

Summary
Microtubule targeting agents are among the most widely used chemotherapeutics for both solid and hematological malignancies. This study characterizes the diaryl-oxazole based anticancer agent PC-046, which was originally identified for development based on selective activity in deleted in pancreas cancer locus 4 (DPC4/SMAD4) deficient tumors. PC-046 has growth inhibitory activity in a variety of tumor types in vitro, and efficacy in SCID mice was shown in human tumor xenografts of MV-4-11 acute myeloid leukemia, MM.1S multiple myeloma, and DU-145 prostate cancer. Pharmacokinetic studies demonstrated relatively high oral bioavailability (71%) with distribution to both plasma and bone marrow. No myelosuppression was seen in non-tumor bearing SCID mice given a single dose just under the acute lethal dose. The COMPARE algorithm in the NCI-60 cell line panel demonstrated that PC-046 closely correlated to other known tubulin destabilizing agents (correlation coefficients ≈ 0.7 for vincristine and vinblastine). Mechanism of action studies showed cell cycle arrest in metaphase and inhibition of tubulin polymerization. Overall, these studies show that PC-046 is a synthetically-derived, small molecule microtubule destabilizing agent. Advantages over existing microtubule destabilizing agents include ease of synthesis, lack of MDR cross-resistance, good oral bioavailability and the lack of acute myelotoxicity.
Keywords: PC-046, Diaryl oxazole, microtubule inhibitor, metaphase arrest
Introduction
Drug discovery based on synthetic lethality is a strategy designed to exploit characteristics that distinguish tumor cells from their normal counterparts [1]. Using isogenic models that differ by only a single mutation, this strategy can be used to identify agents that are highly selective for tumors bearing either gain-of-function or loss of function driver mutations. However, the majority of cell models and human tumors contain additional passenger mutations, and the selective activity of the agent may be unrelated to the intended genotype [2]. We have previously reported on a series of agents that were selected for cytotoxic activity in pancreatic adenocarcinoma cells with the DPC4 (deleted in pancreas cancer locus 4) deletion of the SMAD 4 tumor suppressor gene. A series of over 80 novel biaryl-oxazole-based compounds was synthesized and evaluated for anti-tumor activity. Within this group of compounds, a lead analog, PC-046 [5-(4-methoxyphenyl)pyridine-4-yl], was identified. This analog was quite potent in vitro but produced only a 1.7-fold selectivity for inhibiting growth of the DPC4 (-/-) BxPC3 pancreatic cancer cell line compared to the DPC4 (+/+) isogenic cell line. PC-046 was active in SCID mice bearing flank tumor xenografts of the human MiaPaCa-2 pancreatic cancer cell line [3]. Mechanistically, PC-046 was shown to inhibit several cancer-relevant kinases in vitro, including TRK-B, PDGF-alpha and -beta, and FLT-1 at pharmacologically-achievable concentrations. However, PC-046 also caused human pancreatic cancer cells to accumulate in G2/M phases, suggesting the mechanism of action was not related to either DPC4 status or kinase inhibition. The fact that PC-046 treated cells were consistently inhibited from progressing through mitosis suggested that other mechanisms of action should be considered, and efficacy studies expanded beyond DPC4 dependent tumors.
In the current report, we evaluate PC-046 in several human tumor cell lines in vitro and determine the mechanism of action in hematopoietic cell lines. In addition, we have extended the observations of in vivo activity to several hematologic-derived human tumors in SCID mice. Pharmacokinetic studies comparing three different routes of administration suggest that oral bioavailability is quite good for the compound, and we believe these features support further development of this compound as a potential therapeutic for treating cancer.
Materials and Methods
Drugs and chemicals
The synthesis of PC-046 is reported elsewhere [3]. For in vitro studies, PC-046 stock is prepared in DMSO and subsequently diluted into aqueous media at a final working concentration of less than 0.1% DMSO. For in vivo studies, PC-046 is prepared and administered in 100% DMSO. Caspase inhibitors were obtained from BioVision (Milpitas, CA).
NCI-60 Developmental Therapeutics Program (DTP) human tumor cell line screen
Additional in vitro screening of PC-046 was performed at NIH using the NCI-60 panel of human tumor cell lines [4]. Briefly, cells are seeded on 96-well plates and one set of controls are incubated for one day to determine a time zero density, which is used to calculate cell kill as well as net growth inhibition. The other plates are exposed to the test drug over a 5-log concentration range (or no drug for the controls), and incubated for 2 further days. The plates are then stained with sulforhodamine B (SRB) to determine the viable cell number. Growth inhibition is measured relative to untreated control cells.
Human tumor cell lines
The following cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA): multiple myeloma RPMI 8226 (CCL-155), NIH-H929 (CRL-9068), and U266 (TIB-196); B myelomonocytic leukemia MV-4-11 (CRL-9591); pancreatic MiaPaCa-2 (CRL-1420), Panc-1 (CRL-1469), and BxPC-3 (CRL-1687); mammary adenocarcinoma MCF7 (HTB-22) and MDA-MB231 (HTB-26); colorectal HCT-116 (CCL-247); and prostate DU-145 (HTB-81) and PC-3 (CRL-1435). Drug resistant variants of parental RPMI 8226 (8226/S, sensitive) included the 8226/IM10 cell line, which was selected for resistance to imexon [5] and the 8226/Dox40 cell line, generously provided by Dr. W.S. Dalton (Moffit Cancer Center, Tampa, FL), which overexpresses the MDR-1 multi-drug resistance protein [6]. The MM.1S myeloma cell line was a gift from Dr. ST Rosen (Robert H. Lurie Comprehensive Cancer Center, Chicago, IL) [7]. The acute myelogenous leukemia line, OCI-AML-3 [8], was provided by the Experimental Mouse Shared Service (EMSS) of the University of Arizona Cancer Center (Tucson, AZ). The HCT-116 DPC4 (-/-) line is an isogenic clone of the HCT-116 colorectal line wherein the DPC4 gene has been deleted by homologous recombination in the Vogelstein laboratory at Johns Hopkins University, Baltimore, MD [9]. The small cell lung cancer cell line H69S and its variant, the multi-drug resistant H69AR (doxorubicin-resistant) cell lines were obtained from ATCC. All cells were cultured in RPMI 1640 (MediaTech Inc., Manassas, VA) supplemented with 10% heat-inactivated bovine calf serum and 2 mM L-glutamine (both from Life Technologies, Grand Island, NY). Additionally, the NIH-H929 cells require 0.05 μM β-mercaptoethanol supplementation (Sigma Chemical Co.).
Genetic authentication of cell line identities
Cell line identities were confirmed by autosomal short tandem repeat (STR) analyses [10] by the University of Arizona Genetics Core (Tucson, AZ) within the last 6 months. All cells were negative for mycoplasma (MycoAlert™, Lonza, Walkersville, MD).
In vitro growth inhibition assays
Growth inhibition was assayed by MTT dye reduction according to the method of Mosmann [11], as previously described.
Tubulin Polymerization Assay
A cell-free tubulin polymerization assay [12, 13] was performed following manufacturer’s instructions (Cytoskeleton, Denver, CO). Briefly, a kinetic assay which measures the characteristic tubulin polymerization phases (nucleation, growth, and steady state) was performed in the presence of 100 nM, 1 μM, 5 μM, or 10 μM PC-046. Paclitaxel, 10 μM (Bristol-Myers Squibb, New York, NY), vincristine, 10 μM (Sigma Chemical Co.) and colchicine, 5 μM (Sigma Chemical Co.), were used as controls. The polymerization reaction was measured every minute for 60m at 340 nm using a Gemini XPS microplate reader (Molecular Devices; Sunnyvale, CA). All time points were plotted using SoftMax Pro® software (Molecular Devices).
Cell cycle analyses by flow cytometry
Cells were incubated with 0, 25, 50 or 100 nM of PC-046 for 24h, collected, washed with phosphate-buffered saline (PBS), and fixed with 70% ethanol. Cells were incubated with 0.04 mg/ml of propidium iodide (Sigma Chemical Co., St. Louis, MO) and 0.5 mg/ml RNAse A (Sigma Chemical Co.) for 30m at 37° C. DNA content was analyzed using a Becton Dickinson FACScan (Becton Dickinson, San Jose, CA) and analyzed using Mod-Fit (Verify Software, Topsham, ME). Data are mean ± SEM (n=3).
Measurement of mitotic index by flow cytometry
The mitotic index, and specifically the population of cells in metaphase (phospho-histone H3 positive cells) was analyzed by flow cytometry using a phospho-histone H3 (Ser10) antibody (Cell Signaling, Danvers, MA) and goat anti-rabbit Alexa 488 secondary (Invitrogen Life Technologies, Grant island, NY). The mitotic index measurement procedure was modified from Muehlbauer and Schuler [14].
Western blot analyses of caspases 3, 8 and 9
Cells were treated for 24h with 0 – 500 nM of PC-046 and probed for the activation of caspases 3, 8, and 9 (Cell Signaling, Danvers, MA). Cell lysates were prepared using M-Per lysis buffer (Thermo-Fisher, Rockford IL) with the addition of protease and phosphatase inhibitors. Proteins were quantitated using bicinchoninic acid (BCA) (Thermo-Fisher, Rockford IL), separated by SDS-PAGE, and immunoblotted using standard techniques. β-actin was used as a loading control (Sigma Chemical Co., St. Louis, MO).
Measurement of apoptosis and necrosis by flow cytometry
Apoptosis and necrosis were measured by flow cytometry using concurrent staining by AnnexinV-Alexa488 (Invitrogen) and propidium iodide (PI) [Sigma Chemical Company] after 24hr exposure to 0 – 200 nM of PC-046. Thirty minutes prior to PC-046 exposure, 5 μM of the caspase 8 inhibitor Z-IETD-FMK (BioVision), the pan-caspase inhibitor Q-VD-OPh (BioVision), or a negative control caspase inhibitor (BioVision) was added. Fluorescence was measured on a Becton Dickinson FACScan using CellQuest Pro® software (Becton Dickinson). Unstained cells are deemed alive, Alexa488 positive cells are deemed apoptotic, and dual Alexa488/PI stained cells are deemed necrotic.
Alkylating Activity Assay
To rule out direct binding to DNA, alkylating activity was assayed using a modification of the Friedman and Bogers’ nitrobenzylpyridine assay [15, 16], as described previously [17].
In vivo anti-tumor efficacy in human tumor xenografts in SCID mice
All studies were done according to the Institutional Animal Care and Use Committee (IACUC) and institutional regulations. Six to seven week old male SCID mice were inoculated subcutaneously in the right flank with either myeloma (8226/S, MM.1S), leukemia (MV-4-11, OCI-AML-3) or prostate (DU-145, PC-3) tumor cells. Once the mean tumor volume reached approximately 100 mm3, PC-046 treatment was initiated, with the exception of the MM.1S study, where drug treatment was initiated the day after tumors were inoculated. PC-046 was administered as described in Table 2. Tumor size, body weight, and general health of each mouse was recorded every third day for the duration of the study. To determine if PC-046 treatment was effective in delaying tumor growth, the area under the curve (AUC) was determined using the trapezoidal rule. The distributions of raw values of AUC were skewed and were log transformed to induce a normalized distribution. Once normalized, the AUC data was analyzed for each experiment by treatment group, using a 2 sample t-test.
Table 2.
Antitumor efficacy of PC-046 against human tumor cell lines
| PC-046 Administration | Human Tumor Cell Line | Mean (SD), mm3 | Tumor AUC | |||
|---|---|---|---|---|---|---|
| Dose (mg/kg) | Schedule | Tumor Type | Control | DPC046 | % Change | p-value |
| 55 | Dailyx5, off 5d, repeat 1× | OCI-AM-3 (Acute Myeloid Leukemia) | 17752.6 (8665.6) | 12433.4 (4647.1) | −29.90% | NS |
| 55 | Dailyx5, off 5d, repeat 2× | MV-4-11 (Acute Myeloid Leukemia) | 18702.7 (7654.1) | 11309.3 (5293.3) | −39.50% | 0.0374 |
| 55 | Dailyx5, off 5d, repeat 1.5× | MM.1S1 (Multiple Myeloma) | 18214.4 (6630.0) | 9342.1 (4514.3) | −48.70% | 0.0013 |
| 50 | Every third day, repeat 10× | 8226/S (Multiple Myeloma) | 27292.6 (13688.8) | 28516.9 (9067.3) | +4.5% | NS |
| 55 | Dailyx5, off 7d, repeat 2× | DU-145 (Prostate Cancer) | 69969.6 (22735.3) | 45801.8 (23344.4) | −34.50% | 0.04886 |
| 55 | Dailyx5, off 5d, repeat 1× | PC-3 (Prostate Cancer) | 10438.6 (4303.9) | 8238.1 (2506.2) | −21.10% | NS |
Drug treatment initiated the day after tumor cells were injected; all others dosing started when average tumor volume reached 100 mm3
Pharmacokinetic studies in normal SCID mice
Pharmacokinetics of a single 100 mg/kg dose of PC-046 in non-tumor bearing SCID mice was analyzed by reversed phase chromatography and tandem mass spectrometry [3]. PC-046 was administered intravenously (IV), intraperitoneally (IP) or orally (PO) to compare bioavailability of different routes of administration. Plasma samples were collected 5, 10, 15, 30, 60, 120, 240, 480 or 1440 minutes after administration of PC-046. The DCP-046 concentration-time data (half-life, area under the plasma concentration-time profile (AUC), systemic clearance, and volume of distribution) were analyzed by the non-compartmental approach using WIN-NONLIN Version 5.2 (Pharsight Corporation, St. Louis, MO). Data are mean (SEM), n = 4. The variance in the AUC was estimated using the method of Yuan [18]. Plasma and bone marrow PC-046 levels were measured 15, 30, 45, and 60m after administration of a single 100 mg/kg IP dose of PC-046. Bone marrow was collected by flushing each femur with 100 μl of 0.9% NaCl. The collected mixture (bone marrow flushings) from bone femurs were combined and reported as total PC-046 (μg/ml) per mouse.
Statistical Methods
The COMPARE analysis performed at the NCI uses data from the patterns of growth inhibition response in the 60 cell line panel for the test agent compared to those same patterns stored in the database for known anti-tumor agents. The statistical comparison is summarized as Pearson Correlation Coefficients. In this analysis, compounds with identical patterns of growth inhibitory response would have a coefficient of 1.00 and exactly opposite agents would have a coefficient of −1.00. Correlation coefficients approaching 0.6 to 0.7 are felt to signify mechanistically related compounds [4].
Results
Cytotoxicity (MTT) results against human cancer cell lines
A total of 17 human cell lines representative of 6 tumor types were incubated with PC-046 for 72h and growth inhibition was measured by MTT dye reduction assay. Multiple tumor types were chosen to expand from the prior work that was limited to three pancreatic adenocarcinoma cell lines: BxPC-3, MiaPaCa-2 and Panc-1 [3]. The results, summarized in Table 1, show that the HCT116 DPC4 (-/-) colon cancer cell line was the most sensitive with an IC 50 of 52.0 nM, compared to the 99.5 nM in the parental HCT116, similar to the 1.74x selectivity index previously reported [3]. The multidrug resistant 8226/Dox40 myeloma cell line showed a slight but non-significant resistance compared to the parental 8226/S (p=0.0544), whereas the MRP over-expressing H69AR cells were more sensitive to PC-046 (Table 1). Importantly, all of the IC50 values in Table 1 are significantly lower than the peak plasma concentrations of 1.7 to 4.0 μg/mL, (4.4 to 11 μM) that are achieved in mice given tolerable PC-046 doses in vivo (see pharmacokinetic results, below).
Table 1.
Comparison of PC-046 Cytotoxicity in Human Cancer Cell Lines, In Vitro
| Cell Type | Cell Line | IC50, nM (Mean ± SEM) | ||||
|---|---|---|---|---|---|---|
| Myeloma: | 8226/S | 134.2 ± 13.8 |
|
p=0.60 |
|
p=0.0544 |
| 8226/IM10 | 102.0 ± 27.5 | |||||
| 8226/Dox40 | 188.4 ± 6.1 | |||||
| NIH-H929 | 163.1 ± 13.9 | |||||
| MM.1S | 162.1 ± 39.6 | |||||
| U266 | 70.1 ± 16.2 | |||||
| Leukemia: | OCI-AML3 | 176.9 ± 31.8 | ||||
| MV-4-11 | 192.6 ± 10.8 | |||||
| Pancreatic: | MiaPaCa-2 | 295.5 ± 72.1 | ||||
| Panc-1 | 214.8 ± 49.4 | |||||
| BxPC-3 | 198.7 ± 48.2 | |||||
| Breast: | MDA-MB231 | 206.2 ± 49.2 | ||||
| MCF-7 | 165.6 ± 26.3 | |||||
| Prostate: | PC-3 | 105.4 ± 30.2 | ||||
| DU-145 | 159.9 ± 39.4 | |||||
| Colon: | HCT-116 | 99.5 ± 15.8 |
|
p=0.0212 | ||
| HCT-116 DPC4 (−/−) | 52.0 ± 5.3 | |||||
| Lung: | H69S | 240.5 ± 10.9 |
|
p=0.0001 | ||
| H69AR | 490.5 ± 3.6 | |||||
COMPARE analysis
The COMPARE algorithm, developed at NCI, is a tool that allows individual cell line data to be used to identify a pattern of response to a drug in the NCI 60 cell lines. The sensitivity pattern is then compared to the patterns in the data bank for known anticancer agents, which can be used to identify similar mechanisms of action [4]. COMPARE analyses of PC-046 showed close correlations with known microtubule inhibitors. For example, for the 50% Growth Inhibition (GI50) endpoint, the top 3 correlation coefficients were 0.624 for vincristine, 0.604 for vinblastine and 0.56 for maytansine. For the Total Growth Inhibition (TGI) endpoint, which measures a drug’s cytostatic activity, the top 3 correlation coefficients with PC-046 were 0.758 for maytansine, 0.70 for vinblastine and 0.695 for vincristine. Overall, these analyses strongly support a mechanistic similarity for PC-046 with established natural product-derived microtubule destabilizing compounds. Complete data is available at http://dtp.cancer.gov (NTP compound: NSC 756784).
Tubulin polymerization
To examine to effect of PC-046 on tubulin, we used an in vitro cell-free tubulin polymerization assay [12, 13]. The effect of PC-046 was to block polymerization which was identical to that of colchicine and vincristine, indicating that PC-046 inhibits tubulin polymerization in vitro (Fig. 1). Control experiments demonstrated that paclitaxel stabilized tubulin polymerization, as expected (Fig. 1). Colchicine, by binding to tubulin, prevents microtubule polymerization, and vincristine, by destabilizing the microtubule, results in a flattened curve, indicating a reduction in the amount of tubulin polymer formed.
Fig. 1.

PC-046 inhibits tubulin polymerization in vitro. A kinetic tubulin polymerization assay was performed in the presence of PC-046 (100 nM, 1 μM, 5 μM, or 10 μM), 10 μM paclitaxel, 10 μM vincristine, 5 μM colchicine, or DMSO. Tubulin polymerization phases (nucleation, growth, or steady state) were measured every minute for 60m at 340 nm. A representative figure showing the inhibition of tubulin polymerization using 5 μM of PC-046 is shown
Cell cycle effects; arrest in metaphase
The previous study of PC-046 in pancreatic cancer cells detected an increase in the proportion of cells in S-phase and in the G2/M-phase fraction [3]. In the current study, flow cytometry was used to determine the DNA content of human multiple myeloma cell lines. Incubation with PC-046 for 24h induces an accumulation of cells in the G2/M-phase of three of the four cell lines examined (Fig. 2a [H929], 2b [MM.1S] and 2c [U266]). In contrast, in the 8226/S myeloma cell line, there was significant accumulation of cells in S-phase (Fig. 2d). In additional studies, we used higher concentrations of PC-046 (150 – 250 nM) for 16h in the MM.1S and 8226/S multiple myeloma cell lines to examine the per cent of phospho-histone H3 positive cells in metaphase. After exposure to 150 nM PC-046 for 16h, the % positive histone H3 cells increased from a mean (SEM) at baseline of 3.31 (0.14) to 34.70 (5.84) in the 8226/S cells (Fig. 2e) and from a baseline of 3.14 (0.15) to 17.3 (1.28) in the MM.1S cells (Fig, 2f). The colcemid-treated 8226/S cells showed a % increase to 39.40 (1.48), and in the MM.1S cells, an increase to 12.54 (0.73). Comparing this data to that presented in Fig. 2 shows that almost all the cells accumulating in the G2/M fraction following drug exposure are halting their cell cycle progression in the metaphase portion of mitosis.
Fig. 2.

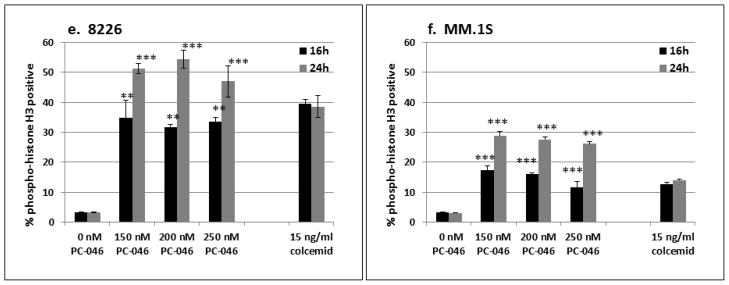
PC-046 causes arrest in G2/M-phase. (a) H929, (b) MM.1S, (c) U266 or (d) 8226/S cells were incubated for 24h with increasing doses of PC-046 and cell cycle measured by propidium iodide staining. Results shown are mean (SEM), n=3.
(e,f) Cell cycle progression is halted in metaphase. 8226/S and MM.1S cells were incubated for 16h with PC-046 and stained for phospho-histone H3 positive cells. The per cent of phospho-histone H3 positive cells in G2/M phase are shown. Results shown are mean (SEM), n=3
** p < 0.01, ***p < 0.001
Western blot of caspase inhibition in MM.1S cells
The cytotoxic activity of microtubule inhibitors is often described as “mitotic catastrophe” which can be caspase independent. To investigate the role of caspase activity in PC-046 mediated cell death, we first examined caspase activity in myeloma cells incubated with PC-046 for 24h. Western blot analysis shows cleavage of three major caspases, 3, 8, and 9, representing both intrinsic and extrinsic apoptotic pathways. This suggested that both the mitochondrial and death receptor (extrinsic) apoptotic pathways might be involved in mediating the apoptotic signal for PC-046. To determine the involvement of each pathway, PC-046-treated 8226/S and MM.1S multiple myeloma cells were co-incubated with 5 μM of the cell permeable pan-caspase inhibitor Q-VD-OPh [19], the cell permeable caspase 8 inhibitor Z-IETD-FMK [18] or a caspase inhibitor negative control for 30m prior to the addition of PC-046. After 24h of PC-046 treatment, cells were evaluated for apoptotic markers using Annexin V and propidium iodide. These studies showed that the pan-caspase inhibitor blocked PC-046-induced apoptosis, reducing the AnnexinV-positive cells from 30.7% to 13.5% (Fig. 3b, 8226/S) and from 32.6% to 12.8% (Fig. 3c, MM.1S). In contrast, the caspase 8 inhibitor did not affect Annexin V-staining, 25.1% (Fig. 3b, 8226/S) and 32.6% (Fig. 3c, MM.1S). This finding suggests that although caspase 8 is modestly activated by PC-046, its activation is not required for propagating the apoptotic signal.
Fig. 3.


PC-046 activates caspases 3, 8 and 9. (a) MM.1S cells were treated for 24h with 0 – 500 nM of PC-046 and probed for the activation of caspases 3, 8 and 9 by western blot. β-actin was used as a loading control. (b, c) 8226/S or MM.1S cells were pre-treated for 30m with 5 μM of the caspase 8 inhibitor Z-IETD-FMK, the pan-caspase inhibitor Q-VD-OPh, or a negative control caspase inhibitor prior to exposure to 0 – 200 nM PC-046 for 24h. Cell death was measured by AnnexinV/PI staining. Results for exposure to 200 nM PC-046 are shown
Efficacy in human hematologic and prostate cancer tumor xenografts in SCID mice
Based on the previous SCID mouse work in human pancreas cancers [3], the dose of IP-administered PC-046 was limited to the range of 50–55 mg/kg, on a schedule of daily × 5 days, then 5 to 7 days off, and repeated for a total of 2–3 courses. On this regimen, the mice routinely lost about 10% of body weight over the repeated courses, but there was no lethality. A total of six human tumor cell lines were studied as flank tumors in SCID mice treated with PC-046. The results, summarized in Table 2, show that statistically-significant reductions in tumor growth were achieved in 3 of the 6 human tumor xenografts. These include 1 of 2 acute myeloid leukemia cell lines (MV-4-11 cells), 1 of 2 multiple myeloma cell lines (MM.1S cells), and 1 of 2 prostate cancer cell lines (DU-145 cells). Growth inhibition averaged about 35% in 5 of the xenograft experiments with the exception of the 8226/S myeloma cell line wherein no growth inhibition was observed in vivo.
Hematologic toxicity SCID mice
Myelosuppression is a common dose-limiting effect for most microtubule inhibitors. To determine the hematologic toxicity of PC-046, blood samples were taken 1, 3, and 5 days after administration from non-tumor bearing mice treated with a single 100 mg/kg or 150 mg/kg dose of PC-046. The samples were analyzed for concentrations of erythrocyte levels and indices, white blood cells, including total WBC, lymphocytes and leukocytes, and the platelet count. In short, there were no significant changes in the levels of any formed blood elements at any of the three time points (data not shown). The time points were chosen to include the typical nadir time of 1–3 days following classic myelosuppressive anti-cancer drugs in mice [20]. Similarly, the drug doses were chosen to include an acutely tolerated dose of 100 mg/kg and a lethal acute dose of 150 mg/kg, wherein mice typically die with 2–5 days after administration. This suggests that the dose-limiting toxicity of PC-046 does not involve acute myelosuppression.
Pharmacokinetics of PC-046 by three routes of administration
To examine the pharmacokinetics of PC-046, non-tumor bearing SCID mice (n =4/time point), were administered 100 mg/kg PC-046 by three different routes of administration: IV, IP and by oral gavage. The mean AUC’s for the IV route (5.02 hr*μg/mL) and the IP route (5.22 hr*μg/ml) were nearly identical. The AUC for the oral route of administration was 3.55 hr* μg/mL which represents surprisingly good oral bioavailability of 71% compared to the IV route. Other pharmacokinetic parameters for the three routes are summarized in Table 3. Because of the relative lack of significant myelosuppression from PC-046, a bone marrow drug distribution study was performed in the femurs of the same non-tumor bearing SCID mice given 100mg/kg IP. The femurs were removed at 30 and 60 minutes following drug injection by surgically removing the femurs, clipping the distal end and then flushing the marrow space with 100 μL of saline. The mean PC-046 concentrations in the femurs at 30 and 60 minutes were approximately 0.07 μg/mL (Supplemental Fig. 1). This compares to mean 15, 30, 45 and 60 minute plasma concentrations of 6.22, 1.75, 1.93 and 0.962 μg/mL, suggesting that drug penetration into the marrow is limited (Supplemental Fig. 1). On the other hand, the femur concentrations at 30 and 60 minutes (0.07 μg/mL or 195 nM), are comparable to the IC50 values for all the cell lines tested in vitro, which ranged from ≈ 50 to 300 nM for a 72 hr exposure (Table 1). Thus, cytotoxic PC-046 concentrations were achieved in the bone marrow compartment, but this caused no significant hematopoietic toxicity, at least after a single dose of the drug.
Table 3.
Pharmacokinetic parameters in mice given 100mg/kg of PC-046 by 3 routes of administration
| Pharamacokinetic Parameter (units) | Mean values by route of administration | ||
|---|---|---|---|
| IV | IP | PO | |
|
| |||
| Cmax (μg/mL) | 1.66 | 4.01 | 1.55 |
| Tmax (hr) | 0.17 | 0.08 | 0.08 |
| T ½ (hr) | 8.40 | 5.17 | 3.17 |
| AUC (hr* μg/mL) | 5.02 (0.29) # | 5.22(0.68) # | 3.55(0.75) # |
| Clearance (L/hr/kg) | 19.9 | --- | --- |
| Vol. Dist. (L/kg) | 240.3 | --- | --- |
| F (bioavailabilty) | --- | 1.04 | 0.71 |
Variance calculated as per Yuan [18]
Discussion
Microtubule inhibitors are a mainstay of clinical oncology practice, with commercial availability of three vinca alkaloids (vincristine, vinblastine, vinorelbine), four taxanes (paclitaxel, nab-paclitaxel, docetaxel and cabazitaxel) and one each, a Halichondrin B analog (eribulin mesylate) and an epothilone (ixabepilone) [21]. These agents are routinely used for the treatment of solid tumors and hematological malignancies. Within this broad group of microtubule inhibitors there are two basic mechanisms of action resulting from tubulin binding: (a) compounds that inhibit tubulin assembly, termed microtubule destabilizing agents such as the vinca alkaloids, and (b) compounds that prevent microtubule disassembly, termed microtubule stabilizers, such as the taxanes [21]. Despite these mechanistic differences there is significant overlap in terms of the dose-limiting clinical toxicities, which include myelosuppression and cumulative-dose related peripheral neuropathy. None of the existing microtubule inhibitors can be reliably delivered orally, necessitating intravenous administration. Another common undesirable feature of microtubule inhibitors is intrinsic or acquired resistance, most often mediated by the MDR1 gene product. This membrane-associated ATP-binding cassette transporter operates as an active drug efflux pump with broad overlap for substrates derived from natural sources, including the vinca alkaloids and the taxanes [22].
Another significant problem with existing natural-product based microtubule inhibitors involves the lack of simple, high-yield, chemical synthesis methods, since all of the existing natural product-derived agents have very complex structures. This necessitates isolation of precursor molecules from natural sources followed by complex isolation, purification and multistep chemical modifications to yield the final active pharmaceutical ingredient. In addition, many of these agents lack good water solubility and thereby require complex and sometimes toxic solvent systems to facilitate intravenous administration.
In the current report, we have described the mechanism of action, pharmacokinetics and anti-tumor efficacy for a novel small-molecule inhibitor of microtubule assembly. Differentiating properties of PC-046 from existing agents include many of the features lacking in the microtubule inhibitor class, as outlined above. First, the synthesis of PC-046 is a 4-step process with reasonable yields at each step [3]. Further, the structure of the drug is quite distinct from that of existing vinca alkaloids and colchicine-like molecules, such as colcemid, which all have prototypic trimethoxy benzyl moieties not found in PC-046. As such, PC-046 does not appear to be a substrate for the classic multidrug resistance protein ABCB1 (Pgp/MDR1) which confers approximately 200 fold resistance to vincristine in the 8226/Dox40 cell line. PC-046 was similarly unaffected by overexpression of the alternate multidrug resistance protein (MRP, ABCC-2 or c-MOAT). The drug also adheres to Lipinski’s “rule of five” for chemicals that have drug-like properties [23]: (a) the lipophilicity or cLog P of 3.28 is < 5, (b) the molecular weight, at 358.13, is < 500 daltons, (c) there are less than ten oxygen and nitrogen atoms (1 O and two N atoms), (d) there are < 5 hydrogen donors. Finally, (e) we have demonstrated that oral absorption, pharmacokinetic distribution, and lack of myelosuppessive properties are favorable.
The most responsive human tumor cell line to PC-046 in the current study was the HCT-116 colon cancer cell line which has the DPC4 (SMAD4) homozygous (-/-) deletion (Table 1). This is not surprising because this agent was synthesized and selected for further study following a campaign to identify novel new structures that had selective activity for Bx-PC3 pancreatic cancer cells with the DPC4 deletion [3]. The roughly 2-fold increased activity for the DPC4 (-/-) HCT-116 cell line, compared to the wild type HCT-116 cells, is relatively low, similar to the previous results with the DPC4 deficient pancreatic cells [3]. The molecular or mechanistic explanation for this DPC4 (-/-) selective action is not known. However, the modest degree of selectivity does not suggest that this agent should be specifically targeted to DPC4 (-/-) tumors in future development.
The current report substantially refines the mechanism of action for PC-046 following the prior work that showed inhibition of some cancer-relevant kinases, but no firm mechanism of action at the molecular level. The new data shows that PC-046 is a microtubule destabilizing agent. The overall effect of microtubule depolymerization comports well with: (a) flow cytometry data showing that PC-046 stops cell cycle progression in the metaphase portion of mitosis, and (b) with the observation that the primary mechanism of cell death with PC-046 is apoptosis mediated through the intrinsic or mitochondrial pathway. It is of interest that there appeared to be activation of both the mitochondrial apoptosis pathway and to a lesser extent the extrinsic, death receptor pathway mediated by caspase 8. However, pharmacologic inhibition of caspase 8 did not protect cells from PC-046-induced apoptosis, whereas the addition of a pan-caspase inhibitor efficiently blocked PC-046-induced apoptosis. This suggests that caspase 8 may not be directly activated by the drug, but rather, may be involved in feedback loop amplification of the death signal.
The effects of PC-046 in SCID mice showed that the drug produced statistically significant tumor growth inhibition in 2 of 4 human hematologic cancer cell lines (MV-4-11 acute myeloid leukemia and MM.1S multiple myeloma) and in one of two prostate cancer cell lines (DU-145). It is important to note that drug dosing was only begun after tumors were palpable, so the effects can be construed as representing reductions in tumor growth rather than the prevention of tumor development. Importantly, the activity seen in the three human tumors in the SCID mice parallels the type of tumors known to be sensitive to microtubule inhibiting drugs in human patients. Perhaps the most interesting mouse work with PC-046 was the observation of surprisingly good, 71%, oral bioavailability, with a simple oral formulation, and the fact that myelosuppression was not seen after acute high doses of the drug were administered by the IP route. The additional PK studies of drug distribution into mouse bone marrow suggests that the marrow is exposed to active drug levels, which is not surprising given the relative lipophilicity of the molecule. Therefore, the lack of acute bone marrow toxicity with PC-046 marks a significant improvement over other microtubule inhibitors such as vinblastine and the taxanes [21]. Overall, these features augur well for further preclinical development of PC-046 as a unique microtubule destabilizing small molecule and the compound is currently being evaluated at the NCI in the hollow fiber in vivo anti-tumor assay.
Supplementary Material
Distribution of PC-046 in (a) plasma or (b) bone marrow after a single 100 mg/kg dose by the IP route. Total bone marrow PC-046 concentration is presented (combined right and left bone marrow flushings). Results shown are mean (SEM), n=4
{kind=link}
Acknowledgments
This work was supported by grants PO1 CA109552 (DD Von Hoff) and P30 CA23074 from the National Cancer Institute, National Institutes of Health, Bethesda MD, USA. We would like to thank Vijay Gokhale, PhD., for synthesis and validation of PC-046. We would like to acknowledge the following University of Arizona Cancer Center core services: Flow Cytometry, Analytical Core, Experimental Mouse Shared Service, and the Biostatistical Core, and the University of Arizona Genetics Core for their services. The NCI-60 screening and COMPARE analysis was performed by the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
Contributor Information
Terry H. Landowski, Email: tlandowski@uacc.arizona.edu.
Betty K. Samulitis, Email: bsamulitis@uacc.arizona.edu.
Robert T. Dorr, Email: bdorr@email.arizona.edu.
