asthma associate with a childhood-onset phenotype and advance asthma genetics beyond the original GWAS-discoveries in three ways: (1) We show that genetic risks predict which childhood-onset asthma cases remit and which become life-course-persistent cases, although these predictions are not sufficiently sensitive or specific to support immediate clinical translation; (2) We elucidate a biological profile of the asthma that arises from these genetic risks: asthma characterized by atopy and airway hyperresponsiveness and leading to incompletely reversible airflow obstruction; and (3) We describe the real-life impact of GWAS-discoveries by quantifying genetic associations with missed school and work and hospitalization.
INTRODUCTION
Asthma is a heterogeneous syndrome with multiple causes.1 Research is beginning to uncover variations in the molecular roots and clinical manifestations of asthma.2 One research stream has discovered molecular-genetic markers of asthma risk.3,4 A second research stream has characterized developmental and biological features of asthma that may indicate “endotypes,” subtypes of disease with distinct pathobiology.5,6 We brought these two streams of research together to test how recently-discovered genetic risks relate to developmental and biological characteristics of asthma across the life course.
Our research seeks to address the question: When hypothesis-free methods like genome-wide association studies (GWAS)7 make discoveries about a heterogeneous syndrome like asthma, what are these discoveries about? To achieve a sample numbering in the tens of thousands, asthma GWAS reduced a developmentally and biologically complex syndrome to the dichotomous outcome of whether or not a physician had diagnosed asthma. Follow-up research is now needed to understand GWAS discoveries. One approach to follow-up is to work “from the bottom up,” by tracing the path from variation in DNA sequence to differences in RNA transcription, subsequent protein production, and onwards up through disease pathogenesis to identify a process or molecule that can be targeted for intervention. A complementary approach is to work “from the top down,” by characterizing how genetic discoveries relate to individual differences in developmental and biological phenotypes, and then working backward to devise interventions that can mitigate genetic risk.8 Here we take the top-down approach to identify developmental and biological signatures of high-genetic-risk cases. Once identified, these signatures can help to identify subjects for bottom-up molecular research.
Our study leveraged data from an unselected cohort followed from birth to mid-life9 with extensive asthma phenotyping across 9 assessments spanning ages 9–38 years. We derived a multi-locus profile of genetic predisposition to asthma--a “genetic risk score”--from a recent comprehensive case-control genome-wide association study (GWAS) of asthma.10 We then tested how genetic risks related to the development, persistence, and biological characteristics of asthma in the general population.
METHODS
To test whether genetic risk was associated with the timing-of-onset, course, and persistence of asthma, we analyzed longitudinal data on asthma phenotypes derived from standardized interviews. To test whether genetic risk was associated with asthma’s biological characteristics, we combined interview data with information derived from assessments of allergy and lung function. We examined 3 biological characteristics: (1) atopy, which, especially in pediatric populations, is associated with a persistent and severe course of disease;11,12 (2) airway hyperresponsiveness, which provides indirect confirmation of active airway inflammation;13,14 and (3) incompletely reversible airflow obstruction, which identifies deleterious changes to the airways resulting from chronic asthma.15 To accompany these etiological analyses, we conducted two additional tests that relate to the concerns of patients and clinicians. To test whether genetic risk was associated with disruptions to daily life, we tested genetic associations with the incidence of days of school and work missed due to asthma, and with asthma-related hospitalizations. To put the magnitude of genetic-risk effects in context and to determine whether molecular-genetic measurements provided novel information about risk, we compared molecular-genetic information to family-history information.16
Sample
Participants are members of the Dunedin Multidisciplinary Health and Development Study, a longitudinal investigation of health and behavior in a complete (unselected) birth cohort. Study members (N=1,037; 91% of eligible births; 52% male) were all individuals born between April 1972 and March 1973 in Dunedin, New Zealand, who were eligible for the longitudinal study based on residence in the province at age 3 and who participated in the first follow-up assessment at age 3. The cohort represents the full range of socioeconomic status in the general population of New Zealand’s South Island and is primarily white.17 Assessments were carried out at birth and ages 3, 5, 7, 9, 11, 13, 15, 18, 21, 26, 32, and, most recently, 38 years, when 95% of the 1,007 surviving Study members took part. At each assessment wave, study members are brought to the Dunedin research unit for a full day of interviews and examinations. The Otago Ethics Committee approved each phase of the study and informed consent was obtained from all study members.
Measures
Genetic Risk Score
A challenge for developmental research following-up GWAS discoveries is that effect sizes for individual asthma-associated single-nucleotide polymorphisms (SNPs) are small,10 leaving longitudinal studies with data necessary to investigate developmental phenotypes of asthma underpowered to test single-SNP effects.18 However, there is evidence that asthma-associated loci make additive contributions to risk, recommending aggregating risk alleles.19,20 Summing risk alleles across GWAS-identified SNPs to compute a “genetic risk score” (GRS) yields a quantitative index of genetic risk with a normal distribution21 and a potentially larger effect size.
We derived our GRS from the largest asthma GWAS to date, published in 2010.10 We selected all SNPs associated with asthma at p<5×10−8. This set of SNPs was then pruned using an R2 threshold of 0.60 to derive the SNP set for the GRS. This process yielded 17 SNPs located in or near the genes IL18R1, IL13, HLA-DQ, IL33, SMAD3, ORMDL3, GSDMB, GSDMA, IL2RB.
