

Abstract
Live-cell imaging of mRNA yields important insights into gene expression, but it has generally been limited to the labeling of one RNA species and has never been used to count single mRNAs over time in yeast. We demonstrate a two-color imaging system with single-molecule resolution using MS2 and PP7 RNA labeling. We use this methodology to measure intrinsic noise in mRNA levels and RNA polymerase II kinetics at a single gene.
The complex lifecycle of mRNAs requires regulation at every step1, 2, 3, and a growing body of evidence suggests that these steps are interconnected4. A comprehensive view of such complex regulatory systems depends on our ability to interrogate gene expression at the most fundamental level: single molecules. Our basic understanding of gene expression has come from ensemble biochemical approaches including northern blotting, reverse-transcriptase PCR, microarrays and high-throughput sequencing5. These approaches provide a measurement of the average mRNA abundance in a population but fail to illuminate certain aspects of gene expression, such as cell-to-cell variation in expression levels or the behaviors of single particles. The advantage of single-molecule FISH lies in the ability to detect single transcripts and distinguish multiple mRNA species using differentially labeled probe sets. This technique has been used to study the stochastic nature of transcription that produces variation within a clonal population6, 7. However, FISH methods fail to provide the temporal resolution needed to accurately study dynamic cellular processes, a major limitation that must be overcome by live-cell imaging. The MS2 system, based on the high-affinity interaction between the MS2 bacteriophage coat protein and its cognate RNA hairpin, has been used for fluorescent labeling of mRNA to address a diversity of biological questions in many different model organisms8. More recently, a similar interaction from the Pseudomonas aeruginosa PP7 bacteriophage has been characterized9 and used to analyze transcriptional activity10. In both cases, RNA hairpin sequences are inserted in tandem into a gene of interest to tag it, and coexpression of a fluorescent coat protein allows for mRNA detection11. We describe here the first example, to our knowledge, of single-mRNA counting of endogenously expressed yeast mRNAs using MS2 and PP7 in living cells, and we demonstrate that tagging does not perturb endogenous expression levels of MDN1, an essential yeast gene (Supplementary Results). Importantly, MS2 and PP7 systems can be used simultaneously for two-color imaging: in this case, to quantify expression of two MDN1 alleles over time. We further demonstrate the utility of the methodology using an intramolecular dual tagging that enables direct observation of RNA polymerase II (pol II) kinetics. The advances represented by this work bridge the gap between FISH and in vivo RNA imaging methodologies and provide the basis for future applications.
MDN1 is the longest yeast gene (14.7 kb) and encodes an essential ATPase involved in ribosome assembly12. It is constitutively expressed at low steady-state numbers and remains transcriptionally active in nearly all cells6. We generated MDN1-tagged stains by genomic integration of a cassette containing 24 copies of either PP7 or MS2 hairpins into the 3′ UTR (Supplementary Fig. 1) and screened for site-specific integration by PCR (Supplementary Fig. 2). To visualize MDN1 mRNAs, we transformed into each strain plasmids that encode the appropriate fluorescent coat protein (MCP for MS2 strains and PCP for PP7 strains). We imaged fluorescent spots representing MDN1 mRNAs (Supplementary Fig. 3) using z-sweep acquisition (Supplementary Fig. 4) and quantified expression levels over time. Notably, neither tag appeared to perturb MDN1 expression levels (Supplementary Fig. 5).
We confirmed that PP7 and MS2 interactions retain binding specificity when combined in vivo (Supplementary Fig. 6). We then generated two-color yeast strains by mating MDN1-24PP7 and MDN1-24MS2 haploids (Fig. 1a) and expressed PCP-2yEGFP and MCP-mCherry such that transcription of each individually tagged MDN1 allele resulted in differentially labeled populations of mRNAs (Fig. 1b–d). Examination of the expression of two alleles provides an experimental system for exploring the stochastic nature of gene expression as cells progress through the cell cycle. Both alleles are subject to the same regulation, transcribed within the same nucleus and exposed to the same environmental variables; therefore, any variability in transcript number provides a measurement of intrinsic noise. Although single-molecule FISH has been used to correlate the expression of two alleles13, it has so far been impossible to investigate this in vivo over time. We used the change in steady-state levels between time points to calculate transcript fluctuations for each allele.
Figure 1. Expression of MDN1 alleles fluctuate independently over time.
