

Abstract
Tth111II is a thermostable Type IIGS restriction enzyme that recognizes DNA sites CAARCA (R = A or G) and cleaves downstream at N11/N9. Here, the tth111IIRM gene was cloned and expressed in E. coli, and Tth111II was purified. The purified enzyme contains internally-bound S-adenosylmethionine (SAM). When the internal SAM was removed, the endonuclease activity was stimulated by adding SAM or its analog sinefungin. The cleavage intermediate is mostly top-strand nicked DNA on a single-site plasmid. Addition of duplex oligos with a cognate site stimulates cleavage activity of the one-site substrate. Tth111II cleaves a two-site plasmid DNA with equal efficiency regardless of site orientation. We propose the top-strand nicking is carried out by a Tth111II monomer and bottom-strand cleavage is carried out by a transient dimer. Tth111II methylates cleavage product-like duplex oligos CAAACAN9, but the modification rate is estimated to be much slower than the top-strand nicking rate. We cloned and sequenced a number of Tth111II star sites which are 1-bp different from the cognate sites. A biochemical pathway is proposed for the restriction and methylation activities of Tth111II.
A large number of restriction endonucleases (REases) have been discovered from bacterial and viral sources and over 650 are commercially available for recombinant DNA applications1. REases are divided into four major types: Type I (R2M2S1 complex, ATP-dependent DNA translocase/helicase activity, S subunit with two target-recognizing domains recognizing bipartite sequences 3–4 bp long separated by a short unspecified DNA), Type II (4–8 bp target sites, no ATP/GTP requirement), Type III (Mod2Res2 or Mod2Res1 complexes with 5–7 bp target sites, usually cleaving downstream of the recognition sequence at N25/N27, ATP-dependent helicase activity, phage or prophage encoded), and Type IV (modification-dependent REases)2. Based on enzyme properties and differences in cleavage sites, the Type II REases are further classified into Type IIC (symmetric or asymmetric target, restriction and modification functions in a single polypeptide), Type IIE (2–3 targets; one site cleaved, one effector site), Type IIG (symmetric or asymmetric target sites; endonuclease activity affected by AdoMet (SAM)), Type IIS (asymmetric target and cleavage sites) among others2. The commonly used EcoRI and BamHI belong to Type IIP with symmetric recognition sites2. Some REases may fall into multiple subtypes. Refer to reference 2 for more information on how restriction enzymes are classified.
Type IIGS enzyme activities are influenced by additional SAM and cleavage takes place outside of their recognition sequences. A few examples of Type IIGS enzymes are: Bce83I (CTTGAG 16/14)3, BslFI (GGGAC 10/14)1, BspKT5I (CTGAAG 16/14)4, BstOZ616I (GGGAC 10/14)1, EciI (GGCGGA 11/9)1, FaqI (GGGAC 10/14)1, GsuI (CTGGAG 16/14)5, StsI (GGATG 10/14)6,7, TaqII (GACCGA 11/9), TsoI (TARCCA 11/9)1,8, TspGWI (ACGGA 11/9)9, and TspDTI (ATGAA 11/9)10,11. Many putative Type IIGS enzymes are present in sequenced microbial genomes12 and the number of putative Type IIGS enzymes is estimated to be over 40001, which represent a significant portion of the Type II REases from native microbial sources.
Since the specificity domain is shared by the endonuclease and methylase activities, it is relatively easier to engineer new enzyme specificities. For example, MmeI (TCCRAC 20/18) was successfully engineered into MmeI(6G) (TCCRAG 20/18), MmeI(4G)(TCCGAC 20/18), MmeI(4C)(TCCCAC 20/18), MmeI(4G6G) (TCCGAG 20/18), and MmeI(4C6G) (TCCCAG 20/18) by semi-rational design and site-directed mutagenesis13. Due to the relatively long cleavage distance, Type IIGS enzymes such as BpmI, BsgI, BsmFI and MmeI have been used in the Serial Analysis of Gene Expression (SAGE)14, Cap Analysis Gene Expression (CAGE)15, vector tag integration analysis16, Parallel Analysis of RNA Ends (PARE)17, paired end sequence reads18 and genome-wide mapping technique (GMAT)19. Despite the importance of Type IIGS enzymes in technology development, only one crystal structure, RM.BpuSI (GGGAC 10/14), is known20. Little is known about the SAM-stimulated catalytic mechanism and cleavage intermediates among this group of enzymes.
RM.Tth111II (Tth111II) is one of the thermostable Type IIGS enzymes with the recognition and cleavage site of CAARCA N11/N9 (R = A or G). The native strain Thermus thermophilus 111 produced approximately 50 units Tth111II per gram of wet cells21. In addition, the specific activity of Tth111II is rather low (one unit per microgram protein for the native Tth111II21). Other similar enzymes TsoI (TARCCA 11/9)1 8, TaqII (GACCGA 11/9)22, TspDTI (ATGAA 11/9))10 and TspGWI (ACGGA 11/9))23, along with Tth111II, form a group of thermostable Type IIGS enzymes with similarities in enzyme properties such as recognition sites, cleavage patterns, thermostability, gene organization, enzyme oligomerization state in solution. The molecular mass of Tth111II in SDS-PAGE was estimated to be ~120 kDa, and the native molecular mass estimated from Sephadex G-120 column was ~120 ± 10 kDa, indicating that Tth111II is a monomer in solution10.
In this work, we describe the cloning, expression, and purification of recombinant Tth111II. We also report the nicking intermediate of the Tth111II cleavage reaction on a single- site substrate. We also tested Tth111II activity on plasmids with two sites (same orientation-head to tail or inverse orientation-tail to tail). In addition, we characterized a number of Tth111II star sites, the modified base in Tth111II site, and stimulation of activity by addition of duplex oligos.
Results
Cloning, expression, and purification of Tth111II
N-terminal protein sequencing of the native Tth111II revealed the first 15 amino acid residues as NWIDLYTHLKQEVP. Inverse PCR and sequencing were carried out to obtain the complete gene coding sequence. The tth111IIRM fusion gene is 3321 bp, coding for a protein with predicted molecular mass of 126 kDa. There is one partial ORF upstream of the tth111IIRM gene that encodes a putative DNA integrase/transposase, suggesting that the tth111IIRM gene might be part of a mobile genetic element. The DNA sequence has been deposited in GenBank (accession number AY726624).