Dunedin cohort genotyping was conducted with a commercially available array (BeadPlex Array; Illumina Inc.) using DNA extracted from whole blood (93%) or buccal swabs (7%). Two SNPs near the gene HLA-DQ could not be assayed due to the complex repetitive structure of the proximal sequence. Thus, the final GRS included 15 SNPs (Supplemental Table 1). GRS SNPs were called successfully in >95% of European-descent cohort members. These n=880 individuals formed the analysis sample. All SNPs met Hardy-Weinberg equilibrium criteria (p>0.10 for all tests). Out of the possible 30 risk alleles, cohort members carried a mean of 13.74 (SD=4.36). Risk allele counts were normally distributed (Supplemental Figure 1). For analyses, we standardized risk allele counts to have a mean=0 and SD=1 (the “genetic risk score”). Effect sizes presented for a 1 SD change in genetic risk reflect the effect of an increase of 4.36 risk alleles. Sensitivity analyses that selectively dropped SNPs from the score one by one indicated that no single SNP accounted for a disproportionate share of the genetic-risk-score association with asthma (Supplemental Table 2). Cohort members’ sex and socioeconomic status were unrelated to their genetic risk (Pearson’s r=0.01 for sex and 0.04 for socioeconomic status).
Family History of Asthma
Family histories for asthma were available for 99% of the cohort, namely reports of asthma provided by study members and both parents for study members’ biological siblings, parents, and grandparents. Family history was summarized as the proportion of family members in the pedigree who had asthma, adjusted for genetic relatedness to the proband of first- and second-degree relatives (i.e. first-degree relatives were counted as 1 and second- degree relatives were counted as 0.5).22
Asthma Phenotypes
Assessments of asthma phenotypes and definitions of biological parameters including atopy, airway hyperresponsiveness and incompletely reversible airflow obstruction have been described in previous publications from this cohort9,23–33 and are summarized in Table 1. We used in-person standardized-interview data to determine the timing-of-onset and persistence of asthma, and to identify life-course-persistent cases (Table 1 Panel A). We used skin-test and lung-function assessments to measure atopy, airway hyperresponsiveness, and incompletely reversible airflow obstruction (Table 1 Panel B). We used additional interview data to determine the number of days of school and work missed due to asthma and asthma-related hospitalizations (Table 1 Panel C).
Table 1.
| Panel A. Developmental Phenotypes of Asthma | |
|---|---|
|
Asthma Onset 35% of Cohort (N=305) |
Current asthma status was determined at ages 9, 11, 13, 15, 18, 21, 26, 32, and 38 years from standardized interviews of study members (or their mothers prior to the age 13 assessment) conducted by pulmonary specialists.9 Current asthma was defined as a diagnosis of asthma in addition to positive symptoms within the past 12 months, including asthma attack, recurrent wheeze (excluding study members reporting only 1 or 2 episodes lasting less than 1 hour), or medical treatment for asthma.23–25 Age-at-asthma-onset was defined as the earliest age at which wheezing symptoms or diagnosis by a physician were recorded. Panel A of Figure 1 shows Kaplan-Meier survival curves illustrating timing of onset of asthma in boys and girls in the Dunedin cohort. |
| Asthma Persistence | Asthma persistence was measured as the count of assessments at which a cohort member manifested current asthma (range=0–9). |
|
Life-Course-Persistent Asthma 11% of Cohort (n=96) |
Life-course-persistent asthma was defined as having current asthma at 2 or more assessments up to puberty (age 13 years) and at 3 or more assessments thereafter (ages 15 through 38 years). |
| Panel B. Biological Characteristics of Asthma | |
|---|---|
|
Atopy 77% of Asthma Cases (n=236) |
Atopy was assessed at one time in childhood (age 13 years) and twice in adulthood (ages 21 and 32 years) through standardized skin tests26 for 12 common allergens: house dust mite (dermatophagoides pteronyssinus) grass, cat, dog, horse, kapok, wool, aspergillus fumigatus, alternaria, penicillium, cladosporum, cockroach (ages 21 and 32 only). A weal diameter 2 mm greater than saline control was considered positive for epidemiological purposes. Atopy was defined as a positive response to one or more allergens.27,28 Asthma cases who tested positive for atopy were classified as having atopic asthma. In addition to allergy testing, total serum immunoglobulin E (IgE), a blood-biomarker of atopy, was measured at one time in childhood (age 11 years) and twice in adulthood (ages 21 and 32 years).29 IgE values were log transformed for analysis. |
|
Airway Hyperre-sponsiveness 52% of Asthma Cases (n=160) |
Airway hyperresponsiveness is an indirect measure of airway inflammation. During the childhood years (ages 9–13), and at ages 15 and 21 years, spirometry and methacholine challenge was conducted using a Godart water spirometer. As described previously,28,30,31,32 a concentration of methacholine of 8 mg/ml or less causing a 20% fall in FEV1(PC20) was used to identify airway hyperresponsiveness. In those in whom a methacholine challenge was not safe or practical to perform, an increase in FEV1 of 10% or more after albuterol was regarded as approximately equivalent to a PC20 of 8 mg/ml or less. This same method was used to define bronchodilator responsiveness at ages 18, 26, 32, and 38 years. At these ages, spirometry was conducted using an Ohio computerized spirometer (Ohio Instruments; age 18) and a Sensormedics body plethysmograph (Sensormedics Corporation, Yorba Linda, CA; ages 26, 32 and 38 years). Asthma cases who met criteria for airway hyperresponsiveness to methacholine or albuterol were classified as having asthma with airway hyperresponsiveness. |
|
Incompletely reversible airflow obstruction 25% of Asthma Cases (n=76) |