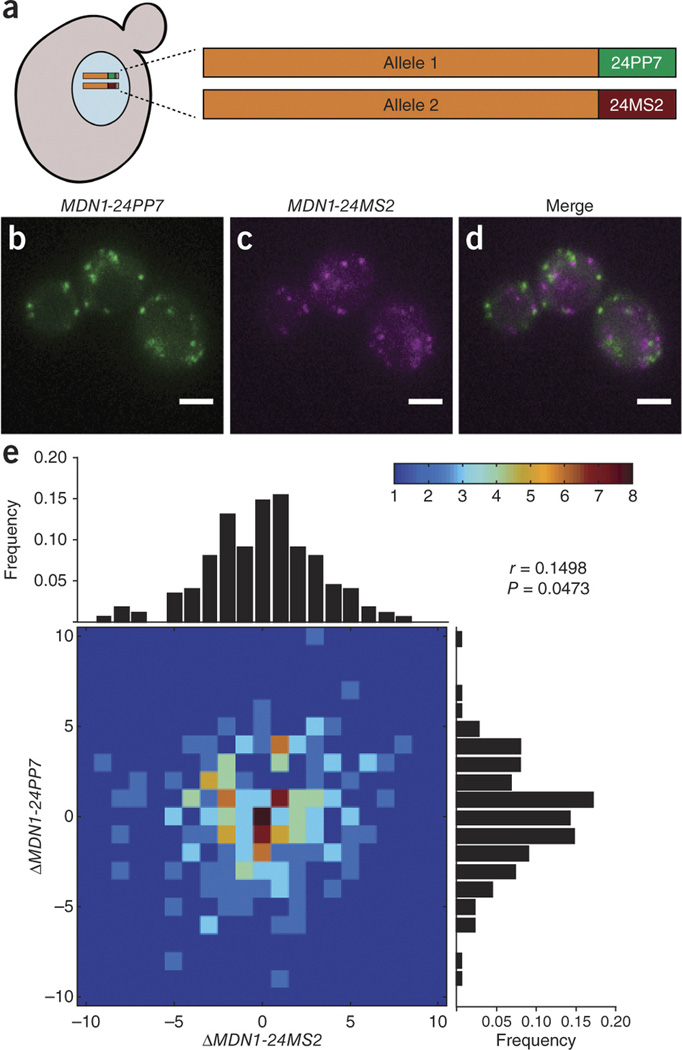
(a) A diploid strain carries one PP7-tagged MDN1 allele and one MS2-tagged MDN1 allele. Coexpression of PCP-2yEGFP and MCP-mCherry results in allele-specific labeling of mRNA. (b,c) Transcription of the indicated allele produces green or magenta fluorescent spots. (d) Overlay of mRNA fluorescence for both alleles. (e) MDN1 mRNAs are counted in single cells every 20 min. Fluctuations were determined as the change in steady-state levels for a given allele between time points, and a heat map was generated to represent these fluctuations. Peripheral histograms represent the distribution of fluctuations for each allele. Pearson's correlation coefficient (r) is generated from the data set (P = 0.0473, n = 210 time points in 33 cells). Scale bars, 3 µm.
Because the half-life of MDN1 mRNAs is 26 min (ref. 14), we imaged cells at 20-min intervals until they entered mitosis, which we determined using cell and nuclear morphology (Supplementary Figs. 7 and 8 and Supplementary Videos 1,2,3,4). The majority of time points analyzed (77%) revealed changes in steady-state levels over time that ranged from −3 to +3 mRNAs, consistent with previous characterizations of MDN1 (ref. 6). Notably, expression of each allele appeared to be largely independent over time (Fig. 1e), with a Pearson's correlation coefficient of 0.1498. In vivo results support the notion that transcript number, even for identical alleles, is stochastic and intrinsically noisy13, and they demonstrate the utility of this approach for studying the coordination of gene expression with single-molecule resolution in living cells. This approach also provides insights into the different expression profiles that cells can maintain over time (Supplementary Fig. 9 and Supplementary Results).
Although the λN system (based on the interaction between protein N of the bacteriophage lambda and boxB RNA) has been used in parallel with MS2 to study the transport of two yeast mRNAs15, the inability to resolve single molecules precluded the types of analyses described here. Overall, the ability to count mRNAs in vivo provides a quantitative tool for measuring many aspects of mRNA metabolism over time. For example, assessment of the role of randomness in cellular processes is important for understanding how cells maintain homeostasis. Previously described in vivo two-color systems have been used to study noise in gene expression16, 17, but at the level of proteins, representing the combined contributions of transcription and translation. We show that mRNAs transcribed from different alleles of the same gene exhibited uncorrelated fluctuations in expression over time. One possible explanation for this is described by the transcription factor binding model10, in which the assembly of the pre-initiation complex cannot be predicted because it depends on the binding of a diffusible factor present at low abundance. Hence, despite the fact that both alleles are indistinguishable in their environment and regulatory mechanism, their transcription initiation is no more correlated than any other two genes. This two-color approach can be extended to further study the coordination of many gene expression regulatory networks.