To express the tth111IIRM gene in E. coli, the fusion gene was cloned into an expression vector pET28a. The yield of Tth111II in cell extracts was ~4,000 units/g wet cells. Compared to the native strain, Tth111II was over-expressed ~80-fold. The recombinant Tth111II was purified to over 95% purity and had a similar molecular weight compared to the native protein (Figure 1A). The Tth111II amino acid sequence contains amino-methyltransferase (MTase, belonging to the γ group) motifs: FGG (234–239: FARGMG) and NPPW (614–620: VVGNPPW) (see below for MTase activity assay). Tth111II contains both endonuclease (R) and methylase (M) domains and a shared specificity domain (S). An α–helical domain connecting the R and M domains is predicted to regulate the endonuclease activity upon binding to specific sites in the presence of SAM. The secondary structure prediction by PROMALS3D and the approximate boundaries of functional domains in Tth111II are shown in Supplementary Figure 1.
Figure 1. SDS-PAGE analysis of recombinant and native Tth111II, and endonuclease activity assays.

(A). Lane 1, protein size marker; Lane 2, purified recombinant Tth111II; lane 3, purified native Tth111II plus BSA. (B). Tth111II endonuclease activity in the absence of additional SAM. Lanes 1, 1 kb DNA marker. Lanes 2–11: 2-fold serial dilution of Tth111II in digestion of 0.5 μg pBR322 (16 units (365 pmole) in lane 2); lane 12, uncut pBR322. Reactions were carried out in Tth Buffer at 65°C for 30 min. (C). Tth111II endonuclease activity in the presence of additional 320 μM SAM.
Tth111II endonuclease activity in the presence of additional SAM
The predicted cleavage products of pBR322 are 1939 bp, 1256 bp, and 1127 bp. Tth111II digestion could not reach completion even at high enzyme concentration where star activity was detected (Figure 1B, lanes 2–4). Some of the cleavage products were bound by the enzyme and caused band shift at high enzyme concentration (lanes 2 and 3). Partial digestion was also observed for Type IIG enzymes such as Eco57I24, MmeI25 and TspGWI23, which is probably due to the simultaneous modification and restriction activities. One unit of Tth111II is defined as the minimal amount of enzyme to digest 1 μg pBR322 to a relatively stable pattern as seen in lane 7. At low enzyme concentration, a nicked open circular species was detected (Lanes 10 and 11). The nicked open circular DNA is a mixture of pBR322 with one or more nicks that are not adjacent to each other.
To examine the effect of additional SAM in the reaction, 320 μM SAM was added and no significant changes were observed in the 1 h digestion of pBR322 (Figure 1C). However, when the two reactions were compared in a time course, addition of SAM significantly enhanced Tth111II activity (Figure 2A, no additional SAM; 2B, +SAM). Although the final digestion patterns were similar, the cleavage reaction with additional SAM was considerably faster than the one without. The reaction with additional SAM is noticeably faster than the reaction without SAM in the initial phase as seen in the graphic representation of one cleavage product in Figure 2C (the fragment indicated by *).
Figure 2. Time course study of Tth111II digestion in the absence or presence of additional SAM.

The reaction conditions were same as described in Figure 1 legend, but with 4 units per lane for various times. Panel (A): no additional SAM added. Lane 1, 1 kb DNA ladder; Lane 2–8: 0, 1, 2, 4, 8, 16, 32 min digestion. Panel (B), 320 μM SAM added; lane 2, uncut pBR322; lanes 3–9: 0, 1, 2, 4, 8, 16, 32 min digestion. (C): A graphic representation of one cleavage product band indicated by “*” as the result of Tth111II digestion with or without additional SAM.
Effects of additional SAM and sinefungin on SAM-depleted Tth111II
The internally bound SAM can be removed by overnight incubation with duplex oligos with the Tth111II sites (methylation of the oligos depleted internally bound SAM). Presumably, after purification of the enzyme away from the modified oligos, the enzyme has now become SAM-depleted. After the internal SAM was removed, Tth111II endonuclease activity was diminished (Figure 3A). The low partial activity may result from left-over internally bound SAM, but we cannot rule out the possibility that the enzyme has a minor activity without SAM. When the reaction was supplemented with 320 μM SAM (Figure 3B) or 1 mM sinefungin (SAM analog which can substitute for binding but not methyl group transfer) (Figure 3C), Tth111II endonuclease activity was restored. Untreated Tth111II (Figure 3D) displayed more partial digestion than the enzyme with additional SAM or sinefungin did. It was concluded that the endonuclease activity of Tth111II required SAM or sinefungin as a stimulatory cofactor. Addition of sinefungin in Tth111II digestion can enhance the endonuclease activity even in the absence of site modification.
Figure 3. Effects of SAM and sinefungin cofactor on SAM-depleted Tth111II endonuclease activity.

Panel (A), SAM-depleted Tth111II digestion of 1 μg pBR322. Panel (B), same as in (A), but with addition of 320 μM SAM. Panel (C), same as in (A), plus 1 mM sinefungin. (D), untreated Tth111II with internally bound SAM. Reaction was carried out in Tth Buffer, 65°C for 30 min. The triangle on top of lanes represents a 2-fold serial dilution of the enzyme (starting at 11.4 pmole). NC, nicked circular; L, linear, SC, supercoiled DNA.
Tth111II star sites
Tth111II shows star activity in the presence of high enzyme concentration, high glycerol concentration, low pH or long incubation (data not shown). The star fragments were cloned and sequenced. The cognate site CAAACA was also confirmed by sequencing. All Tth111II star sites are 1-bp different from the cognate site CAARCA. They are CAAGTA, CTAACA, TAAACA, CAGACA, CAAATA, TAAGCA, CAAACT, CAAACG, CATACA and AAAACA. Though not all possibilities have yet been examined, it is very likely that sites with 1-bp difference from the cognate site could be frequent Tth111II star sites. We have not tested the star activity in the presence of high concentration of glycerol and DMSO. TaqII and TspGWI extreme star activities in the presence of SAM or sinefungin have been utilized to digest genomic DNA for library constructions26.