Incompletely reversible airflow obstruction was defined at ages 18, 26, 32, and 38 years as postbronchodilator FEV1/FVC ratio below age- and sex-specific lower limits of “normal.”32 The lower limit of normal was defined from cohort members with no previous history of asthma, no history of smoking, and no reported wheeze within the previous year as follows: the mean FEV1/FVC ratio minus 1.96 standard deviations, delimiting 2.5% of the normal population with the lowest post-bronchodilator FEV1/FVC ratios.33 Asthma cases who manifested incompletely reversible airflow obstruction by the end of follow-up were classified as having asthma with incompletely reversible airflow obstruction. In addition to the dichotomous measure of incompletely reversible airflow obstruction described above, we also analyzed post-bronchodilator FEV1/FVC ratio, a continuously distributed biomarker of incompletely reversible airflow obstruction. |
| Panel C. Asthma-Related Disruptions to Daily Life | |
|---|---|
|
Days of school or work missed due to asthma 18% of Cohort (n=158 cohort members; n=2,398 days off) |
At ages 9, 11, 13, 15, 18, 21, 26, 32, and 38 years, cohort members (or their mothers prior to the age 13 assessment) reported missed school or work due to asthma. |
|
Hospitalization for asthma or wheezing 8% of Cohort (n=72) |
At each assessment, cohort members (or their mothers prior to the age 13 assessment) reported whether they had been hospitalized due to asthma or wheeze.23 |
Analysis
We analyzed dichotomous outcomes (e.g. asthma case status) using Poisson regression models to derive relative risks (RRs). We analyzed count phenotypes (e.g. the number of assessments at which cohort members manifested current asthma) using negative binomial regression models to account for over-dispersion. Effect sizes from negative binomial models are reported in terms of incident rate ratios (IRRs). We analyzed survival data (e.g. time-to- asthma onset) using Cox regression models to derive hazard ratios (HRs). We analyzed longitudinal repeated-measures data on the biological characteristics of asthma using generalized estimating equations to account for the non-independence of repeated observations.34 These analyses treated each assessment of a biological characteristic as an outcome, allowing us to differentiate cases on the basis of persistence. All analyses were adjusted for sex. All analyses used the continuous genetic risk score (standardized to have mean=0, standard deviation =1). Effect sizes are reported for a one-standard-deviation increase in genetic risk.
RESULTS
Asthma Onset and Persistence
Boys and girls in the Dunedin cohort who had higher genetic risk scores experienced a greater hazard of developing asthma over 38 years of follow-up as compared to their lower-genetic-risk peers (Hazard Ratio (HR)=1.12 [1.01–1.26]). Figure 1 Panel A shows Kaplan-Meier survival curves for asthma onset. Panels B and C show the same data stratified by genetic risk. For illustrative purposes, high genetic risk is defined as a genetic risk score above the cohort median. Genetic associations with asthma onset did not differ by sex (p-value for difference=0.917).
Figure 1. Cohort members at higher genetic risk were more likely to develop asthma ever and did so earlier in life.

Children at higher genetic risk were more likely to develop asthma through age 38 years and did so earlier in life (Hazard Ratio (HR)=1.12 [1.01–1.26]). Hazards were estimated using a Cox regression model testing association between the continuous genetic risk score and timing of asthma onset. The Cox model was adjusted for sex. Panel A plots Kaplan-Meier Survival Curves for asthma onset among girls and boys in the Dunedin cohort. Panels B and C plot the same data separately for girls and boys with genetic risk scores below the cohort mean (low genetic risk) and cohort members with genetic risk scores above the cohort mean (high genetic risk). Risk tables provide number at risk at birth and ages 7, 15, 21, 26, 32, and 38 years.
Cohort members at higher genetic risk met criteria for current asthma at more assessments between ages 9 and 38 years as compared to their lower-genetic-risk peers (Incident Rate Ratio (IRR)=1.16 [1.03–1.30]). Children at higher genetic risk were also more likely to develop “life-course-persistent asthma” defined by onset in childhood (before age 13 years) with recurrent symptoms through age 38 years (n=96, 11% of the cohort) (RR=1.36 [1.14–1.63], Figure 2, Panel A; Supplemental Figure 2 documents the shift in the genetic risk distribution that underlies this effect). Specifically, among the subset of cohort members with asthma onset in childhood (n=187), those at higher genetic risk onset earlier in life (HR=1.17 [1.01–1.34] and were more likely to become life-course-persistent cases (RR=1.20 [1.05–1.38], Figure 2, Panel B; Supplemental Figure 3 presents a ROC curve analysis showing that the genetic risk score modestly improves individual-level predictions of which childhood-onset asthma cases became life-course-persistent cases). We conducted sensitivity analyses that restricted the life-course- persistent case group to cases with asthma onset before age 9 years (n=93) to test if results were affected by cases in the life-course-persistent group who had onset with asthma around the time of puberty. Removing the three life-course-persistent cases who onset with asthma between ages 9 and 13 did not change results (effect-sizes for genetic associations with life-course-persistent asthma were RR=1.37 [1.14–1.65] in the full cohort and RR=1.20 [1.05–1.39] among asthma cases with onset in childhood).
Figure 2. Cohort members at higher genetic risk were more likely to develop life-course-persistent asthma.

Panel A graphs the genetic association with risk of developing life-course-persistent asthma in the full cohort (N=880; RR=1.36 [1.14–1.63]). Panel B graphs the genetic association with life-course-persistent asthma among the subset of cohort members who onset with asthma by age 13 years (n=187; RR=1.20 [1.05–1.38]). Error bars reflect 95% confidence intervals for point estimates. The genetic risk score is the count of risk alleles standardized to have mean=0 and standard deviation=1.
Biological Characteristics of Asthma
We tested genetic associations with 3 biological characteristics of asthma: atopy (assessed at ages 11, 13, 21, and 32), airway hyperresponsiveness (assessed at ages 9–38), and incompletely reversible airflow obstruction (assessed at ages 18, 26, 32, and 38).