Engineering both tags in different regions of the same gene allows other aspects of gene expression to be investigated, such as polymerase dynamics at single genes or conformational changes within mRNA molecules as they are transported. To demonstrate this, we engineered a two-color intramolecular system to investigate pol II dynamics by inserting 24PP7 into the 5′ UTR of MDN1 and 24MS2 into the 3′ UTR (Fig. 2a). Dual tagging of MDN1 was confirmed by FISH (Supplementary Fig. 10), and this gene was placed under the control of a GAL promoter so that expression was inducible and the generation of 5′ and 3′ signals could be followed over time. Upon induction, pol II complexes initiate transcription, and the time measured between the appearance of 5′ signal and 3′ signal provides a direct measurement of pol II elongation rates.
Figure 2. Two-color transcriptional induction as a readout for polymerase dynamics.
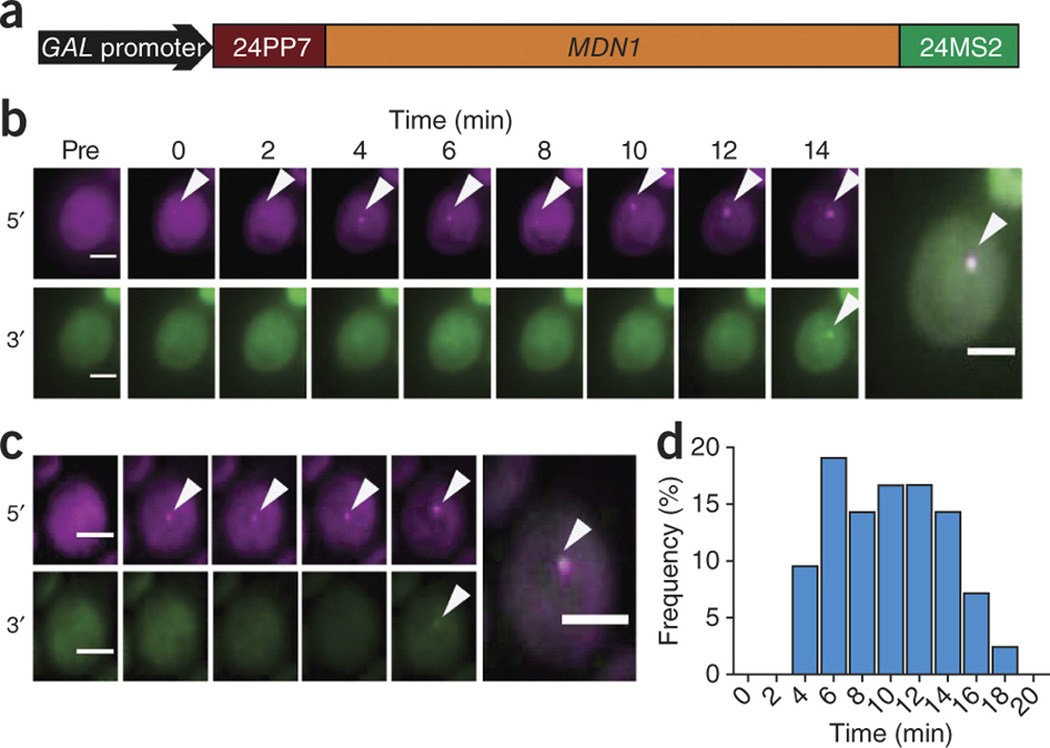
(a) Schematic depicting the intramolecular tagging of MDN1 placed under the control of a GAL-inducible promoter. (b,c) Appearance of 5′ (magenta) and 3′ (green) transcription site signals at indicated times after induction. Arrowheads denote transcription sites, and final frames show an overlay of both signals. (d) Histogram of the time delay between 5′ and 3′ signals, representing variability in pol II elongation rates (n = 42 cells). Scale bars, 3 µm.