Tth111II cleavage intermediate and top-strand nicking kinetics
When a single-site plasmid pZZT1 was digested by Tth111II under the standard condition (+SAM) a nicked circular form was prevalent as the digestion product (Figure 4A). To find out the nicking strand specificity, the nicked product was subjected to DNA run-off sequencing. An extra A peak appears when the DNA template is broken by the terminal transferase activity of Taq DNA polymerase used in the sequencing. There was no apparent run-off of the sequence from the top sequencing primer. But there is a small extra “A” peak (*) in the sequencing result (Figure 4B, top sequencing panel), which indicates a small portion of nicked DNA is at the bottom strand. The actual bottom-strand nicked species was estimated to be below 10% of the total nicked DNA. There was significant run-off from the bottom sequencing primer, which indicates that the major cut (nick) was on the top strand (Figure 4B, bottom sequencing panel). The nicked DNA that appeared in the Tth111II reaction on the single-site substrate is a top-strand nicked intermediate (CAAACA N11↓). To test any non-specific nicking by Tth111II enzyme, we also mutated the single Tth111II site by localized random mutagenesis and eliminated the Tth111II site, generating plasmid pZZT0 (CAAACA to CAAACC). Figure 4C shows that pZZT0 was poorly cleaved/nicked by Tth111II (lane 2), but cleaved or nicked by BspQI and Nt.BspQI (lanes 3 and 4). As a positive control, Tth111II was able to cleave pBR322 (five sites) under the same condition (lane 6). It was concluded that the nicking of the single-site plasmid is site-specific and strand-specific.
Figure 4. Tth111II activity assay on a single-site substrate and run-off sequencing of the nicked products.

(A). Tth111II digestion of pZZT1 with a single Tth111II site plus 320 μM SAM. Lane 2–11; 220 ng pZZT1 was digested with Tth111II in a 2-fold enzyme serial dilution (lane 2 enzyme = 91 pmole); Lane 12, uncut pZZT1. (B). Run-off sequencing of the nicked pZZT1. The template sequence and the sequencing primers and nicking sites are shown. Top sequencing panel, sequence read from the top sequencing primer. Bottom sequencing panel, sequence read from bottom sequencing primer. “*” indicates extra “A” peak in the run-off reactions. (C). Digestion of pZZT0 (no Tth111II site) by Tth111II endonuclease. (D). Stepwise methylase and endonuclease activity assays for RM.Tth111II in the absence of or presence of Mg2+. Lane 1, DNA marker; lanes 2, partially modified pZZT1 after methylation in the digestion buffer in the absence of Mg2+; lane 3, endonuclease digestion of the partially modified pZZT1 in the presence of Mg2+ and SAM (note, the remaining supercoiled DNA is resistant to digestion/nicking due to the methylation of Tth111II site. Thus the remaining supercoiled DNA is an indication of methylase activity of RM.Tth111II); lane 4, Mock-modified (unmethylated) pZZT1; lane 5, mock-modified pZZT1 DNA digested by Tth111II in the presence of Mg2+. N, nicked circular; L, linear DNA; S, supercoiled DNA.
Estimate the modification rate by the methylase activity of RM.Tth111II
We found that Tth111II endonuclease activity is dependent on the presence of Mg2+, i.e. if we leave out the Mg2+ in the reaction, the endonuclease activity is minimized, and only the modification reaction by the methylase activity can take place. The single-site plasmid pZZT1 was incubated with Tth111II and SAM in the absence of Mg2+ at 65°C for 30 min. The partially modified DNA was purified by spin column purification, and then digested again by Tth111II in the presence of SAM and Mg2+ (Figure 4D, lane 3). The remaining supercoiled DNA (methylated in the modification reaction) resistant to Tth111II digestion was quantified. It was divided by time and enzyme concentration. The modification rate was estimated at 0.00028/min.
A time course was conducted for the Tth111II digestion of pZZT1 (Figure 5A). The nicked species was measured against time of incubation (Figure 5B). From the run-off sequencing result, the nicked circular plasmid could be considered as top-strand nicked DNA. So the kinetics of the nicked circular pZZT1 is mostly the kinetics of the Tth111II top-strand nicking activity. The initial rate for the appearance of nicked circular pZZT1 was analyzed (Figure 5C). The rate was directly proportional to the concentration of Tth111II, and the slope was 0.0058/min. The linear correspondence is characteristic for a first-order enzyme reaction, suggesting that only one molecule of Tth111II was required to catalyze this step. As previously reported Tth111II is a monomer in solution10. It is very likely that a monomeric Tth111II enzyme is responsible for the top-strand nicking. Complete nicking of pZZT1 started in the third lane at 16 min through 64 min, and the enzyme to DNA molar ratio was 45:1. This high enzyme to DNA ratio indicates very little turn over for the nicking reaction. Despite the competing methylase activity of the fusion enzyme, the single-site plasmid could be nicked completely. When the nicking reaction rate (0.0058/min) and the dsDNA methylation activity (0.00028/min) were compared, the methylation activity is estimated at only 5%, indicating that Tth111II has slow methylation activity and fast top-strand nicking activity in binding to a single-site substrate. The regulation of methylase and endonuclease activity of Type IIGS fusion enzyme may have significant biological consequences when foreign viral DNA enters the host cells. It is necessary to cleave/nick the invading viral DNA first rather than to modify the DNA.
Figure 5. Kinetics study of Tth111II top-strand nicking.

(A). pZZT1 digested by Tth111II (+320 μM SAM) in 2-fold serial dilutions of the enzyme and in different times: 0, 1, 2, 4, 8, 16, 32 and 64 min. (B). Graphic representation of the nicked circular DNA as percentage of total DNA in the time course. (C). The linear relationship of the apparent nicking rate with Tth111II enzyme concentration.