Atopy
Asthma cases at higher genetic risk were at increased risk to manifest atopy (RR=1.07 [1.01–1.14] across age-13, -21, and -32 assessments). Asthma cases at higher genetic risk also manifested elevated serum IgE levels (r= 0.13, p=0.02 across age-11, -21 and -32 assessments).
Airway Hyperresponsiveness
Asthma cases at higher genetic risk were at increased risk to manifest airway hyperresponsiveness to methacholine or albuterol (RR=1.16 [1.03–1.32] across 9 assessments spanning ages 9–38 years).
Incompletely Reversible Airflow Obstruction
Asthma cases at higher genetic risk were at increased risk to manifest incompletely reversible airflow obstruction defined by a post-bronchodilator FEV1/FVC ratio >2SD below the mean of non-asthmatic subjects (RR=1.28 [1.04–1.57] across age-18, -26, -s32, and -38 assessments). Asthma cases at higher genetic risk also manifested poorer lung function as measured by post-albuterol FEV1/FVC ratio (r=−0.13, p=0.03 across age-18, -26, -32, and -38 assessments).
Supplemental Figure 4 graphs genetic associations with lifetime risk for atopy, airway hyperresponsiveness, and incompletely reversible airflow obstruction among asthma cases.
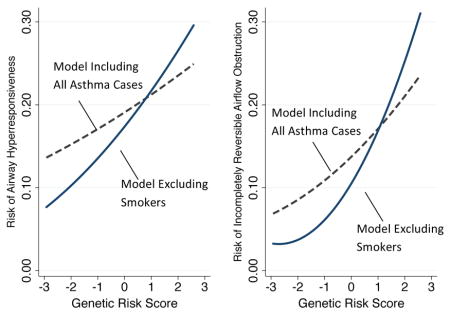
Because many members of the cohort smoked (e.g 34% at age 21; 20% at age 38) and because smoking affects lung function characteristics that we analyzed,35 we conducted sensitivity analyses that excluded smokers (Box 1). Excluding smokers increased the magnitudes of associations between genetic risk and airway hyperresponsiveness (RR=1.38 [1.16–1.64]) and incompletely reversible airflow obstruction (RR=1.63 [1.10–2.41]).
Box 1. Genetic associations with lung function in asthma among non-smokers.
70% of asthma cases smoked at some point during follow-up. We previously constructed developmental phenotypes of smoking behavior in the Dunedin cohort that identify when cohort members initiated smoking.42 We used these data in our sensitivity analysis testing genetic associations with lung-function phenotypes of asthma among non-smokers. We selectively excluded observations of asthma cases that were made after those cases had begun smoking. For example, for a child who onset with asthma at age 11 years and who took up smoking at age 18 years, observations at ages 11, 13, and 15 were included in the analysis of airway hyperresponsiveness, whereas all observations from age 18 on were excluded. Asthma cases with onset after smoking initiation were excluded from analysis entirely. For airway hyperresponsiveness, analyses of nonsmokers included data from n=188 asthma cases. For incompletely reversible airflow obstruction, analyses of nonsmokers included data from n=105 asthma cases. The figure plots the predicted probabilities of airway hyperresponsiveness (Panel A) and incompletely reversible airflow obstruction (Panel B) estimated from models including all asthma cases (dashed gray line; n=305) and models including only non-smoking asthma cases (blue line; n=188 for airway hyperresponsiveness; n=105 for incompletely reversible airflow obstruction). The genetic risk score is the count of risk alleles standardized to have mean=0 and standard deviation=1.
Systematic Review
To measure polygenic risk for asthma, we constructed a genetic risk score. We used databases including PubMed and the National Human Genome Research Institute GWAS Catalog to identify the largest and most comprehensive genome-wide association studies of asthma. From this GWAS, we selected SNPs that were genome-wide significant (p<5×10−8) and that were at least partially independent of one another (R2<0.6) We used these SNPs to construct the genetic risk score. We phenotyped developmental and biological characteristics of asthma using longitudinal data about asthma symptoms, atopy, and lung function (including pharmacological challenge) spanning ages 9–38 years.
Interpretation
Our results advance asthma genetics beyond the original GWAS discoveries in three ways: (1) We show that genetic risks predict which childhood-onset asthma cases remit and which become life-course-persistent cases; (2) We elucidate a biological profile of the asthma that arises from these genetic risks: asthma characterized by atopy, airway hyperresponsiveness and leading to incompletely reversible airflow obstruction; and (3) We describe the real-life impact of GWAS-discoveries by quantifying genetic associations with missed school and work and hospitalization. Because the effect-sizes we observed were small, the predictions offered by genetic risk scores may be weak. More work is needed to clarify the clinical significance of the small increments in risk that can be predicted using SNPs.
There was substantial overlap among atopy, airway hyperresponsiveness, and incompletely reversible airflow obstruction among asthma cases. For example, 72% of asthma cases with incompletely reversible airflow obstruction also manifested atopy and airway hyperresponsiveness during follow-up (Figure 3, Panel A). Asthma cases who did not manifest atopy had genetic risk scores similar to non-cases. Among asthma cases with atopy, those who manifested airway hyperresponsiveness and incompletely reversible airflow obstruction had higher genetic risk scores. Asthma cases with atopy who manifested both airway hyperresponsiveness and incompletely reversible airflow obstruction had the highest genetic risk scores (Figure 3, Panel B).
Figure 3. Asthma cases at higher genetic risk were more likely to manifest atopy, airway hyperresponsiveness, and incompletely reversible airflow obstruction.


