

Abstract
The pre-clinical and clinical development of viral vehicles for gene transfer increased in recent years, and a recombinant adeno-associated virus (rAAV) drug took center stage upon approval in the European Union. However, lack of standardization, inefficient purification methods and complicated retargeting limit general usability. We address these obstacles by fusing rAAV-2 capsids with two modular targeting molecules (DARPin or Affibody) specific for a cancer cell-surface marker (EGFR) while simultaneously including an affinity tag (His-tag) in a surface-exposed loop. Equipping these particles with genes coding for prodrug converting enzymes (thymidine kinase or cytosine deaminase) we demonstrate tumor marker specific transduction and prodrug-dependent apoptosis of cancer cells. Coding terminal and loop modifications in one gene enabled specific and scalable purification. Our genetic parts for viral production adhere to a standardized cloning strategy facilitating rapid prototyping of virus directed enzyme prodrug therapy (VDEPT).
Viral gene therapy and the subcategory of virus-directed enzyme prodrug therapy (VDEPT) provide many opportunities and have been heralded as universal tools for therapy. Despite the potential, clinical success has been delayed due to difficulties in managing the many factors coming into play in the design, production and application of such complex systems. Gene therapy relies on an externally provided transgene also named ‘gene of interest’ (GOI) which is delivered to a target tissue or a specific cell type. Upon reaching the target the goals diverge. Gene sublementation therapy typically aims at maintaining the gene and its expression in the target cell as long as possible to replace a defective gene and/or alter the phenotype of the target cell. In contrast, VDEPT, as presented here, may also be used to kill tumor cells. This two-step method first delivers the gene for a non-endogenous enzyme to a target tissue via a viral vector. Following transduction, the enzyme is expressed, and the ability to specifically activate subsequently administered and otherwise inert prodrugs to potent drugs allows for a cell- or tissue- specific therapy with reduced systemic side effects1. So far, a large variety of delivery methods have been developed including non-viral and viral, genome-integrating or non-integrating systems2,3. Among the non-integrating viral systems usage of the adeno-associated virus (AAV) has been extensively studied. Serotype 2 (AAV-2) has gained significant interest since its great potential for gene therapy has been demonstrated in a successful clinical trial involving retinal infusion for patients with Leber's congenital amaurosis4,5. Most importantly, in 2012 a recombinant AAV serotype 1 became an approved drug in the European Union for treating patients with lipoprotein lipase (LPL) deficiency6.
The AAV-2 is a small, non-enveloped virus belonging to the family of Parvoviridae, and more specifically to the genus Dependovirus. It relies on a helper virus for replication such as adenovirus or herpes simplex virus. The 4.7 kb single-stranded DNA genome harbors the replicative (rep) and capsid forming (cap) genes. The capsid is built from 60 subunits of the viral coat proteins VP1, VP2 and VP3, whereby VP1 and VP2 include the VP3 sequence. Remarkable progress has been made in tailoring this virus to the needs of a gene therapy vector. The need of a helper virus has been eliminated by providing the required genes on an external plasmid7. Moreover, the initially favored broad natural tropism8,9,10,11 was later not only diminished12 but also successfully changed towards specific targets12,13,14,15. Consequently, recombinant adeno-associated viruses (rAAV) are increasingly becoming the vector of choice for a wide range of gene therapy approaches16. However, manufacturing AAV-2 particles for cancer gene therapy still has several drawbacks. Most strategies guiding these viral vectors to specified cell surface receptors involve either insertion of small, directly binding peptides12 or the use of adaptors that mediate between ligand and receptor15,17. To our knowledge, only one recent retargeting-approach used direct genetic coupling of a protein moiety (designed ankyrin repeat protein, DARPin), which is directed towards the Her2 (ErbB2) receptor14.
An additional hindrance for using rAAV particles as gene therapy carriers are unmodified, e. g. wild-type capsids, which emerge from the production method and tend to accumulate predominantly in the liver and bone marrow18 rendering a systemic prodrug approach unfavorable and toxic for off-target cells. Furthermore, the preferred prodrug and enzyme choice for tumor therapy has yet to be defined in the context of the rAAV strategy, the tumor marker, and the tumor cell type, which requires more detailed cell-based studies. For example, the combination of thymidine kinase and ganciclovir is well established for selection, but cytosine deaminase and 5-fluoro-cytosine offers the potential advantage that the activated drug can pass the membrane and thus elicit a killing bystander effect19,20, which might prove essential to eradicate a tumor.
Starting as a team in the international Genetically Engineered Machine (iGEM, iGEM Foundation, MA, USA) competition, a yearly, worldwide and well-known contest in the field of synthetic biology, we addressed the aforementioned obstacles in rAAV gene therapy. The foundational base of iGEM is a system of so-called BioBricks - standardized and interchangeable genetic elements, coding and non-coding, which share a common pre- and suffix allowing an idempotent BioBrick assembly as a uniform and fast cloning strategy21. We combined the latest achievements of rAAV-2 research using this modular BioBrick approach. For targeting we chose two published binding proteins, the Affibody_ZEGFR:190722 and the DARPin_E0123, which are two different small proteins with high-specificity for the cancer cell-surface marker human epidermal growth factor receptor (EGFR). Designed ankyrin repeat proteins (DARPins) originate from ankyrin repeat proteins which mediate protein-protein interactions throughout the cell24. Consisting of repetitive structural units which form an interaction interface25 they can be easily expressed and selected using high-throughput methods23. Affibody molecules share the latter features with DARPins but originate from the Z-domain scaffold derived from staphylococcal protein A26. Both targeting molecules were selected to bind EGFR which is vastly overexpressed e.g. in breast27, non-small-cell lung28 and bladder29 carcinomas with nanomolar affinity and have already been designated for therapeutic purposes30,31, thus rendering them an ideal tool to guide viral vectors specifically to cancer cells.
Here, we present the results of our so-called all-in-one approach using an rAAV vector equipped with EGFR binding molecules fused to the N-terminus of VP2 and small purification and detection tags encoded at the N-terminus and/or in surface-exposed loops of the respective VP3 domain. Both interaction motifs were efficiently displayed on the capsid surface allowing for highly -specific differential targeting and for affinity purification of the rAAV capsids of which the tropism towards heparan sulfate proteoglycan (HSPG) was knocked down32. To our knowledge, this is the first comprehensive toolkit for the production of rAAV-2 particles, which addresses extensively the demands of gene therapy33. Bottom-up construction from standardized parts enables easy adaption to new test requirements combined with fast and efficient purification possibilities. Using these particles armed with genetic constructs which facilitate the expression of prodrug-converting enzymes, we were able to distinguish between control and malignant cells and were thus able to specifically target and eradicate human cancer cells. At the same time, we used a deeply modular approach for the genetic assembly, where all relevant targeting and arming parts can easily be exchanged. Consequently, some of the currently most limiting issues in rAAV construction can be overcome by using a system which enables highly specific targeting of cell surface markers.