Tth111II requires multiple sites for efficient dsDNA cleavage
Tth111II was used to digest the following plasmids: 220 ng pZZT1 (Figure 6A), 500 ng pZZ2TF (two sites, same orientation. Figure 6B), 500 ng pZZ2TR (two sites, inverse orientation, Figure 6C), 500 ng pUC19 (Figure 6D) and 500 ng pBR322 (Figure 6E). The dsDNA cleavage activity of pZZ2TF, pZZ2TR or pBR322 was much higher (~16 times) than pZZT1, but the appearance of nicked circular DNA was similar (no more than 2-fold difference), suggesting that the top-strand nicking activity is not significantly affected by the site numbers, but the bottom-strand nicking activity is greatly affected. Tth111II displayed similar cleavage activity on pZZ2TF (6B) and pZZ2TR (6C), indicating there is no orientation requirement for cut sites. Plasmid pBR322 (5 sites, 6E) was cleaved more efficiently than the single-site plasmid. At low enzyme concentration, nicked circular DNA intermediate was accumulated in digestion of pBR322 (Figure 6E). It was concluded that Tth111II requires more than one site for efficient cleavage and the orientation of two sites made little difference in cleavage efficiency. MmeI also did not show strong preference of site orientation as long as two sites are present in the substrate, i.e. pBR322 has 4 sites, all in the same orientation, and it is cleaved efficiently by MmeI25. In contrast, it was suggested that some discrepancies in cleavage rates of different two-site substrates by FokI could be due to different site orientation27,28.
Figure 6. Tth111II digestion of five plasmid substrates in the presence of 320 μM SAM.

(A). Tth111II digestion of 220 ng pZZT1 with a single site. (B). Tth111II digestion of 500 ng pZZ2TF with two sites in the same orientation. (C). Tth111II digestion of 500 ng pZZ2TR with two sites in the reverse orientation. (D). Tth111II digestion of 500 ng pUC19 with three closely located sites. (E). Tth111II digestion of 500 ng pBR322 with five sites. (A), open circular nicked DNA; (b), dsDNA cut product; (c), supercoiled DNA; (d), linear DNA from double-strand cuts or two sites with nicks on opposite strands. Enzyme in lane 2 of ABCDE = 91 pmole.
Tth111II endonuclease activity is stimulated by addition of duplex oligos with cognate site
Some restriction enzyme activity can be stimulated by addition of duplex oligos with the cognate site. Tth111II was used in digestion of pZZT1 with the addition of different concentration of oligo duplex 1 (one site CAAGCA), duplex 2 (no Tth111II site), duplex 3 (one star site CAAACT) or duplex 4 (Tth111II cleavage product-like) plus SAM (Figure 7). The dsDNA cleavage activity (the appearance of linear DNA) was stimulated by duplex oligo 1 (Figure 7A, 7E) or duplex oligo 4 (Figure 7E), but not from duplex oligo 2 (Figure 7C) or duplex oligo 3 (Figure 7D). Duplex oligo 1 is substrate-like and duplex oligo 4 is product-like, and both stimulated Tth111II dsDNA cleavage activity (Figure 7E). Thus only the cognate recognition site was required for enzyme activity stimulation (the cleavage distance is fixed and the cleavage site has random sequence). When the duplex oligo has no Tth111II binding site, there was no apparent stimulation (Figure 7C); duplex oligo 3 contains a star site CAAACT, but it was not sufficient to stimulate dsDNA cleavage (Figure 7D). The dsDNA cleavage ratio was measured and plotted against the duplex oligo 1 concentration (Figure 7B). The dependence of DNA cleavage rate on enzyme concentration was described by equation (1) in a single-binding model:
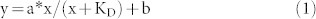 |
y = ds DNA cleavage ratio; x = oligo duplex 1 concentration; a = increase in cleavage ratio, b = base level of cleavage ratio (without additional duplex oligos).
Figure 7. Oligonucleotide stimulation of dsDNA cleavage activity.

(A). Tth111II digestion of pZZTB1 (Nb.BsmI pre-nicked) with the addition of different concentrations of oligos in 30 min at 65°C (+320 μM SAM). Lane 1, 1 kb DNA ladder; Lane 2–9; Tth111II digestion of pZZTB1 (Nb.BsmI pre-nicked) in the presence of 0, 0.067, 0.13, 0.27, 0.53, 1.1, 2.1, 4.3 μM of duplex oligo 1 (one Tth111II site). The final cleavage product is linear DNA. (B). Graphic representation of the relationship of dsDNA cut ratio and concentration of the duplex oligo 1 as shown in panel A. (C). Same reaction condition as in A. Lane 2, uncut pZZTB1; lane 3–12: Tth111II digestion of pZZTB1 (Nb.BsmI pre-nicked) in the presence of 0.017, 0.033, 0.067, 0.13, 0.27, 0.53, 1.1, 2.1, 4.3 μM of duplex oligos 2 (no Tth111II site). (D). Same as in C except the duplex oligos is #3 with one Tth111II star site. (E). Same reaction condition as in A. Lane 2–7, Tth111II digestion of pZZT1 by decreasing amount of Tth111II enzyme without additional duplex oligos. Lane 8–13, Tth111II digestion of pZZTB1 by decreasing amount of Tth111II enzyme plus duplex oligos 1. Lane 14–19, same as lanes 8–13, except the duplex oligo is #4 (cleavage product-like oligos).
The determined optimal KD value is 0.32 μM.
The Tth111II concentration is at 0.76 μM and the apparent KD is 0.32 μM, nearly half of the Tth111II concentration. Similar to Type IIS enzyme cleavage mechanism, it is speculated that the bottom-strand nicking is enhanced by the oligonucleotides with cognate site (enzyme bound to the duplex oligos facilitating formation of a transient dimer with the Tth111II monomer that made the top-strand nick, and the transient dimer making the bottom-strand nick). Further mechanism study is required to confirm this hypothesis.
As expected, the duplex oligo with a cognate site can only enhance dsDNA cleavage of Tth111II on the single-site substrate (pZZT1), but not on pBR322 with five sites (data not shown). After the dsDNA cleavage of the first site, Tth111II could use the cleavage-product sites as a stimulant for additional site cleavage. It is not clear whether two interacting sites are cleaved by Tth111II simultaneously or sequentially.
The methylase specificity of the RM.Tth111II fusion enzyme
When a specific adenine base is already modified by non-radioactive N6 methylation, the base will be totally blocked in the further transfer of the radioactive methyl group –C3H3. The relative methylase activity of Tth111II on duplex oligos CAAACA is 61%, CmAAACA is 100%, CAmAACA is 24%, and CAAACmA is 0%. It was inferred from the labeling experiment that the methylated site is CAARCmA. We also tested the methylase activity on the product-like oligo (CAAACA N9), oligo duplex 4. This unmodified duplex oligos can be labeled with 74% radioactivity, indicating that Tth111II methylase can efficiently modify the cleavage product. The transfer of radioactivity to the oligo duplex 4 did not require Mg2+cofactor (data not shown).