Panel A illustrates overlap among different biological chracteristics of asthma. Across 3 decades of follow-up (between ages 9 and 38 years), n=236 asthma cases manifested atopy during follow-up; n=160 manifested airway hyperresponsiveness; and n=76 manifested incompletely reversible airflow obstruction. Panel B graphs mean genetic risk within subgroups of asthma cases defined by atopic status, airway hyperresponsiveness, and incompletely reversible airflow obstruction. Asthma cases with no atopy who manifested airway hyperresponsiveness or incompletely reversible airflow obstruction are grouped together due to the small number of cases in each cell. Error bars reflect standard errors for subgroup means. The genetic risk score is the count of risk alleles standardized to have mean=0 and standard deviation=1.
Asthma-Related Disruptions to Daily Life
Following asthma onset, cases at higher genetic risk reported increased incidence of days off school or work due to asthma over three decades of follow-up (IRR=1.38 [1.02–1.86]). Asthma cases at higher genetic risk were also at increased hazard to be hospitalized for breathing problems (HR=1.38 [1.07–1.79]).
Molecular Genetic Risk vs. Family History of Asthma
Genotype and family history provided similar amounts of information about asthma risk; effect sizes were similar for the genetic risk score and the family history score in models predicting developmental phenotypes of asthma, airway hyperresponsiveness, and asthma-related disruptions to daily life (Table 2). Family history was not associated with asthma cases’ risks of manifesting atopy or incompletely reversible airflow obstruction.
Table 2.
Effect sizes for the genetic risk score and family history score.
Effect sizes that are not statistically significant at α =0.05 are presented in gray text. Effect sizes are presented for a 1 standard deviation increase in genetic risk and a 1 standard deviation increase in family history. HR denotes a hazard ratio estimated from a Cox regression model; IRR denotes an incident rate ratio estimated from a negative binomial regression model; RR denotes a relative risk estimated from a Poisson regression model. r denotes a standardized coefficient (interpreted as Pearson’s r) from a linear regression model. Analyses of biological characteristics of asthma used generalized estimating equations to account for the non-independence of repeated observations of individuals. All models were adjusted for sex.
| Genetic Risk Score | Family History Score | ||||
|---|---|---|---|---|---|
| Effect Size [95% CI] | |||||
| Developmental Phenotypes of Asthma (Analysis in N=880 Cohort Members) | |||||
| Time to Asthma Onset | HR | 1.12 | [1.01, 1.26] | 1.21 | [1.10, 1.34] |
| Asthma Persistence | IRR | 1.16 | [1.03, 1.30] | 1.25 | [1.13, 1.37] |
| Life Course Persistent Asthma | RR | 1.36 | [1.14, 1.63] | 1.33 | [1.19, 1.48] |
| Among asthma cases with onset by age 13 (n=187) | RR | 1.20 | [1.05, 1.38] | 1.09 | [1.00, 1.18] |
| Biological Characteristics of Asthma (Analysis in N=305 Asthma Cases) | |||||
| Atopy | |||||
| Positive Skin Test (ages 13, 21, 32) | RR | 1.07 | [1.01, 1.14] | 1.03 | [0.98, 1.08] |
| Serum IgE (ages 11, 21, 32) | r | 0.14 | [0.02, 0.25] | 0.10 | [0.00, 0.20] |
| Airway Hyperresponsiveness | |||||
| Case Status (ages 9, 11, 13, 15, 18, 21, 26, 32, 38) | RR | 1.16 | [1.03, 1.32] | 1.13 | [1.05, 1.21] |
| Incompletely Reversible Airflow Obstruction | |||||
| Case Status (ages 18, 26, 32, 38) | RR | 1.28 | [1.04, 1.57] | 1.06 | [0.90, 1.23] |
| Post-albuterol FEV1/FVC 18, 26, 32, 38) | r | −0.13 | [−0.25, −0.02] | −0.07 | [−0.16, 0.02] |
| Disruptions to Daily Life (Analysis in N=305 Asthma Cases) | |||||
| Days Off School/Work Due to Asthma | IRR | 1.38 | [1.02, 1.86] | 1.23 | [1.00, 1.51] |
| Hospitalized for Asthma | HR | 1.38 | [1.07, 1.79] | 1.21 | [1.05, 1.38] |
Genotype- and family history-based measures provided different information about asthma risk. Cohort members’ genetic risk scores were unrelated to their family history scores (Pearson’s r=0.05, Supplemental Figure 5). Adding statistical adjustments for family history to regression models testing genetic associations with asthma phenotypes and complications did not change the effect-size or statistical significance estimated for the genetic risk score. The one exception was that after adjustment for family history, the genetic association with incidence of days of school/work missed due to asthma was no longer statistically significant (IRR=1.32 [0.98–1.79], p=0.07).
DISCUSSION
We tested how newly-discovered genetic risks associated with asthma case status were related to developmental and biological characteristics of asthma. Results from our population-level analyses advance asthma genetics beyond the original GWAS discoveries in three ways: (1) We show that genetic risks predict which childhood-onset asthma cases remit and which become life-course-persistent cases, although these predictions are not sufficiently sensitive or specific to support immediate clinical translation; (2) We elucidate a biological profile of the asthma that arises from these genetic risks: atopic asthma characterized by airway hyperresponsiveness and leading to incompletely reversible airflow obstruction; and (3) We describe the real-life impact of GWAS-discoveries by quantifying genetic associations with missed school and work and hospitalization.