Results
Design, modularization, and production
Several different approaches have been reported to either introduce larger proteins into the viral AAV-2 capsid34,35 or insert small peptides into surface-exposed loops36. In order to specifically redirect rAAV-2 particles to cancer cell surface markers, we alternatively fused two different targeting molecules to the N-terminus of the viral coat protein VP2 and incorporated at the same time a small purification tag into a surface-exposed loop. Based on previous publications, both methods likely neither interfere with capsid assembly, genome packaging, nor infection14. However, so far all alterations of the viral capsid or its encapsidated genome involved tedious cloning steps and the different genetic elements were difficult to exchange. We therefore adapted the helper virus free AAV-2 system to the BioBrick RFC 10 standard21. As a result, all exchangeable genes were cloned on separate plasmids flanked by specific prefix and suffix sequences which provide specific restriction enzyme recognition sequences (Figure 1a). These BioBricks can be assembled using a standardized idempotent cloning strategy (Figure 1b). Next to the wild-type similars, we also constructed our viral modifications and extensions in the same assembly standard creating a highly modular system in which the targeting motives, the loop-integrated purification tag, as well as all elements of the encapsidated genome can be easily exchanged (details will be published elsewhere).
Figure 1. Idempotent genetic assembly strategy.

(a) Schematic representation of the restriction enzyme recognition patterns that build up the Prefix and Suffix, respectively. Flanking every BioBrick, these two elements represent the fundamental structure of the idempotent cloning strategy. Note that NgoMIV and AgeI are optional. (b) Genetic elements constructed using the standard BioBrick assembly for the expression of modified VP2 fusion proteins. (c) Plasmids used for production cell line transfection. Names are listed next to the graphical representation of genetic elements. Modified virus-like particles (right) are formed upon combined expression of all elements, see text for description.
For targeting, we used a mosaic virus approach, in which the VP2 capsid protein is knocked out from the RepCap plasmid with a point mutation in the start codon. A modified VP2 is then provided on an additional plasmid under the control of the CMV promoter. Next to simplifying cloning, this method ensures that the modifications in the VP2 construct, which comprises also the reading frame of VP3, only occur in the capsid at the frequency of the VP2 protein, and that the N-terminal modifications of VP2 are strictly coupled with the loop modifications. Using this approach, we created three different rAAV-2 particles carrying two previously described modifications in the exposed 587-loop. The first modification diminishes the natural tropism towards heparan sulfate proteoglycan (ΔHSPG) due to mutation of two interaction mediating arginine residues (R585A and R588A)12. The second modification is the insertion of a His6 purification tag at amino acid position 587, an exposed position known to tolerate insertion of small peptides37. Since ΔHSPG and His6 are always combined, these modification are denoted as AAV-2ΔHSPG and the resulting viral particle served as the control for the targeting experiments. Including these two modifications, two further particles were created which additionally display EGF receptor targeting molecules – either the Affibody_ZEGFR:190722 or the DARPin_E0123 – fused to the N-terminus of VP2, and which are named AAV-2_Affibody and AAV-2_DARPin, respectively, because a full name such as AAV-2ΔHSPG-VP2587His-Affibody_ZEGFR:1907 is barely readable (Figure 1c). These rAAV variants were produced in HEK-293 cells, harvested from serum-free cell culture supernatant, purified and analyzed for their correct assembly and targeting properties.
Assembly analysis
First, we determined whether the fusion proteins of VP2-DARPin or VP2-Affibody, respectively, had been incorporated into the viral capsids. Western blots of viral particle samples were visualized using the B1 antibody, which binds a linear epitope common to all three VP proteins38, and protein bands of expected size were detected either corresponding to VP2-Affibody-Flag (Figure 2a, lane A_2, approx. 75 kDa) or VP2-DARPin-Flag (Figure 2b, lane B_2, approx. 85 kDa). Comparison with AAV-2ΔHSPG (Figure 2a, b, lanes A_1, B_1) confirmed that the expression of wild-type VP2 (approx. 67 kDa) was prevented (VP2-KO) via the start codon mutations in plasmids pSB1C3_001-RepCapΔHSPG-VP2-KO, pSB1C3_001VP2ΔHSPG587His-DARPin-Flag and pSB1C3_001VP2ΔHSPG587His-Affibody-Flag. Blotted samples from the two targeting viral particles were additionally probed with the anti-Flag antibody (Figure 2a, b, lanes A_3, B_3) confirming correct sizes of both VP2 fusion proteins. Rough estimations of the band size and density using ImageJ (ImageJ 1.47, NIH) revealed that the modified VP2 fusion proteins represented approximately 11–14% of the total band intensity (corrected for protein mass; in detail: AAV-2_Affibody: VP1 29%, VP2_Affibody 14%, VP3 57%; AAV-2_DARPin: VP1 27%, VP2_DARPin 11%, VP3 62%).
Figure 2. Western blots of viral capsids equipped with targeting modules, concentrated by centrifugal ultrafiltration.

Viral particles were separated on a 7.5%–15% SDS gel and blotted onto a PVDF membrane. Particles with deleted HSPG motive served as controls (lanes A_1, B_1) using B1 antibody. (a) Western blot analysis of AAV-2_Affibody. Capsid proteins were detected using B1 antibody (lane A_2). VP2-Affibody was specifically detected via anti-Flag antibody (lane A_3) (b) Western blot analysis of AAV-2_DARPin. Capsid proteins were detected using B1 antibody (lane B_2). VP2-DARPin was specifically detected via anti-Flag antibody (lane B_3).