Discussion
In theory, the Tth111II methylase activity is independent of restriction digestion and continues modification even after the digestion is complete, i.e. longer reaction time would generate more modified product. In reality, if the Tth111II digestion reaction was prolonged, however, the DNA could be digested into a smear by strong star activity (data not shown). The possible Tth111II cleavage/nicking and modification pathway is shown in Figure 8.
Figure 8. Possible reaction steps for RM.Tth111II endonuclease and methylase activities.

“*” indicates top-strand (CAARCA) nicking; “Δ” indicates bottom-strand nicking; solid arrows, endonuclease activity; dashed arrow, methylase activity; solid blocks: Tth111II enzyme; solid line, target DNA; dotted block and dotted line, a second enzyme molecule which may be bound to duplex oligos with one site or cleavage product oligos with a cognate site. Blue color endonuclease domain indicates possible conformation changes after binding to the cognate site and SAM after allosteric activation.
We recently found that another Type IIGS enzyme RM.BpuSI forms a unique enzyme conformation when bound to SAM and cognate DNA29. This enzyme conformation is different from the one without SAM. SAM binding in conjunction of cognate site binding may alter the enzyme structure and the α-helical region connecting the endonuclease catalytic domain and the methylase domain as the result of allosteric activation. As demonstrated in Figure 2, addition of SAM cofactor influenced the cleavage rate of Tth111II endonuclease activity.
The biological function of restriction-modification systems is to restrict/cleave foreign invading DNA (reviewed in30). But the bi-functional Type IIGS fusion enzymes such as Tth111II face a decision problem when it encounters unmodified (foreign) DNA: either to cleave or modify the target DNA. Our data indicated that dsDNA cleavage rate is faster in the presence of SAM or sinafungin, and top-strand nicking reaction is ~20-times faster than the modification rate. Since only one strand is modified in CAARCmA sequence, how the host cells repair the DNA incision by Tth111II following DNA replication is unknown. Before DNA replication, the strand in all CAARCmA sites are methylated, the complement strand TGYTTG is not modified. After one round of DNA replication, the nascent strand opposite of TGYTTG is not modified, which requires immediate methylation (otherwise this unmodified site would be subjected to nicking or cleavage). Our data indicated that two-site interactions are required for efficient cleavage by Tth111II and single-site substrate only suffered from top-strand nicking (nicks can be easily repaired/ligated by DNA ligase). The orientation of Tth111II sites made very little difference in the cleavage efficiency. It is not clear what mechanism(s) to prevent two-site interactions (Tth111II or MmeI) at the replication fork immediate following DNA replication. Macromolecular crowding is one possibility.
A number of Tth111II homologs are known. We have cloned and expressed TthHB27I and demonstrated its endonuclease activity (SYX, unpublished result). Many more homologs are listed in REBASE, including TnaRKUORF1766P, TkoORF1158P31, DkaORF1203P32, ParDORF1516P, PaeIMORF242P33, Cma167ORF1062P, PstA1501ORF660P, HauORF544P, PluTORF233P34, AspB510ORF19910P, Sru1385ORF27P35, CthGBORF1068P, CtaORF6P, CglpCGR2ORFAP, SgrTORFCP. Interestingly, two other thermophilic REases, TspGWI and TaqII, show low amino acid sequence identity to Tth111II in the R and S domains, and have their own homologous group with over 60 different members. When the amino acid sequences in the specificity domains among a large number of homologs of related recognition sequences are compared, it is possible to identify the amino acid residues involved in particular base recognition by bioinformatics approach and structure-guided alignment. Semi-rational design can generate variant enzymes with altered specificities13. More studies are needed to understand the cleavage/nicking mechanism and to engineer the specificities of Tth111II-family enzymes.
Conclusion
The tth111IIRM gene has been cloned and expressed in E. coli. Recombinant Tth111II enzyme has been purified to near homogeneity. Tth111II displays star activities at high enzyme and glycerol concentration. Most of the star sites are one base variation from the cognate site. A single-site plasmid can be efficiently nicked by Tth111II, generating a top-strand nicked intermediate. On a single-site substrate, top-strand nicking reaction is much faster than methylation of one adenine in the same site. This long distance nicking property (CAARCA N11↓) and its enzyme thermostability may be utilized in diagnostic applications such as nicking endonuclease signal amplification (NESA). Cleavage product-like duplex oligos (CAAACAN9) can be efficiently modified by RM.Tth111II. Cleavage of the single-site plasmid can be stimulated by addition of duplex oligos with one Tth111II site. The Tth111II-family enzymes may provide a platform for engineering variants with altered specificities.
Methods
Purification of native Tth111II
Tth111II was purified by chromatography through heparin Sepharose column, Q-HP column, heparin TSK column, and polycation A column. The enzyme was stored in a storage buffer with 100 mM NaCl, 10 mM Tris-HCl (pH 7.4), 1 mM DTT, 0.1 mM EDTA, 50% glycerol.
Cloning of tth111IIRM gene
Purified Tth111II protein was sequenced by the Edman degradation method to obtain the N-terminal amino acid sequence (aa). Inverse PCR and primer walking was used for cloning the RM gene. A pair of degenerate inverse PCR primers were designed from the N-terminal aa sequence:
5′- ACCCATCTAAAACATGATGTNCCNTGGTTT -3′(286-192)
5′- TGTTTTAGATGGGTRTANAGRTCDATCCA - 3′(286-244)
Restriction fragments of the Thermus thermophilus 111 genomic DNA were self-ligated and used as templates of inverse PCR. PCR products were purified from low-melting agarose gel and sequenced using the initial degenerate primers. The full-length tth111IIRM gene was obtained through multiple rounds of inverse PCR walking and sequencing.
Expression of Tth111II in E. coli
The tth111IIRM gene was amplified in PCR and cloned into pET28a (EMD Bioscience, Germany) (insert flanked by NdeI and BamHI sites). The insert of the final clone was confirmed to be wild-type (WT) sequence.
Tth111II endonuclease activity assay
Unless otherwise specified, Tth111II endonuclease activity was assayed in 20 mM Tris-HCl (pH 7.5), 120 mM NaCl, 10 mM MgSO4 (Tth Buffer) with 320 μM SAM at 65°C. In some cases sinefungin, a SAM analog, was used. One unit of Tth111II was defined as the amount of enzyme to digest 1 μg pBR322 at 65°C in 1 hour.