We analyzed data from a prospective life-course study of a 1,037-member birth cohort that included in-person assessments of asthma phenotypes at ages 9, 11, 13, 15, 18, 21, 26, 32, and 38 years. Cohort members at higher genetic risk onset with asthma earlier in life, confirming GWAS reports of early-onset asthma as the developmental phenotype most strongly associated with discovered genetic risks.10,36–38 We refine this developmental phenotype by showing that discovered genetic risks are specifically associated with a life-course-persistent developmental phenotype of asthma: Among childhood-onset asthma cases, those at higher genetic risk were more likely to suffer persistent asthma through age 38 years. Higher genetic risk asthma cases were also more likely to be atopic, confirming preliminary GWAS evidence that at least some asthma-associated loci are specific to atopic asthma.39 We further show that among atopic cases, higher genetic risk predisposes to airway hyperresponsiveness, and the development of incompletely reversible airflow obstruction. Collectively, these findings describe a biological profile of asthma associated with GWAS discoveries. Research is now needed to trace molecular pathways from these variants to atopy, airway hyperresponsiveness, and incompletely reversible airflow obstruction in asthma.
Genetic associations with developmental and biological characteristics of asthma translated to an increased burden of asthma-related disruptions to daily life. Asthma cases at higher genetic risk missed more days of school and work over three decades of follow-up and were more likely to be hospitalized for asthma. Magnitudes of genetic effects were small, with each standard deviation increase in genetic risk score predicting an increase in risk for these disruptions to daily life by about one third. Nevertheless, results indicate that full mitigation of genetic risk could reduce the burden asthma imposes on patients, with potential benefits accruing to society through reductions in school and work absenteeism, and in health-care costs.
Finally, we tested whether recent GWAS discoveries captured information that might otherwise be obtained through a family history.40,41 GWAS discoveries for asthma provided novel information about inherited risk that was not captured by family history of asthma; the genetic risk score made independent, additive contributions to prediction models over and above family history. Both genotype- and family-based risk measures predicted asthma onset and persistence, but genotype was a more consistent predictor of biological characteristics of asthma. Surprisingly, genotypic risk was only weakly related to family history of asthma, with some family-history-negative cohort members carrying high genetic risk for asthma. This lack of concordance between genotypic and family-based risk assessments is not unique to asthma. We previously observed this phenomena in the cases of obesity and smoking problems,42,43 and others have reported similar results for some cancers.40 In studies of diabetes and cancer, taking family history into account did not change effect sizes estimated for GWAS-discovered genetic risks,44,45 suggesting low correlation between genotypic and family history risk measures. In turn, this lack of correlation suggests that family history captures more than the additive contributions of common variants. Replication is needed of the non-association between genetic risk and family history, using whole-genome scores.46 If replicated, future genetic-discovery research in asthma could focus on genetic features of family history not detectable in GWAS, including rare variants, interactions among genes (epistasis) and, because family histories capture shared environments as well as genes, interactions between genes and environmental factors.8,47–49 Our findings also suggest that GWAS discoveries may help to identify which childhood-onset asthma cases are most likely to become life-course-persistent cases, even after accounting for differences in family histories of asthma. Because the effect-sizes we observed were small, the predictions offered by genetic risk scores may be weak. More work is needed to clarify the clinical significance of the small increments in risk that can be predicted using SNPs.50
We acknowledge several limitations. First, our analyses were based on cohort members of European descent and for whom follow-up has extended only to age 38 years, limiting generalizability. Replication of findings in non-white cohorts and extension of findings beyond mid-life are critical next steps. Nevertheless, this is one of the longest-running birth cohorts with detailed clinical and objective measurements pertinent to asthma and the cohort is representative of the population studied; it originated as an un-selected birth cohort and has been followed-up with 95% retention. Second, because our study did not include medical records, some cases of asthma and hospitalization may have been missed or misclassified, leading to underestimating genetic effects. Third, our genetic risk score included only SNPs meeting genome-wide significance criteria in GWAS,10 and omitted 2 of these due to technical limitations of the genotyping array. The genotyped SNPs represent only a subset of asthma-causing variants, many of which remain undiscovered.51 Further, because the relative effect-sizes of these SNPs on the developmental and biological characteristics of asthma that we analyzed remain unknown, we did not weight the SNPs in our score. Whole-genome genetic-risk-score approaches that include hundreds or thousands of variants that meet more liberal significance criteria and that apply empirically defined weighting schemes may yield more comprehensive measures of genetic risk.52 Such whole-genome scores remain a work in-progress in the case of asthma; effect sizes reported in a recent whole-genome score study of asthma19 were comparable to those in our study. Finally, because our genetic-risk-score approach studied the aggregate effect of multiple asthma-associated SNPs, we cannot address the question of molecular mechanism. Larger samples and meta-analyses will be required to assess the specific contributions of individual risk variants to developmental and biological characteristics of asthma. The contribution of our study is to guide such investigations by pointing to target phenotypes: early-onset, life-course-persistent asthma, and atopic asthma characterized by airway hyperresponsiveness and incompletely reversible airflow obstruction.
Supplementary Material
Acknowledgments
Role of Funding Source
Funding sources had no role in the design or conduct of this study. DWB and AC had complete access to all data.
Footnotes
Conflict of Interest Statement. The authors declared no conflict of interest.
