

Abstract
In this review, voltage-gated proton channels are considered from a mainly teleological perspective. Why do proton channels exist? What good are they? Why did they go to such lengths to develop several unique hallmark properties such as extreme selectivity and ΔpH-dependent gating? Why is their current so minuscule? How do they manage to be so selective? What is the basis for our belief that they conduct H+ and not OH–? Why do they exist in many species as dimers when the monomeric form seems to work quite well? It is hoped that pondering these questions will provide an introduction to these channels and a way to logically organize their peculiar properties as well as to understand how they are able to carry out some of their better-established biological functions.
Keywords: HV1, HVCN1, ion channels, voltage sensing, permeation, selectivity
1. Introduction
Voltage-gated proton channels (HV) are membrane proteins that mediate the rapid movement of protons (H+) across cell membranes while excluding all other ions. Like other ion channels, the proton channel adopts two functional configurations. When ‘open’, each channel allows up to 105 H+ to cross the membrane per second but when they are ‘closed’, they do not allow any ions to cross the membrane. They are ‘voltage-gated’ because they open or close in response to changes in the membrane potential of the cell. Because H+ carries a positive charge, its movement across the membrane comprises an electrical current that can be recorded experimentally. A voltage-gated proton channel gene was not identified until 2006 [1,2]. Since then, some of the working parts of the molecule have been described, for example the conduction pathway (figure 1), but no crystal structure exists. Although a large number of enzymes, including many crucial to bioenergetics, contain proton pathways (or channels) that are essential to the operation of the molecule, the voltage-gated proton channel is a distinct category of protein and has not been reported to exist in mitochondria or chloroplasts. In many cells, proton channels are present in the plasma membrane; in a few instances (dinoflagellates and phagosomes in white blood cells), they have been shown to exist in organelle membranes.
Figure 1.
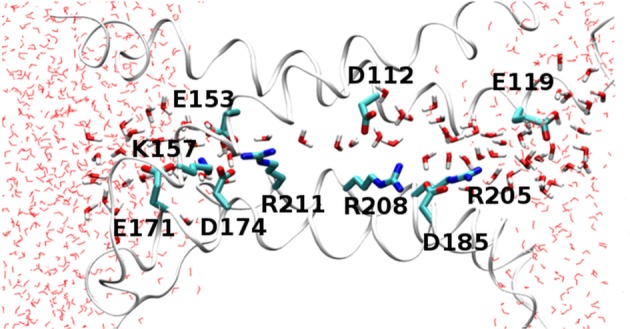
A snapshot from a molecular dynamics simulation of the open state of the human voltage-gated proton channel, hHV1. The four transmembrane helices are shown as ribbons with the extracellular end of the channel to the right. Charged amino acids (labelled) and water molecules in the pore are shown in licorice representation. This snapshot illustrates a configuration that is present only 10% of the time, in which there is a continuous water wire; usually the water wire is interrupted by a D112–R208 salt bridge. The external and internal charge networks are thought to stabilize the open conformation of the channel. (Adapted from [3].)
When these words were written, this was the 100th review written entirely (n = 75) or partially (n = 25) about voltage-gated proton channels. The number of original research papers, 157, was less than double that number1. From these statistics, one might conclude that this review needs only to discuss the two most recent original papers. Instead, we will step back and attempt to view the subject from a distance. Why do voltage-gated proton channels exist? What good are they? Why do they have so many unique properties? What kind of a world would it be if there were no proton channels? We describe the properties of proton channels teleologically and anthropomorphically; we do not pretend to understand the evolutionary forces that produced this protein, but simply describe it as though it were a sentient being whose properties serve the purposes we impute to it.
1.1. The origin of HV species
The abbreviation for voltage-gated proton channels is HV (H for H+, the conducted ionic species, subscript V for voltage-gated). To date no more than one gene has been identified in any species, thus all HV are HV1, with a prefix that indicates species (e.g. hHV1 for human, EhHV1 for the coccolithophore Emiliania huxleyi). The official numbering of other channels is often more complex, including subtypes, e.g. KV3.2. The HV family tree (figure 2) continues to grow, but already includes several kingdoms (or supergroups, if one prefers this classification system), including unconfirmed HV in Chlorella (Protista kingdom) and in the common wine grape, Vitis vinifera (Plantae kingdom), confirming the Bacchanalian proclivities of HV. Taylor et al. [5] noted that HV appear to be absent in land plants and suggested an explanation: these species tend to have a large inward pH gradient, which seems fundamentally incompatible with the exquisite design of the proton channel (in most species) as a passive acid extrusion mechanism. This argument may apply to plasma membranes, but HV could still exist in plant vacuole membranes. Recent BLAST searches produce a handful of likely candidates among algae, fungi and a few higher plants. The branch length in figure 2 reflects the extent of difference in amino acid sequence between the branch-dweller and its neighbours. Mammalian HV1 are all very similar to each other, but as we branch out to invertebrates, diversity blossoms and single-celled organisms have wildly diverse sequences. Sequence differences, in turn, might be expected to presage functional differences, nicely illustrated by the novel properties of the kHV1 in the dinoflagellate Karlodinium veneficum [4], which has the longest branch of all. This is the only known proton channel that opens to allow inward proton current; all other HV open only when there is an outward electrochemical proton gradient, and consequently their predominant function is acid extrusion from cells (see ‘Why do proton channels have ΔpH-dependent gating?’). Because kHV1 opens negative to the Nernst potential for protons, EH, it allows inward current, and is thus capable of mediating an action potential [4], just as are voltage-gated sodium channels in more traditional excitable cells such as nerve and muscle [6]. As the HV family tree grows, and especially as the properties of HV in the 30 not-yet-studied species in figure 2 are explored, further diversity in function will likely be found.
Figure 2.

Phylogenetic tree of confirmed and probable voltage-gated proton channels. Branch length is proportional to the degree of difference from neighbouring proteins. For the eight starred species, the gene has been expressed heterologously and confirmed electrophysiologically to encode a voltage-gated proton channel. (Adapted from [4].)
Based on sequence homology analysis [4,7], HV are closely related to two other groups of proteins that also contain voltage-sensing domains (VSDs), the voltage-sensing phosphatases (VSP) and c15orf27 (molecules of unknown function). These groups are in turn related by common ancestry to Na+ and Ca2+ channels, and appear to have diverged from the K+ channel family over one billion years ago [3]. Figure 3 illustrates the molecular structure and membrane topology of three classes of VSD-containing molecules.
Figure 3.

Molecular architecture and orientation in the membrane of three classes of molecules that contain VSDs. (a) Illustrates the monomer of each, with its transmembrane segments numbered (S1, S2, etc.) starting from the intracellular N terminus. In each, the VSD includes the first four segments, S1–S4, in particular S4 which contains a series of cationic residues (Arg or Lys) that are thought to move outward upon membrane depolarization. The VSDs from these molecules are homologous. (b) Each molecule in its functional oligomeric state. The voltage-gated potassium (K+) channel is a tetramer, with four VSDs surrounding a single central pore, formed by the four S5–S6 segments, through which K+ crosses the membrane. The proton channel is a dimer in mammals and many other species, but each monomer has its own conduction pathway [8,9] and can function independently. Phosphorylation of Thr29 in the intracellular N terminus enhances the gating of HV in phagocytes and other cells [10,11]. The VSP is a monomer whose VSD senses membrane potential and accordingly regulates the activity of the attached intracellular phosphatase [12]. (Adapted from [13].)
1.2. Why do voltage-gated proton channels exist?
A flippant answer to this question is ‘for the same reason that other ion channels exist!’ Cells need to regulate the flow of ions across their membranes, and the most sensible way of doing this is to produce an array of proteins that selectively transport individual ionic species in a tightly controlled fashion. Because the consequences of allowing ions to cross membranes differ drastically for each species of ion, we must delve a bit deeper for a more informative answer. Reflecting the importance of the conducted ion species, traditionally ion channels have received names that indicate the type of ion they transport, sometimes modified by a salient characteristic (inwardly rectifying K+ channel; voltage-gated Na+ channel; Ca2+-activated K+ channel, stretch-activated non-selective cation channel). When a channel opens in a cell membrane, it conducts ions passively down the electrochemical gradient for the conducted ion. The chemical part of an electrochemical gradient simply reflects that the concentration of one ionic species is often greater on one side of the membrane. Ions tend to flow down the gradient, from high to low concentration, other things being equal (‘other things’ being the electrical contribution discussed in the next paragraph). Usually K+ current is outward, because the K+ concentration is much greater inside the cell, [K+]i, than outside, [K+]o. Most cells have large inward Na+ and Ca2+ gradients, meaning that the concentrations of these cations are much higher outside the cell than inside; hence Na+ and Ca2+ channels almost always carry inward current.
1.2.1. A digression on the Nernst potential
In addition to the chemical gradient, we also need to consider the electrical potential across the cell membrane, the ‘membrane potential’. Because ions are charged, they are affected by electrical fields. For example, [K+] is approximately 160 mM inside cells and 4.5 mM outside, so in the absence of electrical forces, a K+ channel would carry outward current. Electrical current is defined in terms of the movement of positive charge, so this means K+ leaves the cell. This definition makes the behaviour of anions less intuitively accessible; outward Cl– current is recorded electrically when there is inward Cl– flux (‘flux’ simply means the physical movement of ions from one place to another). The effects of a concentration gradient can be modified or overridden if we control the membrane potential, which can be done experimentally by means of a circuit called a ‘voltage clamp’. Membrane potential is defined as the potential inside the cell relative to outside, which is defined as the reference voltage, 0 mV. As the membrane is hyperpolarized2, the increasingly negative charge inside the cell tends to prevent the positively charged K+ from leaving, despite the outward concentration gradient. We eventually reach a special voltage called the Nernst potential for K+, EK, at which the electrical force perfectly balances the chemical potential and no net current will flow:
| 1.1 |
where R is the gas constant, T is temperature, F is Faraday's constant, z is the charge of the ion (+1 in this case), [K+]o is the extracellular (outside) concentration of K+, and [K+]i is the intracellular concentration of K+. The constant 2.303(RT/zF) is approximately 58 mV at 20°C so when [K+]o = [K+]i, EK = 0 mV and when there is a 10-fold outward K+ gradient, e.g. [K+]i = 160 mM and [K+]o = 16 mM, EK is −58 mV. The Nernst potentials for other ions are calculated similarly, with the caveat that the correct z must be used for anions or divalent ions. If we consider our favourite channel, the voltage-gated proton channel, its permeant ion is H+ alone. The concentration of H+ is traditionally presented as pH, the negative logarithm of the concentration, so 10−7 M H+ is pH 7. We define the pH gradient ΔpH as pHo − pHi, and from the Nernst equation (equation (1.1)), we see that for ΔpH = 1 unit, EH is −58 mV. A positive ΔpH means there is an outward H+ (chemical) gradient; which means that at 0 mV (no applied voltage), H+ will tend to exit the cell through any H+ selective pathway. The Nernst potential is very useful in characterizing ion channels. The selectivity of a channel (which ions it allows to pass) can be determined by varying ion concentrations and measuring the reversal potential (the voltage where no net current flows), and comparing this to the Nernst potential calculated for each ionic species present.
When a channel opens and conducts current, one result is a change in the membrane potential. The rule is that each channel drives the membrane potential toward its own Nernst potential. Thus, when Na+ and Ca2+ channels open, they depolarize the membrane, whereas open K+ channels tend to hyperpolarize the membrane. There are also a number of non-selective ion channels. When they open, they drive the membrane potential toward their Nernst potential, which is 0 mV; hence, they usually depolarize the membrane, because for the most part cells have negative resting membrane potentials. A cell with a large number of open non-selective ion channels bears a disturbing resemblance to an electrophysiologically dead cell, exhibiting a familiar Gestalt that is witnessed by electrophysiologists at the end of every experiment—a leaky membrane with no reaction to radical changes in bath solutions, and a relentless approach toward no membrane resistance and a 0 mV membrane potential. Even so, there are situations in which brief opening of non-selective ion channels serves a useful purpose. For example, non-selective endplate channels in skeletal muscle open in response to acetylcholine released from a motor neuron, depolarizing the muscle membrane enough to initiate an action potential that propagates along the membrane and produces contraction [16].
The raison d’être for proton channels differs in different species. A clue to their general function in multicellular species, in fact in all species except dinoflagellates [4], is that they are uniquely regulated by the pH gradient, ΔpH (defined as pHo − pHi), in such a manner that they open only when there is an outward electrochemical gradient for H+ (i.e. ΔpH > 0) [17]. As is discussed below in ‘Why do proton channels have ΔpH-dependent gating?’, when HV open, they always carry outward current. As a result, they also tend to hyperpolarize the membrane. In certain situations, this effect provides identifiable benefit to the cell, but perhaps more frequently, the primary purpose appears to be regulating pH. When HV open, H+ leave the cell. In other words, HV extrude acid, thereby increasing pHi. One may speculate that HV exist in a large number of species because all cells engage in metabolism, which leads to a net production of protons that must be extruded to maintain a pHi compatible with cellular activities. Numerous other functions have been proposed that are specific to particular cells in particular situations [17]; full discussion of these is beyond the scope of this review.
Proton channels are extremely efficient proton extruders, capable of changing pHi an order of magnitude faster than other H+ equivalent transporters [18–20], at rates up to 6 pH units min−1 in small cells [18], more slowly in large cells [21,22]. Because they do not require ATP [23], they can extrude acid at minimal metabolic cost to the cell. Under some circumstances, the concomitant extrusion of charge could incur some cost owing to the necessity of charge compensation. In one intensely studied case, however—the respiratory burst in phagocytes (figure 4)—the cell is already enthusiastically extruding negative charge in the form of superoxide anion produced by NADPH oxidase, so the activation of HV simultaneously extrudes acid and compensates charge. During the respiratory burst, proton channels set the membrane potential to precisely the value at which H+ current is equal and opposite to the electron current [24]. The extraordinarily efficient activity of HV also includes two additional simultaneous functions. H+ efflux compensates the massive electron flux without causing undue osmotic imbalance, because the biochemical products of H+ inside the phagosome include a variety of membrane permeable species (H2O, H2O2 and HOCl) which can dissipate an osmotic gradient simply by diffusing through the membrane. Finally, H+ flux into the phagosome is crucial because H+ is required in large quantities as a substrate for the major reaction products, H2O2 and HOCl [24].
Figure 4.

Proton pathways and proton transport proteins in phagocytes during the ‘respiratory burst’. Part of a white blood cell (for example, a neutrophil) is depicted as its plasma membrane engulfs a bacterium. The enzyme NADPH oxidase (a.k.a. ‘NOX2’) assembles and becomes active in the membrane of the phagosome, the newly forming intracellular compartment enclosing the invader. NOX2 activity is so intense in neutrophils that it consumes orders-of-magnitude more oxygen as a substrate to form O2·− than is used by the rest of cell metabolism combined, hence the misnomer ‘respiratory burst’ [24–26]. NOX2 is electrogenic, because it moves electrons from cytoplasmic NADPH across the membrane to reduce O2 to O2·− [27]. The prodigious electron flux permits NOX2 activity to be measured directly as electron current [28,29], which without compensation would depolarize the membrane by approximately 11 kV min−1 [23]. Voltage-gated proton channels compensate electrically for the electron flux, prevent large pH changes in cytoplasm or phagosome, minimize the osmotic consequences of charge compensation, and are a necessary substrate for producing H2O2 and HOCl [24]. (Adapted from [13].)
The teleological benefits of opening proton channels in different cells tend to fall into four general categories, each of which is exemplified in the phagocyte respiratory burst: (i) electrical—charge compensation, setting the membrane potential or generating action potentials, (ii) control of pHi (intracellular pH), (iii) control of pHo (extracellular pH) and (iv) osmotic—e.g. cell or organelle volume regulation.
(i) Charge compensation or regulating membrane potential by HV has been proposed for neutrophils [27,30–35], eosinophils [32,36–41], monocytes [42], macrophages [20], osteoclasts [43], basophils [44], microglia [45,46], dendritic cells [47], sperm [48], B lymphocytes [49,50] and cardiac fibroblasts [51]. Producing more dramatic electrical effects than simply setting the resting membrane potential, proton channels in dinoflagellates exhibit properties consistent with the suggestion that they mediate the action potential that triggers a luciferase-induced bioluminescent flash [4,52–54].
(ii) Regulating pHi is a general function, but has highly specific consequences in sperm capacitation [55], breast cancer cells [56], egg maturation [57,58], basophils during histamine release [44], neutrophils and eosinophils during phagocytosis [34,41,59–61] and coccolithophores during calcification [62].
(iii) Regulating pHo is considered an important function of hHV1 in human airway epithelium [63] and in exacerbating the invasiveness of breast cancer cells [64]. In the latter situation, HV benefit the cells to the detriment of the organism.
(iv) Finally, osmotic effects are thought to be involved in HV1 activity in microglia [65]. H+ flux via HV in phagosomes likely minimizes the volume changes that would occur if charge were compensated by other means [24].
In summary, the consequences of proton channel activity are highly diverse and depend strongly on the precise situation in each type of cell or organelle.
1.3. Why do proton channels need to be so selective?
Ion channels are often selective for one ion over others present in physiological solutions. That said, biology tends to be parsimonious—Na+ and K+ channels are selective, but not extremely so, and in fact, they do not need to be. If the ‘wrong’ ion permeates 1% of the time, this will have a negligible impact on the cell. After all, Na+ and K+ gradients are being dissipated continually, particularly in excitable cells, and the Na+/K+-ATPase works continuously to restore these gradients. Na+ channels still manage to mediate action potentials even though their error rate is substantial: PK/PNa is 0.05–0.1 [66–69] and as high as 0.23–0.30 when K+ is inside and Na+ outside [70], as usually is the case. Selectivity is often quantified as relative permeability (for example PK/PNa is the permeability to K+ relative to that of Na+), which can be calculated from measured reversal potentials (Vrev) using the Goldman–Hodgkin–Katz voltage equation [71–73]
| 1.2 |
Permeability means the facility with which a particular ion crosses the membrane. This will depend mainly on which ion transport proteins are present, because small ions cannot permeate biological membranes (lacking proteins) at physiologically relevant rates. If the membrane contains only one type of ion channel, permeability indicates how efficiently the ion permeates this type of channel. Note that permeability is a property of the ion and the channel in combination; increasing the concentration of ion X increases the conductance (gX) calculated from the measured current (IX) divided by the driving voltage (V-EX), but does not change the permeability (PX). The Goldman–Hodgkin–Katz equation (equation (1.2)) says that the ion with the highest permeability will dominate Vrev, at least when its concentration is reasonably high. In practice, measuring Vrev in solutions containing as few ion species as possible simplifies the calculation. K+ channels tend to be more selective than Na+ channels, with PNa/PK approximately 0.001–0.1 [73,74]. On the other hand, many K+ channels are highly permeable to Tl+ and Rb+, and often PNH4/PK and PCs/PK are a modest, but respectable 0.1–0.2. Ca2+ channels are more selective, with a relative permeability to physiological monovalent cations, PK/PCa or PNa/PCa of just 10−3 to 10−4 [73], although essentially all Ca2+ channels conduct certain other divalent cations (Sr2+ or Ba2+) relatively indiscriminately [73] and often better than Ca2+. Teleologically, Ca2+ channels need to be highly selective against monovalent cations because the Ca2+ concentration is substantially lower than that of K+ or Na+. All of these traditional ion channels can still do their jobs without being meticulously selective. Other channels are even less selective. Cl– channels tend to be very poorly selective and conduct fairly large organic anions [75,76]; their virtue depends on the virtual absence of other anions in physiological solutions. Acetylcholine receptor channels at the neuromuscular junction are altogether non-selective among cations—their job is to depolarize the membrane just enough to initiate an action potential in the skeletal muscle membrane [16]. An open non-selective channel will drive the membrane potential toward 0 mV, and this rather imprecise effort is still entirely adequate to depolarize the membrane to the threshold (Vthreshold) for opening voltage-gated Na+ channels.
Proton channels, on the other hand, must be extraordinarily selective in order to accomplish anything useful. The proton concentration is literally a million times smaller than that of other ions, so a truly impressive relative permeability of PH/PK = 106 would still allow similar numbers of K+ (present inside cells at approx. 10−1 M) and H+ (present inside cells at approx. 10−7 M) to permeate. In fact there is no evidence that any other ion permeates at all, which means HV may be considered to be proton specific. Replacing the predominant cation or anion in the bath solution does not change Vrev detectably, once liquid junction potentials are corrected [23]. In some studies, changes in pH shift Vrev by an amount indistinguishable from the predicted shift in EH [4,22,36,77–79]. More frequently, the measured Vrev deviates from EH, but the explanation is simply that it is very difficult to control pH perfectly, especially when the measurement of Vrev itself requires activating large H+ fluxes across the membrane. Reducing the ionic strength (decreasing the concentrations of all cations and anions, except for H+ and OH–) by 90%, a strategy intended to distinguish cation from anion permeable channels [80], does not change Vrev, independently confirming H+ selectivity [7]. It is clear that proton channels both need and exhibit extreme selectivity. The question is—how do they accomplish this? One key to understanding proton selectivity lies in the mechanism by which protons move from point A to point B within proteins, and how this differs from the movement of other ions. This is discussed in the following section, before we return to the question of selectivity.
1.4. Proton transfer in liquid water and in proteins
The discussion below seeks to provide a qualitative description of the various chemistries used by nature to transfer protons within proteins. We purposefully exclude the important topics of the energetics and the mechanics of proton transfer. For entry to these topics, we suggest one of many reviews [81]. We first consider proton transfer in liquid (bulk) water, then proton transfer through a single file of waters and finally the roles of protein side chains.
The key to all of the proton transfer events discussed here is movement of the proton through a well-aligned hydrogen bond [82–84]. It seems a loosely kept secret that hydrogen bonds abound in liquid water, therefore the mechanism of proton transfer should be simple (!). The ion at the centre of proton transfer through water is hydronium (H3O+), with the slight complication that hydronium never exists as a free ion in bulk water, only as an idealized structure [85]. Most of the time, protons in liquid water are contained within Eigen cations (H9O4+), composed of a hydronium ion with hydrogen bonds to a primary hydration sphere of three waters [86,87]. At any instant, the hydronium is closer to one of its primary sphere waters than it is to the other two; the hydronium and its closest water are designated the ‘special pair’ [88]. A new special pair forms, on average, every 40 fs. After several picoseconds, one of these special pairs becomes a Zundel cation (H5O2+; H2O–H+ · · · OH2) and achieves the optimum hydrogen bond geometry that allows the proton to be shared between two waters (H2O–H+–OH2), and then transferred to the acceptor water (H2O · · · H+–OH2) [88]. This event is often termed ‘proton hopping’ because the proton has, in essence, hopped from a lone pair of electrons on one oxygen to a lone pair on the acceptor oxygen. Following the proton transfer event in the Zundel cation, the Eigen cation re-forms. A recent study produced a somewhat different picture: the proton moves rapidly (less than picosecond) along a short wire of two to three hydrogen-bonded waters, but rests as a Zundel cation for longer periods (2–5 ps) between these concerted hops [89].
The through-bond character of proton hopping enables protons to effectively diffuse through water rather than around it, as other ions must. In 1936, Huggins [90] noted that ‘the charge shifts (through hydrogen-bonded waters) with little actual motion of the atoms’. In fact, the mobility of H+ through water is 4.5–7 times greater than that of other common physiological ions (K+, Na+, Ca2+ or Cl–) [91]. In 1905, Danneel [92] explained the anomalously high mobility of protons in water as the Grotthuss mechanism, in recognition of work a century earlier by Freiherr von Grotthuss [93,94]. Grotthuss-type proton transfer in water also appears to be a component of Brønsted acid–base chemistry. Acids dissolved in water, for example acetic acid, transfer their proton to a base through a hydronium intermediate (actually the hydronium within the Eigen cation) [95].
Now we turn our attention to proton transfer within proteins, using lessons learned from liquid water as well as studies of H+ channels, H+ pumps and enzymes that include H+ pathways. A proton transfer pathway or proton transfer chain (PTC) is a sequential series of proton carriers that follow a structurally defined route within a protein. In enzymes, a PTC leads from the proton source (e.g. bulk water) to the proton sink (e.g. the active site or a redox centre). The reverse direction applies when protons are released by the chemistry within the protein. In transmembrane proton pumps, one PTC leads from solvent to the internal proton-pumping element, while a second PTC leads from the pumping element to solvent on the other side of the membrane. The proton carriers of a PTC are either side chains of amino acids or internal water molecules; in most known PTCs within proteins both of these components are involved. As in liquid water, a key requirement is hydrogen bonding. Each component of a PTC must be capable of hydrogen bonding both the preceding and the next proton carrier, although this may not occur simultaneously, as discussed below.
A PTC composed solely of a single file of waters, a water wire, is perhaps the simplest pathway to visualize (figure 5a). A proton enters a water file by forming a covalent bond with the first water to form a hydronium ion. The hydronium ion may hydrogen bond to the next water to form the Zundel cation, H5O2+ [97]. In the confined channels of PTCs in proteins, formation of the yet larger Eigen cation is precluded by geometrical constraints when the channel diameter does not exceed 4 Å [98]. The mechanism of proton transfer in a water wire is likely to be modified from that of bulk water because the waters of the linear water file can form a maximum of three hydrogen bonds, one fewer than waters in bulk [99]. Nevertheless, successive proton hops between waters in the file can rapidly move the proton from the source to the sink. Several of the waters may align their hydrogen bonds for concerted hopping, and the proton may rapidly hop back and forth along these groups of waters prior to reaching the proton sink [99,100]. In order to move a second proton from source to sink, it is necessary for each of the waters to turn 180°, thus presenting an open lone pair on each water O to the incoming proton [101,102]. The overall speed of proton transfer through a transmembrane water file is rapid, and can occur in less than 500 ps in gramicidin channels, because single-channel currents corresponding to more than 2 × 109 H+ s−1 have been reported [103]. This is a record for a selective ion channel, eclipsing the BK Ca2+-activated K+ channel, which conducts a mere 109 K+ s−1 at +300 mV in 3.4 M K+ [104]. When a single file of waters is confined in a hydrophobic channel with a diameter of 4.0 Å, the calculated proton transfer rate is several-fold faster than in bulk water [98]. Evidently, even at atomic dimensions, laminar flow is more efficient than turbulent flow. In a linear water chain, the proton can hop forward or backward along the file, but not sideways or up and down. Moreover, a protein may construct a channel that optimizes the positions of the waters for rapid proton transfer [99]; branching in a hydrogen-bonded water network slows proton conduction [105].
Figure 5.

Conduction of H+ (a) and OH– (b) occurs by protons hopping (a) by the classical Grotthuss mechanism as an excess proton hopping from donor H3O+ to acceptor H2O, or (b) as proton ‘hole’ migration, respectively. In the latter case, the net leftward migration of OH– results from a series of retrograde hops of H+ to the right. The net result in both cases is the transfer of one proton from left to right, and these processes are indistinguishable electrophysiologically. Reprinted with permission from [96]. Copyright © 2006 American Chemical Society.
The quintessential example of a purely aqueous proton pathway through a protein spanning the membrane is the gramicidin channel. This short peptide (a dimer of 15-amino acid monomers) produced by Bacillus brevis forms a cylindrical tube filled with water [106]. The interior is hydrophilic; peptide bond carbonyl groups coordinate a single-file row of a dozen water molecules [107]. Titratable groups, for example carboxyls, are absent. Gramicidin is permeable to a variety of monovalent cations, but it excels at conducting protons [108]. Normalized to the permeant ion concentration, the proton conductance of gramicidin is 100-fold greater than that of other ions [109]. The facility of proton permeation arises from their ability to hop along the water file without displacing the waters, whereas other cations must wait for the water molecules to diffuse through the pore ahead of them [109]. As mentioned above, H+ flux at rates exceeding 2 × 109 s−1 have been recorded in gramicidin channels [110]. The example of gramicidin shows clearly that a narrow pore filled with water is capable of conducting protons with high efficiency, but the transfer is not selective, because other cations pass, albeit more slowly.
1.4.1. A digression on why proton channels carry such tiny currents
The human proton channel hHV1, has an estimated conductance of 78 fS at physiological pH and human body temperature [111]. Given a large driving force, say 100 mV, one channel would carry 7.8 fA of H+ current, which translates into 5 × 104 H+ s−1. Does this mean that HV are not efficient proton channels, in comparison with gramicidin, for example? Perish the thought! The gramicidin value mentioned above (2 × 109 H+ s−1) was obtained at +160 mV in 5 M HCl! Our hHV1 can withstand a pH range beyond anything they encounter in the human body, but at pH < 4.5 or pH > 8.5 they, or the membranes they reside in, do not survive long enough to permit study. By contrast, when incorporated into synthetic lipid bilayers, gramicidin continues to function at pH −1, in 10 M HCl [110]! It is revealing to note that the single-channel proton current through gramicidin is almost directly proportional to H+ concentration over a wide pH range from 0 to 4.5 [23,110,112]. Extrapolated to the pH range where HV1 single-channel measurements are possible (pH 5.0–6.5) [111], the gramicidin H+ current is actually smaller [23]. We conclude that HV are very respectable proton channels that carry small currents simply because the concentration of their permeant ion (H+) is extremely low in physiological solutions.
1.4.2. Involvement of amino acid side chains in proton transfer within proteins
Long water wires do exist within functional PTCs in proteins. The ‘D’ pathway of the mitochondrial enzyme cytochrome c oxidase (CcO) is proposed to be a file of 8–10 hydrogen-bonded waters that transfers protons from a surface-exposed aspartic acid residue to an internal glutamate residue [113,114]. One fascinating aspect of the D pathway is that the continuity of the water file is constantly being broken and restored at one point by an amide side chain that moves in and out of the water file [115]. Nevertheless, proton transfer through the waters remains rapid [115,116]. The functional pathway for proton transfer to the quinone-binding site of Escherichia coli succinate dehydrogenase may involve up to 13 waters [117].
With the possible exception of the succinate dehydrogenase pathway [117], native proton transfer pathways in proton pumps and enzymes involve side chains of certain amino acids (particularly carboxylates) along with internal waters. The chemistries of the various side chains increase the number of mechanisms available for proton transfer. For example, in the 1970s, Nagle and others [101,102,118] recognized that certain side chains of amino acids, for example hydroxyls, could participate in proton transfer without ionization, i.e. without the net dissociation of a proton. In such a mechanism, the hydroxyl group uses hydrogen bonds to simultaneously acquire a proton from an upstream proton carrier and release its proton to a downstream proton carrier. Chemically, a lone pair of electrons on the hydroxyl O acquires a H+ while another pair releases a H+. Throughout the process the hydroxyl remains as –OH, avoiding ionization to either −O− or –OH2+. In theory, a long chain of hydrogen-bonded hydroxyl groups could carry out concerted proton transfer over a considerable distance. This is not seen in known or proposed PTCs in proteins, perhaps because the precise alignment of hydrogen bonds is difficult to sustain over a long series of hydroxyl groups. In fact, when the hydroxyl groups of serine, threonine and tyrosine do appear in the proton transfer pathways of proteins, they do so individually, and they are bracketed by carriers capable of forming a stable ion, such as a carboxyl or water [23]. For example, a recently identified proton pathway in Clostridium pasteurianum [FeFe]-hydrogenase comprises a five-member motif with Ser bracketed by two Glu residues (Glu282, Ser319, Glu279, H2O, Cys299) [119].
The acidic and basic side chains of amino acids add conventional acid–base chemistry to the realm of proton transfer. Most significantly, the fact that proton acquisition and proton release are separable events for an acid or base brings the aspect of rapid side chain movements into play. For example, at the entrance to the D pathway for proton transfer of CcO, a carboxylate side chain accepts a proton from solvent (water), then the protonated carboxyl group rotates 180° about the terminal C–C bond of the side chain, establishes a hydrogen bond to the oxygen of the first water of the D pathway and transfers the proton to this water [120]. Just the rotation of the protonated carboxyl moves the proton approximately 2.2 Å along the trajectory of the transfer path. (The approximate centre-to-centre distance of the O atoms of a carboxyl group is 2.2 Å; a proton cannot hop from O to O of the carboxyl because the geometry of the atoms precludes an intramolecular hydrogen bond.) In CcO, the rotation of this D pathway carboxyl moves the proton from ‘outside’ to ‘inside’ [120]. The crystal structures of CcO show no space for hydronium or other ions to move around the carboxyl, so its rotation constitutes a proton-specific gate. The chemistry of such carboxyl rotation has been studied in detail in a bacterial ferredoxin, where a critical aspartic acid residue also moves a proton from outside to inside [121]. Examination of other protein structures suggests that carboxyl rotation may be common in proton transfer pathways.
The imidazole group of histidine is analogous to a carboxyl, in that it contains two atoms (Nδ and Nε) that can be protonated, but a proton cannot hop between these two sites because there is no intramolecular hydrogen bond. Similarly to carboxyls, neutral imidazole can be protonated on the open N, rotate 180°, and then deprotonate to the next proton carrier [122–125]. In this case, the rotation moves the proton approximately 2.1 Å, the centre-to-centre distance of the N atoms of the imidazole ring.
Transfer of protons can occur via larger, but still rapid, movements of whole protein side chains (rather than just carboxyl or imidazole ends of side chains), e.g. the swinging motion of a lysine within the K pathway of CcO [126,127].
1.5. How can one design a proton selective channel?
With the basics of proton transfer in mind, let us take up the question how a proton channel may select for protons. A proton channel requires two features to be selective for protons. First, there must be a proton transfer pathway, using any of the options described above for the transfer of protons through proteins. As we have seen, the mechanisms of proton transfer in proteins are closely related to proton acid–base chemistry in solution, and therefore a PTC in a channel protein is inherently selective for protons. Based on PTCs in other proteins, we might expect the PTCs of ion channels to include both waters and amino acid side chains. Although the channel must supply a pathway for the proton that spans the entire membrane, the proton selective section of the pathway may be shorter. In principle, selectivity can be achieved by directing all of the protons to pass through a single titratable side chain, such as a carboxyl or an imidazole group, at some point in the channel. As is the case with many ion channels, HV1 has large aqueous vestibules (figure 1), separated by a narrow region approximately 10–12 Å long in which water is mainly single file [3] (figure 1). This short, narrow region is the section of the transmembrane proton pathway that is thought to confer selectivity for protons.
Second, proton selectivity requires some mechanism to prevent other ions from also flowing through that region of the channel that contains the PTC. A priori, it would seem that a straightforward design strategy would be close protein packing. The passage of monovalent cations, for example, requires an open space, a pore, through which the ions can move. By contrast, proton transfer requires no open space, because the H+ may be transferred as part of the protein. As it moves from carrier to carrier through the protein via H-bonds, the H+ is covalently associated with lone pairs of electrons on internal waters or protein side chains. Given this fundamental difference in the structural requirements for proton transfer versus the transfer of other ions, it would seem that evolution could produce a highly selective proton channel simply by filling in the space around the PTC component of the channel.
1.5.1. Of water wires and Na+
Water wires (water files) have been proposed for part or all of the PTC component of some H+ channels. The H+ selectivity of these channels varies. At this time, HV1 is the only one that appears to be completely proton selective (Table 1 of [17]). The influenza virus M2 channel is highly proton selective, but also conducts K+ and Na+ [128–130]. A synthetic hydrophobic α-helix composed of leucine and serine (i.e. lacking formal charges) oligomerizes to form a channel containing a water wire that conducts H+ more than 40-fold better than Li+, based on the presence of detectable single-channel currents [131].
There is no doubt that a water file forms an excellent pathway for protons; the question is how to enforce proton selectivity. A relevant paradigm is the transmembrane H+ or Na+ channel of the Fo–F1-type ATP synthases. In some bacterial versions of these enzymes, the transmembrane channel of the Fo rotor transfers Na+, but in related enzymes (including mitochondrial ATP synthase), the channel is specific for H+ [132,133]. The transmembrane channel of Fo is constructed of two water-filled half channels leading to and from a controlling group located in the middle of the membrane [134,135]. In the Na+ channels, the central group is a multi-valent coordination site for Na+, while in the H+ channels the central group appears to be a single carboxyl side chain [136]. For the H+-conducting ATP synthases of Spirulina platensis and Bacillus pseudofirmus, spectacularly selective binding (108–109 higher for H+ than Na+) is demonstrated by the Fo rotor being driven by H+ at pH 9 and 200 mM Na+ [137,138]. This does not prove that Na+ cannot reach the input half-channel; simply that it does not bind and catalyze enzyme activity. However, the working enzyme translocates H+ across the entire membrane with superb selectively. Hence, nature appears to have conferred H+ selectivity upon the transmembrane channel of many F-type ATP synthases by replacing a Na+-binding site with a single carboxyl side chain. The elucidation of further structural factors that confer H+ selectivity in the F-type ATP synthases should be highly instructive for other H+ channels, and vice versa.
Other more subtle and difficult to validate mechanisms have been proposed for H+ selectivity by water wires. One such is the ‘frozen water’ hypothesis, some form of which has been proposed for the M2 viral proton channel (figure 6b) discussed below [140–142], for the synthetic proton channel of Lear et al. mentioned above [131,143], and for HV1 [144,145]. In this mechanism, one or more waters at a narrow region in an aqueous pore are constrained by the pore walls, essentially frozen in place in such a way that they can still translocate a proton by the Grotthuss mechanism, while simultaneously preventing other cations from permeating. Although nominally frozen, the waters need to retain enough mobility that they can reorient after each proton conduction event [101,102].
Figure 6.
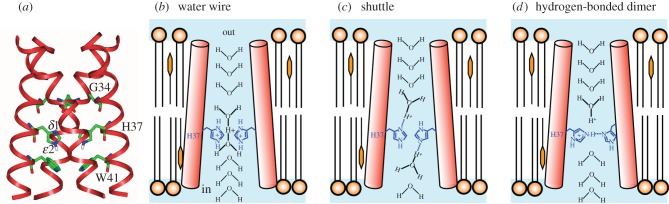
The well-studied M2 proton channel of influenza A virus is a homotetramer; (a) shows the high-resolution crystal structure with key residues labelled. Three proposals for the proton selective conduction are illustrated in (b–d), with only two of the four protomers shown for clarity. In the water wire model (b), the proton pathway is exclusively aqueous, with protonation of multiple His37 serving to open the pore by electrostatic repulsion. Proton selectivity in the shuttle model (c) is achieved by successive protonation and deprotonation of His37, with a ring flip completing each conduction event. In (d) protonated and unprotonated His37 form hydrogen-bonded dimers that are broken during H+ conduction. (Adapted from [139].)
Another way to enforce proton selectivity still envisions a water wire, but one that is continuous only transiently. Molecular dynamics simulations of hHV1 in the open state indicate that a continuous water wire exists just 10% of the time (figure 1), and it persists only for a nanosecond or so [3]. One could speculate that protons, speedy little devils that they are, could zip through such a pathway, whereas much bulkier and slower ordinary cations could not permeate within such a short-time window. Such a situation is proposed for the normal function of the D pathway of CcO, although not as a solution to the problem of ion selectivity. Although the D pathway transfers protons at rates at least up to 104 s−1 [116], high-resolution structures show a clear discontinuity in the water file of this pathway in the form of two amide side chains of Asn139 and Asn121 [127,146]. Molecular dynamics simulations suggest that the amide side chain of Asn139 spontaneously swings out of the way to allow formation of a continuous water file [115]. The rate of H+ transfer remains rapid because the movements of protein side chains and internal waters are even more rapid.
Yet another hypothetical mechanism for proton selectivity postulates a continuous water wire that is energetically or electrostatically unfavourable to ions. We have seen how a proton can move back and forth along a file of waters. This reduces the effective charge of the proton at any specific location, i.e. the charge is delocalized [85,97,99,147,148]. By contrast, the charge on an ion, for example Na+, exists at a discrete location; so it becomes possible for a protein structure to purposefully exclude such a charge. A delocalized-charge mechanism has been proposed for a synthetic proton channel [149].
1.5.2. Aquaporin: a water pathway that excludes protons
A paradigmatic example of a water-filled channel that excludes protons is aquaporin (AQP), a membrane protein that provides a pathway for rapid water permeation across the membrane [150], but excludes all ions including protons [151,152]. The structure of AQP revealed a narrow region 20 Å long at the centre of which is a pair of NPA (Asn–Pro–Ala) motifs from nearby short helices [153]. It was proposed that the two Asn amido groups formed hydrogen bonds with a single water molecule, preventing the water from hydrogen bonding with adjacent waters, thereby precluding proton conduction [153]. Shortly after this, a nearby ‘constriction region’ with conserved Arg, His and Phe residues was identified [154], later called the ‘selectivity filter’. A number of molecular dynamics studies ensued to evaluate the structural and physical basis for blockage. Initial simulations in the absence of H+ were interpreted to support the water orientation hypothesis [155], but subsequent calculations showed that the free energy barrier opposing the movement of an excess proton through the pore peaks at the NPA motif [156–159]. Several of the latter studies concluded that ion blockage results from electrostatic effects combining the ion's desolvation penalty and interactions with charged and polar groups of the channel. A simpler model was proposed by Burykin & Warshel [160,161], who argued that AQP with all of the ionizable groups in the channel neutralized, and even a 4 Å hole in the membrane exhibited a large energy barrier to proton permeation, and concluded that the desolvation penalty for protons to leave bulk water and cross the centre of the membrane was itself sufficient to exclude protons. However, point mutations of the selectivity filter region, R195 V or R195 V/H180A, were identified that enabled proton conduction through AQP1 [162,163]! These results showed that neutralizing the charge of R195 in the selectivity filter suffices to confer proton permeability, despite the continued presence of the NPA region and the desolvation penalty for inserting protons into the narrow pore.
AQP can be mutated to conduct protons, whereas HV1 can be mutated to exclude protons. In the human HV1 channel (figure 1), the Asp112 crucial to proton selectivity is salt-bridged with Arg208 about 90% of the time, so their charges are neutralized. When Asp112 is neutralized by mutation (e.g. to Ser or Ala) molecular dynamics simulations reveal that the charge owing to the now-unpaired Arg208 produces a 10 kcal mol−1 barrier to cations [3]. It was shown recently that Asp112 can be shifted one turn of the helix outward to position 116 without loss of proton selectivity [164]. Molecular dynamics simulations reveal that at its new location, Asp116 is generally paired with Arg208 or Arg205. There is an electrostatic barrier to cation permeation when Arg208 is unpaired, but not when it interacts with Asp185 or Asp116 [164]. Evidently, proton exclusion can be achieved by an unpaired cationic group in a critical location. It is well established that the charge selectivity of anion-selective, ligand-gated channels can be switched to cation selectivity by introducing negatively charged Glu at key locations [165,166], and that cation selective channels can be made to conduct anions by various mutations that include neutralization of Glu [167,168]. These rules of charge selectivity can be used to destroy proton selectivity, but producing proton selectivity is a subtler task.
1.6. The extensively studied M2 viral proton channel as a model
The M2 channel of the influenza A virus [169] is a favourite example of a highly proton selective ion channel [170–172]. This channel is relatively simple, being a homotetramer of 97-amino acid monomers (figure 6). Four transmembrane helices of M2 proteins associate to form a single channel that transfers protons into an endosome containing a virus that has been taken up by a cell. Proton uptake initiates the loss of the viral protein coat. The M2 channel is not as selective for protons as the HV1 channel, and was shown recently to exhibit measurable permeability to Na+ and K+ [128–130,173], which tarnishes it somewhat as a paradigm of perfect proton permeability. Even so, the M2 proton channel selects for H+ over Na+ or K+ by a factor of approximately 106 or more [128–130,170,173]. As discussed below, this is achieved by proton transfer through a His37 cluster, combined with physical occlusion of Na+ or K+ transfer through the His37 cluster. The rate of H+ transfer from exterior to interior is relatively slow at 100 s−1 or less by most accounts [128–130,171,173–176]; a gate composed of a ring of Trp41 side chains [177,178] slows reverse proton transfer even more.
From the analysis of site-directed mutants and high-resolution structures, the key to H+ selective conductance is His37 [179,180]. As the channel assembles as a tetramer of four identical α helices, there is a tetrad of His37 whose side chains face the centre of the pore [181]. His37 plays key roles both in activating the channel and in proton selectivity. Protonation of two His37 opens the channel [141,176,182,183]. H+ conduction is mediated by the third His37 to be protonated [139,169,184], and its relatively high pKa may limit H+ flux [139,185]. Mutagenic alteration of His37 (for example H37G) abolishes proton selectivity [179]. The mechanism by which His37 imparts proton selectivity (figure 6c) was originally suggested to be protonation of the imidazole δ-nitrogen from the extracellular side of the membrane, deprotonation of the imidazole ε-nitrogen to the intracellular side, followed by a ring flip to restore the orientation of the singly protonated state [186]. This shuttle mechanism can be described quantitatively by simple mathematical models [184,187,188]. Because each H+ conduction event entails conformational changes of His37 and Trp41, one could reasonably define M2 as a carrier rather than a channel. The essentials of this model remain, i.e. protonation and de-protonation of His37 plus a flipping motion of the imidazole ring. Further details of the mechanism are currently debated, as discussed below.
High-resolution crystal structures (figure 6a) and NMR structures have provided considerable structural information about the rings of His37 and Trp41 residues and the positions of internal waters [169,176]. In addition, the existence of hydrogen bonds and the protonation state of the His37 can be determined using NMR spectroscopy [139,189–191]. These data allow for detailed models of proton transfer through His37 [169,176,192]. In two current models, the selectivity filter for protons involves just three components and two proton transfer steps: a hydronium from the exterior aqueous region of the channel transfers a proton to the His37 cluster (which is organized differently in each model), and then the His37 cluster transfers the proton to a water below the ring of Trp41 side chains, when this Trp41 gate is open to allow a hydrogen bond to form between a component of the His37 cluster and the acceptor water molecule.
In the mechanism proposed by Zhou, Cross and co-workers [176,183,192], based primarily on calculations on an NMR structure and further spectroscopy, the deprotonated His37 tetrad is organized as two hydrogen-bonded imidazole–imidazolium dimers (figure 6d). The introduction of a proton from an upstream hydronium breaks the H-bond of one of these dimers (either one, apparently) producing two imidazolium side chains that rotate and swing away from each other. One swings down to transfer a proton to a downstream water revealed by the opening of the Trp41 gate. The de-protonation of this His37 allows re-formation of an imidazole–imidazolium dimer.
The mechanism (figure 6c) of Hong, DeGrado, and co-workers [169,193] is primarily based upon a 1.65 Å crystal structure of the transmembrane channel [194] combined with solid-state NMR dynamics data. In this structure, the orientation of the side chains of the ring of four His37 roughly resembles that seen in the NMR structure used by Zhou, Cross, et al. but the side chains are evenly spaced around the ring and direct H-bonds between side chains are absent. Rather, the four imidazole/imidazoliums form a hydrogen-bonded network with associated waters, which are located atop the His37 ring and between the histidines and the Trp41 ring [194]. The histidine side chains and the waters are capable of extensive delocalization of an added proton; continuous ring flips of the histidines facilitate the sharing of the added proton with the waters between the His37 and Trp41 rings. The three components of the PTC in the Hong–DeGrado model are a hydronium from the aqueous channel above the His37/water cluster, the His37/water cluster itself and a water to receive the proton below the Trp41 gate. The rate-limiting step in H+ conduction is imidazolium ring reorientation [169].
A principal difference in the two models is the nature of the His37 structure that is protonated by a single proton from the exterior side of the channel and then de-protonated by water beyond the Trp41 gate. In the Zhou–Cross model, the His37 proton transfer group cycles between an imidazole–imidazolium dimer and two imidazolium side chains, while in the Hong–DeGrado model, the His37 structure is a His37/water cluster in both its protonated and de-protonated forms, whose degree of hydration increases as the degree of protonation is increased. Evidence that the hydrogen-bonding partner of His37 is water instead of another His37 was obtained from 1H NMR chemical shifts [193]. A swinging movement of the histidine side chains appears less required in the Hong–DeGrado model, because either of the waters located between His37 and Trp41 in the His37/water cluster can mediate proton transfer to the water beyond Trp41.
1.7. What makes hHV1 selective?
Returning to our main interest, the voltage-gated proton channel, our current explanation of proton selectivity consists mainly of describing which parts of the molecule are involved. Because no crystal structure exists for the channel in any species, what we believe about structure is derived from homology modelling based on crystal structures of voltage-gated Na+ and K+ channels (specifically, their homologous VSDs) and on molecular dynamics simulations in conjunction with experimental data. As illustrated in the snapshot in figure 1, hHV1 narrows to a region approximately 10 Å long containing more or less a single file of waters that most of the time is interrupted at an Asp112–Arg208 salt bridge [3]. Experimentally, the PTC within HV appears to consist of more than water. For example, a number of characteristics of proton permeation through HV led to the idea that the rate-limiting step occurred inside the pore, not in the approach through bulk solution [195–198]. Postulating a PTC including one or more titratable groups [18,197,198] could explain why the high selectivity, deuterium isotope effect and temperature dependence of conduction differed drastically from proton conduction through the water-filled gramicidin channel [109,199,200], but were similar to the corresponding properties of the M2 channel [170,171,201,202], in which proton conduction entails protonation/deprotonation of His residues [186].
The H+ selectivity of voltage-gated proton channels in both humans [7] and dinoflagellates [4] requires an aspartate residue in the middle of the S1 transmembrane segment, at a narrow region of the pore (figure 1), Asp112 and Asp51, respectively. Certainly, H+ selectivity could be achieved by a mechanism in which Asp112 is protonated and deprotonated by every proton transferred. When Asp112 is replaced with a neutral amino acid, HV not only lose their proton specificity but become preferentially permeable to anions. Phenomenologically speaking, that this Gestalt is identical in both species indicates that the selectivity mechanism is strongly conserved evolutionarily, given that there is only 14.6% identity between the two proteins [4,17]. Replacing Asp with Glu preserved proton specificity, but other mutants, including the His mutant, D112H or D51H, respectively, were permeable to anions [4,7]. Recent demonstration that the selectivity of hHV1 is preserved when Asp is moved from position 112 to 116, one turn of the S1 helix outwards (but not to a dozen other positions tested) reiterates the role of Asp, but indicates some structural latitude in the placement of the H+ selective carboxyl group [164].
There exists a clear preference for carboxyl side chains (Glu or Asp) in proton transfer pathways of pumps and enzymes [120,121,203–220], often as the group that introduces protons into the pathway. The D pathway of CcO has been established as an experimental system for examining the ability of side chains to sustain steady-state proton transfer [120]. In the normal D pathway, the initial proton acceptor is the carboxyl group of a conserved aspartic acid. Histidine cannot replace Asp as the initial proton acceptor at the normal position [221]. The thiol of Cys can function in this system as an initial proton acceptor, but only when it is moved to a point in the pathway with significant structural constraints that limit rotamer possibilities [120,221]. The preference for a carboxyl group for proton uptake is likely due to several factors: its structure is analogous to two waters connected by a carbon, its functional pKa is readily modified by the surrounding protein, and it has the capability to rapidly rotate end-over-end—like a turnstile—through a compact protein structure in order to transfer a proton to the next element in the PTC [120].
Nevertheless, simply locating an Asp in a pore-lining position is not enough to produce proton selectivity. In the c subunit of H-ATPase, the key carboxyl of Asp61 can be moved to a different helix (D61G/A24D), but not to nearby positions 58, 60 or 62 on the same helix [203,222]. Asp185 also faces the pore in hHV1 [3,144,145,223] but can be neutralized by mutation (D185S, D185N, D185V) without affecting proton selectivity [7]; conversely, Asp185 is still present in D112x mutants (i.e. mutants in which Asp112 is changed to His, Lys, Asn, Ser, Ala or Phe) that lack H+ selectivity [7]. As shown in figure 1, the pore is likely wider at the level of Asp185 [3]; evidently Asp must reside at a narrow point to produce selectivity. A compact protein structure around the carboxyl will prevent the passage of other ions.
That the His mutant of hHV1, D112H, was permeable to anions [7] was initially surprising, in view of the strong similarity of the transmembrane domain of HV1 to the VSD of K+ and Na+ channels [1,2]. The VSD includes the first four transmembrane helices (S1–S4), which are thought to act as the voltage-sensing component of the channel; they mechanically couple to the pore domain, which comprises the four S5–S6 segments (figure 3). Although the VSD of K+ or Na+ channels does not normally conduct current, when the outermost of a series of Arg residues in the S4 helix is replaced by His, a voltage-gated proton conductance appears [224–226]. This result supports the view that the VSD is roughly hourglass-shaped with aqueous vestibules that access both sides of the membrane and focus onto a short occluded region [224]. When His is located at this constriction, it selectively translocates protons [224–228]. Why does His introduced at a comparable region in hHV1 not shuttle protons? A crucial difference is that in the mutant K+ channel VSD, His replaces a cationic Arg, whereas in the D112H mutant of hHV1, His replaces a negatively changed Asp. In hHV1, Asp112 normally apposes the cationic Arg208 (figure 1); mutation of Asp results in uncompensated positive charge that excludes protons and produces anion selectivity [3]. Intriguingly, in aquaporin, proton exclusion occurs at the ‘ar/R region’, in which Arg195 apposes His180 [163].
In ATP synthase, the crucial acidic group Asp61 (in E. coli) exists in proximity to a highly conserved Arg210 residue, called the ‘stator charge’ whose function is thought to be to ensure release of the proton from the acidic binding site [133,136,203,229,230]. In an intriguing parallel, the proton selectivity element in hHV1, both at its native position, Asp112, and when repositioned to Asp116, is found by MD simulations to be mainly engaged in salt linkages with one or more Arg residues in the S4 helix [3,164].
1.8. What is the difference between H+ and OH– channels?
It turns out that distinguishing H+ and OH– channels is very difficult. The electrochemical gradient that drives conduction of either species is always identical. For example, at pHo 7.5, pHi 6.0, the Nernst potentials are EH = EOH = −87 mV (at 20°C). At symmetrical pH, applying +100 mV to the inside of a cell (or an isolated membrane) drives H+ out and/or OH– in, resulting in currents that are indistinguishable to a voltage-clamp circuit. The effects on pH on either side of the membrane are also identical; moving H+ out increases pHi and decreases pHo by the same amount as moving OH– in. It is likely that most conduction of H+ and OH– occurs by closely related mechanisms involving proton hopping (figure 5) by a Grotthuss-like mechanism [89,92–94,231–234]. In the chemical systems in figure 5, H+ is more likely to carry the current when donor and acceptor are acidic (pKa < 7), whereas OH– conduction (as a proton ‘hole’) is more likely when they are basic (pH > 7) [96].
As discussed above, the human voltage-gated proton channel, hHV1, is proton selective because of a specially located aspartic acid residue (Asp112) [7], which is almost certainly negatively charged in situ, with a low pKa. When this Asp112 is mutated to a neutral amino acid (e.g. Ser, Ala and Asn), the channel becomes anion selective. This charge reversal is understandable if we consider that, in all likelihood, the negatively charged Asp was compensated in the WT channel by one or more nearby cationic charges, so that elimination of the Asp leaves unpaired cationic charges. However, although Cl– is clearly permeant in D112x mutants, the preferred charge carrier appears to be OH– by several orders of magnitude (in terms of relative permeability, POH/PCl, although not necessarily in real numbers) [7]. Why this should be is not obvious. The pathway in WT hHV1, comprising an aqueous pore that most of the time is interrupted at an Asp112–Arg208 salt bridge [3], supports a high rate of H+ flux. Mutation of Asp112 alters the electrostatic environment drastically, by replacing a net-neutral salt bridge with an uncompensated cationic charge, producing a 10 kcal mole−1 barrier for cations [3]. Facile OH– permeation should not be too surprising, because the mechanism of OH– conduction also involves H+ hopping and occurs rapidly. Because OH– permeates by means of proton hopping (figure 5b), it has tremendous mobility advantages over Cl–, both because of its small effective size and the rapidity of proton hopping (discussed above).
In several tissues that exhibit H+/OH– flux at extremely high pH, such as Chara australis at pH 10.5 [235,236] or frog skeletal muscle at pH 10 [237], OH– flux seems more likely than H+ flux simply because there are vastly more OH– than H+ to act as charge carriers.
In summary, the consequences of H+ or OH– permeation are similar, the mechanism of transport is similar, and which species is transported may depend on the pKa of critical groups in the pathway or on the net charge at a narrow part of the channel.
1.9. Why do proton channels have ΔpH-dependent gating?
A great many ion channels are voltage gated, which means that they have a high probability of being open (and conducting their particular favourite ion) at certain membrane voltages and a high probability of being closed (not conducting anything) at other voltages. Most voltage-gated ion channels, including our favourite, HV, open when the cell membrane is depolarized. For most ion channels, the voltage dependence is absolute, subject to modulation by permeant ion effects discussed below. A K+ channel that opens at −40 mV in physiological solutions will carry outward current. The same channel exposed to elevated external K+ concentration will still open at −40 mV but will carry inward current. Channel opening depends only on voltage; once a channel is open, the direction of current flow depends only on the electrochemical driving force (V − Eion). There are rather subtle effects of permeant ion species and concentration on the gating of several channels [238–240], most famously the ‘occupancy hypothesis’ of Clay Armstrong in which occupancy of the pore by a permeating ion slows the closing of the channel, also called the ‘foot-in-the-door’ effect [241]. ClC−0 Cl– channels [242,243] and inwardly rectifying K+ channels [244,245] have more pronounced permeant ion effects on their gating than most other channels. In contrast with HV, however, these effects are exerted by permeant ions mainly from just one side of the membrane.
One of the most extraordinary properties of every voltage-gated proton channel identified to date is ΔpH-dependent gating [17,18,23,246,247]. The effect of proton concentration (pH) on the gating of proton channels is not only pronounced, but it occurs equally on both sides of the membrane [248]. The consequence is that the position of the proton conductance–voltage (gH–V) relationship is determined by the pH gradient, ΔpH, defined as pHo − pHi [248].
Figure 7 illustrates the practical consequences of this unique ΔpH dependence. Here, we indicate the position of the gH–V relationship in a convenient format in terms of Vthreshold, the ‘threshold’ voltage at which proton channels first start to open. Data collected from studies of 15 different cell types are all described well by the upper line. Vthreshold is plotted as a function of the reversal potential, Vrev. One could equally replace the experimentally determined Vrev with the more theoretical value of the proton Nernst potential, EH, because the perfect selectivity of HV means that Vrev should be very close to EH. In practice, however, it is not so easy to establish pH accurately, even when one uses such high buffer concentrations that buffer is practically the only anion in the solution. The proton channel is a superb pH meter that reports precisely the pH gradient across the membrane. For this reason, we prefer to believe what HV tells us about pH, which is contained in Vrev. Returning to figure 7, we see that Vthreshold is always positive to Vrev—the dashed line shows the point at which they are equal. Finally, the punch line: proton channels do not open unless the electrochemical gradient is outwards, as it is for all the symbols on the graph (except the solid circles). Therefore, when the proton channel first opens, it will conduct acid out of the cell. Voila! The primary function of proton channels is acid extrusion!
Figure 7.

The property of ΔpH-dependent gating is universally conserved in proton channels, but exists in two forms. In most cells (15 varieties are illustrated here as various symbols), proton channels open only when there is an outward electrochemical gradient, and this results in acid leaving the cell. This pattern is seen over a very wide range of pHi and pHo; Vthreshold is always above the dashed line of equality. In the single case of the dinoflagellate K. veneficum (filled circles), Vthreshold for the kHV1 channel varies with identical slope with changes in pHi and pHo; but at any given ΔpH, Vthreshold is 60 mV more negative than in other HV1; this means that opening kHV1 produces inward proton current under all conditions. Most cells use HV1 to eliminate excess acid, but dinoflagellates have other goals in mind, such as conducting action potentials and triggering a bioluminescent flash. (Adapted from [17].)
Why do cells need to extrude acid? (Here, we will be kingdomcentric and consider mainly animal cells.) As was cogently stated by Roos & Boron [249] in their historic 1981 review, ‘the central problem of pHi regulation [is] the neutralization of intracellular acid derived from a variety of sources’ [249, p. 365]. Cells produce acid continuously during metabolism and are constantly looking for ways to get rid of it. From this perspective, the worst possible thing is a pathway that would allow H+ to leak into cells! The ΔpH dependence of proton channels ensures that this disaster will never happen.
Nevertheless, for the proton channel to be most useful, it does need to open reasonably near EH so that as soon as enough acid builds up, proton channels will open and release it. A quibbler might argue that the ΔpH dependence illustrated in figure 7 results in Vthreshold changing only 40 mV unit−1 change in ΔpH, insufficient to parallel EH which changes 58 mV unit−1. This point is technically valid, but arguably may not reflect a design flaw. Perhaps the approach of the upper solid and dashed lines in figure 7 means that Vthreshold is closest to EH when there is a large outward gradient, i.e. when the cell has become very acidic and is in dire need of relief from acid build-up.
A persistent quibbler might point out that Vthreshold is an artificial concept, because the probability that proton channels open (Popen) does not have a true threshold, but decreases exponentially with hyperpolarization. From this perspective, a larger cushion between Vthreshold and EH might be a prudent strategy in the negative voltage range, where cells may sit at their resting potential for days on end, and even a tiny H+ influx might have untoward cumulative consequences.
Fine, but what about the solid dots in figure 7? In the phylogenetic tree in figure 2, we saw that the proton channel that differs most in amino acid sequence from all the others is kHV1, a dinoflagellate proton channel. This sequence difference is reflected in a crucial functional difference, which is obvious from the dots in figure 7. Although kHV1 has ΔpH-dependent gating with precisely the same slope as all other proton channels, the absolute position is offset by −60 mV [4]. The consequence of this unique set-point is that when kHV1 first opens, it conducts inward current. The biological effects of channel opening could not be more different! Acid enters the cell instead of leaving. Why would this ever be desirable?
One answer is that this property would allow the proton channel to mediate an action potential. Conducting an action potential requires a conductance that activates negative to its Nernst potential [250,251]. As is discussed above, two familiar examples are voltage-gated Na+ and Ca2+ channels, both of which activate negative to their Nernst potentials and do in fact conduct action potentials [252–254]. The proton channel identified recently in a non-bioluminescent dinoflagellate species, K. veneficum, was found to activate well negative to EH over a wide range of pH (figure 7), precisely the behaviour needed to mediate an action potential [4]. These properties strongly support suspicions that proton channels mediate the action potential that triggers the bioluminescent flash [4,23,53,255].
As it turns out, several bioluminescent dinoflagellates, when stimulated mechanically by waves or other disturbances of the seawater, emit a rapid flash that can be very impressive when plankton blooms concentrate many individuals in one location [256]. Forty years ago, voltage-gated proton channels were first conceived by J. Woodland Hastings, who proposed that they trigger the flash [52]. This response occurs when a stimulus evokes an action potential in the large intracellular flotation vacuole [257–260]. The action potential invades the scintillons [261,262], numerous evaginations of membrane that form small vesicles [263] that are packed with luciferin, luciferin-binding protein and luciferase [255,264–266]. Because the vacuole has very low pH of 3.5–4.5 [54,267], an open proton channel will conduct protons very rapidly into the scintillon, where the pH is near neutral [268]. Luciferase is quiescent at high pH, but is activated by a drop in pH to approximately 6.5 [255]. Thus, the influx of protons through HV rapidly lowers the pH of the scintillon and thereby activates luciferase. An additional parallel mechanism is that luciferin-binding protein releases luciferin (the substrate for luciferase) at low pH [255,265,269].
In order for HV to carry out their function, they must open negative to EH and conduct inward current. Figure 7 shows that this is precisely what they do. Any other proton channel would just sit there, refusing to conduct inward current, because the vacuole interior is the topological equivalent of extracellular space. The discovery of the unique properties of kHV1 [4] confirms Woody Hastings's hypothesis, nearly 40 years later. It turns out that having a Vthreshold negative to EH is required not only to mediate the action potential, but also to activate luciferase in the scintillons!
1.10. Why do proton channels in mammals exist as dimers?
In most ‘higher’ species, HV1 exist as dimers [270], as illustrated in figure 3, with the peculiar property that monomeric constructs exhibit all of the main physiological features of the dimer [8,9,223,271,272]. This situation raises the question, why bother to dimerize? Proteins assemble into multimers for a variety of reasons. Heteromultimers obviously need to assemble all their parts to make a complete, functioning complex. Some membrane proteins are more stably expressed when they form multimers; as monomers they are rapidly degraded. For example, two key membrane-bound components of the NADPH oxidase complex, gp91phox and p22phox reside in the membrane as a heterodimer, but do not remain in the phagocyte membrane when mutation prevents one from being expressed [273]. However, monomeric HV constructs appear content to function indefinitely in cell membranes.
The most consistent functional difference between monomeric and dimeric HV is in gating kinetics. Monomeric constructs open approximately five times faster than dimeric channels [8,223,271,274,275], as is evident by comparison of figure 8a,b. This distinct difference in speed might seem to be important in some situations, until we remember that among species, activation kinetics varies over several orders of magnitude. HV in snail neurons open within a few milliseconds [246], whereas mammalian HV require seconds or even minutes [31]. It would appear that a species with a need for rapidly opening proton channels would be better served by making a channel that is intrinsically fast, rather than tinkering with stoichiometry. This perspective reveals no obvious advantage of dimeric over monomeric architecture.
Figure 8.

Kinetics of proton currents generated by the normal dimeric human proton channel, hHV1 (a) and a monomeric construct produced when the C terminus was truncated, hHV1ΔC (b). The dimeric channel, which occurs in native cells [276], opens with sigmoid kinetics and approximately five times slower than monomeric constructs (note different time scales). Conditions are the same, pHi = pHo = 7.5, 23°C, voltage pulses to +50 mV. In (c), the S4 helix of the Ciona intestinalis proton channel was labelled with a fluorescent tag that moves when the channel opens upon depolarization (upper noisy traces), preceding the activation of current (black line). In a Hodgkin–Huxley type cooperative gating mechanism [252], both subunits move before either conducts and the predicted time course is sigmoidal, the square of the motion time course (lower noisy traces, superimposed on line) (a,b adapted from [223]; c from [272]).
A more subtle property of dimeric HV1 channels is that they gate cooperatively [272,275]. Gonzalez et al. [272] attached a fluorescent tag on the S4 transmembrane helix, which could be seen to move upon depolarization with an exponential time course (upper noisy traces in figure 8c) that preceded the turn-on of H+ current (smooth lines in figure 8c). The implication is that both monomers must respond to voltage before either one can conduct. In such a system, the effective gating charge that moves when the channel opens is doubled. Opening a tetrameric K+ channel requires movement of all four of its VSDs, and the charge moved is quadruple that of a single VSD. The upshot is that by requiring both protomers to move before either can conduct, HV1 has twice as steep a dependence on membrane potential [223,272,277].
What good is this? Consider the plight of a neutrophil that has just engulfed a bacterium (figure 4). In order to produce sufficient ROS to kill the microbe, thousands of NADPH oxidase complexes quickly assemble in the phagosome membrane, and begin to transfer electrons from NADPH in the cytoplasm across the membrane to reduce O2 to O2·−. The separation of charge, electrons moving across the membrane and protons left behind, causes rapid depolarization. In fact, if this process were not compensated, the membrane potential of a human eosinophil would depolarize at a rate of approximately 18 V s−1 [23], and membrane dielectric breakdown would occur in a tiny fraction of a second. Clearly, something must be done.
To limit the depolarization, cations must leave or anions must enter. As we saw in figure 4, phagocytes have determined that the best solution to this problem is to open proton channels and extrude H+ to balance the transmembrane electron transfer. It turns out that NADPH oxidase is sensitive to membrane potential; it is inhibited by depolarization, and turns off altogether at about +200 mV [32]. Of course, this problem is self-imposed—it is the electrogenic activity of NADPH oxidase [27] that causes the depolarization. Fortunately, depolarization opens proton channels, and the greater the depolarization, the more channels open (and the greater the driving force for H+ efflux), until the electron efflux is perfectly balanced by proton efflux. In this remarkable system, NADPH oxidase activity causes depolarization and lowers pHi and both of these factors open proton channels, which in turn have the effects of limiting the depolarization as well as minimizing changes in pHi. Despite the efficiency of HV, the membrane potential still depolarizes enough, roughly to +50 mV in human neutrophils [278–280], to impair NADPH oxidase activity by approximately 15–20% [24]. Matters would be worse if hHV1 were not dimeric, however, because the weaker sensitivity to membrane potential of a monomeric hHV1 would mean that greater depolarization would be needed to open enough proton channels, and consequently, more self-inhibition of NADPH oxidase activity would ensue [223]. That hHV1 is dimeric makes NADPH oxidase more efficient.
Although this explanation applies specifically to HV1 in phagocytes, it could also apply in a more general way for other mammalian cells. Steeper voltage dependence means that a smaller depolarization is required to open enough H+ channels to do any job. This makes HV1 more efficient, and less of a drain on the resources of the cell.
Finally, there are several unicellular species in which HV are probably monomers [4,270], because the protein lacks the coiled-coil domain in the C terminus that is the main enforcer of dimerization [8,9,223,271,272,281,282]. The original question was ‘Why are some HV dimers?’ We will ignore the obverse question ‘Why are some HV monomers?’ because peer pressure seems inadequate as a basis for evolution.
1.11. Concluding thoughts
The field of voltage-gated proton channels is expanding rapidly in all directions: its presence in new species, its presence in new cell types, novel functions, involvement in normal physiology as well as in disease, and advances in structure–function relationships. The function of HV in all situations depends on its selectivity for protons as well as a unique ΔpH-dependent gating mechanism. Both properties are subjects of intense study. The mechanism of proton selectivity involves an aspartate in the centre of the pore; further details are unclear. The mechanism of ΔpH-dependent gating is remains obscure. The proton channel field has benefited from work on other proton-conducting molecules and on VSDs of other voltage-gated ion channels. As our understanding of HV develops, one hopes that proton channels will return the favour.
Acknowledgements
The authors appreciate helpful comments and discussions with Timothy A. Cross, William F. DeGrado, Mei Hong, Casey R. Law, Régis Pomès and Susan M.E. Smith. The content is solely the responsibility of the authors and does not necessarily represent the views of the National Institute of General Medical Sciences or the National Institutes of Health.
Endnotes
The total includes 10 papers that are only partly about proton channels but excludes seven papers whose authors thought they were studying proton channels, but in all probability were not.
Since the dawn of electrophysiology over two centuries ago in the heyday of Luigi Galvani, cells have been known to have an electrical potential across their plasma membranes. During an action potential, or nerve impulse, this ‘polarization’ was reduced or eliminated, a process described as ‘depolarization’ (literally removal of polarization), which was thought to reflect a transient breakdown of the membrane potential. Similarly, ‘hyperpolarization’ described an increase in polarization beyond the normal resting membrane potential. Once it became clear that the membrane potential transcended 0 mV during the action potential [14,15], it was necessary to redefine the terminology. Today depolarization simply means making the membrane potential more positive, and hyperpolarization means making it more negative.
Funding statement
This work was supported in part by NSF award MCB-0943362 and NIH R01-GM087507.
References
- 1.Ramsey IS, Moran MM, Chong JA, Clapham DE. 2006. A voltage-gated proton-selective channel lacking the pore domain. Nature 440, 1213–1216 (doi:10.1038/nature04700) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sasaki M, Takagi M, Okamura Y. 2006. A voltage sensor-domain protein is a voltage-gated proton channel. Science 312, 589–592 (doi:10.1126/science.1122352) [DOI] [PubMed] [Google Scholar]
- 3.Kulleperuma K, Smith SME, Morgan D, Musset B, Holyoake J, Chakrabarti N, Cherny VV, DeCoursey TE, Pomès R. 2013. Construction and validation of a homology model of the human voltage-gated proton channel hHV1. J. Gen. Physiol. 141, 445–465 (doi:10.1085/jgp.201210856) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith SME, Morgan D, Musset B, Cherny VV, Place AR, Hastings JW, DeCoursey TE. 2011. Voltage-gated proton channel in a dinoflagellate. Proc. Natl Acad. Sci. USA 108, 18 162–18 167 (doi:10.1073/pnas.1115405108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor AR, Brownlee C, Wheeler GL. 2012. Proton channels in algae: reasons to be excited. Trends Plant Sci. 17, 675–684 (doi:10.1016/j.tplants.2012.06.009) [DOI] [PubMed] [Google Scholar]
- 6.Hodgkin AL, Huxley AF. 1952. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J. Physiol. 116, 449–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musset B, Smith SME, Rajan S, Morgan D, Cherny VV, DeCoursey TE. 2011. Aspartate 112 is the selectivity filter of the human voltage-gated proton channel. Nature 480, 273–277 (doi:10.1038/nature10557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koch HP, Kurokawa T, Okochi Y, Sasaki M, Okamura Y, Larsson HP. 2008. Multimeric nature of voltage-gated proton channels. Proc. Natl Acad. Sci. USA 105, 9111–9116 (doi:10.1073/pnas.0801553105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tombola F, Ulbrich MH, Isacoff EY. 2008. The voltage-gated proton channel Hv1 has two pores, each controlled by one voltage sensor. Neuron 58, 546–556 (doi:10.1016/j.neuron.2008.03.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musset B, Capasso M, Cherny VV, Morgan D, Bhamrah M, Dyer MJS, DeCoursey TE. 2010. Identification of Thr29 as a critical phosphorylation site that activates the human proton channel Hvcn1 in leukocytes. J. Biol. Chem. 285, 5117–5121 (doi:10.1074/jbc.C109.082727) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan D, Cherny VV, Finnegan A, Bollinger J, Gelb MH, DeCoursey TE. 2007. Sustained activation of proton channels and NADPH oxidase in human eosinophils and murine granulocytes requires PKC but not cPLA2α activity. J. Physiol. 579, 327–344 (doi:10.1113/jphysiol.2006.124248) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y. 2005. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243 (doi:10.1038/nature03650) [DOI] [PubMed] [Google Scholar]
- 13.DeCoursey TE. 2010. Voltage-gated proton channels find their dream job managing the respiratory burst in phagocytes. Physiology 25, 27–40 (doi:10.1152/physiol.00039.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodgkin AL, Huxley AF. 1945. Resting and action potentials in single nerve fibres. J. Physiol. 104, 176–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodgkin AL, Huxley AF. 1939. Action potentials recorded from inside a nerve fibre. Nature 144, 710–711 (doi:10.1038/144710a0) [Google Scholar]
- 16.Fatt P, Katz B. 1951. An analysis of the end-plate potential recorded with an intracellular electrode. J. Physiol. 115, 320–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeCoursey TE. 2013. Voltage-gated proton channels: molecular biology, physiology, and pathophysiology of the HV family. Physiol. Rev. 93, 599–652 (doi:10.1152/physrev.00011.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeCoursey TE, Cherny VV. 1994. Voltage-activated hydrogen ion currents. J. Membr. Biol. 141, 203–223 (doi:10.1007/BF00235130) [DOI] [PubMed] [Google Scholar]
- 19.Demaurex N, Grinstein S, Jaconi M, Schlegel W, Lew DP, Krause KH. 1993. Proton currents in human granulocytes: regulation by membrane potential and intracellular pH. J. Physiol. 466, 329–344 [PMC free article] [PubMed] [Google Scholar]
- 20.Kapus A, Romanek R, Qu AY, Rotstein OD, Grinstein S. 1993. A pH-sensitive and voltage-dependent proton conductance in the plasma membrane of macrophages. J. Gen. Physiol. 102, 729–760 (doi:10.1085/jgp.102.4.729) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meech RW, Thomas RC. 1987. Voltage-dependent intracellular pH in Helix aspersa neurones. J. Physiol. 390, 433–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas RC, Meech RW. 1982. Hydrogen ion currents and intracellular pH in depolarized voltage-clamped snail neurones. Nature 299, 826–828 (doi:10.1038/299826a0) [DOI] [PubMed] [Google Scholar]
- 23.DeCoursey TE. 2003. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 83, 475–579 (doi:10.1152/physrev.00028.2002) [DOI] [PubMed] [Google Scholar]
- 24.Murphy R, DeCoursey TE. 2006. Charge compensation during the phagocyte respiratory burst. Biochim. Biophys. Acta 1757, 996–1011 (doi:10.1016/j.bbabio.2006.01.005) [DOI] [PubMed] [Google Scholar]
- 25.Baldridge CW, Gerard RW. 1932. The extra respiration of phagocytosis. Am. J. Physiol. 103, 235–236 [Google Scholar]
- 26.Sbarra AJ, Karnovsky ML. 1959. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J. Biol. Chem. 234, 1355–1362 [PubMed] [Google Scholar]
- 27.Henderson LM, Chappell JB, Jones OTG. 1987. The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochem. J. 246, 325–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeCoursey TE, Cherny VV, Zhou W, Thomas LL. 2000. Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proc. Natl Acad. Sci. USA 97, 6885–6889 (doi:10.1073/pnas.100047297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schrenzel J, Serrander L, Bánfi B, Nüsse O, Fouyouzi R, Lew DP, Demaurex N, Krause KH. 1998. Electron currents generated by the human phagocyte NADPH oxidase. Nature 392, 734–737 (doi:10.1038/33725) [DOI] [PubMed] [Google Scholar]
- 30.Henderson LM, Chappell JB, Jones OTG. 1988. Superoxide generation by the electrogenic NADPH oxidase of human neutrophils is limited by the movement of a compensating charge. Biochem. J. 255, 285–290 [PMC free article] [PubMed] [Google Scholar]
- 31.DeCoursey TE, Cherny VV. 1993. Potential, pH, and arachidonate gate hydrogen ion currents in human neutrophils. Biophys. J. 65, 1590–1598 (doi:10.1016/S0006-3495(93)81198-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeCoursey TE, Morgan D, Cherny VV. 2003. The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature 422, 531–534 (doi:10.1038/nature01523) [DOI] [PubMed] [Google Scholar]
- 33.Ramsey IS, Ruchti E, Kaczmarek JS, Clapham DE. 2009. Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc. Natl Acad. Sci. USA 106, 7642–7647 (doi:10.1073/pnas.0902761106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El Chemaly A, Okochi Y, Sasaki M, Arnaudeau S, Okamura Y, Demaurex N. 2010. VSOP/Hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J. Exp. Med. 207, 129–139 (doi:10.1084/jem.20091837) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okochi Y, Sasaki M, Iwasaki H, Okamura Y. 2009. Voltage-gated proton channel is expressed on phagosomes. Biochem. Biophys. Res. Commun. 382, 274–279 (doi:10.1016/j.bbrc.2009.03.036) [DOI] [PubMed] [Google Scholar]
- 36.Gordienko DV, Tare M, Parveen S, Fenech CJ, Robinson C, Bolton TB. 1996. Voltage-activated proton current in eosinophils from human blood. J. Physiol. 496, 299–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeCoursey TE, Cherny VV, DeCoursey AG, Xu W, Thomas LL. 2001. Interactions between NADPH oxidase-related proton and electron currents in human eosinophils. J. Physiol. 535, 767–781 (doi:10.1111/j.1469-7793.2001.00767.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bankers-Fulbright JL, Gleich GJ, Kephart GM, Kita H, O'Grady SM. 2003. Regulation of eosinophil membrane depolarization during NADPH oxidase activation. J. Cell Sci. 116, 3221–3226 (doi:10.1242/jcs.00627) [DOI] [PubMed] [Google Scholar]
- 39.Bánfi B, Schrenzel J, Nüsse O, Lew DP, Ligeti E, Krause KH, Demaurex N. 1999. A novel H+ conductance in eosinophils: unique characteristics and absence in chronic granulomatous disease. J. Exp. Med. 190, 183–194 (doi:10.1084/jem.190.2.183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Femling JK, et al. 2006. The antibacterial activity of human neutrophils and eosinophils requires proton channels but not BK channels. J. Gen. Physiol. 127, 659–672 (doi:10.1085/jgp.200609504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu X, Mose E, Zimmermann N. 2013. Proton channel HVCN1 is required for effector functions of mouse eosinophils. BMC Immunol. 14, 24 (doi:10.1186/1471-2172-14-24) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Musset B, Cherny VV, DeCoursey TE. 2012. Strong glucose dependence of electron current in human monocytes. Am. J. Physiol. Cell Physiol. 302, C286–C295 (doi:10.1152/ajpcell.00335.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mori H, Sakai H, Morihata H, Kawawaki J, Amano H, Yamano T, Kuno M. 2003. Regulatory mechanisms and physiological relevance of a voltage-gated H+ channel in murine osteoclasts: phorbol myristate acetate induces cell acidosis and the channel activation. J. Bone Miner. Res. 18, 2069–2076 (doi:10.1359/jbmr.2003.18.11.2069) [DOI] [PubMed] [Google Scholar]
- 44.Musset B, Morgan D, Cherny VV, MacGlashan DW, Jr, Thomas LL, Ríos E, DeCoursey TE. 2008. A pH-stabilizing role of voltage-gated proton channels in IgE-mediated activation of human basophils. Proc. Natl Acad. Sci. USA 105, 11 020–11 025 (doi:10.1073/pnas.0800886105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eder C, DeCoursey TE. 2001. Voltage-gated proton channels in microglia. Prog. Neurobiol. 64, 277–305 (doi:10.1016/S0301-0082(00)00062-9) [DOI] [PubMed] [Google Scholar]
- 46.Wu LJ, Wu G, Sharif MR, Baker A, Jia Y, Fahey FH, Luo HR, Feener EP, Clapham DE. 2012. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat. Neurosci. 15, 565–573 (doi:10.1038/nn.3059) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szteyn K, Yang W, Schmid E, Lang F, Shumilina E. 2012. Lipopolysaccharide-sensitive H+ current in dendritic cells. Am. J. Physiol. Cell Physiol. 303, C204–C212 (doi:10.1074/jbc.M111.275925) [DOI] [PubMed] [Google Scholar]
- 48.Musset B, et al. 2012. NOX5 in human spermatozoa: expression, function and regulation. J. Biol. Chem. 287, 9376–9388 (doi:10.1074/jbc.M111.314955) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Capasso M, et al. 2010. HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat. Immunol. 11, 265–272 (doi:10.1038/ni.1843) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schilling T, Gratopp A, DeCoursey TE, Eder C. 2002. Voltage-activated proton currents in human lymphocytes. J. Physiol. 545, 93–105 (doi:10.1113/jphysiol.2002.028878) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.El Chemaly A, Guinamard R, Demion M, Fares N, Jebara V, Faivre JF, Bois P. 2006. A voltage-activated proton current in human cardiac fibroblasts. Biochem. Biophys. Res. Commun. 340, 512–516 (doi:10.1016/j.bbrc.2005.12.038) [DOI] [PubMed] [Google Scholar]
- 52.Fogel M, Hastings JW. 1972. Bioluminescence: mechanism and mode of control of scintillon activity. Proc. Natl Acad. Sci. USA 69, 690–693 (doi:10.1073/pnas.69.3.690) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nawata T, Sibaoka T. 1979. Coupling between action potential and bioluminescence in Noctiluca: effects of inorganic ions and pH in vacuolar sap. J. Comp. Physiol. 134, 137–149 (doi:10.1007/BF00610472) [Google Scholar]
- 54.Nicolas MT, Nicolas G, Johnson CH, Bassot JM, Hastings JW. 1987. Characterization of the bioluminescent organelles in Gonyaulax polyedra (dinoflagellates) after fast-freeze fixation and antiluciferase immunogold staining. J. Cell Biol. 105, 723–735 (doi:10.1083/jcb.105.2.723) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lishko PV, Botchkina IL, Fedorenko A, Kirichok Y. 2010. Acid extrusion from human spermatozoa is mediated by flagellar voltage-gated proton channel. Cell 140, 327–337 (doi:10.1016/j.cell.2009.12.053) [DOI] [PubMed] [Google Scholar]
- 56.Wang Y, Li SJ, Pan J, Che Y, Yin J, Zhao Q. 2011. Specific expression of the human voltage-gated proton channel Hv1 in highly metastatic breast cancer cells, promotes tumor progression and metastasis. Biochem. Biophys. Res. Commun. 412, 353–359 (doi:10.1016/j.bbrc.2011.07.102) [DOI] [PubMed] [Google Scholar]
- 57.Baud C, Barish ME. 1984. Changes in membrane hydrogen and sodium conductances during progesterone-induced maturation of Ambystoma oocytes. Dev. Biol. 105, 423–434 (doi:10.1016/0012-1606(84)90299-9) [DOI] [PubMed] [Google Scholar]
- 58.Humez S, Fournier F, Guilbault P. 1995. A voltage-dependent and pH-sensitive proton current in Rana esculenta oocytes. J. Membr. Biol. 147, 207–215 (doi:10.1007/BF00233548) [DOI] [PubMed] [Google Scholar]
- 59.Henderson LM, Chappell JB, Jones OTG. 1988. Internal pH changes associated with the activity of NADPH oxidase of human neutrophils. Further evidence for the presence of an H+ conducting channel. Biochem. J. 251, 563–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morgan D, Capasso M, Musset B, Cherny VV, Ríos E, Dyer MJS, DeCoursey TE. 2009. Voltage-gated proton channels maintain pH in human neutrophils during phagocytosis. Proc. Natl Acad. Sci. USA 106, 18 022–18 027 (doi:10.1073/pnas.0905565106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morgan D, Cherny VV, Murphy R, Katz BZ, DeCoursey TE. 2005. The pH dependence of NADPH oxidase in human eosinophils. J. Physiol. 569, 419–431 (doi:10.1113/jphysiol.2005.094748) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taylor AR, Chrachri A, Wheeler G, Goddard H, Brownlee C. 2011. A voltage-gated H+ channel underlying pH homeostasis in calcifying coccolithophores. PLoS Biol. 9, e1001085 (doi:10.1371/journal.pbio.1001085) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iovannisci D, Illek B, Fischer H. 2010. Function of the HVCN1 proton channel in airway epithelia and a naturally occurring mutation, M91T. J. Gen. Physiol. 136, 35–46 (doi:10.1085/jgp.200910379) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y, Li SJ, Wu X, Che Y, Li Q. 2012. Clinicopathological and biological significance of human voltage-gated proton channel Hv1 over-expression in breast cancer. J. Biol. Chem. 287, 13 877–13 888 (doi:10.1074/jbc.M112.345280) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morihata H, Nakamura F, Tsutada T, Kuno M. 2000. Potentiation of a voltage-gated proton current in acidosis-induced swelling of rat microglia. J. Neurosci. 20, 7220–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hille B. 1972. The permeability of the sodium channel to metal cations in myelinated nerve. J. Gen. Physiol. 59, 637–658 (doi:10.1085/jgp.59.6.637) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Campbell DT. 1976. Ionic selectivity of the sodium channel of frog skeletal muscle. J. Gen. Physiol. 67, 295–307 (doi:10.1085/jgp.67.3.295) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chandler WK, Meves H. 1965. Voltage clamp experiments on internally perfused giant axons. J. Physiol. 180, 788–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ebert GA, Goldman L. 1976. The permeability of the sodium channel in Myxicola to the alkali cations. J. Gen. Physiol. 68, 327–340 (doi:10.1085/jgp.68.3.327) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ravindran A, Kwiecinski H, Alvarez O, Eisenman G, Moczydlowski E. 1992. Modeling ion permeation through batrachotoxin-modified Na+ channels from rat skeletal muscle with a multi-ion pore. Biophys. J. 61, 494–508 (doi:10.1016/S0006-3495(92)81854-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goldman DE. 1943. Potential, impedance, and rectification in membranes. J. Gen. Physiol. 27, 37–60 (doi:10.1085/jgp.27.1.37) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hodgkin AL, Katz B. 1949. The effect of sodium ions on the electrical activity of giant axon of the squid. J. Physiol. 108, 37–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hille B. 2001. Ion channels of excitable membranes, 3rd edn Sunderland, MA: Sinauer Associates, Inc [Google Scholar]
- 74.LeMasurier M, Heginbotham L, Miller C. 2001. KcsA: it's a potassium channel. J. Gen. Physiol. 118, 303–314 (doi:10.1085/jgp.118.3.303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bretag AH. 1987. Muscle chloride channels. Physiol. Rev. 67, 618–724 [DOI] [PubMed] [Google Scholar]
- 76.Vaughan PC, French AS. 1989. Non-ligand-activated chloride channels of skeletal muscle and epithelia. Prog. Biophys. Mol. Biol. 54, 59–79 (doi:10.1016/0079-6107(89)90009-6) [DOI] [PubMed] [Google Scholar]
- 77.Cherny VV, Thomas LL, DeCoursey TE. 2001. Voltage-gated proton currents in human basophils. Biol. Membr. 18, 458–465 [Google Scholar]
- 78.DeCoursey TE, Cherny VV. 1996. Voltage-activated proton currents in human THP-1 monocytes. J. Membr. Biol. 152, 131–140 (doi:10.1007/s002329900092) [DOI] [PubMed] [Google Scholar]
- 79.Kapus A, Romanek R, Grinstein S. 1994. Arachidonic acid stimulates the plasma membrane H+ conductance of macrophages. J. Biol. Chem. 269, 4736–4745 [PubMed] [Google Scholar]
- 80.Barry PH. 2006. The reliability of relative anion–cation permeabilities deduced from reversal (dilution) potential measurements in ion channel studies. Cell Biochem. Biophys. 46, 143–154 (doi:10.1385/CBB:46:2:143) [DOI] [PubMed] [Google Scholar]
- 81.Wraight CA. 2006. Chance and design: proton transfer in water, channels and bioenergetic proteins. Biochim. Biophys. Acta 1757, 886–912 (doi:10.1016/j.bbabio.2006.06.017) [DOI] [PubMed] [Google Scholar]
- 82.Huggins ML. 1931. The role of hydrogen bonds in conduction by hydrogen and hydroxyl ions. J. Am. Chem. Soc. 53, 3190–3191 (doi:10.1021/ja01359a511) [Google Scholar]
- 83.Latimer WM, Rodebush WH. 1920. Polarity and ionization from the standpoint of the Lewis theory of valence. J. Am. Chem. Soc. 42, 1419–1433 (doi:10.1021/ja01452a015) [Google Scholar]
- 84.Pauling L. 1939. The hydrogen bond. In The nature of the chemical bond and the structure of molecules and crystals: an introduction to modern structural chemistry (ed. Pauling L.), pp. 264–314 Ithaca, NY: Cornell University Press [Google Scholar]
- 85.Marx D, Tuckerman ME, Hutter J, Parrinello M. 1999. The nature of the hydrated excess proton in water. Nature 397, 601–604 (doi:10.1038/17579) [Google Scholar]
- 86.Agmon N. 1995. The Grotthuss mechanism. Chem. Phys. Lett. 244, 456–462 (doi:10.1016/0009-2614(95)00905-J) [Google Scholar]
- 87.Eigen M. 1964. Proton transfer, acid–base catalysis, and enzymatic hydrolysis. Part I: elementary processes. Angew. Chem. Int. Ed. 3, 1–19 (doi:10.1002/anie.196400011) [Google Scholar]
- 88.Markovitch O, Chen H, Izvekov S, Paesani F, Voth GA, Agmon N. 2008. Special pair dance and partner selection: elementary steps in proton transport in liquid water. J. Phys. Chem. B. 112, 9456–9466 (doi:10.1021/jp804018y) [DOI] [PubMed] [Google Scholar]
- 89.Hassanali A, Giberti F, Cuny J, Kühne TD, Parrinello M. 2013. Proton transfer through the water gossamer. Proc. Natl Acad. Sci. USA 110, 13 723–13 728 (doi:10.1073/pnas.1306642110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huggins ML. 1936. Hydrogen bridges in ice and liquid water. J. Phys. Chem. 40, 723–731 (doi:10.1021/j150375a004) [Google Scholar]
- 91.Robinson RA, Stokes RH. 1959. Electrolyte solutions. London, UK: Butterworths [Google Scholar]
- 92.Danneel H. 1905. Notiz über Ionengeschwindigkeiten. Z. Elektrochem. Angew. Phys. Chem. 11, 249–252 (doi:10.1002/bbpc.19050111603) [Google Scholar]
- 93.de Grotthuss CJT. 1806. Mémoire sur la décomposition de l'eau et des corps qu'elle tient en dissolution à l'aide de l’électricité galvanique. Ann. Chim. LVIII, 54–74 [Google Scholar]
- 94.de Grotthuss CJT. 2006. Memoir on the decomposition of water and of the bodies that it holds in solution by means of galvanic electricity. 1805. Biochim. Biophys. Acta 1757, 871–875 (doi:10.1016/j.bbabio.2006.07.004) [DOI] [PubMed] [Google Scholar]
- 95.Mohammed OF, Pines D, Dreyer J, Pines E, Nibbering ET. 2005. Sequential proton transfer through water bridges in acid–base reactions. Science 310, 83–86 (doi:10.1126/science.1117756) [DOI] [PubMed] [Google Scholar]
- 96.Riccardi D, Konig P, Prat-Resina X, Yu H, Elstner M, Frauenheim T, Cui Q. 2006. ‘Proton holes’ in long-range proton transfer reactions in solution and enzymes: a theoretical analysis. J. Am. Chem. Soc. 128, 16302–16311 (doi:10.1021/ja065451j) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pomès R, Roux B. 1996. Theoretical study of H+ translocation along a model proton wire. J. Phys. Chem. 100, 2519–2527 (doi:10.1021/jp9525752) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brewer ML, Schmitt UW, Voth GA. 2001. The formation and dynamics of proton wires in channel environments. Biophys. J. 80, 1691–1702 (doi:10.1016/S0006-3495(01)76140-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shepherd LMS, Morrison CA. 2010. Simulating proton transport through a simplified model for transmembrane proteins. J. Phys. Chem. B 114, 7047–7055 (doi:10.1021/jp910262d) [DOI] [PubMed] [Google Scholar]
- 100.Hassan SA, Hummer G, Lee YS. 2006. Effects of electric fields on proton transport through water chains. J. Chem. Phys. 124, 204510 (doi:10.1063/1.2198820) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagle JF, Morowitz HJ. 1978. Molecular mechanisms for proton transport in membranes. Proc. Natl Acad. Sci. USA 75, 298–302 (doi:10.1073/pnas.75.1.298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nagle JF, Tristram-Nagle S. 1983. Hydrogen bonded chain mechanisms for proton conduction and proton pumping. J. Membr. Biol. 74, 1–14 (doi:10.1007/BF01870590) [DOI] [PubMed] [Google Scholar]
- 103.Cukierman S, Quigley EP, Crumrine DS. 1997. Proton conduction in gramicidin A and in its dioxolane-linked dimer in different lipid bilayers. Biophys. J. 73, 2489–2502 (doi:10.1016/S0006-3495(97)78277-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brelidze TI, Magleby KL. 2004. Protons block BK channels by competitive inhibition with K+ and contribute to the limits of unitary currents at high voltages. J. Gen. Physiol. 123, 305–319 (doi:10.1085/jgp.200308951) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mikulski R, West D, Sippel KH, Avvaru BS, Aggarwal M, Tu C, McKenna R, Silverman DN. 2013. Water networks in fast proton transfer during catalysis by human carbonic anhydrase II. Biochemistry 52, 125–131 (doi:10.1021/bi301099k) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wallace BA, Ravikumar K. 1988. The gramicidin pore: crystal structure of a cesium complex. Science 241, 182–187 (doi:10.1126/science.2455344) [DOI] [PubMed] [Google Scholar]
- 107.Levitt DG, Elias SR, Hautman JM. 1978. Number of water molecules coupled to the transport of sodium, potassium and hydrogen ions via gramicidin, nonactin or valinomycin. Biochim. Biophys. Acta 512, 436–451 (doi:10.1016/0005-2736(78)90266-3) [DOI] [PubMed] [Google Scholar]
- 108.Hladky SB, Haydon DA. 1972. Ion transfer across lipid membranes in the presence of gramicidin A. I. Studies of the unit conductance channel. Biochim. Biophys. Acta 274, 294–312 (doi:10.1016/0005-2736(72)90178-2) [DOI] [PubMed] [Google Scholar]
- 109.Myers VB, Haydon DA. 1972. Ion transfer across lipid membranes in the presence of gramicidin A. II. The ion selectivity. Biochim. Biophys. Acta 274, 313–322 (doi:10.1016/0005-2736(72)90179-4) [DOI] [PubMed] [Google Scholar]
- 110.Cukierman S. 2000. Proton mobilities in water and in different stereoisomers of covalently linked gramicidin A channels. Biophys. J. 78, 1825–1834 (doi:10.1016/S0006-3495(00)76732-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cherny VV, Murphy R, Sokolov V, Levis RA, DeCoursey TE. 2003. Properties of single voltage-gated proton channels in human eosinophils estimated by noise analysis and by direct measurement. J. Gen. Physiol. 121, 615–628 (doi:10.1085/jgp.200308813) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eisenman G, Enos B, Hägglund J, Sandblom J. 1980. Gramicidin as an example of a single-filing ionic channel. Ann. NY Acad. Sci. 339, 8–20 (doi:10.1111/j.1749-6632.1980.tb15964.x) [DOI] [PubMed] [Google Scholar]
- 113.Hosler JP, Ferguson-Miller S, Mills DA. 2006. Energy transduction: proton transfer through the respiratory complexes. Annu. Rev. Biochem. 75, 165–187 (doi:10.1146/annurev.biochem.75.062003.101730) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhu J, Han H, Pawate A, Gennis RB. 2010. Decoupling mutations in the D-channel of the aa3-type cytochrome c oxidase from Rhodobacter sphaeroides suggest that a continuous hydrogen-bonded chain of waters is essential for proton pumping. Biochemistry 49, 4476–4482 (doi:10.1021/bi100344x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Henry RM, Yu CH, Rodinger T, Pomès R. 2009. Functional hydration and conformational gating of proton uptake in cytochrome c oxidase. J. Mol. Biol. 387, 1165–1185 (doi:10.1016/j.jmb.2009.02.042) [DOI] [PubMed] [Google Scholar]
- 116.Johansson AL, Högbom M, Carlsson J, Gennis RB, Brzezinski P. 2013. Role of aspartate 132 at the orifice of a proton pathway in cytochrome c oxidase. Proc. Natl Acad. Sci. USA 110, 8912–8917 (doi:10.1073/pnas.1303954110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Horsefield R, Yankovskaya V, Sexton G, Whittingham W, Shiomi K, Omura S, Byrne B, Cecchini G, Iwata S. 2006. Structural and computational analysis of the quinone-binding site of complex II (succinate–ubiquinone oxidoreductase): a mechanism of electron transfer and proton conduction during ubiquinone reduction. J. Biol. Chem. 281, 7309–7316 (doi:10.1074/jbc.M508173200) [DOI] [PubMed] [Google Scholar]
- 118.Dunker AK, Marvin DA. 1978. A model for membrane transport through α-helical protein pores. J. Theor. Biol. 72, 9–16 (doi:10.1016/0022-5193(78)90015-2) [DOI] [PubMed] [Google Scholar]
- 119.Ginovska-Pangovska B, Ho MH, Linehan JC, Cheng Y, Dupuis M, Raugei S, Shaw WJ. 2013. Molecular dynamics study of the proposed proton transport pathways in [FeFe]-hydrogenase. Biochim. Biophys. Acta 1837, 131–138 (doi:10.1016/j.bbabio.2013.08.004) [DOI] [PubMed] [Google Scholar]
- 120.Varanasi L, Hosler JP. 2012. Subunit III-depleted cytochrome c oxidase provides insight into the process of proton uptake by proteins. Biochim. Biophys. Acta 1817, 545–551 (doi:10.1016/j.bbabio.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen K, Hirst J, Camba R, Bonagura CA, Stout CD, Burgess BK, Armstrong FA. 2000. Atomically defined mechanism for proton transfer to a buried redox centre in a protein. Nature 405, 814–817 (doi:10.1038/35015610) [DOI] [PubMed] [Google Scholar]
- 122.Meyer E. 1992. Internal water molecules and H-bonding in biological macromolecules: a review of structural features with functional implications. Protein Sci. 1, 1543–1562 (doi:10.1002/pro.5560011203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li S, Hong M. 2011. Protonation, tautomerization, and rotameric structure of histidine: a comprehensive study by magic-angle-spinning solid-state NMR. J. Am. Chem. Soc. 133, 1534–1544 (doi:10.1021/ja108943n) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tu CK, Silverman DN, Forsman C, Jonsson BH, Lindskog S. 1989. Role of histidine 64 in the catalytic mechanism of human carbonic anhydrase II studied with a site-specific mutant. Biochemistry 28, 7913–7918 (doi:10.1021/bi00445a054) [DOI] [PubMed] [Google Scholar]
- 125.Silverman D, McKenna R. 2007. Solvent-mediated proton transfer in catalysis by carbonic anhydrase. Acc. Chem. Res. 40, 669–675 (doi:10.1021/ar7000588) [DOI] [PubMed] [Google Scholar]
- 126.Brändén M, Sigurdson H, Namslauer A, Gennis RB, Ädelroth P, Brzezinski P. 2001. On the role of the K-proton transfer pathway in cytochrome c oxidase. Proc. Natl Acad. Sci. USA 98, 5013–5018 (doi:10.1073/pnas.081088398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Koepke J, Olkhova E, Angerer H, Muller H, Peng G, Michel H. 2009. High resolution crystal structure of Paracoccus denitrificans cytochrome c oxidase: new insights into the active site and the proton transfer pathways. Biochim. Biophys. Acta 1787, 635–645 (doi:10.1016/j.bbabio.2009.04.003) [DOI] [PubMed] [Google Scholar]
- 128.Leiding T, Wang J, Martinsson J, DeGrado WF, Årsköld SP. 2010. Proton and cation transport activity of the M2 proton channel from influenza A virus. Proc. Natl Acad. Sci. USA 107, 15 409–15 414 (doi:10.1073/pnas.1009997107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Moffat JC, Vijayvergiya V, Gao PF, Cross TA, Woodbury DJ, Busath DD. 2008. Proton transport through influenza A virus M2 protein reconstituted in vesicles. Biophys. J. 94, 434–445 (doi:10.1529/biophysj.107.109082) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Peterson E, Ryser T, Funk S, Inouye D, Sharma M, Qin H, Cross TA, Busath DD. 2011. Functional reconstitution of influenza A M2(22–62). Biochim. Biophys. Acta 1808, 516–521 (doi:10.1016/j.bbamem.2010.10.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lear JD, Wasserman ZR, DeGrado WF. 1988. Synthetic amphiphilic peptide models for protein ion channels. Science 240, 1177–1181 (doi:10.1126/science.2453923) [DOI] [PubMed] [Google Scholar]
- 132.Feniouk BA, Kozlova MA, Knorre DA, Cherepanov DA, Mulkidjanian AY, Junge W. 2004. The proton-driven rotor of ATP synthase: ohmic conductance (10 fS), and absence of voltage gating. Biophys. J. 86, 4094–4109 (doi:10.1529/biophysj.103.036962) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.von Ballmoos C, Wiedenmann A, Dimroth P. 2009. Essentials for ATP synthesis by F1Fo ATP synthases. Annu. Rev. Biochem. 78, 649–672 (doi:10.1146/annurev.biochem.78.081307.104803.) [DOI] [PubMed] [Google Scholar]
- 134.Angevine CM, Herold KA, Fillingame RH. 2003. Aqueous access pathways in subunit a of rotary ATP synthase extend to both sides of the membrane. Proc. Natl Acad. Sci. USA 100, 13 179–13 183 (doi:10.1073/pnas.2234364100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dong H, Fillingame RH. 2010. Chemical reactivities of cysteine substitutions in subunit a of ATP synthase define residues gating H+ transport from each side of the membrane. J. Biol. Chem. 285, 39 811–39 818 (doi:10.1074/jbc.M110.175844) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dimroth P. 2000. Operation of the Fo motor of the ATP synthase. Biochim. Biophys. Acta 1458, 374–386 (doi:10.1016/S0005-2728(00)00088-8) [DOI] [PubMed] [Google Scholar]
- 137.Krah A, Pogoryelov D, Langer JD, Bond PJ, Meier T, Faraldo-Gómez JD. 2010. Structural and energetic basis for H+ versus Na+ binding selectivity in ATP synthase Fo rotors. Biochim. Biophys. Acta 1797, 763–772 (doi:10.1016/j.bbabio.2010.04.014) [DOI] [PubMed] [Google Scholar]
- 138.Preiss L, Yildiz O, Hicks DB, Krulwich TA, Meier T. 2010. A new type of proton coordination in an F1Fo-ATP synthase rotor ring. PLoS Biol. 8, e1000443 (doi:10.1371/journal.pbio.1000443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hu F, Schmidt-Rohr K, Hong M. 2012. NMR detection of pH-dependent histidine-water proton exchange reveals the conduction mechanism of a transmembrane proton channel. J. Am. Chem. Soc. 134, 3703–3713 (doi:10.1021/ja2081185) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Forrest LR, Kukol A, Arkin IT, Tieleman DP, Sansom MSP. 2000. Exploring models of the influenza A M2 channel: MD simulations in a phospholipid bilayer. Biophys. J. 78, 55–69 (doi:10.1016/S0006-3495(00)76572-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sansom MSP, Kerr ID, Smith GR, Son HS. 1997. The influenza A virus M2 channel: a molecular modeling and simulation study. Virology 233, 163–173 (doi:10.1006/viro.1997.8578) [DOI] [PubMed] [Google Scholar]
- 142.Chen H, Wu Y, Voth GA. 2007. Proton transport behavior through the influenza A M2 channel: insights from molecular simulation. Biophys. J. 93, 3470–3479 (doi:10.1529/biophysj.107.105742) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Randa HS, Forrest LR, Voth GA, Sansom MSP. 1999. Molecular dynamics of synthetic leucine–serine ion channels in a phospholipid membrane. Biophys. J. 77, 2400–2410 (doi:10.1016/S0006-3495(99)77077-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ramsey IS, Mokrab Y, Carvacho I, Sands ZA, Sansom MSP, Clapham DE. 2010. An aqueous H+ permeation pathway in the voltage-gated proton channel Hv1. Nat. Struct. Mol. Biol. 17, 869–875 (doi:10.1038/nsmb.1826) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wood ML, Schow EV, Freites JA, White SH, Tombola F, Tobias DJ. 2012. Water wires in atomistic models of the Hv1 proton channel. Biochim. Biophys. Acta 1818, 286–293 (doi:10.1016/j.bbamem.2011.07.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. 2006. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc. Natl Acad. Sci. USA 103, 16 117–16 122 (doi:10.1073/pnas.0606149103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wicke E, Eigen M, Ackermann T. 1954. Über den Zustand des Protons (Hydroniumions) in wässriger Lösung. Z. Phys. Chem. 1, 340–364 (doi:10.1524/zpch.1954.1.5_6.340) [Google Scholar]
- 148.Scheiner S. 1981. Proton transfers in hydrogen-bonded systems: cationic oligomers of water. J. Am. Chem. Soc. 103, 315–320 (doi:10.1021/ja00392a012) [Google Scholar]
- 149.Wu Y, Ilan B, Voth GA. 2007. Charge delocalization in proton channels, II: the synthetic LS2 channel and proton selectivity. Biophys. J. 92, 61–69 (doi:10.1529/biophysj.106.091942) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Agre P, King LS, Yasui M, Guggino WB, Ottersen OP, Fujiyoshi Y, Engel A, Nielsen S. 2002. Aquaporin water channels: from atomic structure to clinical medicine. J. Physiol. 542, 3–16 (doi:10.1113/jphysiol.2002.020818) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pohl P, Saparov SM, Borgnia MJ, Agre P. 2001. Highly selective water channel activity measured by voltage clamp: analysis of planar lipid bilayers reconstituted with purified AqpZ. Proc. Natl Acad. Sci. USA 98, 9624–9629 (doi:10.1073/pnas.161299398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zeidel ML, Ambudkar SV, Smith BL, Agre P. 1992. Reconstitution of functional water channels in liposomes containing purified red cell CHIP28 protein. Biochemistry 31, 7436–7440 (doi:10.1021/bi00148a002) [DOI] [PubMed] [Google Scholar]
- 153.Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. 2000. Structural determinants of water permeation through aquaporin-1. Nature 407, 599–605 (doi:10.1038/35036519) [DOI] [PubMed] [Google Scholar]
- 154.Sui H, Han BG, Lee JK, Walian P, Jap BK. 2001. Structural basis of water-specific transport through the AQP1 water channel. Nature 414, 872–878 (doi:10.1038/414872a) [DOI] [PubMed] [Google Scholar]
- 155.Tajkhorshid E, Nollert P, Jensen MO, Miercke LJ, O'Connell J, Stroud RM, Schulten K. 2002. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science 296, 525–530 (doi:10.1126/science.1067778) [DOI] [PubMed] [Google Scholar]
- 156.Chakrabarti N, Tajkhorshid E, Roux B, Pomès R. 2004. Molecular basis of proton blockage in aquaporins. Structure 12, 65–74 (doi:10.1016/j.jmb.2003.08.003) [DOI] [PubMed] [Google Scholar]
- 157.de Groot BL, Frigato T, Helms V, Grubmüller H. 2003. The mechanism of proton exclusion in the aquaporin-1 water channel. J. Mol. Biol. 333, 279–293 (doi.org/10.1016/j.jmb.2003.08.003) [DOI] [PubMed] [Google Scholar]
- 158.Ilan B, Tajkhorshid E, Schulten K, Voth GA. 2004. The mechanism of proton exclusion in aquaporin channels. Proteins 55, 223–228 (doi:10.1002/prot.20038) [DOI] [PubMed] [Google Scholar]
- 159.Miloshevsky GV, Jordan PC. 2004. Water and ion permeation in bAQP1 and GlpF channels: a kinetic Monte Carlo study. Biophys. J. 87, 3690–3702 (doi.org/10.1529/biophysj.104.043315) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Burykin A, Warshel A. 2003. What really prevents proton transport through aquaporin? Charge self-energy versus proton wire proposals. Biophys. J. 85, 3696–3706 (doi.org/10.1016/S0006-3495(03)74786-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Burykin A, Warshel A. 2004. On the origin of the electrostatic barrier for proton transport in aquaporin. FEBS Lett. 570, 41–46 (doi.org/10.1016/j.febslet.2004.06.020) [DOI] [PubMed] [Google Scholar]
- 162.Beitz E, Wu B, Holm LM, Schultz JE, Zeuthen T. 2006. Point mutations in the aromatic/arginine region in aquaporin 1 allow passage of urea, glycerol, ammonia, and protons. Proc. Natl Acad. Sci. USA 103, 269–274 (doi:10.1073/pnas.0507225103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wu B, Steinbronn C, Alsterfjord M, Zeuthen T, Beitz E. 2009. Concerted action of two cation filters in the aquaporin water channel. EMBO J. 28, 2188–2194 (doi:10.1038/emboj.2009.182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Morgan D, Musset B, Kulleperuma K, Smith SME, Rajan S, Cherny VV, Pomès R, DeCoursey TE. 2013. Peregrination of the selectivity filter delineates the pore of the human voltage gated proton channel hHV1. J. Gen. Physiol. 142, 625–640 (doi:10.1085/jgp.201311045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Keramidas A, Moorhouse AJ, Pierce KD, Schofield PR, Barry PH. 2002. Cation-selective mutations in the M2 domain of the inhibitory glycine receptor channel reveal determinants of ion-charge selectivity. J. Gen. Physiol. 119, 393–410 (doi:10.1085/jgp.20028552) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wotring VE, Weiss DS. 2008. Charge scan reveals an extended region at the intracellular end of the GABA receptor pore that can influence ion selectivity. J. Gen. Physiol. 131, 87–97 (doi:10.1085/jgp.200609701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Galzi JL, Devillers-Thiéry A, Hussy N, Bertrand S, Changeux JP, Bertrand D. 1992. Mutations in the channel domain of a neuronal nicotinic receptor convert ion selectivity from cationic to anionic. Nature 359, 500–505 (doi:10.1038/359500a0) [DOI] [PubMed] [Google Scholar]
- 168.Gunthorpe MJ, Lummis SCR. 2001. Conversion of the ion selectivity of the 5-HT3A receptor from cationic to anionic reveals a conserved feature of the ligand-gated ion channel superfamily. J. Biol. Chem. 276, 10 977–10 983 (doi:10.1074/jbc.M009575200) [PubMed] [Google Scholar]
- 169.Hong M, Degrado WF. 2012. Structural basis for proton conduction and inhibition by the influenza M2 protein. Protein Sci. 21, 1620–1633 (doi:10.1002/pro.2158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chizhmakov IV, Geraghty FM, Ogden DC, Hayhurst A, Antoniou M, Hay AJ. 1996. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J. Physiol. 494, 329–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lin TI, Schroeder C. 2001. Definitive assignment of proton selectivity and attoampere unitary current to the M2 ion channel protein of influenza A virus. J. Virol. 75, 3647–3656 (doi:10.1128/JVI.75.8.3647-3656.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mould JA, Drury JE, Frings SM, Kaupp UB, Pekosz A, Lamb RA, Pinto LH. 2000. Permeation and activation of the M2 ion channel of influenza A virus. J. Biol. Chem. 275, 31 038–31 050 (doi:10.1074/jbc.M003663200) [DOI] [PubMed] [Google Scholar]
- 173.Ma C, et al. 2009. Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc. Natl Acad. Sci. USA 106, 12 283–12 288 (doi:10.1073/pnas.0905726106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Holsinger LJ, Nichani D, Pinto LH, Lamb RA. 1994. Influenza A virus M2 ion channel protein: a structure–function analysis. J. Virol. 68, 1551–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pielak RM, Chou JJ. 2010. Kinetic analysis of the M2 proton conduction of the influenza virus. J. Am. Chem. Soc. 132, 17 695–17 697 (doi:10.1021/ja108458u) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath D, Zhou H, Cross T. 2010. Insight into the mechanism of the influenza A proton channel from structure in a lipid bilayer. Science 330, 509–512 (doi:10.1126/science.1191750) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tang Y, Zaitseva F, Lamb RA, Pinto LH. 2002. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J. Biol. Chem. 277, 39 880–39 886 (doi:10.1074/jbc.M206582200) [DOI] [PubMed] [Google Scholar]
- 178.Okada A, Miura T, Takeuchi H. 2001. Protonation of histidine and histidine–tryptophan interaction in the activation of the M2 ion channel from influenza A virus. Biochemistry 40, 6053–6060 (doi:10.1021/bi0028441) [DOI] [PubMed] [Google Scholar]
- 179.Venkataraman P, Lamb RA, Pinto LH. 2005. Chemical rescue of histidine selectivity filter mutants of the M2 ion channel of influenza A virus. J. Biol. Chem. 280, 21 463–21 472 (doi:10.1074/jbc.M412406200) [DOI] [PubMed] [Google Scholar]
- 180.Wang C, Lamb RA, Pinto LH. 1995. Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue. Biophys. J. 69, 1363–1371 (doi:10.1016/S0006-3495(95)80003-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shuck K, Lamb RA, Pinto LH. 2000. Analysis of the pore structure of the influenza A virus M2 ion channel by the substituted-cysteine accessibility method. J. Virol. 74, 7755–7761 (doi:10.1128/JVI.74.17.7755-7761.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kass I, Arkin IT. 2005. How pH opens a H+ channel: the gating mechanism of influenza A M2. Structure 13, 1789–1798 (doi.org/10.1016/j.str.2005.08.022) [DOI] [PubMed] [Google Scholar]
- 183.Miao Y, et al. 2012. M2 proton channel structural validation from full-length protein samples in synthetic bilayers and E. coli membranes. Angew. Chem. Int. Ed. Engl. 51, 8383–8386 (doi:10.1002/anie.201204666) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhou HX. 2011. A theory for the proton transport of the influenza virus M2 protein: extensive test against conductance data. Biophys. J. 100, 912–921 (doi:10.1016/j.bpj.2011.01.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hu J, Fu R, Nishimura K, Zhang L, Zhou HX, Busath DD, Vijayvergiya V, Cross TA. 2006. Histidines, heart of the hydrogen ion channel from influenza A virus: toward an understanding of conductance and proton selectivity. Proc. Natl Acad. Sci. USA 103, 6865–6870 (doi:10.1073/pnas.0601944103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. 1997. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc. Natl Acad. Sci. USA 94, 11 301–11 306 (doi:10.1073/pnas.94.1.11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lear JD. 2003. Proton conduction through the M2 protein of the influenza A virus; a quantitative, mechanistic analysis of experimental data. FEBS Lett. 552, 17–22 (doi:10.1016/S0014-5793(03)00778-6) [DOI] [PubMed] [Google Scholar]
- 188.Polishchuk AL, Lear JD, Ma C, Lamb RA, Pinto LH, DeGrado WF. 2010. A pH-dependent conformational ensemble mediates proton transport through the influenza A/M2 protein. Biochemistry 49, 10 061–10 071 (doi:10.1021/bi101229m) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hu F, Luo W, Hong M. 2010. Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR. Science 330, 505–508 (doi:10.1126/science.1191714) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mandel M. 1965. Proton magnetic resonance spectra of some proteins. I. Ribonuclease, oxidized ribonuclease, lysozyme, and cytochrome c. J. Biol. Chem. 240, 1586–1592 [PubMed] [Google Scholar]
- 191.Markley J. 1975. Observation of histidine residues in proteins by means of nuclear magnetic resonance spectroscopy. Acc. Chem. Res. 8, 70–80 (doi:10.1021/ar50086a004) [Google Scholar]
- 192.Dong H, Yi M, Cross TA, Zhou HX. 2013. Ab initio calculations and validation of the pH-dependent structures of the His37-Trp41 quartet, the heart of acid activation and proton conductance in the M2 protein of Influenza A virus. Chem. Sci. 4, 2776–2787 (doi:10.1039/C3SC50293G) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hong M, Fritzsching KJ, Williams JK. 2012. Hydrogen-bonding partner of the proton-conducting histidine in the influenza M2 proton channel revealed from 1H chemical shifts. J. Am. Chem. Soc. 134, 14 753–14 755 (doi:10.1021/ja307453v) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Acharya R, et al. 2010. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl Acad. Sci. USA 107, 15 075–15 080 (doi:10.1073/pnas.1007071107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.DeCoursey TE, Cherny VV. 1996. Effects of buffer concentration on voltage-gated H+ currents: does diffusion limit the conductance? Biophys. J. 71, 182–193 (doi:10.1016/S0006-3495(96)79215-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.DeCoursey TE, Cherny VV. 1995. Voltage-activated proton currents in membrane patches of rat alveolar epithelial cells. J. Physiol. 489, 299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.DeCoursey TE, Cherny VV. 1997. Deuterium isotope effects on permeation and gating of proton channels in rat alveolar epithelium. J. Gen. Physiol. 109, 415–434 (doi:10.1085/jgp.109.4.415) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.DeCoursey TE, Cherny VV. 1998. Temperature dependence of voltage-gated H+ currents in human neutrophils, rat alveolar epithelial cells, and mammalian phagocytes. J. Gen. Physiol. 112, 503–522 (doi:10.1085/jgp.112.4.503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Akeson M, Deamer DW. 1991. Proton conductance by the gramicidin water wire. Model for proton conductance in the F1Fo ATPases? Biophys. J. 60, 101–109 (doi:10.1016/S0006-3495(91)82034-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Chernyshev A, Cukierman S. 2002. Thermodynamic view of activation energies of proton transfer in various gramicidin A channels. Biophys. J. 82, 182–192 (doi:10.1016/S0006-3495(02)75385-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Mould JA, Li HC, Dudlak CS, Lear JD, Pekosz A, Lamb RA, Pinto LH. 2000. Mechanism for proton conduction of the M2 ion channel of influenza A virus. J. Biol. Chem. 275, 8592–8599 (doi:10.1074/jbc.275.12.8592) [DOI] [PubMed] [Google Scholar]
- 202.Zhou HX. 2011. Mechanistic insight into the H2O/D2O isotope effect in the proton transport of the influenza virus M2 protein. J. Membr. Biol. 244, 93–96 (doi:10.1007/s00232-011-9402-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Fillingame RH. 1990. Molecular mechanics of ATP synthesis by F1Fo–Type H+-transporting ATP synthases. In The bacteria (ed. Krulwich TA.), pp. 345–390 New York, NY: Academic Press [Google Scholar]
- 204.Lanyi JK. 1993. Proton translocation mechanism and energetics in the light-driven pump bacteriorhodopsin. Biochim. Biophys. Acta 1183, 241–261 (doi:10.1016/0005-2728(93)90226-6) [DOI] [PubMed] [Google Scholar]
- 205.Freier E, Wolf S, Gerwert K. 2011. Proton transfer via a transient linear water-molecule chain in a membrane protein. Proc. Natl Acad. Sci. USA 108, 11 435–11 439 (doi:10.1073/pnas.1104735108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lórenz-Fonfría VA, et al. 2013. Transient protonation changes in channelrhodopsin-2 and their relevance to channel gating. Proc. Natl Acad. Sci. USA 110, E1273–E1281 (doi:10.1073/pnas.1219502110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Paddock ML, Ädelroth P, Chang C, Abresch EC, Feher G, Okamura MY. 2001. Identification of the proton pathway in bacterial reaction centers: cooperation between Asp-M17 and Asp-L210 facilitates proton transfer to the secondary quinone (QB). Biochemistry 40, 6893–6902 (doi:10.1021/bi010280a) [DOI] [PubMed] [Google Scholar]
- 208.Ruivo R, Bellenchi GC, Chen X, Zifarelli G, Sagné C, Debacker C, Pusch M, Supplisson S, Gasnier B. 2012. Mechanism of proton/substrate coupling in the heptahelical lysosomal transporter cystinosin. Proc. Natl Acad. Sci. USA 109, E210–E217 (doi:10.1073/pnas.1115581109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cornish AJ, Gärtner K, Yang H, Peters JW, Hegg EL. 2011. Mechanism of proton transfer in [FeFe]-hydrogenase from Clostridium pasteurianum. J. Biol. Chem. 286, 38 341–38 347 (doi:10.1074/jbc.M111.254664) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Vedovato N, Gadsby DC. 2013. Proton current through stalled Na/K-ATPase pumps: three residues point to a possible route and mechanism. Biophys. J. 104, 22a (doi:10.1016/j.bpj.2012.11.158) [Google Scholar]
- 211.Lee HJ, Gennis RB, Ädelroth P. 2011. Entrance of the proton pathway in cbb3-type heme-copper oxidases. Proc. Natl Acad. Sci. USA 108, 17 661–17 666 (doi:10.1073/pnas.1107543108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Cheng VWT, Johnson A, Rothery RA, Weiner JH. 2008. Alternative sites for proton entry from the cytoplasm to the quinone binding site in Escherichia coli succinate dehydrogenase. Biochemistry 47, 9107–9116 (doi:10.1021/bi801008e) [DOI] [PubMed] [Google Scholar]
- 213.Zhou J, Sharp LL, Tang HL, Lloyd SA, Billings S, Braun TF, Blair DF. 1998. Function of protonatable residues in the flagellar motor of Escherichia coli: a critical role for Asp 32 of MotB. J. Bacteriol. 180, 2729–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Nishizawa T, et al. 2013. Structural basis for the counter-transport mechanism of a H+/Ca2+ exchanger. Science 341, 168–172 (doi:10.1126/science.1239002) [DOI] [PubMed] [Google Scholar]
- 215.Lim HH, Shane T, Miller C. 2012. Intracellular proton access in a Cl−/H+ antiporter. PLoS Biol. 10, e1001441 (doi:10.1371/journal.pbio.1001441) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Siegbahn PEM. 2013. Water oxidation mechanism in photosystem II, including oxidations, proton release pathways, O–O bond formation and O2 release. Biochim. Biophys. Acta 1827, 1003–1019 (doi:10.1016/j.bbabio.2012.10.006) [DOI] [PubMed] [Google Scholar]
- 217.Unal ES, Zhao R, Goldman ID. 2009. Role of the glutamate 185 residue in proton translocation mediated by the proton-coupled folate transporter SLC46A1. Am. J. Physiol. Cell Physiol. 297, C66–C74 (doi:10.1152/ajpcell.00096.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Klimacek M, Brunsteiner M, Nidetzky B. 2012. Dynamic mechanism of proton transfer in mannitol 2-dehydrogenase from Pseudomonas fluorescens: mobile Glu292 controls proton relay through a water channel that connects the active site with bulk solvent. J. Biol. Chem. 287, 6655–6667 (doi:10.1074/jbc.M111.289223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kaback HR, Sahin-Tóth M, Weinglass AB. 2001. The kamikaze approach to membrane transport. Nat. Rev. Mol. Cell Biol. 2, 610–620 (doi:10.1038/35085077) [DOI] [PubMed] [Google Scholar]
- 220.Flock U, Thorndycroft FH, Matorin AD, Richardson DJ, Watmough NJ, Ådelroth P. 2008. Defining the proton entry point in the bacterial respiratory nitric-oxide reductase. J. Biol. Chem. 283, 3839–3845 (doi:10.1074/jbc.M704615200) [DOI] [PubMed] [Google Scholar]
- 221.Varanasi L, Hosler J. 2011. Alternative initial proton acceptors for the D pathway of Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry 50, 2820–2828 (doi:10.1021/bi102002v) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Miller MJ, Oldenburg M, Fillingame RH. 1990. The essential carboxyl group in subunit c of the F1Fo ATP synthase can be moved and H+-translocating function retained. Proc. Natl Acad. Sci. USA 87, 4900–4904 (doi:10.1073/pnas.87.13.4900) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Musset B, Smith SM, Rajan S, Cherny VV, Sujai S, Morgan D, DeCoursey TE. 2010. Zinc inhibition of monomeric and dimeric proton channels suggests cooperative gating. J. Physiol. 588, 1435–1449 (doi:10.1113/jphysiol.2010.188318) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Starace DM, Bezanilla F. 2004. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature 427, 548–553 (doi:10.1038/nature02270) [DOI] [PubMed] [Google Scholar]
- 225.Struyk AF, Cannon SC. 2007. A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J. Gen. Physiol. 130, 11–20 (doi:10.1085/jgp.200709755) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Gosselin-Badaroudine P, Keller DI, Huang H, Pouliot V, Chatelier A, Osswald S, Brink M, Chahine M. 2012. A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS ONE 7, e38331 (doi:10.1371/journal.pone.0038331) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Starace DM, Bezanilla F. 2001. Histidine scanning mutagenesis of basic residues of the S4 segment of the Shaker K+ channel. J. Gen. Physiol. 117, 469–490 (doi:10.1085/jgp.117.5.469) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Starace DM, Stefani E, Bezanilla F. 1997. Voltage-dependent proton transport by the voltage sensor of the Shaker K+ channel. Neuron 19, 1319–1327 (doi:10.1016/S0896-6273(00)80422-5) [DOI] [PubMed] [Google Scholar]
- 229.Cain BD, Simoni RD. 1989. Proton translocation by the F1FoATPase of Escherichia coli. Mutagenic analysis of the a-subunit. J. Biol. Chem. 264, 3292–3300 [PubMed] [Google Scholar]
- 230.Fillingame RH, Girvin ME, Fraga D, Zhang Y. 1992. Correlations of structure and function in H+ translocating subunit c of F1Fo ATP synthase. Ann. NY Acad. Sci. 671, 323–333; discussion 333–324 (doi:10.1111/j.1749-6632.1992.tb43806.x) [DOI] [PubMed] [Google Scholar]
- 231.Eigen M, De Maeyer L. 1958. Self-dissociation and protonic charge transport in water and ice. Proc. R. Soc. Lond. A 247, 505–533 (doi:10.1098/rspa.1958.0208) [Google Scholar]
- 232.Bernal JD, Fowler RH. 1933. A theory of water and ionic solution, with particular reference to hydrogen and hydroxyl ions. J. Chem. Phys. 1, 515–548 (doi:10.1063/1.1749327) [Google Scholar]
- 233.Conway BE, Bockris JO'M, Linton H. 1956. Proton conductance and the existence of the H3O· ion. J. Chem. Phys. 24, 834–850 (doi:10.1063/1.1742619) [Google Scholar]
- 234.Deng Y, Josberger E, Jin J, Rousdari AF, Helms BA, Zhong C, Anantram MP, Rolandi M. 2013. H+-type and OH−-type biological protonic semiconductors and complementary devices. Sci. Rep. 3, 2481 (doi:10.1038/srep02481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Al Khazaaly S, Beilby MJ. 2012. Zinc ions block H+/OH− channels in Chara australis. Plant Cell Environ. 35, 1380–1392 (doi:10.1111/j.1365-3040.2012.02496.x) [DOI] [PubMed] [Google Scholar]
- 236.Lucas WJ. 1979. Alkaline band formation in Chara corallina: due to OH− efflux or H+ influx? Plant Physiol. 63, 248–254 (doi:10.1104/pp.63.2.248) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Venosa RA, Kotsias BA, Horowicz P. 1994. Frog striated muscle is permeable to hydroxide and buffer anions. J. Membr. Biol. 139, 57–74 (doi:10.1007/BF00232675) [DOI] [PubMed] [Google Scholar]
- 238.Goodchild SJ, Xu H, Es-Salah-Lamoureux Z, Ahern CA, Fedida D. 2012. Basis for allosteric open-state stabilization of voltage-gated potassium channels by intracellular cations. J. Gen. Physiol. 140, 495–511 (doi:10.1085/jgp.201210823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Richard EA, Miller C. 1990. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science 247, 1208–1210 (doi:10.1126/science.2156338) [DOI] [PubMed] [Google Scholar]
- 240.Shapiro MS, DeCoursey TE. 1991. Permeant ion effects on the gating kinetics of the type L potassium channel in mouse lymphocytes. J. Gen. Physiol. 97, 1251–1278 (doi:10.1085/jgp.97.6.1251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Swenson RP, Jr, Armstrong CM. 1981. K+ channels close more slowly in the presence of external K+ and Rb+. Nature 291, 427–429 (doi:10.1038/291427a0) [DOI] [PubMed] [Google Scholar]
- 242.Pusch M, Ludewig U, Rehfeldt A, Jentsch TJ. 1995. Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature 373, 527–531 (doi:10.1038/373527a0) [DOI] [PubMed] [Google Scholar]
- 243.Chen TY, Miller C. 1996. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 108, 237–250 (doi:10.1085/jgp.108.4.237) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Hagiwara S, Yoshii M. 1979. Effects of internal potassium and sodium on the anomalous rectification of the starfish egg as examined by internal perfusion. J. Physiol. 292, 251–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Horowicz P, Gage PW, Eisenberg RS. 1968. The role of the electrochemical gradient in determining potassium fluxes in frog striated muscle. J. Gen. Physiol. 51, 193–203 [PMC free article] [PubMed] [Google Scholar]
- 246.Byerly L, Meech R, Moody W, Jr, 1984. Rapidly activating hydrogen ion currents in perfused neurones of the snail, Lymnaea stagnalis. J. Physiol. 351, 199–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Doroshenko PA, Kostyuk PG, Martynyuk AE. 1986. Transmembrane outward hydrogen current in intracellularly perfused neurones of the snail Helix pomatia. Gen. Physiol. Biophys. 5, 337–350 [PubMed] [Google Scholar]
- 248.Cherny VV, Markin VS, DeCoursey TE. 1995. The voltage-activated hydrogen ion conductance in rat alveolar epithelial cells is determined by the pH gradient. J. Gen. Physiol. 105, 861–896 (doi:10.1085/jgp.105.6.861) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Roos A, Boron WF. 1981. Intracellular pH. Physiol. Rev. 61, 296–434 [DOI] [PubMed] [Google Scholar]
- 250.Jack JJB, Noble D, Tsien RW. 1975. Chapter 8. Nonlinear properties of excitable membranes. In Electric current flow in excitable cells, pp. 225–260 Oxford, UK: Clarendon Press [Google Scholar]
- 251.Moore JW. 1959. Excitation of the squid axon membrane in isosmotic potassium chloride. Nature 183, 265–266 (doi:10.1038/183265b0) [DOI] [PubMed] [Google Scholar]
- 252.Hodgkin AL, Huxley AF. 1952. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hagiwara S, Naka KI. 1964. The initiation of spike potential in barnacle muscle fibers under low intracellular Ca++. J. Gen. Physiol. 48, 141–162 (doi:10.1085/jgp.48.1.141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Ashcroft FM, Stanfield PR. 1981. Calcium dependence of the inactivation of calcium currents in skeletal muscle fibers of an insect. Science 213, 224–226 (doi:10.1126/science.213.4504.224) [DOI] [PubMed] [Google Scholar]
- 255.Schmitter RE, Njus D, Sulzman FM, Gooch VD, Hastings JW. 1976. Dinoflagellate bioluminescence: a comparative study of in vitro components. J. Cell Physiol. 87, 123–134 (doi:10.1002/jcp.1040870115) [DOI] [PubMed] [Google Scholar]
- 256.de Quatrefages A. 1850. Observations sur les noctiluques. Ann. Sci. Nat. Zool. (Ser. 3) 14, 226–235 [Google Scholar]
- 257.Chang JJ. 1960. Electrophysiological studies of a non-luminescent form of the dinoflagellate Noctiluca miliaris. J. Cell. Comp. Physiol. 56, 33–42 (doi:10.1002/jcp.1030560106) [DOI] [PubMed] [Google Scholar]
- 258.Eckert R. 1965. II. Asynchronous flash initiation by a propagated triggering potential. Science 147, 1142–1145 (doi:10.1126/science.147.3662.1142) [DOI] [PubMed] [Google Scholar]
- 259.Eckert R. 1965. I. Specific nature of triggering events. Science 147, 1140–1142 (doi:10.1126/science.147.3662.1140) [DOI] [PubMed] [Google Scholar]
- 260.Hisada M. 1957. Membrane resting and action potentials from a protozoan, Noctiluca scintillans. J. Cell. Comp. Physiol. 50, 57–71 (doi:10.1002/jcp.1030500105) [DOI] [PubMed] [Google Scholar]
- 261.Eckert R, Reynolds GT. 1967. The subcellular origin of bioluminescence in Noctiluca miliaris. J. Gen. Physiol. 50, 1429–1458 (doi:10.1085/jgp.50.5.1429) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Eckert R, Sibaoka T. 1968. The flash-triggering action potential of the luminescent dinoflagellate Noctiluca. J. Gen. Physiol. 52, 258–282 (doi:10.1085/jgp.52.2.258) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Harvey EB. 1917. A physiological study of Noctiluca, with special reference to light production, anaesthesia and specific gravity. Proc. Natl Acad. Sci. USA 3, 15–16 (doi:10.1073/pnas.3.1.15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Fogel M, Schmitter RE, Hastings JW. 1972. On the physical identity of scintillons: bioluminescent particles in Gonyaulax polyedra. J. Cell Sci. 11, 305–317 [DOI] [PubMed] [Google Scholar]
- 265.Fogel M, Hastings JW. 1971. A substrate-binding protein in the Gonyaulax bioluminescence reaction. Arch. Biochem. Biophys. 142, 310–321 (doi:10.1016/0003-9861(71)90289-X) [DOI] [PubMed] [Google Scholar]
- 266.Nicolas MT, Sweeney BM, Hastings JW. 1987. The ultrastructural localization of luciferase in three bioluminescent dinoflagellates, two species of Pyrocystis, and Noctiluca, using anti-luciferase and immunogold labelling. J. Cell Sci. 87, 189–196 [DOI] [PubMed] [Google Scholar]
- 267.Nawata T, Sibaoka T. 1976. Ionic composition and pH of the vacuolar sap in marine dinoflagellate Noctiluca. Plant Cell Physiol. 17, 265–272 [Google Scholar]
- 268.Hastings JW. 2001. Bioluminescence. In Cell physiology sourcebook: a molecular approach (ed. Sperelakis N.), pp. 1115–1131, 3rd edn San Diego, CA: Academic Press [Google Scholar]
- 269.Krieger N, Hastings JW. 1968. Bioluminescence: pH activity profiles of related luciferase fractions. Science 161, 586–589 (doi:10.1126/science.161.3841.586) [DOI] [PubMed] [Google Scholar]
- 270.Smith SME, DeCoursey TE. 2013. Consequences of dimerization of the voltage-gated proton channel. Prog. Mol. Biol. Transl. Sci. 117, 335–360 (doi:10.1016/B978-0-12-386931-9.00012-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Musset B, Smith SM, Rajan S, Cherny VV, Morgan D, DeCoursey TE. 2010. Oligomerization of the voltage gated proton channel. Channels 4, 260–265 (doi:10.4161/chan.4.4.12789) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Gonzalez C, Koch HP, Drum BM, Larsson HP. 2010. Strong cooperativity between subunits in voltage-gated proton channels. Nat. Struct. Mol. Biol. 17, 51–56 (doi:10.1038/nsmb.1739) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Roos D, et al. 1996. Mutations in the X-linked and autosomal recessive forms of chronic granulomatous disease. Blood 87, 1663–1681 [PubMed] [Google Scholar]
- 274.Fujiwara Y, Kurokawa T, Takeshita K, Kobayashi M, Okochi Y, Nakagawa A, Okamura Y. 2012. The cytoplasmic coiled-coil mediates cooperative gating temperature sensitivity in the voltage-gated H+ channel Hv1. Nat. Commun. 3, 816 (doi:10.1038/ncomms1823) [DOI] [PubMed] [Google Scholar]
- 275.Tombola F, Ulbrich MH, Kohout SC, Isacoff EY. 2010. The opening of the two pores of the Hv1 voltage-gated proton channel is tuned by cooperativity. Nat. Struct. Mol. Biol. 17, 44–50 (doi:10.1038/nsmb.1738) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Petheő GL, Orient A, Baráth M, Kovács I, Réthi B, Lányi A, Rajki A, Rajnavölgyi E, Geiszt M. 2010. Molecular and functional characterization of HV1 proton channel in human granulocytes. PLoS ONE 5, e14081 (doi:10.1371/journal.pone.0014081) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Gonzalez C, Rebolledo S, Perez ME, Larsson HP. 2013. Molecular mechanism of voltage sensing in voltage-gated proton channels. J. Gen. Physiol. 141, 275–285 (doi:10.1085/jgp.201210857) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Geiszt M, Kapus A, Nemet K, Farkas L, Ligeti E. 1997. Regulation of capacitative Ca2+ influx in human neutrophil granulocytes. Alterations in chronic granulomatous disease. J. Biol. Chem. 272, 26 471–26 478 (doi:10.1074/jbc.272.42.26471) [DOI] [PubMed] [Google Scholar]
- 279.Jankowski A, Grinstein S. 1999. A noninvasive fluorimetric procedure for measurement of membrane potential. Quantification of the NADPH oxidase-induced depolarization in activated neutrophils. J. Biol. Chem. 274, 26 098–26 104 (doi:10.1074/jbc.274.37.26098) [DOI] [PubMed] [Google Scholar]
- 280.Rada BK, Geiszt M, Káldi K, Tímár C, Ligeti E. 2004. Dual role of phagocytic NADPH oxidase in bacterial killing. Blood 104, 2947–2953 (doi:10.1182/blood-2004-03-1005) [DOI] [PubMed] [Google Scholar]
- 281.Lee SY, Letts JA, Mackinnon R. 2008. Dimeric subunit stoichiometry of the human voltage-dependent proton channel Hv1. Proc. Natl Acad. Sci. USA 105, 7692–7695 (doi:10.1073/pnas.0803277105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Li SJ, Zhao Q, Zhou Q, Unno H, Zhai Y, Sun F. 2010. The role and structure of the carboxyl-terminal domain of the human voltage-gated proton channel Hv1. J. Biol. Chem. 285, 12 047–12 054 (doi:10.1074/jbc.M109.040360) [DOI] [PMC free article] [PubMed] [Google Scholar]