Purification of recombinant Tth111II from E. coli source
E.coli [pET28a-Tth111IIRM] was grown in LB + Km overnight without IPTG induction. The cells were collected by centrifugation, resuspended in 10 mM Tris-HCl (pH 7.8), 50 mM NaCl buffer and sonicated. The lysate was then heat-treated at 65°C for 1 hour. After re-centrifugation, the supernatant was loaded on a heparin 6 Fast-Flow column equilibrated with 10 mM Tris-HCl (pH 7.8), 50 mM NaCl. After washing with 10 column-volumes of the same buffer, the column was eluted in 10 mM Tris-HCl (pH 7.8), with a gradient of 50 mM to 1 M NaCl. Tth111II was eluted between 0.65 M and 0.8 M NaCl. The Tth111II was then dialyzed against 20 mM Tris-HCl (pH 7.5), 240 mM NaCl. Remaining DNA was removed by chromatography through a DEAE Sepharose column.
DNA substrates for Tth111II digestion: pUC19 and pBR322
Plasmid substrates pUC19 and pBR322 were used for Tth111II digestion. Three Tth111II sites in pUC19 are present in close proximity (1936, 1403, 1435 bp, underlined sequences):
5′ CAAACAAACCACCGCTGGTAGCGGTGGTTTTTTGTTTGCAAGCAG 3′
3′ GTTTGTTTGGTGGCGACCATCGCCACCAAAAAACAAACGTTCGTC 5′
The Tth111II sites in pBR322 are located at 1936, 3063, 3070, 3102 and 4358 bp. But only three large fragments were detected in 1% agarose gel.
Construction of a single site plasmid pZZT1
Since the three Tth111II sites in pUC19 are located in the replication origin, we used the random mutagenesis method to mutate two Tth111II sites (CAAGCA and CAAACA, two sites shown in italic above). A pair of primers was designed as following:
5′-TAGCGGTGGTTTTTTTGTTNNNNAGCAGCAGATTACGCGCAG-3′(309-159)
5′-CTGCGCGTAATCTGCTGCTNNNNAACAAAAAAACCACCGCTA-3′(309-160)
Inverse PCR was performed using pUC19 template with the above primers and PCR product was digested by DpnI and transformed into ER2502 and plated on an Amp plate. Eighteen colonies were grown and the plasmids were extracted and sequenced. One plasmid retained the high copy number feature and mutated the two Tth111II sites to TGTTCC (complement strand GGAACA) and CCAGCA:
5′-CAAACAAACCACCGCTGGTAGCGGTGGTTTTTTTGTTCC CCAGCAG-3′.
This pUC19 derivative was named pZZT1 with one Tth111II site and used for nicking intermediate study.
Construction of pZZ2TF, pZZ2TR, pZZTB1, and pZZT0
Two other plasmids were constructed from pZZT1 by creating additional sites via inverse PCR. pZZ2TF is a pZZT1 derivative with two Tth111II sites at 1396 bp and 462 bp positioned in the same orientation (head to tail); pZZ2TR has two Tth111II sites at 1396 and 436 bp positioned in the opposite orientation (tail to tail). Plasmid pZZTB1 carries an engineered BsmI site in the following sequence:
 |
Nb.BsmI (GAATGC/−1)) can be used to cleave (nick) the nicked intermediate of Tth111II digestion, which resulted in dsDNA breaks (blunt ends).
To mutate the single Tth111II site in pZZT1, a mutagenic primer with random bases replacing the CAAACA sequence was used in inverse PCR mutagenesis. AmpR transformants were selected on LB + Amp agar plates and individual plasmids were prepared and sequenced. One plasmid containing the following sequence CAAACC was named pZZT0 and used for the detection of non-specific nicking activity.
The effect of additional SAM on Tth111II activity
The effect of additional SAM on Tth111II activity was measured by two methods. First, Tth111II activity was compared in cleavage of pBR322 in the presence or absence of 320 μM SAM. Tth111II enzyme was diluted in 2-fold serial dilution. The amount of pBR322 used and the reaction time were fixed. Second, a time course of reaction at a fixed amount of Tth111II enzyme was compared in cleavage of pBR322 with or without SAM.
Tth111II methylase activity
Plasmid pBR322 was treated with 6 units of Tth111II in the presence or absence of 320 μM SAM in 20 mM Tris-HCl (pH 7.5), 120 mM NaCl at 65°C for 1 hour. The DNA was then purified through a Qiagen spin column, which was digested again by Tth111II in Tth buffer and 320 μM SAM, 30 min at 65°C.
Stimulation of Tth111II activity by duplex oligonucleotides (oligos)
The activity of Tth111II on pBR322 or pZZT1 with additional duplex oligos at different concentrations was compared. The duplex oligos used were:
Oligo duplex1 (one Tth111II site CAAGCA)
ACCTGCAGGCATGCAAGCATGGCGTAATCATGGTCATAG
TGGACGTCCGTACGTTCGTACCGCATTAGTACCAGTATC
Oligo duplex 2 (no Tth111II site)
GGATTTAAACCTAGAGGAGCAGATAATCGACCGGATAGA
CCTAAATTTGGATCTCCTCGTCTATTAGCTGGCCTATCT
Oligo duplex 3 (one Tth111II star site CAAACT)
AATAGTGATAGATCTAGAGCAAACTTGAGATTCCCAAAT
TTATCACTATCTAGATCTCGTTTGAACTCTAAGGGTTTA
Oligo duplex 4 (one Tth111II site CAAACA, cleavage product-like)
AGTTGGTAGCTCTTGATCCGGCAAACAAACCACCGC
TCAACCATCGAGAACTAGGCCGTTTGTTTGGTGGCG
The oligo duplex 1 contains one Tth111II site (CAAGCA) and 20 bp downstream. Oligo duplex 2 does not contain any Tth111II site. Oligo duplex 3 has a Tth111II-related site (CAAACT; designated as a Tth111II star site). Oligo duplex 4 has one Tth111II site (CAAACA) and 9 bp downstream the recognition sequence, which is two bp shorter than the standard product on the top strand and the same length in the bottom strand. Thus, oligo duplex 4 could be regarded as binding only oligos. Tth111II digestion of pZZT1 in the presence of different concentrations of oligo duplexes was measured (30 min at 65°C in the presence of Mg2+ and 320 μM SAM). Digestion of pBR322 was also measured with or without oligo duplex 1.