Author Contributions:
Literature Search DWB AC MRS RJH
Study Design DWB MRS TEM RP AC
Data Collection TEM MRS RJH RP AC
Data Analysis DWB HLH RH TEM KS BW AC
Data Interpretation DWB TEM MRS RJH AC
Writing DWB TEM and AC drafted the manuscript; DWB TEM AC MRS RJH RP KS BW RH and HLH provided critical revision
References
- 1.Sears MR. Descriptive epidemiology of asthma. Lancet. 1997;350(Suppl 2):SII1–4. doi: 10.1016/s0140-6736(97)90028-3. [DOI] [PubMed] [Google Scholar]
- 2.Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18:716–25. doi: 10.1038/nm.2678. [DOI] [PubMed] [Google Scholar]
- 3.Zhang YM, Moffatt MF, Cookson WOC. Genetic and genomic approaches to asthma: new insights for the origins. Curr Opin Pulm Med. 2012;18:6–13. doi: 10.1097/MCP.0b013e32834dc532. [DOI] [PubMed] [Google Scholar]
- 4.Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242:10–30. doi: 10.1111/j.1600-065X.2011.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonfield TL, Ross KR. Asthma heterogeneity and therapeutic options from the clinic to the bench. Curr Opin Allergy Clin Immunol. 2012;12:60–7. doi: 10.1097/ACI.0b013e32834edb5b. [DOI] [PubMed] [Google Scholar]
- 6.Agache I, Akdis C, Jutel M, Virchow JC. Untangling asthma phenotypes and endotypes. Allergy. 2012;67:835–46. doi: 10.1111/j.1398-9995.2012.02832.x. [DOI] [PubMed] [Google Scholar]
- 7.Pearson TA, Manolio TA. How to interpret a genome-wide association study. JAMA. 2008;299:1335–44. doi: 10.1001/jama.299.11.1335. [DOI] [PubMed] [Google Scholar]
- 8.Belsky DW, Moffitt TE, Caspi A. Genetics in population health science: Strategies and opportunities. Am J Public Health. doi: 10.2105/AJPH.2012.301139. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sears MR, Greene JM, Willan AR, et al. A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. N Engl J Med. 2003;349:1414–22. doi: 10.1056/NEJMoa022363. [DOI] [PubMed] [Google Scholar]
- 10.Moffatt MF, Gut IG, Demenais F, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–21. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chipps BE. Determinants of asthma and its clinical course. Ann Allergy, Asthma Immunol. 2004;93:309–15. doi: 10.1016/S1081-1206(10)61388-9. quiz 15–8, 80. [DOI] [PubMed] [Google Scholar]
- 12.Cowan K, Guilbert TW. Pediatric asthma phenotypes. Curr Opin Pediatr. 2012;24:344–51. doi: 10.1097/MOP.0b013e32835357ab. [DOI] [PubMed] [Google Scholar]
- 13.Joos GF, O’Connor B, Anderson SD, et al. Indirect airway challenges. Eur Respir J. 2003;21:1050–68. doi: 10.1183/09031936.03.00008403. [DOI] [PubMed] [Google Scholar]
- 14.Anderson SD. Indirect challenge tests: Airway hyperresponsiveness in asthma: its measurement and clinical significance. Chest. 2010;138:25S–30S. doi: 10.1378/chest.10-0116. [DOI] [PubMed] [Google Scholar]
- 15.Brightling CE, Gupta S, Gonem S, Siddiqui S. Lung damage and airway remodelling in severe asthma. Clin Exp Allergy. 2012;42:638–49. doi: 10.1111/j.1365-2222.2011.03917.x. [DOI] [PubMed] [Google Scholar]
- 16.Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health. 2010;31:69–87. doi: 10.1146/annurev.publhealth.012809.103621. 1 p following. [DOI] [PubMed] [Google Scholar]
- 17.Moffitt TE, Caspi A, Rutter M, Silva PA. Sex differences in antisocial behavior: Conduct disorder, delinquency, and violence in the Dunedin longitudinal study. Cambridge: Cambridge University Press; 2001. [Google Scholar]
- 18.Sullivan PF. Spurious genetic associations. Biol Psychiatry. 2007;61:1121–6. doi: 10.1016/j.biopsych.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Spycher BD, Henderson J, Granell R, et al. Genome-wide prediction of childhood asthma and related phenotypes in a longitudinal birth cohort. J Allergy Clin Immunol. 2012;130:503–9. e7. doi: 10.1016/j.jaci.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joubert BR, Reif DM, Edwards SW, et al. Evaluation of genetic susceptibility to childhood allergy and asthma in an African American urban population. BMC Med Genet. 2011;12:25. doi: 10.1186/1471-2350-12-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plomin R, Haworth CMA, Davis OSP. Common disorders are quantitative traits. Nat Rev Genet. 2009;10:872–8. doi: 10.1038/nrg2670. [DOI] [PubMed] [Google Scholar]
- 22.Milne BJ, Moffitt TE, Crump R, et al. How should we construct psychiatric family history scores? A comparison of alternative approaches from the Dunedin Family Health History Study. Psychol Med. 2008;38:1793–802. doi: 10.1017/S0033291708003115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones DT, Sears MR, Holdaway MD, et al. Childhood Asthma in New-Zealand. Br J Dis Chest. 1987;81:332–40. doi: 10.1016/0007-0971(87)90181-1. [DOI] [PubMed] [Google Scholar]
- 24.Taylor DR, Cowan JO, Greene JM, Willan AR, Sears MR. Asthma in remission - Can relapse in early adulthood be predicted at 18 years of age? Chest. 2005;127:845–50. doi: 10.1378/chest.127.3.845. [DOI] [PubMed] [Google Scholar]
- 25.Sutherland TJ, Sears MR, McLachlan CR, Poulton R, Hancox RJ. Leptin, adiponectin, and asthma: findings from a population-based cohort study. Ann Allergy, Asthma Immunol. 2009;103:101–7. doi: 10.1016/S1081-1206(10)60161-5. [DOI] [PubMed] [Google Scholar]
- 26.Sears MR, Herbison GP, Holdaway MD, Hewitt CJ, Flannery EM, Silva PA. The relative risks of sensitivity to grass pollen, house dust mite and cat dander in the development of childhood asthma. Clin Exp Allergy. 1989;19:419–24. doi: 10.1111/j.1365-2222.1989.tb02408.x. [DOI] [PubMed] [Google Scholar]