References
- 1.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–8. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 2.Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011;10:351–64. doi: 10.1038/nrd3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaw AY, Henderson MC, Flynn G, Samulitis B, Han H, Stratton S, et al. Characterization of novel diaryl oxazole-based compounds as potential agents to treat pancreatic cancer. J Pharmacol Exp Ther. 2009;331:636–647. doi: 10.1124/jpet.109.156406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holbeck SL, Collins JM, Doroshow JH. Analysis of Food and Drug Administration-approved anticancer agents in the NCI-60 panel of human tumor cell lines. Mol Cancer Ther. 2010;9:1451–1460. doi: 10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dvorakova K, Payne CM, Tome ME, Briehl MM, Vasquez MA, Waltmire CR, et al. Molecular and cellular characterization of imexon-resistant RPMI 8226/I myeloma cells. Mol Cancer Ther. 2002;1:185–195. [PubMed] [Google Scholar]
- 6.Dalton WS, Durie BG, Alberts DS, Gerlach JH, Cress AE. Characterization of a new drug-resistant human myeloma cell line that express P-glycoprotein. Cancer Res. 1986;46:5125–5130. [PubMed] [Google Scholar]
- 7.Goldman-Leikin RE, Salwen HR, Herst CV, Variakojis D, Bian ML, Le Beau MM, et al. Characterization of a novel myeloma cell line, MM. 1. J Lab Clin Med. 1989;113:335–345. [PubMed] [Google Scholar]
- 8.Rayappa C, McCulloch EA. A cell culture model for the treatment of acute myeloblastic leukemia with fludarabine and cytosine arabinoside. Leukemia. 1993;7:992–999. [PubMed] [Google Scholar]
- 9.Zhou S, Buckhaults P, Zawel L, Bunz F, Riggins G, Dai JL, et al. Targeted deletion of Smad4 shows it is required for transforming growth factor b and activin signaling in colorectal cancer cells. Proc Natl Acad Sci USA. 1998;95:2412–2416. doi: 10.1073/pnas.95.5.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins PJ, Hennessy LK, Leibelt CS, Roby RK, Reeder DJ, Foxall PA. Developmental validation of a single-tube amplification of the 13 CODIS STR loci, D2S1338, D19S433, and amelogenin: the AmpFℓSTR® Identifiler PCR Amplification Kit. J Forensic Sci. 2004;49:1265–1277. [PubMed] [Google Scholar]
- 11.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 12.Shelanski ML, Gaskin F, Cantor CR. Microtubule assembly in the absence of added nucleotides. Proc Natl Acad Sci USA. 1973;70:765–768. doi: 10.1073/pnas.70.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JC, Timasheff SN. In vitro reconstitution of calf brain microtubules: effects of solution variable. Biochemistry. 1977;16:1754–1762. doi: 10.1021/bi00627a037. [DOI] [PubMed] [Google Scholar]
- 14.Muehlbauer PA, Schuler MJ. Measuring the mitotic index in chemically-treated human lymphocyte cultures by flow cytometry. Mutat Res. 2003;537:117–130. doi: 10.1016/s1383-5718(03)00076-7. [DOI] [PubMed] [Google Scholar]
- 15.Friedman DM, Boger E. Colorimetric estimation of nitrogen mustards in aqueous media. Anal Chem. 1961;33:906–910. [Google Scholar]
- 16.Christian RA, Chaffee SK, Hovick CJ, Steele WJ. A stable colorimetric assay for cyclophosamide and its alkylating metabolites based on the alkylation of 4-(4′-nitrobenzyl)-pyridine. Life Sci. 1980;27:2595–2599. doi: 10.1016/0024-3205(80)90545-7. [DOI] [PubMed] [Google Scholar]
- 17.Dorr RT, Wisner L, Samulitis BK, Landowski TH, Remers WA. Anti-tumor activity and mechanism of action for a cyanoaziridine-derivative, AMP423. Cancer Chemother Pharmacol. 2012;69:1039–1049. doi: 10.1007/s00280-011-1784-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan J. Estimation of variance for AUC in animal studies. J Pharm Sci. 1993;82:761–763. doi: 10.1002/jps.2600820718. [DOI] [PubMed] [Google Scholar]
- 19.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8:345–352. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- 20.Gregoli PA, Bondurant MC. Function of caspases in regulating apoptosis caused by erythropoietin deprivation in erythroid progenitors. J Cell Physiol. 1999;178:133–143. doi: 10.1002/(SICI)1097-4652(199902)178:2<133::AID-JCP2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 21.Lord BI, Woolford LB, Molineux G. Kinetics of neutrophil production in normal and neutropenic animals during the response to filgrastim (r-metHu G-CSF) or filgrastim SD/01 (PEG-r-metHU G-CSF) Clin Cancer Res. 2001;7:2085–2090. [PubMed] [Google Scholar]
- 22.Perez EA. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol Cancer Ther. 2009;8:2086–2095. doi: 10.1158/1535-7163.MCT-09-0366. [DOI] [PubMed] [Google Scholar]
- 23.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10:194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 24.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of PC-046 in (a) plasma or (b) bone marrow after a single 100 mg/kg dose by the IP route. Total bone marrow PC-046 concentration is presented (combined right and left bone marrow flushings). Results shown are mean (SEM), n=4