Preparation of Tth111II without SAM (SAM-depleted Tth111II)
Ninety μl of Tth111II (0.96 mg/ml) (70 pmol) was incubated with 96 pmol of oligo duplex 1 in Tth Buffer, 22 hours at 65°C. Oligos were then removed by chromatography through a DEAE column. This resulted in the removal of the enzyme-bound SAM. The SAM depleted Tth111II apo enzyme was tested for endonuclease activity on pBR322 with or without 320 μM SAM or 1 mM sinefungin.
Determination of Tth111II modified base
Four labeled duplexes were constructed by combining the following oligo (top strand)
5′-Fluorescein-CCACCGCTACCAGCGGTGGTTTGTTTGCCGGATCAAG-3′(317-013)
with each of the following 4 oligos (bottom strand):
5′-CTTGATCCGGCAAACAAACCACCGCTGGTAGCGGTGG-3′(317-014)
5′-CTTGATCCGGCmAAACAAACCACCGCTGGTAGCGGTGG-3′(317-033)
5′-CTTGATCCGGCAmAACAAACCACCGCTGGTAGCGGTGG-3′(317-034)
5′-CTTGATCCGGCAAACmAAACCACCGCTGGTAGCGGTGG-3′(317-035).
Four oligo duplexes were used for methylation study (oligo duplex 5 (317-013/317-014), oligo duplex 6 (317-013/317-033), oligo duplex 7 (317-013/317-034) and oligo duplex 8 (317-013/317-035)). Since Tth111II site is degenerate at the fourth position, CAARCA, the third A is unlikely a candidate for the methylation base. Four nmol of each oligo duplex (1 through 8) was incubated separately with 1 unit of Tth111II in a buffer of 120 mM NaCl, 20 mM Tris-HCl, pH 7.5, 37°C for 24 hours with 4 nmol3H-SAM. The oligo duplexes were purified via Qiagen spin columns and the radioactivity of the eluted oligonucleotides was counted. The methylation of cleavage product-like duplex (CAAACA N9) was also compared with or without Mg2+.
Estimate of modification rate by RM.Tth111II
Step 1, methyltransferase activity assay in the absence of Mg2+ ions: plasmid pZZT1 with a single Tth111II site was incubated with RM.Tth111II in the presence of 0.32 mM SAM in 20 mM Tris-HCl (pH 7.5), 120 mM NaCl, 65°C for 30 min. Step 2, DNA purification: pZZT1 was purified by Qiagen spin column. Step 3, the partially-modified DNA was then digested by RM.Tth111II in 20 mM Tris-HCl (pH 7.5), 120 mM NaCl, 10 mM MgSO4, 0.32 mM SAM, 65°C for 30 min. The modification rate was estimated from the remaining supercoiled DNA resistant to Tth111II nicking/digestion (unmethylated DNA would be converted to nicked circular after endonuclease digestion). Thus, methylation rate can be expressed as remaining supercoiled DNA divided by time and enzyme concentration. To simplify the analysis, only supercoiled and nicked circular DNAs were quantified. The small fraction of linear DNA was not included in the calculation.
Determination of Tth111II star sites
Plasmid pBR322 was extensively digested with 200 units of Tth111II in 5% glycerol at 65°C for 2 h. The digested fragments were blunted with Klenow fragment of E. coli DNA polymerase I and ligated to EcoRV-cleaved LITMUS28 vector. The ligated pool was transformed into ER2683. Plasmid extracted from cultures of several colonies was sequenced using primers flanking the insert to determine the original cut sites.
Tth111II nicking strand determination
One μg of pZZT1 was digested by Tth111II in a 2-fold serial dilution and 0.25 μg of the digested DNA was analyzed on a low-melting agarose gel. The nicked DNA was purified and sequenced by the following primers on both strands:
5′-GCTACAGAGTTCTTGAAGTGGTGG-3′(309-141)
5′-CAAAATCCCCTTAACGTGAGTTTC-3′(309-142)
A second method of determining the nicking site on pZZTB1 made use of a second digestion using the nicking enzyme Nb.BsmI. One μg pZZTB1 was digested by Tth111II in a 2-fold serial dilution; the nicked product was purified and digested again with 10 units of Nb.BsmI for 30 min at 65°C. The single- and double-digested DNAs were analyzed. Nicking by Nb.BsmI will convert nicked (top-strand) open circles to linear DNA.
Kinetics of Tth111II cleavage on pZZT1 with a single site
Two hundred and twenty ng of pZZT1 was digested by different concentrations of Tth111II (+SAM). The reactions were stopped at different times: 0, 1, 2, 4, 8, 16, 32, and 64 min. The density of the resulting DNA bands were measured and quantified by scanning. The data on substrate disappearance, intermediate appearance and product appearance and enzyme concentration were then analyzed using Sigma-Plot software (Systat Software, San Jose, CA).
Author Contributions
Z.Z. and S.Y.X. analyzed data and wrote the manuscript; D.R. purified the native and recombinant Tth111II enzyme; Z.Z., S.G., H.F., A.Q. contributed experimental data.
Supplementary Material
Supplementary Materials
Acknowledgments
We thank Rich Roberts, Bill Jack, and Lise Raleigh for critical comments; Laurie Mazzola, Beth Ann Cantin and Barton Slatko for DNA sequencing; Siu-Hong Chan and Rick Morgan for helpful discussion; Jim Ellard and Donald Comb for their support. This project was funded by New England Biolabs, Inc.