- 27.Sears MR, Burrows B, Flannery EM, Herbison GP, Holdaway MD. Atopy in childhood. I. Gender and allergen related risks for development of hay fever and asthma. Clin Exp Allergy. 1993;23:941–8. doi: 10.1111/j.1365-2222.1993.tb00279.x. [DOI] [PubMed] [Google Scholar]
- 28.Hancox RJ, Milne BJ, Poulton R, et al. Sex differences in the relation between body mass index and asthma and atopy in a birth cohort. Am J Respir Crit Care Med. 2005;171:440–5. doi: 10.1164/rccm.200405-623OC. [DOI] [PubMed] [Google Scholar]
- 29.Sears MR, Burrows B, Flannery EM, Herbison GP, Hewitt CJ, Holdaway MD. Relation between airway responsiveness and serum IgE in children with asthma and in apparently normal children. N Engl J Med. 1991;325:1067–71. doi: 10.1056/NEJM199110103251504. [DOI] [PubMed] [Google Scholar]
- 30.Sears MR, Jones DT, Holdaway MD, et al. Prevalence of bronchial reactivity to inhaled methacholine in New Zealand children. Thorax. 1986;41:283–9. doi: 10.1136/thx.41.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sears MR, Burrows B, Herbison GP, Holdaway MD, Flannery EM. Atopy in childhood. II. Relationship to airway responsiveness, hay fever and asthma. Clin Exp Allergy. 1993;23:949–56. doi: 10.1111/j.1365-2222.1993.tb00280.x. [DOI] [PubMed] [Google Scholar]
- 32.Rasmussen F, Taylor DR, Flannery EM, et al. Risk factors for airway remodeling in asthma manifested by a low postbronchodilator FEV1/vital capacity ratio: a longitudinal population study from childhood to adulthood. Am J Respir Crit Care Med. 2002;165:1480–8. doi: 10.1164/rccm.2108009. [DOI] [PubMed] [Google Scholar]
- 33.European Respiratory Society. Standardized lung function testing. Official statement of the European Respiratory Society. Eur Respir J. 1993;16:1–100. [PubMed] [Google Scholar]
- 34.Zeger SL, Liang KY, Albert PS. Models for longitudinal data: A generalized estimating equation approach. Biometrics. 1988;44:1049–60. [PubMed] [Google Scholar]
- 35.Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379:1341–51. doi: 10.1016/S0140-6736(11)60968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forno E, Lasky-Su J, Himes B, et al. Genome-wide association study of the age of onset of childhood asthma. J Allergy Clin Immunol. 2012;130:83–90. e4. doi: 10.1016/j.jaci.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bouzigon E, Corda E, Aschard H, et al. Effect of 17q21 variants and smoking exposure in early- onset asthma. N Engl J Med. 2008;359:1985–94. doi: 10.1056/NEJMoa0806604. [DOI] [PubMed] [Google Scholar]
- 38.Halapi E, Gudbjartsson DF, Jonsdottir GM, et al. A sequence variant on 17q21 is associated with age at onset and severity of asthma. Eur J Hum Genet. 2010;18:902–8. doi: 10.1038/ejhg.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferreira MA, Matheson MC, Duffy DL, et al. Identification of IL6R and chromosome 11q13. 5 as risk loci for asthma. Lancet. 2011;378:1006–14. doi: 10.1016/S0140-6736(11)60874-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heald B, Edelman E, Eng C. Prospective comparison of family medical history with personal genome screening for risk assessment of common cancers. Eur J Hum Genet. 2012;20:547–51. doi: 10.1038/ejhg.2011.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Do CB, Hinds DA, Francke U, Eriksson N. Comparison of family history and SNPs for predicting risk of complex disease. PLoS Genet. 2012;8:e1002973. doi: 10.1371/journal.pgen.1002973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belsky DW, Moffitt TE, Baker TB, et al. Polygenic risk accelerates the developmental progression to heavy, persistent smoking and nicotine dependence: Evidence from a 4-decade longitudinal study. Arch Gen Psychiatry. doi: 10.1001/jamapsychiatry.2013.736. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belsky DW, Moffitt TE, Houts R, et al. Polygenic risk, rapid childhood crowth, and the development of obesity: Evidence from a 4-decade longitudinal study. Arch Pediatr Adolesc Med. 2012;166:515–21. doi: 10.1001/archpediatrics.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meigs JB, Shrader P, Sullivan LM, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359:2208–19. doi: 10.1056/NEJMoa0804742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.So HC, Kwan JS, Cherny SS, Sham PC. Risk prediction of complex diseases from family history and known susceptibility loci, with applications for cancer screening. Am J Hum Genet. 2011;88:548–65. doi: 10.1016/j.ajhg.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2011;13:135–45. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.von Mutius E. Gene-environment interactions in asthma. J Allergy Clin Immunol. 2009;123:3–11. doi: 10.1016/j.jaci.2008.10.046. [DOI] [PubMed] [Google Scholar]
- 49.Guerra S, Martinez FD. Asthma genetics: from linear to multifactorial approaches. Annu Rev Med. 2008;59:327–41. doi: 10.1146/annurev.med.59.060406.213232. [DOI] [PubMed] [Google Scholar]
- 50.Khoury MJ, Gwinn M, Bowen MS, Dotson WD. Beyond Base Pairs to Bedside: A Population Perspective on How Genomics Can Improve Health. Am J Public Health. 2011 doi: 10.2105/AJPH.2011.300299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wjst M, Sargurupremraj M, Arnold M. Genome-wide association studies in asthma: what they really told us about pathogenesis. Curr Opin Allergy Clin Immunol. 2013;13:112–8. doi: 10.1097/ACI.0b013e32835c1674. [DOI] [PubMed] [Google Scholar]
- 52.Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17:1520–8. doi: 10.1101/gr.6665407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.