In vivo analysis of transcription site fluorescence has been demonstrated previously to estimate transcription rates10, 18, but it has not been possible to directly observe the movement of polymerases through two different regions of a single gene. We induced MDN1 transcription by shifting to galactose-containing medium and imaged with low laser intensity every 2 min. An average elongation rate of 25 ± 2 bases per s, determined by the time it took for polymerases to transverse the 14.7-kb gene, was consistent with other measurements derived from modeling of one-color data10. Most surprisingly, we observed a high degree of cell-to-cell variability (Fig. 2b,c and Supplementary Videos 5,6,7,8) in elongation rates, ranging from 14 bases per s to 61 bases per s (Fig. 2d), and these cell-to-cell differences were not dependent on cell-cycle stage.
These findings suggest that elongation may not be as processive as previously believed. Differences in transcription factor abundance, alternate chromatin states and other variables likely underlie such cell-to-cell variation, and further studies will be required to uncover the mechanistic details behind this observation. This intramolecular assay will be useful in answering additional questions regarding transcription, such as the extent to which downstream processes can feed back to influence transcription4. Furthermore, similar intramolecular assays using these reagents will provide insights into other stages of an mRNA's life cycle. For example, mRNA-containing granules are believed to function in transport and translation in neurons19, and FISH experiments in neurons suggest that differential trafficking occurs depending on the mRNA species20. These tools allow for identification of the mRNA constituents of neuronal granules and measurement of differences in trafficking kinetics for different mRNA species, all within the same cell. We have used yeast to demonstrate the validity of this approach, but we emphasize that it will find applications in all model systems.
Methods
Strain preparation
W303 yeast cells were first transformed with pSH47 to allow induction of the CRE recombinase. Diploids were tagged as described previously11. Plasmids containing 24× hairpins were generated from multimerizing 2xPP7V3 (PP7SL on Addgene) or 2xMS2ORF (MS2SL-stable on Addgene) sequences (Supplementary Table 1). Primers with homology to the MDN1 3′ UTR were used to amplify an integration cassette containing 24 PP7V3 or MS2ORF hairpins (pDZ416 and pDZ415) and the KANR marker flanked by loxP sites (Supplementary Table 2a). Cells were transformed by homologous recombination using 1 µg PCR product, and positive integrants were selected for by growth on G418/YPD plates and confirmed by PCR (Supplementary Table 2b). Integrants were grown in 2% Gal −Ura minimal synthetic medium overnight for removal of the KANR marker. Colonies that grew on YPD but not G418/YPD plates were screened by PCR analysis to confirm both KANR removal (Supplementary Table 2b) and PP7/MS2 hairpin copy number (Supplementary Table 2c). Cells were transformed with 300 ng PMet25PCP-2yEGFP (pDZ270) or PMet25MCP-2yEGFP (pDZ274) and visualized by epifluorescence to confirm expression and labeling of MDN1 mRNAs. Yeast enhanced GFP, or yEGFP, represents a GFP variant that has been codon optimized for enhanced fluorescence in Candida albicans. We have had success labeling mRNA by fusing two yEGFP molecules to the C-terminal end of the coa protein10. Tetrads for both PP7 and MS2 integrants were dissected and plated on selective medium, and haploid mating type was determined by single-colony PCR (Supplementary Table 2d,e). Haploids were again visualized by epifluorescence to confirm expression and labeling of MDN1 mRNAs. All two-color experiments were carried out by coexpressing PCP-2yEGFP and MCP-mCherry. Fluorescent haploids strains (MDN1-24PP7 + PMet25PCP-2yEGFP and MDN1-24MS2 + PMet25MCP-mCherry) were mated on selective plates to produce diploids that contain two 3′-tagged MDN1 alleles and express two sets of uniquely labeled MDN1 mRNAs. The intramolecular strain containing a dual tagging of MDN1 was constructed by genomic insertion by homologous recombination of a DNA fragment (from pDZ570) containing a histidine selectable marker, GAL promoter and 24PP7 into the 5′ end of a diploid MDN1-24MS2 strain.
FISH
MDN1 expression levels were measured by single-molecule FISH according to the procedure previously detailed6, 21. Cells were grown at 30 °C in liquid medium to an optical density (OD600) of 0.6–0.8 and fixed by addition of 32% (v/v) paraformaldehyde to the culture (final concentration of 4% paraformaldehyde) for 45 min at room temperature. Yeast cell walls were digested at 30 °C with lyticase (Sigma), and resulting spheroblasts were deposited on poly-L-lysine–coated (Sigma) cover slips and stored in 70% (v/v) ethanol at −20 °C. Cells were then rehydrated in 2× SSC followed by 2× SSC/40% (v/v) formamide. Coverslips were inverted onto 20 µL hybridization solution containing 0.5 ng of dye-conjugated DNA probes (Supplementary Table 3) and left at 37 °C overnight. Coverslips were then washed in 2× SSC/40% (v/v) formamide for 15 min at 37 °C followed by 2× SSC/0.1% (v/v) Triton X-100 at room temperature for 15 min and 1× SSC at room temperature for 15 min. Nuclei were stained in 1× PBS containing 0.5 µg/mL DAPI, and coverslips were mounted onto slides using ProLong Gold antifade reagent (Invitrogen) and sealed with nail polish.
FISH imaging
FISH samples were imaged with an Olympus BX61 epifluorescence microscope using an Olympus UPlanApo 100×/1.35-NA oil-immersion objective. Illumination was achieved using an X-Cite 120 PC (EXFO) light source with 31000 (DAPI) and SP-102v1 (cy3) filter sets (Chroma Technology). Images were acquired using IPLab (BD Biosciences) software for instrument control and a CoolSNAP HQ camera (Photometrics) with a 6.4-µm pixel size CCD. Serial z sections were collected every 0.2 µm over a vertical range of 4 µm and max projected before data analysis.
Live-cell imaging
Yeast were inoculated at very low OD and grown in synthetic minimal medium for 12–16 h at 30 °C to a final OD600 of 0.1–0.2. Cells were centrifuged at 4,000 r.p.m. for 3 min, and pellets were resuspended in 2 mL fresh medium. We prepared 35-mm glass-bottom microwell dishes (MatTek) for adhesion by coating them with 1 mg/mL filter-sterilized Concanavalin A (Sigma) for 5 min and then subjecting them to a brief wash. 400 µL cell suspension was added to the dish, and cells were allowed to settle and adhere for 5 min. The suspension was then aspirated off and immediately replaced with 3 mL fresh medium containing 1% (v/v) OxyFluor (Oxyrase) antibleach reagent and 20 mM sodium DL-lactate (Sigma), added slowly to the periphery of the dish so as not to disrupt adhered cells. Dishes were taken to the microscope and placed on the stage maintained in a 30 °C/60% Rh enclosure (Precision Plastics) for 30 min to ensure stable imaging conditions. Cells were imaged on a custom-built epifluorescence inverted microscope (Olympus) using a 150×/1.45-NA UApo objective (Olympus). Fluorescence excitation sources include a 488-nm Ar+ laser (Melles Griot), a 561-nm diode-pumped solid-state laser (Cobalt) and a 436-nm diode laser (Power Technologies) for imaging GFP, mCherry and CFP, respectively. Fluorescence was filtered using band-pass emission filters (Semrock) and imaged using an iXon back-illuminated EMCCD camera (Andor). Images were acquired using MetaMorph (Molecular Devices). Visualization of single molecules was achieved using 90-ms exposures and 1.9-mW laser intensity for GFP-labeled mRNAs and 2.4-mW laser intensity for mCherry-labeled mRNAs (as measured from the back focal plane). Nuclei were visualized by low-power excitation of the H2A2-yECFP histone fusion protein. Z stacks were collected as the stage performed a continuous 'z sweep' over 4.5 µm, with the EMCCD reading out every 0.75 µm (stack size = 7). These z sweeps were critical to achieving high-intensity yet rapid acquisition through the cell so as to minimize blurring of diffusing mRNAs and prevent oversampling of mRNAs within the cell volume. Time-lapse two-color movies were imaged in this manner every 20 min, and z stacks were max projected before analysis. Intramolecular two-color experiments were imaged using the same z-sweep parameters. Cells were shifted from raffinose to galactose-containing medium and imaged every 2 min for 28 min to visualize the appearance of 5′ and 3′ signals. Low-intensity laser illumination was used to limit photobleaching and because resolution of single transcripts was not required.
Data analysis
Three-dimensional stacks underwent maximum-intensity projection along the z axis. A previously described two-dimensional Gaussian mask algorithm22 was implemented through custom-made software for the IDL platform (ITT Visual Information Solutions). Diffraction-limited fluorescence signal was used to identify and count single mRNAs for both FISH and live-cell images. Cellular boundaries were defined by a hand-drawn mask using a custom script in ImageJ and combined with detection data using custom-made software in IDL to compute the total number of mRNA molecules per cell. MDN1 expression kinetics were evaluated using the change in mRNA copy number for each allele between frames acquired 20 min apart; Pearson's correlations were determined using GraphPad Prism (GraphPad Software), and the fluctuation heat map and histograms were generated using MATLAB (MathWorks). At each time frame, cell-cycle stage was assessed using nuclear H2A2-yECFP fluorescence signal in conjunction with cellular morphology. The histogram for the time delay between the appearance of 5′ and 3′ signal in intramolecular experiments was also generated using GraphPad Prism. Calculations for elongation rates were done by dividing the length of the gene by this time for 42 cells.
Supplementary Material
MDN1-24PP7 labeled by 2xyEGFP (allele 1)
MDN1-24MS2 labeled by mCherry (allele 2)
H2A2-yECFP fusion (nucleus)
Merge
Induced cell 1: 5′ PP7
Induced cell 1: 3′ MS2
Induced cell 2: 5′ PP7
Induced cell 2: 3′ MS2
Acknowledgements
We thank H.Y. Park, S.J. Gandhi, T. Lionnet and D.R. Larson for helpful discussions and feedback. This work was supported by the US National Institutes of Health (GM57071 to R.H.S. and National Research Service Award individual fellowship F32GM083430 to J.A.C.), Canadian Institutes of Health Research (MOP-BMB-232642 to D.Z.), Fonds de recherche du Québec–Santé (Chercheur Boursier to D.Z.) and funding from the Canada Foundation for Innovation.
Footnotes
AUTHORCONTRIBUTIONS
Experiments and data analysis were performed by S.H. and P.R. Tagging constructs and coat protein expression vectors were created by D.Z., J.A.C., S.H. and P.R. The manuscript was written by S.H. Project consultation and guidance were provided by D.Z., J.A.C. and R.H.S.
COMPETINGFINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Garneau NL, Wilusz J, Wilusz CJ. Nat. Rev. Mol. Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 2.Sexton T, et al. Nat. Struct. Mol. Biol. 2007;14:1049–1055. doi: 10.1038/nsmb1324. [DOI] [PubMed] [Google Scholar]
- 3.Terry LJ, Shows EB, Wente SR. Science. 2007;318:1412–1416. doi: 10.1126/science.1142204. [DOI] [PubMed] [Google Scholar]
- 4.Hocine S, Singer RH, Grunwald D. Cold Spring Harb. Perspect. Biol. 2010;2:a000752. doi: 10.1101/cshperspect.a000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lockhart DJ, Winzeler EA. Nature. 2000;405:827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- 6.Zenklusen D, Larson DR, Singer RH. Nat. Struct. Mol. Biol. 2008;15:1263–1271. doi: 10.1038/nsmb.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raj A, van Oudenaarden A. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyagi S. Nat. Methods. 2009;6:331–338. doi: 10.1038/nmeth.1321. [DOI] [PubMed] [Google Scholar]
- 9.Lim F, Downey TP, Peabody DS. J. Biol. Chem. 2001;276:22507–22513. doi: 10.1074/jbc.M102411200. [DOI] [PubMed] [Google Scholar]
- 10.Larson DR, et al. Science. 2011;332:475–478. doi: 10.1126/science.1202142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haim L, et al. Nat. Methods. 2007;4:409–412. doi: 10.1038/nmeth1040. [DOI] [PubMed] [Google Scholar]
- 12.Nissan TA, et al. Mol. Cell. 2004;15:295–301. doi: 10.1016/j.molcel.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 13.Gandhi SJ, et al. Nat. Struct. Mol. Biol. 2011;18:27–34. doi: 10.1038/nsmb.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holstege FC, et al. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 15.Lange S, et al. Traffic. 2008;9:1256–1267. doi: 10.1111/j.1600-0854.2008.00763.x. [DOI] [PubMed] [Google Scholar]
- 16.Elowitz MB, et al. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 17.Raser JM, O’Shea EK. Science. 2004;304:1811–1814. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darzacq X, et al. Nat. Struct. Mol. Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson P, Kedersha N. J. Cell Biol. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cajigas IJ, et al. Neuron. 2012;74:453–466. doi: 10.1016/j.neuron.2012.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MDN1-24PP7 labeled by 2xyEGFP (allele 1)
MDN1-24MS2 labeled by mCherry (allele 2)
H2A2-yECFP fusion (nucleus)
Merge
Induced cell 1: 5′ PP7
Induced cell 1: 3′ MS2
Induced cell 2: 5′ PP7
Induced cell 2: 3′ MS2