Viral particle purification and surface display
Since the viral particles harvested from the supernatant of serum-free cultured HEK-293 producer cells are diluted and contain some cell debris as well as unassembled capsid proteins, which both interfere with cellular assays, concentration and purification measures are required. Using ultrafiltration devices with a molecular weight cut-off (MWCO) of 1 MDa we enriched rAAV-2 capsids about 27-fold (Figure 3a) and at the same time, the high MWCO facilitated depletion of unassembled viral coat proteins. A slight enrichment of single capsid proteins in the filtrate might have emerged either from broken capsids or cells during ultrafiltration (Figure 3b). Given the fact that the coat protein VP2 is non-essential39 for viral assembly, a small population of produced viral particles lacks the VP2 fusion proteins. These unmodified capsids can cause negative side effects such as unspecific transduction14. To easily deplete vector preparations from these unwanted particles, we took advantage of the His6-purification tag inserted into the VP2-Affibody and the VP2-DARPin. Concentrated preparations of the targeted viruses displaying (His)6-tags were incubated with Ni-NTA resin. Unmodified particles were removed by washing and the targeting molecule displaying capsids were eluted with imidazole. To regain physiological conditions, the imidazole containing buffer was exchanged for phosphate buffered saline (PBS) using centrifugal concentrators. Using this method we were able to deplete concentrated vector preparations from unmodified rAAV-2 particles which represented approximately 20% of the total capsid number (Figure 3c).
Figure 3. Viral particle concentration, depletion of unassembled capsid proteins, affinity purification and surface display of targeting modules.

(a) Four days post transfection the supernatant from HEK-293 producer cells was harvested (supernatant) and concentrated via centrifugal ultrafiltration (concentrate, filtrate). The number of packed viral particles was determined via qPCR (n = 3, mean ± SD). (b) Samples from viral particle producing HEK-293 cells (supernatant), ultrafiltration filtrate and concentrate and fresh cell medium (neg. control) were incubated on an ELISA plate. Assembled particles or individual capsid proteins were detected using the A20 or the B1 antibody, followed by incubation with HRP-coupled anti-mouse antibody (n = 3, mean ± SD) (c) Ultrafiltration concentrated viral particles were incubated with Ni-NTA resin washed and capsids were eluted with PBS containing 500 mM imidazole. Subsequently, 4 × 109 purified and non-purified particles were incubated in an ELISA plate, coated with anti-His antibody. Viral particles were detected using the capsid specific A20 antibody, followed by HRP-coupled anti-mouse antibody (n = 4, mean ± SD) (d) 1.8 × 109 His-affinity purified AAV-2_AffibodyFlag and AAV-2_DARPinFlag particles were incubated on an ELISA plate coated with anti-Flag antibody. Assembled capsids were detected using the A20 antibody, followed by biotin-coupled isotype specific IgG3 antibody and streptavidin-coupled horseradish peroxidase (n = 3, mean ± SD).
Successful guidance of viral vectors to EGFR-overexpressing cancer cells requires accessible targeting moieties. To assess surface display of the Affibody and the DARPin, the respective modified rAAV-2 samples were subjected to an enzyme-linked immunosorbent assay (ELISA). ELISA plates were coated with anti-Flag-antibody and incubated with equal capsid titers of ultrafiltration-purified AAV-2ΔHSPG, AAV-2_Affibody-Flag and AAV-2-DARPin-Flag preparations followed by detection of bound particles with the A20 antibody, which is specific for assembled capsids. While the signal for AAV-2ΔHSPG remained at background level, both targeted viruses showed a comparable high display of the Affibody or DARPin (Figure 3d). These results demonstrate incorporation of the Affibody_ZEGFR:1907 and the DARPin_E01 into AAV-2 capsids and their display based on fusions to VP2.
Target cell binding and infectivity
The key feature of tumor-tissue guided viral vectors is the coupling of infection to the binding of markers expressed on the cell surface. First we investigated binding of viral targeting vectors to EGFR-overexpressing A431 cells in an ELISA-type experiment. Cell-bound viral particles were detected using the AAV-2 capsid specific antibody A20. As shown in figure 4a more than twice the amount of rAAV-2 particles equipped with either Affibody or DARPin were bound on fixed A431 target cells compared to control cells. In fact, binding of rAAV-2 particles to HeLa cells, which express an intermediate number of EGF receptor molecules on their surface40, remained at the same level as the binding of control AAV-2ΔHSPG particles. Since binding to target cells represents only the first step in a receptor guided gene therapy, we next assayed the ability of AAV-2_Affibody and AAV-2_DARPin to transduce different cell lines. Three cell types with an EGFR surface density density ranging from very low (approx. 104, MCF7) over intermediate (approx. 2 × 105, HeLa) to very high (approx. 4 × 106, A431)40 were incubated with equal genomic titers of targeting capsids. Since the infection pathway of the AAV-2 involves converting its single-stranded genome into a double-strand, the number of infectious particles is directly proportional to the number of double-stranded viral genomes inside the cells after 24 h. Thus, the infectious titer can be determined via quantitative real-time PCR36. As depicted in figure 4b, AAV-2_Affibody and AAV-2_DARPin very specifically transduced A431 target cells at very high rates (up to 700-fold, compared to MCF7 cells) and HeLa cells to an intermediate level (up to 200-fold). In absolute genome copy numbers, MCF7 cells were barely transduced. At the same time AAV-2ΔHSPG could be detected in A431, HeLa and MCF7 only at negligible low background levels. These results demonstrate that Affibody and DARPin guided viral capsids are suitable for high-specificity targeting towards high-level EGFR displaying cancer cells and that the rate of infection is strongly dependent on the level of EGF receptor expression.
Figure 4. Binding and infection specificity of rAAV-2 particles for different cell types.

(a) 1.8 × 109 viral particles were incubated on fixed 2.0 × 103 A431 or HeLa cells, respectively. Unbound particles were removed by washing and intact capsids were detected via A20 antibody. All values are expressed relative to binding of AAV-2ΔHSPG to HeLa cells, which was set to 100% (n = 5, mean ± SD). (b) 104 A431, HeLa, or MCF7 cells were incubated with 3 × 108 genomic viral particles for 24 h. Subsequently, the cells were harvested, digested with proteinase K and incubated with S1 nuclease. Double-stranded viral DNA was then quantified using qPCR (n = 3, mean ± SD).
In vitro tumor cell killing
In a second step, the ability of the targeting/prodrug activation system to kill target cells was exemplarily evaluated by testing the AAV-2_Affibody in an MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) cell viability assay of transduced cells. The assay (Figure 5) revealed that more than 80% of the target cells were efficiently killed after four days utilizing cytosine deaminase (CD)-armed AAV-2_Affibody particles in combination with 500 μM 5-fluorocytosine (5-FC). With 250 μM 5-FC the cell viability was reduced by more than 50%. In contrast, AAV-2ΔHSPG particles without binding moiety, or incubation with the prodrug alone affected the cell viability at most to 20%, but typically only to an insignificant level (Figure 5, AAV-2ΔHSPG and cells only).
Figure 5. Cell viability assay.