References
- Roberts R. J., Vincze T., Posfai J. & Macelis D. REBASE--a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 38, D234–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R. J. et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31, 1805–1812 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matvienko N. N., Kramarov V. M., Ivanov L. & Matvienko N. I. Bce83I, a restriction endonuclease from Bacillus cereus 83 which recognizes novel nonpalindromic sequence 5′-CTTGAG-3′ and is stimulated by S-adenosylmethionine. Nucleic Acids Res 20, 1803 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapovalova N. I., Zheleznaia L. A., Matvienko N. N. & Matvienko N. I. [A new site-specific endonuclease BspKT51]. Biokhimiia 59, 485–493 (1994). [PubMed] [Google Scholar]
- Janulaitis A., Bitinaite J. & Jaskeleviciene B. A new sequence-specific endonuclease from Gluconobacter suboxydans. FEBS Lett 151, 243–247 (1983). [DOI] [PubMed] [Google Scholar]
- Kita K., Suisha M., Kotani H., Yanase H. & Kato N. Cloning and sequence analysis of the StsI restriction-modification gene: presence of homology to FokI restriction-modification enzymes. Nucleic Acids Res 20, 4167–4172 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita K., Kotani H., Ohta H., Yanase H. & Kato N. StsI, a new FokI isoschizomer from Streptococcus sanguis 54, cleaves 5′ GGATG(N)10/14 3′. Nucleic Acids Res 20, 618 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowron P. M. et al. Three-stage biochemical selection: cloning of prototype class IIS/IIC/IIG restriction endonuclease-methyltransferase TsoI from the thermophile Thermus scotoductus. BMC molecular biology 14, 17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylicz-Stachula A., Bujnicki J. M. & Skowron P. M. Cloning and analysis of a bifunctional methyltransferase/restriction endonuclease TspGWI, the prototype of a Thermus sp. enzyme family. BMC Mol Biol 10, 52 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowron P. M. et al. A new Thermus sp. class-IIS enzyme sub-family: isolation of a ‘twin' endonuclease TspDTI with a novel specificity 5′-ATGAA(N(11/9))-3′, related to TspGWI, TaqII and Tth111II. Nucleic Acids Res 31, e74 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylicz-Stachula A. et al. Related bifunctional restriction endonuclease-methyltransferase triplets: TspDTI, Tth111II/TthHB27I and TsoI with distinct specificities. BMC molecular biology 13, 13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela D. A. et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc Natl Acad Sci U S A 105, 18964–18969 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R. D. & Luyten Y. A. Rational engineering of type II restriction endonuclease DNA binding and cleavage specificity. Nucleic Acids Res 37, 5222–5233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velculescu V. E., Zhang L., Vogelstein B. & Kinzler K. W. Serial analysis of gene expression. Science 270, 484–487 (1995). [DOI] [PubMed] [Google Scholar]
- Shiraki T. et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc Natl Acad Sci U S A 100, 15776–15781 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani J., Holic N., Martinez K., Danos O. & Perea J. A high throughput method for genome-wide analysis of retroviral integration. Nucleic Acids Res 34, e134 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- German M. A., Luo S., Schroth G., Meyers B. C. & Green P. J. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat Protoc 4, 356–362 (2009). [DOI] [PubMed] [Google Scholar]
- Ng P. et al. Multiplex sequencing of paired-end ditags (MS-PET): a strategy for the ultra-high-throughput analysis of transcriptomes and genomes. Nucleic Acids Res 34, e84 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh T. Y. & Zhao K. High-resolution, genome-wide mapping of chromatin modifications by GMAT. Methods Mol Biol 387, 95–108 (2008). [DOI] [PubMed] [Google Scholar]
- Shen B. W. et al. Characterization and crystal structure of the type IIG restriction endonuclease RM.BpuSI. Nucleic Acids Res 39, 8223–8236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinomiya T., Kobayashi M. & Sato S. A second site specific endonuclease from Thermus thermophilus 111, Tth111II. Nucleic Acids Res 8, 3275–3285 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker D., Hoff M., Oliphant A. & White R. A second type II restriction endonuclease from Thermus aquaticus with an unusual sequence specificity. Nucleic Acids Res 12, 5567–5581 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylicz-Stachula A., Harasimowicz-Slowinska R. I., Sobolewski I. & Skowron P. M. TspGWI, a thermophilic class-IIS restriction endonuclease from Thermus sp., recognizes novel asymmetric sequence 5′-ACGGA(N11/9)-3′. Nucleic Acids Res 30, e33 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janulaitis A., Petrusyte M., Maneliene Z., Klimasauskas S. & Butkus V. Purification and properties of the Eco57I restriction endonuclease and methylase--prototypes of a new class (type IV). Nucleic Acids Res 20, 6043–6049 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R. D., Bhatia T. K., Lovasco L. & Davis T. B. MmeI: a minimal Type II restriction-modification system that only modifies one DNA strand for host protection. Nucleic Acids Res 36, 6558–6570 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylicz-Stachula A., Zolnierkiewicz O., Jezewska-Frackowiak J. & Skowron P. M. Chemically-induced affinity star restriction specificity: a novel TspGWI/sinefungin endonuclease with theoretical 3-bp cleavage frequency. BioTechniques 50, 397–406 (2011). [DOI] [PubMed] [Google Scholar]
- Vanamee E. S., Santagata S. & Aggarwal A. K. FokI requires two specific DNA sites for cleavage. J Mol Biol 309, 69–78 (2001). [DOI] [PubMed] [Google Scholar]
- Catto L. E., Bellamy S. R., Retter S. E. & Halford S. E. Dynamics and consequences of DNA looping by the FokI restriction endonuclease. Nucleic Acids Res 36, 2073–2081 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrade-Loucheur A., Xu S. Y. & Chan S. H. The Role of the Methyltransferase Domain of Bifunctional Restriction Enzyme RM.BpuSI in Cleavage Activity. PLoS One 8, e80967 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingoud A. & Jeltsch A. Structure and function of type II restriction endonucleases. Nucleic acids research 29, 3705–3727 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui T. et al. Complete genome sequence of the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1 and comparison with Pyrococcus genomes. Genome Res 15, 352–363 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravin N. V. et al. Complete genome sequence of the anaerobic, protein-degrading hyperthermophilic crenarchaeon Desulfurococcus kamchatkensis. J Bacteriol 191, 2371–2379 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz-Gibbon S. T. et al. Genome sequence of the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. Proc Natl Acad Sci U S A 99, 984–989 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchaud E. et al. The genome sequence of the entomopathogenic bacterium Photorhabdus luminescens. Nat Biotechnol 21, 1307–1313 (2003). [DOI] [PubMed] [Google Scholar]
- Mongodin E. F. et al. The genome of Salinibacter ruber: convergence and gene exchange among hyperhalophilic bacteria and archaea. Proc Natl Acad Sci U S A 102, 18147–18152 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials