

Background: Scaffold proteins bring signaling proteins together to enhance signal transduction.
Results: The scaffold state-switching model predicted that scaffolds amplify, accelerate, and insulate localized signaling, and these predictions were validated experimentally using PKC and AKAP7α.
Conclusion: Amplification, acceleration, and insulation can arise directly from scaffold tethering.
Significance: The scaffold state-switching model provides a mechanistic understanding of the kinetic role of scaffold proteins in signal transduction.
Keywords: AKAP, Cell Signaling, Enzyme Kinetics, Mathematical Modeling, Protein Kinase C (PKC), Scaffold Proteins, Anchoring
Abstract
Scaffold proteins localize two or more signaling enzymes in close proximity to their downstream effectors. A-kinase-anchoring proteins (AKAPs) are a canonical family of scaffold proteins known to bind protein kinase A (PKA) and other enzymes. Several AKAPs have been shown to accelerate, amplify, and specify signal transduction to dynamically regulate numerous cellular processes. However, there is little theory available to mechanistically explain how signaling on protein scaffolds differs from solution biochemistry. In our present study, we propose a novel kinetic mechanism for enzymatic reactions on protein scaffolds to explain these phenomena, wherein the enzyme-substrate-scaffold complex undergoes stochastic state switching to reach an active state. This model predicted anchored enzymatic reactions to be accelerated, amplified, and insulated from inhibition compared with those occurring in solution. We exploited a direct interaction between protein kinase C (PKC) and AKAP7α as a model to validate these predictions experimentally. Using a genetically encoded PKC activity reporter, we found that both the strength and speed of substrate phosphorylation were enhanced by AKAP7α. PKC tethered to AKAP7α was less susceptible to inhibition from the ATP-competitive inhibitor Gö6976 and the substrate-competitive inhibitor PKC 20-28, but not the activation-competitive inhibitor calphostin C. Model predictions and experimental validation demonstrated that insulation is a general property of scaffold tethering. Sensitivity analysis indicated that these findings may be applicable to many other scaffolds as well. Collectively, our findings provide theoretical and experimental evidence that scaffold proteins can amplify, accelerate, and insulate signal transduction.
Introduction
Signaling enzymes transduce extracellular cues into cellular responses, often signaling via a wide cohort of effector proteins. The promiscuity of signaling enzymes has led to the evolution of scaffolding proteins and the advancement of the anchoring hypothesis. A growing body of evidence underlies this hypothesis, which states that the spatial sequestration of signaling enzymes with their substrate proteins is an important determinant of the efficacy and specificity of enzyme catalysis, most notably protein phosphorylation (1, 2). Some scaffolds have been shown to accelerate or amplify signal transduction (3, 4), whereas others create specificity, allowing distinct context-dependent responses using the same promiscuous enzyme (5, 6). Although physiological and pathological roles have been identified for an increasing number of scaffolds, there is little mechanistic theory to explain how the co-localization provided by scaffolds modulates cell signaling.
A-kinase anchoring proteins, AKAPs,4 are a family of >50 functionally related yet structurally diverse proteins that have demonstrated many of these scaffolding phenomena (7). Whereas AKAPs were originally characterized by their ability to direct the actions of the cAMP-dependent protein kinase toward specific substrates, significant work has demonstrated that they function as more general scaffolds, integrating the actions of multiple enzymes (8). For example, we have recently described the ability of AKAP7α to localize the actions of PKC to a membrane domain (9). Individual AKAPs have been shown to accelerate (3) or amplify (4) protein phosphorylation, yet it is unclear how these phenomena arise. They are hypothesized to be the result of enhanced enzyme-substrate interactions on a scaffold, but there is little quantitative evidence to explain how these macromolecular complexes actually influence enzyme catalysis.
Enzyme kinetics usually carry the assumption that both the enzyme and substrate are freely diffusing (10), but tethered enzymes and substrates contradict this assumption. In this paper, we propose a novel mechanism for scaffold-tethered enzymatic reactions, the scaffold state-switching model. This model predicts that scaffold tethering of enzymes and substrates can lead to amplification and acceleration of signal transduction. These model predictions are validated experimentally by examining the kinetics of phosphorylation by PKC both on and off AKAP7α. We then investigated how scaffold tethering affected the sensitivity of PKC to different inhibitors. This analysis led to the surprising finding that AKAP7α insulated PKC from ATP- and substrate-competitive inhibitors but not activation-competitive inhibitors. Further, our model demonstrated that insulation arose solely from scaffold tethering. The scaffold state-switching model provides a theoretical framework to study how and when acceleration, amplification, and insulation emerge from scaffold localized reactions.
EXPERIMENTAL PROCEDURES
Expression Constructs
The following constructs were obtained from Addgene: PKCα-FLAG (plasmid 10805), C-kinase activity reporter (CKAR; plasmid 10806), and MyrPalm-CKAR (plasmid 14862). AKAP7α-CKAR was created by flanking AKAP7α (Dr. John Scott, University of Washington) with HindIII restriction sites and subcloning it into the N terminus of CKAR.
Pharmacological Manipulations of PKC
PKC activation was achieved using phorbol-12,13-dibutyrate (PDBu; EMD Millipore 524390) at a concentration of 250 nm. ATP competitive inhibition of PKC was achieved by preincubating transfected cells with Gö6976 (EMD Millipore 365250) at a concentration of 1 μm for 5 min. The substrate competitive inhibitor PKC 20-28 (EMD Millipore 476480) was incubated with cells for 30 min at 37 °C at a concentration of 16 μm. The activation inhibitor calphostin C (EMD Millipore 208725) was preincubated with transfected cells at a concentration of 200 nm for 30 min at 37 °C followed by a 15-min photoactivation via LED illumination.
Fluorescence Resonance Energy Transfer (FRET)
FRET measurements were executed as described previously (9). Briefly, Vero cells were seeded at 50% confluence onto glass coverslips (Warner Instruments) and transfected with 500 ng of plasmid DNA using the Lipofectamine PLUS transfection system (Invitrogen). Cells were maintained at room temperature in imaging buffer (pH 7.3, 172 mm NaCl, 2.4 mm KCl, 10 mm HEPES, 4 mm CaCl2, 4 mm MgCl2, 10 mm glucose) for the duration of the imaging, which began following a 5-min equilibration period on the microscope stage. All images were collected using a Zeiss Pascal confocal microscope and a 40×/1.2 NA objective. Excitation of cyan fluorescent protein (CFP) was carried out using a 440-nm laser (Toptica Photonics). A HQ535/50M and HQ480/40M emission filter with a 510DCLP dichroic were used (Chroma Technology). Channel intensities were quantified using ImageJ software. Individual traces were background-corrected, standardized against their base-line values to put them on a scale of 1, and photobleach-corrected against a linear fit of change in fluorescence for untreated cells. Data presented are composite traces from multiple cells and experiments as indicated within each figure.
Confocal Imaging
Representative images of Myristolated CKAR (MyrCKAR) and AKAP7α-CKAR correspond to emissions collected in the CFP channel. Images were collected under identical conditions and settings, using the same laser (440 nm) and filter configuration as the FRET experiments. Images were processed and arranged using ImageJ.
Model Development
A computational model was developed to describe PKC phosphorylation of CKAR both with and without the AKAP7α scaffold, implemented in MATLAB (MathWorks). These models were based on mass action kinetics of either the standard enzyme mechanism or the scaffold state-switching mechanism. The model parameters were defined using literature values of biochemical rate constants (supplemental Tables S2, S4, and S5). Parameters for phosphatase activity, basal activity, and enzyme concentration were estimated by nonlinear least squares fitting to the CKAR experimental FRET measurements (supplemental Table S3). Confidence intervals (95%) on parameter estimates were calculated using the covariance matrix from the least squares fit. A detailed description of model equations and parameters is in supplemental text (supplemental Fig. S4 and Table S1).
Model Sensitivity Analysis
Model sensitivity analysis was performed by randomly sampling kinetic parameter values from a fixed range of physiological values for the different rate constants as determined by a literature search (see supplemental text). The parameter space was sampled using a MATLAB built in Latin-Hypercube sampling algorithm to ensure complete coverage of the parameter space.
Statistical Analysis
Unpaired t tests were performed using Prism 5.02 (GraphPad Software). p < 0.05 was considered significant.
RESULTS
Scaffold State-switching Model Predicts Amplification and Acceleration of Reactions Occurring on Scaffolds
The standard model of enzyme catalysis assumes that the enzyme and substrate freely diffuse in solution, reversibly associating to form an active intermediate state in which the enzyme catalyzes the conversion of the substrate to product (10). Yet these assumptions are not valid for the tethered “solid-state” interactions that occur within an enzyme-substrate-scaffold complex. Thus we developed a new “scaffold state-switching” model to examine how tethering an enzyme and substrate to a scaffold protein affects enzyme catalysis. The key underlying assumption is that enzymatic reactions occurring on scaffolds exhibit stochastic switching of the enzyme-scaffold-substrate complex between active (E-κ-S) and inactive (E-κ-S) intermediate states (Fig. 1). In this model we assume that the scaffold acts as a simple tether, although some scaffolds have been shown to directly regulate enzyme activity as well (11). We defined a dimensionless number ϵ to compare the rate of enzyme-substrate interaction on the scaffold relative to that in solution. This “scaffold efficiency number” is defined as ϵ = kϵ/(kf·[S]tot), where ϵ >1 indicates faster enzyme-substrate interaction on the scaffold than in solution.
FIGURE 1.
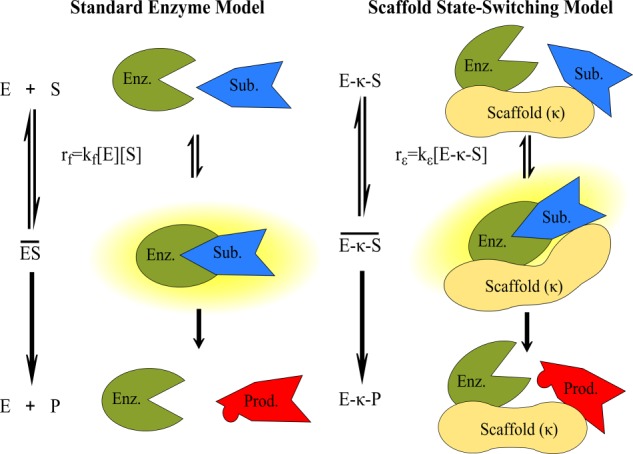
Scaffold state-switching model. Enzymes in solution follow standard enzyme kinetics (left), wherein an enzyme (E) and substrate (S) reversibly associate to form an active intermediate (ES) and catalyze the formation of product (P). The scaffold state-switching model (right), in contrast, describes anchored enzymatic reactions. In this model, both enzyme and substrate are bound to the scaffold (κ), and the complex undergoes stochastic switching between inactive (E-κ-S) and active (E-κ-S) intermediate states through which the catalysis proceeds to form the product (E-κ-P). Note that the enzyme substrate interaction rate on the scaffold (rϵ) is a first-order reaction compared with the second-order reaction in solution (rf).
We next tested whether scaffold state-switching is sufficient to predict amplification and acceleration of signaling, as was observed experimentally for several AKAPs (3, 4, 6, 12). We modeled the activation of PKC and its phosphorylation of CKAR in solution (Free, Fig. 2A) and when tethered to a scaffold (Scaffold, Fig. 2A). The Scaffold model assumes a scaffold state-switching mechanism, where active PKC and CKAR switch stochastically between active and inactive intermediate states. In both models, PKC is activated by PDBu and CKAR is dephosphorylated by phosphatases. Rate constants were curated from the literature as detailed in the supplemental text, with the scaffold efficiency number left as a free unknown parameter.
FIGURE 2.
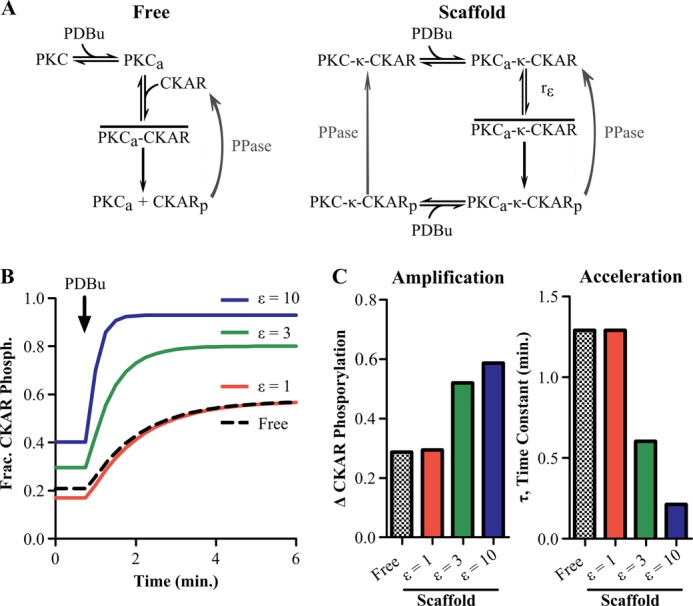
State-switching model predicts acceleration and amplification of substrate phosphorylation. A, network diagram detailing assumptions of the computational models. In the free enzyme model (left), quiescent PKC is activated by PDBu. The active enzyme (PKCa) reversibly associates with its substrate, CKAR, to form the active intermediate (PKCa-CKAR) and then carry out phosphorylation (CKARp). However, in the scaffold state-switching model (right), a preassembled complex containing PKC and CKAR exists on a scaffold (PKC-κ-CKAR), causing PDBu-bound PKC to switch stochastically between inactive (PKCa-κ-CKAR) and active (PKCa-κ-CKAR) intermediate states. The efficiency of this state-switching mechanism is referred to as the scaffold efficiency number (ϵ). B, state-switching model predicting that increased scaffold efficiency results in a concomitant amplification and acceleration of CKAR phosphorylation. C, quantification of amplification and acceleration by the scaffold for increasing scaffold efficiency number.
When the scaffold efficiency number is set to 1, the active intermediate formation rate is the same on the scaffold and in solution, creating similar phosphorylation kinetics in solution and on the scaffold (Fig. 2B). Increasing the scaffold efficiency number resulted in a concomitant increase in both the magnitude and rate of CKAR phosphorylation on the scaffold (Fig. 2B, solid lines). These kinetic increases were quantified by changes in the fraction of phosphorylated CKAR and exponential time constant upon PDBu stimulation (Fig. 2C). These model predictions show that when ϵ is >1, the enhanced rate of active intermediate formation on a scaffold is sufficient to amplify and accelerate PKC signaling.
AKAP7α Amplifies Substrate Phosphorylation by PKC as Predicted by the Scaffold State-switching Model
To validate our model predictions experimentally, we used the FRET biosensor CKAR, a live-cell kinetic reporter of intracellular PKC activity (13). This biosensor undergoes FRET in its dephosphorylated state but loses this ability upon phosphorylation by PKC. We generated an anchored PKC activity reporter, AKAP7α-CKAR, by genetically fusing the PKC scaffold AKAP7α to CKAR (Fig. 3A). We have shown previously that AKAP7α directly binds PKC with a 27 nm affinity (9). As AKAP7α is membrane-targeted in cells via N-terminal lipid modifications, we used MyrCKAR to represent the free substrate. This ensures comparative activation of PKC by PDBu for both biosensors. Validation of AKAP7α-CKAR was confirmed via Western blotting using anti-GFP and anti-AKAP7 antibodies (supplemental Fig. S1). Confocal images of the two biosensors demonstrate the constructs to be appropriately targeted to the plasma membrane with similar expression levels (Fig. 3B). Anchored PKC-mediated phosphorylation, assessed by changes in AKAP7α-CKAR FRET ratio, was 2-fold higher than the response of the free PKC reported by MyrCKAR (7.4% ± 0.7% AKAP7α-CKAR versus 3.7% ± 0.6% MyrCKAR) upon stimulation with PDBu (Fig. 3C). We also observed a 1.8-fold acceleration of the phosphorylation rate of AKAP7α-CKAR, although this trend did not reach statistical significance. The amplified response of AKAP7α-CKAR was not simply due to overexpression of AKAP7α, because co-expression of AKAP7α with MyrCKAR induced responses similar to MyrCKAR alone (supplemental Fig. S2). We also examined whether the larger response of AKAP7α-CKAR was due to a larger dynamic range of this biosensor. Under conditions that maximize phosphorylation (combined PDBu and phosphatase inhibitor calyculin A), AKAP7α-CKAR and MyrCKAR exhibited similar responses (supplemental Fig. S3). Thus, we conclude that the observed increases in PKC-mediated phosphorylation reported by AKAP7α-CKAR is indeed due to scaffold efficiency, and not due to perturbation of the cellular environment.
FIGURE 3.

AKAP7α accelerates and amplifies CKAR phosphorylation. A, schematic diagram of the FRET probes MyrCKAR (top) and AKAP7α-CKAR (bottom). MyrCKAR, characterized previously, contains an N-terminal Myr/Palm sequence sufficient to target it to the plasma membrane. AKAP7α-CKAR, generated by fusing AKAP7α to the CKAR backbone, is also membrane-targeted by virtue of a Myr/Palm domain contained within the N terminus of the AKAP. B, confocal images of MyrCKAR and AKAP7α-CKAR obtained in the CFP channel (440-nm laser). C, cells expressing MyrCKAR (black circles) (n = 11) or AKAP7α-CKAR (red squares) (n = 9) stimulated with 250 nm PDBu and exhibiting increases in the FRET ratio. Both the free model (black line) and scaffold model (red line) were simultaneously fit to the MyrCKAR and AKAP7α-CKAR data, respectively, resulting in a scaffold efficiency number (ϵ) of 5.70 ± 0.37 (95% confidence interval). D, quantification of experimental data (solid bars) and corresponding model fits (checkered bars) reveal amplification and a trend toward acceleration of AKAP7α-CKAR (red) versus MyrCKAR (black). All error bars are S.E.; ***, p < 0.001.
We refined our model parameters by simultaneously fitting the free and scaffold models to the MyrCKAR and AKAP7α-CKAR data, respectively. All parameter values for free and scaffold models were constrained to be equal, with the exception of the rate constant for active intermediate formation on the scaffold (kϵ) and in solution (kf). During the fit, both models were scaled to the measured steady-state PDBu response of MyrCKAR (for further explanation see the supplemental Fig. S5 and Table S3). The fitted model was in good agreement with the experimental data for both MyrCKAR and AKAP7α-CKAR (Fig. 3C). By fitting the model to these data, we inferred a scaffold efficiency number of ϵ = 5.70 ± 0.37 for PKC on AKAP7α. Similar to the experimental data, the model predicts that AKAP7α amplifies PKC signaling in response to PDBu (Fig. 3D, left). The simulations also show a 61% percent decrease in the time constant of CKAR phosphorylation on AKAP7α, consistent with the trend measured experimentally (Fig. 3D, right). Collectively, these data and simulations indicate that the amplification and acceleration of PKC phosphorylation of substrates mediated by AKAP7α are caused by an approximately 5-fold increase in the rate of active intermediate formation.
AKAP7α Insulates PKC from Substrate- and ATP-competitive Inhibitors but Not Activation-competitive Inhibitors
In addition to amplification and acceleration, scaffold tethering may impact other aspects of cell signaling such as the sensitivity to outside regulators. We sought to test whether the scaffold state-switching model can predict whether scaffolds insulate PKC from certain classes of inhibitors, lowering their effective potency in live cells. We hypothesized that the scaffold would cause PKC to interact preferentially with tethered CKAR, insulating PKC from a substrate-competitive inhibitor in solution. Conversely, we hypothesized that activation- and ATP-competitive inhibitors would be similarly potent for PKC with and without the scaffold. To evaluate this hypothesis in silico, we extended our model to incorporate these three different classes of inhibitors (supplemental Fig. S6). We simulated the effect of each inhibitor on the steady-state response of PKC in the presence of PDBu, both with and without scaffold (Fig. 4 and supplemental Fig. S7). Results were normalized to the PDBu-stimulated increase in CKAR phosphorylation without inhibitor.
FIGURE 4.

State-switching model predicts insulation of anchored PKC. Dose responses of PKC to three different classes of inhibitors were simulated, where the PKC activity was quantified by the change in CKAR phosphorylation upon stimulation with PDBu. All responses are normalized to the noninhibited response to PDBu. The kinetic parameters from the fitted models were used in the simulation (ϵ = 5.7). A, activation-competitive inhibitors were predicted to have similar potency either with (ϵ = 5.7) or without (Free) tethering to the scaffold. B, substrate-competitive inhibitors acting on anchored PKC were predicted to have reduced potency (increased IC50) compared with free enzyme. C, ATP-competitive inhibitors were also predicted to exhibit lower potency for anchored PKC versus free PKC.
The activation-competitive PKC inhibitor was modeled by assuming that it competed with the binding of PDBu and endogenous lipid activators of PKC. The model predicted similar potency of activation-competitive inhibitor for both free PKC and scaffold-tethered PKC, consistent with our hypothesis (Fig. 4A). Note that the rise of the normalized inhibitor response curve at low doses of calphostin C is due to a greater effect on basal PKC activity than PDBu-stimulated PKC activity.
The substrate-competitive PKC inhibitor was modeled by assuming that it reversibly associates with active PKC when PKC and the substrate are not in the active intermediate state. The substrate-competitive inhibitor was predicted to have a 3.7-fold lower potency for scaffold-tethered PKC compared with free PKC, consistent with our hypothesis above (Fig. 4B). To understand the mechanism underlying how the scaffold insulated the kinase from substrate-competitive inhibitors, we examined how the amount of enzyme-substrate active intermediate depends on the inhibitor concentration (supplemental Fig. S8). This analysis showed that because scaffold tethering increased the amount of active intermediate, more inhibitor was needed to obtain active intermediate levels (and downstream CKAR phosphorylation) similar to that without scaffold. In contrast, scaffold-induced insulation was not predicted for activation-competitive inhibitors because the amount of PKC that becomes activated is independent of the enzymatic reaction efficiency.
The ATP-competitive PKC inhibitor was modeled by assuming that it reduces the effective PKC catalytic rate constant by competing with ATP in a rapid equilibrium manner. In contrast to our original hypothesis, the model predicted that the ATP-competitive inhibitor would have a surprising 4.2-fold lower potency for scaffold-tethered PKC compared with free PKC (Fig. 4C). To understand why the model predicted insulation for ATP-competitive inhibitors, we examined the effect of changing the catalytic rate constant (kcat) on the magnitude of CKAR phosphorylation (supplemental Fig. S9). This analysis showed that by increasing the efficiency of phosphorylation, the scaffold pushed the system to a state where catalytic rate constant is less rate-limiting to phosphorylation. Thus, by increasing the rate of enzyme-substrate active intermediate formation, scaffolds may create a “catalytic reserve” that requires more inhibitor to obtain a similar amount of inhibition. This counterintuitive insulation of ATP-competitive inhibitors suggests that insulation is a novel property of scaffold tethering. Thus, the scaffold state-switching model predicted that substrate tethering can insulate PKC from ATP- and substrate-competitive inhibitors but not an activation-competitive inhibitor.
To test the model predictions of inhibitor insulation, we performed experiments comparing the sensitivity of MyrCKAR and AKAP7α-CKAR with calphostin C (an activation-competitive PKC inhibitor), Gö6976 (an ATP-competitve PKC inhibitor), and pseudosubstrate oligopeptide PKC 20-28 (a substrate-competitive PKC inhibitor). In agreement with the model predictions, pretreatment with the activation inhibitor calphostin C significantly inhibited PKC substrate phosphorylation elicited by PDBu on both MyrCKAR (109.8% inhibition) and AKAP7α-CKAR (71.5% inhibition) (Fig. 5A). Strikingly, we found that whereas the pseudosubstrate inhibitor 20–28 robustly suppressed MyrCKAR phosphorylation (72.0% inhibition), PKC tethered to AKAP7α was insulated from this substrate-competitive inhibitor (6.4% inhibition). These experimental data indicate that AKAP7α insulates PKC from substrate competitive inhibitors even more than predicted by the computational model (Fig. 5B). Similarly, although the ATP-competitive inhibitor Gö6976 was a potent inhibitor of MyrCKAR phosphorylation (72.2% inhibition), Gö6976 did not significantly decrease PKC activity on AKAP7α-CKAR (Fig. 5C). This AKAP7α-induced insulation of PKC from Gö6976 qualitatively agreed with the lower potency of ATP-competitive inhibitors predicted by the model. Indeed, the experimentally measured insulation was even greater than in the model, perhaps due to underestimation of the scaffold efficiency number. Thus, these experiments successfully validate model predictions of drug sensitivity for PKC tethered to AKAP7α for three distinct classes of inhibitors.
FIGURE 5.

PKC tethered to AKAP7α is insulated from certain classes of pharmacological inhibitors. A, cells expressing either MyrCKAR (n = 17) or AKAP7α-CKAR (n = 12) were pretreated with the activation-competitive PKC inhibitor calphostin C (200 nm). Calphostin C significantly inhibited the response of MyrCKAR to PDBu (solid red bar). Similarly, AKAP7α-CKAR was also significantly inhibited by calphostin C (solid green bar). The difference in the amount of inhibition between MyrCKAR and AKAP7α-CKAR was not significant (p = 0.099). These measurements agree with predictions from the free enzyme model (checkered red bar) as well as in the AKAP7α scaffold state-switching model (ϵ = 5.7, checkered green bar). B, cells expressing either MyrCKAR (n = 8) or AKAP7α-CKAR (n = 8) were pretreated with the substrate-competitive PKC inhibitor PKC 20-28 (16 μm). PKC 20-28 significantly inhibited PDBu-dependent responses of MyrCKAR (solid red bar), yet no significant inhibition was observed for AKAP7α-CKAR (solid green bar). The inhibition of MyrCKAR was significantly greater than that of AKAP7α-CKAR, consistent with predictions from the free (checkered red bar) and the scaffold model (ϵ = 5.7, checkered green bar). C, cells expressing either MyrCKAR (n = 9) or AKAP7α-CKAR (n = 7) were pretreated with the ATP-competitive PKC inhibitor Gö6976 (1 μm). Gö6976 significantly inhibited the PDBu-evoked FRET response of MyrCKAR (solid red bar). In contrast, no significant inhibition was observed for AKAP7α-CKAR (solid green bar). MyrCKAR inhibition was significantly greater than AKAP7α-CKAR, in agreement with the model-predicted lower potency of Gö6976 for the scaffold (checkered green bar) compared with free PKC (checkered red bar). All error bars are S.E.; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Extent of Acceleration, Amplification, and Insulation Varies Depending on Enzyme Kinetics
Here we have focused on how AKAP7α tethering modulates PKC signaling. However, as numerous other enzyme-substrate-scaffold complexes exist within cells, we sought to determine whether these same principles can be applied to other scaffold complexes as well. From the literature we identified biologically plausible ranges of kinetic rate parameters for select protein kinases (e.g. PKA, CaMKII) and phosphatases (e.g. PP1, PP2A; supplemental Tables S5 and S6). Parameters were randomly sampled within these ranges to obtain models for 2000 distinct hypothetical protein complexes. To allow fair comparison between complexes, the scaffold efficiency number was held constant at the level estimated for PKC-AKAP7α (ϵ = 5.7). Each hypothetical protein complex was simulated to quantify the predicted acceleration, amplification, and insulation. The distribution and co-variation of these features were examined to identify the range of possible scaffold “phenotypes” (Fig. 6). Overall, this analysis demonstrated that amplification, acceleration, and insulation are expected to arise for many protein complexes, although their quantitative levels can vary considerably. Amplification and acceleration showed a negative correlation, meaning that there are some cases where a scaffold can greatly amplify the response of a kinase but the rate of the response will have very little acceleration, and vice versa (Fig. 6A). Similarly, insulation and amplification were negatively correlated (Fig. 6B). This strong negative correlation arose because kinases that fully phosphorylated their substrate without the scaffold could not exhibit further amplification when tethered to the scaffold. Yet these kinases that exhibited high activity without the scaffold had a larger catalytic reserve, leading to a larger insulation from inhibitor when tethered to the scaffold. Examples of specific responses can be seen in supplemental Fig. S10. We verified that the trends seen in this analysis are not strictly correlated with any single kinetic parameter but rather a result of the effect of the scaffold on the system as a whole (supplemental Fig. S11). Overall, this sensitivity analysis showed that the extent of acceleration, amplification, and insulation by a scaffold can vary depending on the rate constants of the particular kinase and phosphatase.
FIGURE 6.

Amplification, acceleration, and insulation vary depending on enzyme kinetics. Enzyme kinetic parameters were varied over a physiological range to produce models of 2000 hypothetical enzyme-substrate-scaffold complexes (black circles). For all simulations the scaffold efficiency number was held constant at ϵ = 5.7. The PKC-AKAP7α-CKAR model is indicated as a gray star. A, amplification is negatively correlated with acceleration. Amplification was quantified as the ratio of the steady-state substrate phosphorylation (Sp) for scaffold to solution biochemistry, Sp(scaffold)/Sp(free). Acceleration was quantified as the ratio of half-maximal time, t50, in solution to on the scaffold, t50(free)/t50(scaffold). B, insulation and amplification are also negatively correlated. Insulation from substrate-competitive inhibitors was quantified as the log of the ratio of the IC50 on scaffold to in solution, log10(IC50(scaffold)/IC50(free)).
DISCUSSION
We propose scaffold state switching as a novel mechanism shaping the kinetics of signaling on protein scaffolds. This model assumes that enzymes and substrates tethered to a scaffold switch stochastically between active and inactive intermediate states. Our computational models and subsequent experimental validation showed that the scaffold state-switching model accurately predicted the amplification and acceleration of PKC phosphorylation of a tethered substrate. Reconciling our computational and experimental data, we estimated that AKAP7α tethering increased the PKC rate of association with its substrate by >5-fold. We then extended our model to address how scaffolding proteins could affect the external influence of three classes of pharmacological kinase inhibitors. Both our computational simulations and experimental evidence demonstrated that AKAP7α insulated bound PKC from substrate- and ATP-competitive inhibitors. However, no protection was offered against activation-competitive inhibitors. Finally, model sensitivity analysis indicated that scaffolds may amplify, accelerate, and insulate a broad range of signaling pathways. Overall, this work provides a new kinetic mechanism for scaffold-localized reactions and a theoretical underpinning with which to further understand how scaffold proteins shape cell signaling.
Acceleration and amplification by scaffold proteins have been shown for several different protein complexes (3, 4, 12). Physiological and pathological roles of acceleration and amplification by scaffolds have been demonstrated in multiple tissues including the heart, brain, and pancreas (14–17). Here we have not only shown that AKAP7α can amplify and accelerate PKC signaling, we have also provided a mechanistic explanation of how amplification and acceleration occur. In developing the scaffold state-switching model, we have generated an important tool for understanding how different scaffolds may uniquely modulate signaling enzymes by providing more or less acceleration, amplification, and insulation.
Although others have reported insulation created by scaffolds, the scaffold state-switching model provides new evidence that insulation can result directly from scaffold tethering and the resulting enhanced rate of active intermediate formation. AKAP5, for example, reduces the potency of certain ATP-competitive PKC inhibitors by competing for access to the substrate-binding pocket of the enzyme (4). In contrast, here we provide strong evidence that the insulation of PKC by AKAP7α is an emergent property of anchoring, which is consistent with our previous observation that AKAP7 does not inhibit PKC activity (9, 11). This insulation is distinct from the substrate specificity by scaffold tethering (5), as substrate specificity would have predicted reduced potency only for the substrate-competitive inhibitor. Insulation of PKC from both ATP- and substrate-competitive inhibitors indicates that insulation is a native property of scaffold tethering. Further, the scaffold state-switching model provides a biophysical explanation of how tethering alone can cause insulation on protein scaffolds.
The scaffold state-switching model differs from previous approaches used to model signaling on protein scaffolds. Levchenko et al. (18) and O'Shaunessy et al. (19) developed computational models to examine the role of scaffolds in MAPK signaling. By assuming that the scaffold fixes the enzyme and substrate in an active intermediate state, they predicted amplification and acceleration of the MAPK pathway. However, such an approach cannot predict insulation from competing substrates as shown here with the scaffold state-switching model. Saucerman et al. assumed that AKAPs increase the local concentration of substrates 10-fold (20, 21) based on kinetic experimental data of tethered PKA (12). However, their phenomenological models of AKAPs implicitly assumed enhanced association rates in solution rather than mechanistically representing the solid-state transitions (22) within a protein complex as done in the current study. Others have modeled signaling on scaffolds using spatially explicit stochastic algorithms, which can add further details of cellular anatomy (23, 24). Yet these models again either assumed that the scaffold fixed enzyme and substrate in an active intermediate state (23) or enhanced association rates due to high local concentrations (24). Thus, the scaffold state-switching model has advantages in its mechanistic representation of reactions on a scaffold as well as the prediction of insulation from competing substrates.
In some instances, the relative stoichiometry of scaffold to enzyme or substrate can enhance or suppress signaling, an effect termed combinatorial inhibition (18, 19). Given the high affinity interaction between PKC and AKAP7α (9) and the physical linkage of AKAP7α to CKAR we assumed that the scaffold was fully occupied by enzyme and substrate. However, future extensions of this model exploring other scaffold enzyme permutations may require the incorporation of enzyme or substrate dissociation from the scaffold. For example, phosphorylation of AKAP-Lbc can lead to a decreased association rate of protein kinase D (PKD) for the scaffold which increases phosphorylation of PKD by tethered PKC through substrate turnover (25). Additionally, some scaffolds can directly alter the activity of bound enzymes (26). AKAP5 inhibits PKC by tethering to the PKC catalytic pocket (4). The yeast MAPK scaffold Ste5 directly increases the catalytic rate constant of Ste7 phosphorylation of Fus3-independent of tethering (27). However, direct effects of AKAP7 on PKC have not been documented. Thus, models of other scaffolds may require consideration of both tethering and direct enzyme regulation.
Collectively, the scaffold state-switching model and its experimental validation shed light on the biophysical underpinnings of anchored enzymatic reactions that extend beyond PKC and AKAP7α. Our work has shown how amplification, acceleration, and insulation arise from tethering reactions to scaffold proteins. The wide range of scaffold-enzyme-substrate complexes surveyed in our sensitivity analysis suggests that our findings are broadly applicable and therefore of great interest for many clinical and bioengineering applications. Despite uncertainty in specific kinetic parameters, the model was able to predict a range of experimental data, giving us confidence in its validity. Our mechanistic representation of enzyme-scaffold dynamics provides a quantitative definition of the anchoring hypothesis. Future extensions of this theoretical and experimental framework will allow analysis of more complex signalosomes and further our understanding of how cells use promiscuous enzymes to make specific decisions.
This work was supported, in whole or in part, by National Institutes of Health Grants HL82705 (to K. L. D.-K.), and HL094476 (to J. J. S.). This work was also supported by State of Connecticut Department of Public Health Grant 2011-0142 (to K. L. D.-K.), American Heart Association Predoctoral Fellowship 11PRE7830027 (to J. M. R.), and National Institutes of Health Pharmacological Sciences Training Grant Predoctoral Fellowship at the University of Virginia T32 GM007055 (to E. C. G.).

This article contains supplemental text, Figs. S1–S11, Tables S1–S6, and additional references.
- AKAP
- A-kinase anchoring protein
- CKAR
- C-kinase activity reporter
- Gö6976
- 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole
- MyrCKAR
- myristolated CKAR
- PDBu
- phorbol-12,13-dibutyrate
- CFP
- cyan fluorescent protein.
REFERENCES
- 1. Scott J. D., Dessauer C. W., Taskén K. (2013) Creating order from chaos: cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 53, 187–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Edwards A. S., Scott J. D. (2000) A-kinase anchoring proteins: protein kinase A and beyond. Curr. Opin. Cell Biol. 12, 217–221 [DOI] [PubMed] [Google Scholar]
- 3. Tavalin S. J. (2008) AKAP79 selectively enhances protein kinase C regulation of GluR1 at a Ca2+-calmodulin-dependent protein kinase II/protein kinase C site. J. Biol. Chem. 283, 11445–11452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoshi N., Langeberg L. K., Gould C. M., Newton A. C., Scott J. D. (2010) Interaction with AKAP79 modifies the cellular pharmacology of PKC. Mol. Cell 37, 541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park S. H., Zarrinpar A., Lim W. A. (2003) Rewiring MAP kinase pathways using alternative scaffold assembly mechanisms. Science 299, 1061–1064 [DOI] [PubMed] [Google Scholar]
- 6. Greenwald E. C., Saucerman J. J. (2011) Bigger, better, faster: principles and models of AKAP anchoring protein signaling. J. Cardiovasc. Pharmacol. 58, 462–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pidoux G., Taskén K. (2010) Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J. Mol. Endocrinol. 44, 271–284 [DOI] [PubMed] [Google Scholar]
- 8. Dodge-Kafka K. L., Soughayer J., Pare G. C., Carlisle Michel J. J., Langeberg L. K., Kapiloff M. S., Scott J. D. (2005) The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437, 574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Redden J. M., Le A. V., Singh A., Federkiewicz K., Smith S., Dodge-Kafka K. L. (2012) Spatiotemporal regulation of PKC via interactions with AKAP7 isoforms. Biochem. J. 446, 301–309 [DOI] [PubMed] [Google Scholar]
- 10. Michaelis L., Menten M. L., Johnson K. A., Goody R. S. (2011) The original Michaelis constant: translation of the 1913 Michaelis-Menten paper. Biochemistry 50, 8264–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Faux M. C., Rollins E. N., Edwards A. S., Langeberg L. K., Newton A. C., Scott J. D. (1999) Mechanism of A-kinase-anchoring protein 79 (AKAP79) and protein kinase C interaction. Biochem. J. 343, 443–452 [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang J., Ma Y., Taylor S. S., Tsien R. Y. (2001) Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. U.S.A. 98, 14997–15002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Violin J. D., Zhang J., Tsien R. Y., Newton A. C. (2003) A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J. Cell Biol. 161, 899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patel H. H., Hamuro L. L., Chun B. J., Kawaraguchi Y., Quick A., Rebolledo B., Pennypacker J., Thurston J., Rodriguez-Pinto N., Self C., Olson G., Insel P. A, Giles W. R., Taylor S. S., Roth D. M. (2010) Disruption of protein kinase A localization using a trans-activator of transcription (TAT)-conjugated A-kinase-anchoring peptide reduces cardiac function. J. Biol. Chem. 285, 27632–27640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carnegie G. K., Soughayer J., Smith F. D., Pedroja B. S., Zhang F., Diviani D., Bristow M. R., Kunkel M. T., Newton A. C., Langeberg L. K., Scott J. D. (2008) AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell 32, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sanderson J. L., Dell'Acqua M. L. (2011) AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist 17, 321–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lester L. B. (1997) Anchoring of protein kinase A facilitates hormone-mediated insulin secretion. Proc. Natl. Acad. Sci. U.S.A. 94, 14942–14947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Levchenko A., Bruck J., Sternberg P. W. (2000) Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. U.S.A. 97, 5818–5823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Shaughnessy E. C., Palani S., Collins J. J., Sarkar C. A. (2011) Tunable signal processing in synthetic MAP kinase cascades. Cell 144, 119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saucerman J. J., Brunton L. L., Michailova A. P., McCulloch A. D. (2003) Modeling β-adrenergic control of cardiac myocyte contractility in silico. J. Biol. Chem. 278, 47997–48003 [DOI] [PubMed] [Google Scholar]
- 21. Saucerman J. J., Healy S. N., Belik M. E., Puglisi J. L., McCulloch A. D. (2004) Proarrhythmic consequences of a KCNQ1 AKAP-binding domain mutation: computational models of whole cells and heterogeneous tissue. Circ. Res. 95, 1216–1224 [DOI] [PubMed] [Google Scholar]
- 22. Malbon C. C., Tao J., Shumay E., Wang H. Y. (2004) AKAP (A-kinase anchoring protein) domains: beads of structure-function on the necklace of G-protein signalling. Biochem. Soc. Trans. 32, 861–864 [DOI] [PubMed] [Google Scholar]
- 23. Locasale J. W., Shaw A. S., Chakraborty A. K. (2007) Scaffold proteins confer diverse regulatory properties to protein kinase cascades. Proc. Natl. Acad. Sci. U.S.A. 104, 13307–13312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oliveira R. F., Terrin A., Di Benedetto G., Cannon R. C., Koh W., Kim M., Zaccolo M., Blackwell K. T. (2010) The role of type 4 phosphodiesterases in generating microdomains of cAMP: large scale stochastic simulations. PloS One 5, e11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carnegie G. K., Smith F. D., McConnachie G., Langeberg L. K., Scott J. D. (2004) AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol. Cell 15, 889–899 [DOI] [PubMed] [Google Scholar]
- 26. Good M. C., Zalatan J. G., Lim W. A. (2011) Scaffold proteins: hubs for controlling the flow of cellular information. Science 332, 680–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Good M., Tang G., Singleton J., Reményi A., Lim W. A. (2009) The Ste5 scaffold directs mating signaling by catalytically unlocking the Fus3 MAP kinase for activation. Cell 136, 1085–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]