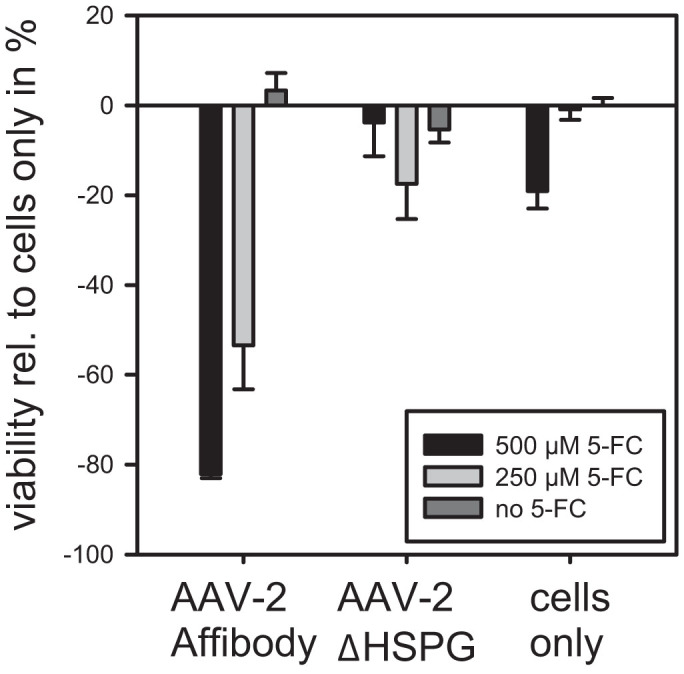
104 A431 or HeLa cells were transduced with 3 × 108 genomic viral particles. After 48 h fresh medium containing 5-FC was added to the cells. On day six an MTT assay was carried out and the amount of reduced formazan quantified. Values were calulated relative to the cells-only signal and then the latter was substracted to reflect the decrease in viability.
Since cell internal DNA damage signals generated by activated genotoxins such as 5-fluorodeoxyuridine monophosphate or ganciclovir-triphosphate (activated forms of 5-FC and GCV) are sensed and ultimately transduced to apoptotic signals41, the activity of the effector caspases 3 and 7 was measured in rAAV-2 treated cells (Figure 6). The ‘Apo-ONE Homogeneous Caspase-3/7 Assay’ revealed that AAV-2_DARPin and AAV-2_Affibody efficiently induce apoptosis in A431 target cells while caspase activity in off-target HeLa cells remained at background levels. The combination of the AAV-2_DARPin together with the cytosine deaminase showed the most potent apoptosis induction in A431 cells, with comparable efficacies for 500 μM and 250 μM 5-FC (Figure 6c, approx. 12 times more caspase activity relative to AAV2ΔHSPG), followed by AAV-2_DARPin particles armed with the mouse guanylate kinase – herpes simplex virus thymidine kinase (mGMK-TK30) (Figure 6d, approx. 8 times more caspase activity relative to AAV2ΔHSPG). Apoptosis induction through Affibody-guided vectors showed a comparable potency to DARPin guided vectors at 500 μM prodrug concentrations, but at 250 μM 5-FC or GCV AAV-2_DARPin superseded AAV-2_Affibody by a factor of 2.5 (Figure 6a, c) and 5 (Figure 6b, d) for 5-FC and GCV, respectively.
Figure 6. Apoptosis induction in A431 relative to HeLa cells.

2 × 103 A431 or HeLa cells, respectively, were transduced with 3 × 108 genomic viral particles on day one. On day three, the culture medium was exchanged and prodrug solutions were added. On day six, caspase-3 and caspase-7 activity was measured using the Apo-ONE Homogeneous Caspase-3/7 Assay (Promega). Samples were transferred to a 96 well microplate, and fluorescence was measured using a microplate reader. The caspase activity ratio of A431 to HeLa cells for each prodrug concentration is given relative to the respective AAV-2ΔHSPG ratio (n = 3, mean ± SD). As labeled, panels (a) to (d) show experiments with combinations of Affibody or DARPin and CD (5-FC) or mGMK-TK30 (ganciclovir), respectively. Note that the scale in panel c differs from the other panels.
Apoptosis induction and targeting specificity in mixed cell culture
In a final step, we evaluated the ability of AAV-2_Affibody and AAV-2_DARPin for differential targeting. EGFR-overexpressing A431 cells were either combined with HeLa or MCF7 cells in a mixed cell culture. In cells undergoing early apoptosis, Annexin V is translocated from the inner side of the plasma membrane to the outer layer and thus becomes surface exposed42. Hence staining with an anti-Annexin V antibody reveals cells in an early apoptotic state. Since experiments showed that A431 cells undergoing apoptosis lose the majority of their EGF receptors (Figure 7, upper right panel, A431 untreated 84% EGFR positive) camptothecin induced apoptotic A431 cells were included as control (Figure 7, lower right panel, A431 + camptothecin 27% EGFR positive). As a consequence, cells were simultaneously infected and treated with 250 μM 5-FC, and flow cytometry analysis was carried out 48 h later. Targeted A431 cells and off-target HeLa or MCF7 cells were mixed in a 1:4 ratio. Flow cytometry analysis with simultaneous staining for EGFR expression and Annexin V revealed specific apoptosis induction in EGF receptor overexpressing A431 cells with comparable efficacies for AAV2-Affibody and AAV-2-DARPin (Figure 7, left panels). Control AAV-2ΔHSPG particles did not mediate prodrug activated apoptosis (Figure 7, middle panels), showing almost the same staining pattern as cells which were not treated with any of the viral particles (Figure 7, middle panel, untreated cells). Taken into account that A431 cells undergoing apoptosis lose most of their EGF receptors and thus stain as apoptotic but EGFR negative, these results demonstrate that retargeted AAV-2_Affibody and AAV-2_DARPin viral particles discriminate between cells depending on EGFR expression and primarily induce EGFR dependent apoptosis. This justifies to further develop the concept in animal models and predict future applications in patients.
Figure 7. Effect of rAAV-2 infection on apoptosis induction in mixed cell cultures.

20 × 103 A431 cells were mixed with 80 × 103 HeLa cells or 80 × 103 MCF7 cells, and the mixture was transduced with either His-tag affinity purified AAV-2_Affibody, AAV-2_DARPin or AAV-2ΔHSPG capsids delivering the cytosine deaminase. Cells only served as a negative control and A431 cells incubated with 20 μM camptothecin for 4 h served as a positive control for apoptotic cells. After addition of the viral particles, cells were incubated in DMEM containing 250 μM 5-FC. 48 h later, cells were detached, washed and incubated with an anti-EGFR-Alexa488 antibody on ice for 1 h. An anti-Annexin-V-PE antibody was added and cells were analyzed by flow cytometry. For every sample 30,000 events were recorded. Dead cells were excluded using initial gating based on forward scatter height versus sideward scatter height. The anti-Annexin-V fluorescence is plotted against the EGFR fluorescence and the percentages of cells in each quadrant are given below each plot. Experiments show that EGFR expression of A431 cells decreases upon apoptosis (right panel). rAAV without targeting does not mediate prodrug activated apoptosis (middle panels). Targeted AAV primarily induces EGFR dependent apoptosis, but a bystander effect might harm nearby cells.
Discussion
In recent years, adverse reactions in patients treated with prodrug strategies had been reduced significantly and currently approximately 10% of worldwide marketed drugs can be classified as prodrugs, albeit almost all of them are activated by endogenous enzymes43. By implementing a two-step process – systemic application of a non-toxic, non-bioactive prodrug and its conversion to a pharmacologically active agent delivered on-site of a therapeutically relevant target – systemic side effects can be minimized. In cancer chemotherapy, most classical drugs target rapidly proliferating cells, thus not only affecting neoplastic cells, but also harming natural regenerative processes. At present, there are two major drawbacks of using viruses as gene shuttles for prodrug converting enzymes: First, the limited knowledge on how to specifically retarget the viral vectors towards cancer cells, and second, the limited knowledge to make the right choice for the enzyme prodrug combination. In addition, rapid prototyping is hindered by laborious cloning steps, which is key to the advance of disease-specific and personalized medicine.
Based on the AAV-2 scaffold, we present two modularly assembled recombinant viral vectors which present diminished natural tropism and rationally designed retargeting via general binding scaffolds towards cancer cell surface markers. These viral particles deliver genes coding for prodrug converting enzymes, which again can easily be swapped to test other enzyme prodrug pairs. In contrast to systems reported by others, our vector design and fast two-step purification process assures specific purification only of targeted viral particles (Figure 1c, 3b, 3c).
As cancer cell surface marker, we chose the epidermal growth factor receptor 1 also named ErbB1. This marker is a clinically validated prototype receptor belonging to the family of receptor tyrosine kinases vastly overexpressed in several different types of cancer27,28,29. The EGF receptor is already exploited for cancer therapy with small molecule inhibitors such as gefitinib (trade name Iressa)44 or erlotinib (trade name Tarceva) and antibodies such as cetuximab (IMC225, trade name Erbitux) in the case of colon as well as squamous cell carcinoma of the head and neck45. For targeting, we tested the small and robustly folding DARPin_E01 and the Affibody_ZEGFR:1907, which both have reported EGFR binding affinities in the nanomolar range and originate either from the ankyrin proteins or from the Z-domain scaffold of the staphylococcal protein A. Although monoclonal antibodies have already been used as targeting devices for either the adeno-associated virus in an adapter mediated way46 or for the adenovirus in a covalently bound way47, both the DARPin and the Affibody outperform them in terms of size, aggregation tendency, lack of disulfide bonds and glycosylation. Expressed as fusions to the viral coat protein VP2, both were readily displayed on the viral surface (Figure 3b) and were available for receptor target binding (Figure 4). Additionally we armed the viral particles with genes coding for two different prodrug converting enzymes: the cytosine deaminase (CD) and the mouse guanylate kinase – herpes simplex virus thymidine kinase (mGMK-TK30) with the latter being a fusion enzyme manufactured to overcome a typical bottleneck in ganciclovir activation48. Following specific transduction by our guided rAAV-2 particles EGFR-overexpressing A431 cells, but not HeLa or MCF7 control cells, underwent apoptosis upon addition of the respective prodrug (Figure 6 and 7). Taken together AAV-2_Affibody, as well as AAV-2_DARPin both with diminished natural tropism and armed with prodrug converting enzymes were able to specifically transduce target but not control cells in mono-, as well as in mixed cell culture. In combination with a recent study from Münch et al.14 who used a DARPin specific for Her2/neu for rAAV mediated delivery of TK in an artificial cell system with HER2/neu stable expressing CHO cells and a tumor-cell xenograft model, we corroborate the versatility of the rAAV approach. In line with demands to avoid animal experiments, we demonstrate that essential data for targeting and prodrug activation can be obtained in cell culture. However, we also acknowledge that clinicians demand animal testing before clinical testing can be initiated. Münch and coworkers observed residual off-targeting assuming unmodified viral particles in the vector stock as a reason. Using our all-in-one approach, which couples a loop exposed His-tag with the presentation of the targeting moiety in one protein, this challenge can be overcome. Following this strategy, modified rAAV-2 particles can specifically and rapidly be purified from serum-free cell culture supernatant with a single step centrifugal ultrafiltration followed by an easy to perform affinity chromatography. By doing so, recombinant vector stocks can not only be depleted from unmodified capsids but at the same time the need for expensive and time consuming density-gradient ultracentrifugation becomes dispensable.
Being the seventh largest entity in gene therapy clinical trials in 2012, adeno-associated vectors are increasingly becoming the vector of choice in gene-directed enzyme prodrug therapy (GDEPT) or more generally in gene therapy49. The recommendation of an rAAV based therapy by the European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP) in 2012 is a hallmark6 underlining their importance. This raises a strong need for standardized sharing of pharmacodynamic, pharmacokinetic, toxicologic, and efficacy data from non-clinical and clinical studies performed by laboratories using different vector – transgene combinations. These efforts are mostly driven by the international AAV-2 Reference Standard Working Group (AAV2RSWG) founded in 200850. We believe that it would be of great advantage to not only unify comparison methods on the protein level but to also achieve standardization on the genetic level. Based on the iGEM BioBrick standard21 which has evolved to the de facto standard for international interchange of genetic elements among young research teams in the field of synthetic biology, we here managed to utilize a standardized set of genetic parts to assemble a true AAV-2 based targeting vector. These so-called BioBricks build up an AAV-2 construction kit for virus-directed enzyme prodrug therapy. It comprises coding and non-coding genetic elements for rAAV-2 vector production, including two different high-specificity EGFR targeting molecules which are expressed as fusions to the viral coat protein VP2. Additionally, we included two genes coding for prodrug convertases which – when packaged into the targeting vectors – will be specifically expressed inside targeted cancer cells and thus facilitate their cell death by conversion of non-toxic prodrugs into toxic compounds.
Taken together, we present here the first example of a standardized, recombinant and easy to purify AAV-2 based viral targeting vector which addresses every demand of a virus-directed enzyme prodrug therapy by combining N-terminal capsid fusion with loop modification employing two modern targeting moieties and two enzyme—prodrug combinations. Using this comprehensive approach might not only help fighting various devastating types of cancer characterized by EGFR overexpression, but also build up a basis for targeting a plethora of other cell surface markers through standardized exchange of the guiding molecules.
Methods
Generation of plasmids
All plasmids used, except pHelper (Stratagene) share the same pSB1C3 backbone which was derived from the iGEM pSB1C3 backbone51 and were assembled using iGEM standard BioBrick assembly19. The rep/cap insert in pSB1C3_001-RepCapΔHSPG-VP2-KO was derived from pAAV-RC (Stratagene). The exchange region around position aa 587 harbors two arginine residues (Arg585, Arg588) which are largely responsible for the natural HSPG-tropism of AAV-2 capsids32. The viral-brick - a short synthetic gene construct - containing a (His)6 tag inserted at this position also contained arginine to alanine mutations which largely abolish the binding of rAAV-2 capsids to the primary AAV-2 receptor on the cell surface12. Targeting plasmids pSB1C3_001VP2ΔHSPG587His-DARPin-Flag and pSB1C3_001VP2ΔHSPG587His-Affibody-Flag are derivatives of the Rep/Cap plasmid, harboring the VP2 open reading frame under control of a CMV promoter. DARPin_E01 and Affibody_ZEGFR:1907 were ordered as synthetic gene constructs flanked by iGEM pre- and suffixes (GeneArt, Life technologies). Vector plasmids pSB1C3-CD and pSB1C3-mGMK-TK30 harbor the AAV-2 left and right inverted terminal repeats (ITRs, from pAAV-MCS, Stratagene), a CMV promoter, a human beta-globin intron, the cytosine-deaminase52 (CD) or the fused genes of mouse guanylate kinase (mGMK) and a herpes simplex virus thymidine kinase mutant (TK30)48, as well as the human growth hormone (hGH) polyA signal. The mGMK-TK30 was kindly provided by M. E. Black (Washington State University, USA). The CD gene was amplified from E. coli. XL1 blue genome using primers CD_prefix 5′-CGTCTAGATGGCCGGCGTGTCGAATAACGCTTTACAAACAATTATTAAC-3′ and CD_suffix 5′-CCAGAAGCCATCGATTACAAACGTACCGGTCTG-3′. Details on the modularization and cloning strategy will be published elsewhere (manuscript in preparation).
Viral particle production and purification
Production of recombinant adeno-associated virus serotype 2 particles (rAAV-2) was generally based on the adenovirus-helper free AAV-packaging strategy7. Viral like particles/targeting vectors were generated/produced by transfection of 4 × 106 HEK-293 cells cultured in seven 10 cm cell culture dishes with four plasmids at a total DNA amount of 15 μg per dish using linear polyethylenimine (25 kDa, Polysciences Inc) as described previously53. Targeting plasmids (pSB1C3_001ΔHSPG-VP2587His-DARPin, pSB1C3_001ΔHSPG-VP2587His-Affibody), RepCap (pSB1C3-RepCap_001ΔHSPG-VP2-KO), vector plasmids (pSB1C3-CD, pSB1C3-mGMK-TK30) and the helper plasmid (pHelper, Stratagene) were transfected in a molar 4:1:5:5 ratio. For the generation of viral vector controls, a triple transfection with pSB1C3_001-RepCapΔHSPG, pHelper and the respective vector plasmid was carried out. 24 h after transfection, the growth medium was exchanged for FreeStyle 293 Expression Medium (Gibco) to allow serum-free production of rAAV-2 particles. Four days post transfection, the supernatants were harvested and twice cleared from cell debris by centrifugation (700 × g, 10 min; 10,000 × g, 10 min). rAAV-2 particles within the supernatant were concentrated and purified using Vivaspin 20 centrifugal concentrators (1 MDa MWCO, Sartorius) and PBS, making use of the high molecular weight cut-off membrane, which allows most contaminating molecules and proteins to pass through, but holds back rAAV-2 particles.
His-tag affinity purification
Modified viral particles were concentrated and clarified as described above, and purified by immobilized metal-ion affinity chromatography (IMAC). For batch binding to the matrix, 5 ml samples were incubated with 500 μl Ni-NTA agarose resin (Qiagen) in binding buffer (PBS, 20 mM imidazole) at 80 rpm for 2 h at 4°C on an orbital shaker. The slurry was transferred to a gravity-flow column and washed with 5 ml PBS containing 50 mM imidazole. Modified viral particles were eluted in 5 ml elution buffer (PBS, 500 mM imidazole) and then concentrated to 2 ml. Buffer was exchanged approx. 105-fold in five serial dilution and concentration steps with PBS using Vivaspin 20 centrifugal concentrators (1 MDa MWCO, Sartorius).
Determination of capsid, genomic and infectious titers
Capsid titers were determined using the AAV Titration ELISA (Progen) according to the manufacturer's instructions. Genomic titers of rAAV-2 particles were determined using quantitative realtime PCR (qPCR) as described previously54. Briefly, an aliquot of purified viral vectors was supplemented with 5 U DNase I and DNase I buffer (Fermentas), incubated for 3 h at 37°C, heat inactivated for 10 min at 65°C, and finally diluted 100-fold. 5 μl of these DNase I resistant particles containing the viral DNA supplemented with 7.5 μl Quantifast SybrGreen MasterMix (Qiagen) and CMV_forward/reverse primers55 (10 μM each) were used as template in a qPCR reaction on a RotorGene cycler (Qiagen). Viral genome titers were calculated from a standard curve of 13–13 × 106 copies of a CMV promoter-containing plasmid. Infectious titers were determined as described by Rohr et. al.55, but using the Quantifast SybrGreen qPCR Kit (Qiagen) on a RotorGene qPCR cycler. Briefly, 104 cells per well in a 24 well cell culture plate were seeded and incubated for 24 h with equal genomic titers of the viral particles. Subsequently, the cells were harvested, digested with proteinase K (Sigma) and residual single-stranded DNA was removed using S1 nuclease (Fermentas). Double-stranded DNA which originated from infectious particles harboring viral ssDNA56 was then quantified using qPCR.
ELISA
Surface display and accessibility of the two targeting molecules DARPin_E01 and Affibody_ZEGFR:1907 were validated by sandwich ELISA. MaxiSorp 96-well plates (Nunc) were coated with an anti-Flag antibody (F1804, Sigma; 1:500 in PBS) for 1.5 h and afterwards blocked with 1 × RotiBlock/PBS (Roth) for 1 h. 1.8 × 109 viral particles were applied and incubated for 1.5 h. Captured and fully assembled viral capsids were detected using the AAV-2-specific antibody A2038 (Progen, 1:1,000 dilution), a biotin-coupled isotype specific IgG3 antibody (SouthernBiotech, 1:2,000 dilution) and self-produced streptavidin-coupled horseradish peroxidase. All antibody dilutions were made in dilution buffer (1 × RotiBlock/PBS, 0.05% Tween 20 (v/v)). Following every incubation step, the plate was washed three times with dilution buffer. Last, 0.5 μl of peroxidase substrate o-Phenylenediamine (1 mM, Sigma) in substrate buffer (50 mM citric acid, 100 mM disodium hydrogen phosphate, 0.01% H2O2, pH 5.5) was added per well. The reaction was stopped after 15 min with 1 M sulfuric acid and 50 mM sodium sulfite and quantified via absorption at 492 nm using a microplate reader (Tecan Sunrise).
Removal of unassembled capsid proteins via ultrafiltration devices was analyzed by ELISA. A 96 well MaxiSorp plate (Nunc) was coated overnight at 4°C with equal total protein amounts (as determined with the NanoOrange Protein Quantitation Kit (Life Technologies)) of AAV-2 producer cell supernatant (from the day of viral particle harvest) and filtrate, as well as concentrated samples taken from the ultrafiltration device. Fresh cell culture medium served as control. Wells were blocked as described above and incubated with A2038 antibody (Progen, 1:1,000 dilution), or B1 antibody (1:10 dilution). Plates were washed three times with dilution buffer and assembled capsids or individual capsid proteins were detected and visualized using an HRP-coupled anti-mouse antibody (sc-2005, SantaCruz, dilution 1:3,000) with peroxidase substrate as described above.
In order to assess the successful removal of unmodified viral vectors via Ni-NTA purification, a 96 well MaxiSorp plate (Nunc) was coated with an anti-His antibody for 1.5 h (HIS-1, Sigma, dilution 1:500 in PBS), followed by blocking with RotiBlock for 1 h. Equal amounts of Ni-NTA-purified and non-purified particles as determined by measurement of genomic titers were applied and incubated for 1.5 h. The subsequent detection and visualization of assembled capsids were done as described above.
Cell culture
HEK-293, HeLa, and A431 (ATCC) cells were cultured in DMEM supplemented with stable glutamine, sodium pyruvate, 10% fetal calf serum, 1% penicillin/streptomycin (v/v), 4.5 g/l glucose (1.5 g/l for MCF7 cells) to a maximum confluence of 80% and then split 1:10 (HEK-293, MCF7, A431), or 1:50 (HeLa), respectively. All cell culture media and chemicals where purchased from PAA.
Prodrug solutions containing Ganciclovir (GCV, Sigma) or 5-fluorocytosine (5-FC, Sigma) were prepared as ten-fold concentrates (2 mM and 5 mM) in PBS followed by sterile filtration.
Western blots
Viral particles were separated on a 7.5%–15% SDS gel and blotted onto a PVDF membrane. After blocking with 5% (w/v) non-fat dry milk in TBST (TBS with 0.05% (v/v) Tween) for 1 h at room temperature, the membrane was either incubated with the B1 (1:50 dilution) or with anti-Flag antibody (Sigma F1804, 1:1000) diluted in TBST-milk (TBST with 3% (w/v) milk powder). Between and after both incubation steps the membranes were washed three times with TBST-milk for 5 min at room temperature. Detection was performed using a goat anti-rabbit antibody coupled to horseradish peroxidase (Santa Cruz; 1:5,000 dilution in TBST-milk) and Pico ECL Western Blotting Substrate (Pierce).
Cell-binding assay
Binding of viral targeting vectors to the surface of A431 epidermoid carcinoma cells overexpressing EGFR57, was analyzed by whole cell ELISA. Cells were harvested, washed twice with PBS and transferred into a 96 well MaxiSorp plate (Nunc) at a concentration of 2,000 cells per well and spun down at 250 × g. The supernatant was removed and the plate dried for 4 h at room temperature. HeLa cells were used as control. Unspecific binding was blocked using 0.8% BSA/PBS for 1 h. Subsequently, cells were incubated with 1.8 × 109 viral particles for 2 h. Unbound capsids were removed via washing twice with PBS/0.05% Tween 20 (v/v). Intact rAVVs were detected as described under ELISA using a HRP-coupled anti-mouse antibody (sc-2005, SantaCruz, dilution 1:3,000) to detect bound A20 antibodies.
Caspase 3/7 assay
In order to assess apoptosis induction, 2,000 A431 or HeLa cells, respectively, were seeded per well of a 96 well culture plate and transduced with 3 × 108 genomic viral particles on day one. On day three, the culture medium was exchanged and prodrug solutions were added to cells. On day six, caspase-3 and caspase-7 activity was measured using the Apo-ONE Homogeneous Caspase-3/7 Assay (Promega) according to the manufacturer's instructions. Briefly, Apo-ONE buffer, supplemented with the Z-DEVD-R110 caspase substrate was added to cell culture medium in a 1:1 ratio. Plates were incubated at room temperature on an ELISA shaker for 4 h. During incubation, active caspase-3, or -7 cleaves the DEVD peptide from the pro-fluorescent substrate, releasing fluorescent rhodamine 110. Samples were transferred into a black 96 well microplate (Nunc) and fluorescence was measured using a microplate reader (Thermo Scientific Varioscan Flash, excitation wavelength 499 nm, emission wavelength 521 nm).
Flow cytometry
Specificity of rAAV-2 infection and apoptosis induction was examined in mixed cell cultures. A431 and HeLa, or A431 and MCF7 cells, respectively, were mixed in a 1:4 ratio and transduced with either AAV-2_DARPin or AAV-2_Affibody. AAV-2ΔHSPG capsids were used as controls. Additionally, A431 cells incubated with 20 μM camptothecin (Sigma) for 4 h served as a positive control for apoptotic cells. All viral capsids were purified as described under His-tag affinity purification. After addition of the viral particles, cells were immediately incubated in DMEM containing 250 μM 5-FC. 48 h later cells were detached using cell scrapers and washed twice in Annexin V binding buffer (BD Biosciences, additional 1% BSA). Subsequently, cells were incubated with an anti-EGFR-Alexa488 antibody (Cell Signaling, 1:20 dilution) on ice for 1 h. After 45 min an Annexin-5-PE antibody was added (Promega, 1:100 dilution). Finally cells were washed once in Annexin V binding buffer and cells were analyzed by flow cytometry (FACS Calibur, Becton Dickinson) using CellQuest software provided by Becton Dickinson. For every sample 30,000 events were recorded. Dead cells were excluded using initial gating with forward scatter height versus sideward scatter height.
MTT assay
In order to access cell viability, 10,000 A431 or HeLa cells, respectively, were seeded per well in a 96-well cell-culture plate and transduced with 3 × 108 genomic viral particles 24 h later. After 48 h, fresh medium supplemented with 5-FC was added to the cells and incubated for three additional days. On day six, cells were incubated in 100 μl fresh DMEM including 25 μl MTT solution (3.65 mg/ml MTT in PBS, Sigma) for 3 h. Subsequently, the culture medium was removed and the reduced formazan was solubilized for 30 min on an ELISA shaker using 200 μl DMSO (Sigma) and 25 μl Sörensen buffer (0.1 M Glycine, 0.1 M NaCl, in H2O, pH 10.5). Absorbance at 570 nm was measured on a Tecan Sunrise microplate reader.
Author Contributions
S.H., T.B., H.J.W., A.F., B.K., V.M., S.B., P.S. and K.M.M. conceived the idea. T.B., H.W., A.F., B.K., V.M., S.B., P.S. cloned constructs and conducted initial experiments. S.H. designed and conducted final experiments and wrote the manuscript with the help of K.M.M., K.M.A. provided scientific input, K.M.M. supervised the project.
Acknowledgments
This study was supported by the Excellence Initiative of the German Federal and State Governments (EXC 294, BIOSS) and by the DFG Priority Program SPP1170. The authors especially want to thank PD Dr. Jürgen Kleinschmidt (DKFZ, Heidelberg, Germany) for providing the A20 and B1 antibodies as well as helpful comments on the project. They are grateful to Prof. Dr. Margaret E. Black (Washington State University, USA) for providing the coding sequence for the mGMK-TK30. Additionally the authors want to thank Dr. Pamela Holzlöhner and Martin Listek (Universität Potsdam, Germany) for help with the flow cytometry experiments, as well as Amor Hajri (Universität Freiburg, Germany), Sebastian Hanke, Franziska Matyssek (Universität Potsdam, Germany) for technical assistance and the members of the Freiburg iGEM team 2010 Achim Mall, Anna Oschowitzer, Kerstin Klingele, Anissa Bender, Jessica Günzle, Kira Meyerovich, and Christian Weingärtner for clonig and testing of initial constructs.
References
- Springer C. J. & Niculescu-Duvaz I. Gene-directed enzyme prodrug therapy (GDEPT): choice of prodrugs. Adv. Drug Deliv. Rev. 22, 351–364 (1996). [DOI] [PubMed] [Google Scholar]
- Bhatia S. et al. Innovative Approaches for Enhancing Cancer Gene Therapy. Discov. Med. 15, 309–317 (2013). [PubMed] [Google Scholar]
- Erbs P. et al. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 60, 3813–22 (2000). [PubMed] [Google Scholar]
- Maguire A. M. et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N. Engl. J. Med. 358, 2240–8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonelli F. et al. Gene therapy for Leber's congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol. Ther. 18, 643–50 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will the floodgates open for gene therapy? Nat. Biotechnol 30, 805 (2012). [DOI] [PubMed] [Google Scholar]
- Xiao X., Li J. & Samulski R. J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 72, 2224–32 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali R. R. et al. Gene transfer into the mouse retina mediated by an adeno-associated viral vector. Hum. Mol. Genet. 5, 591–4 (1996). [DOI] [PubMed] [Google Scholar]
- During M. J. et al. Peroral gene therapy of lactose intolerance using an adeno-associated virus vector. Nat. Med. 4, 1131–5 (1998). [DOI] [PubMed] [Google Scholar]
- Kessler P. D. et al. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc. Natl. Acad. Sci. U. S. A. 93, 14082–7 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberl D. D., Alexander I. E., Halbert C. L., Russell D. W. & Miller A. D. Persistent expression of human clotting factor IX from mouse liver after intravenous injection of adeno-associated virus vectors. Proc. Natl. Acad. Sci. U. S. A. 94, 1426–31 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucas J. et al. Engineering adeno-associated virus serotype 2-based targeting vectors using a new insertion site-position 453-and single point mutations. J. Gene Med. 11, 1103–13 (2009). [DOI] [PubMed] [Google Scholar]
- Grimm D. & Kay M. a. From virus evolution to vector revolution: use of naturally occurring serotypes of adeno-associated virus (AAV) as novel vectors for human gene therapy. Curr. Gene Ther. 3, 281–304 (2003). [DOI] [PubMed] [Google Scholar]
- Münch R. C. et al. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. 21, 109–18 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waehler R., Russell S. J. & Curiel D. T. Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 8, 573–87 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coura R. D. S. & Nardi N. B. The state of the art of adeno-associated virus-based vectors in gene therapy. Virol. J. 4, 99 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ried M. U., Girod A., Leike K., Büning H. & Hallek M. Adeno-associated virus capsids displaying immunoglobulin-binding domains permit antibody-mediated vector retargeting to specific cell surface receptors. J. Virol. 76, 4559–66 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G. et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol. 78, 6381–8 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa T. et al. Cytotoxicity of adenoviral-mediated cytosine deaminase plus 5-fluorocytosine gene therapy is superior to thymidine kinase plus acyclovir in a human renal cell carcinoma model. J. Urol. 162, 949–54 (1999). [DOI] [PubMed] [Google Scholar]
- Deng L.-Y., Wang J.-P., Gui Z.-F. & Shen L.-Z. Antitumor activity of mutant bacterial cytosine deaminase gene for colon cancer. World J. Gastroenterol. 17, 2958–64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty R. P., Endy D. & Knight T. F. Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2, 5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman M. et al. Directed evolution to low nanomolar affinity of a tumor-targeting epidermal growth factor receptor-binding affibody molecule. J. Mol. Biol. 376, 1388–402 (2008). [DOI] [PubMed] [Google Scholar]
- Steiner D., Forrer P. & Plückthun A. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J. Mol. Biol. 382, 1211–27 (2008). [DOI] [PubMed] [Google Scholar]
- Bork P. Hundreds of ankyrin-like repeats in functionally diverse proteins: mobile modules that cross phyla horizontally? Proteins 17, 363–74 (1993). [DOI] [PubMed] [Google Scholar]
- Stumpp M. T., Binz H. K. & Amstutz P. DARPins: a new generation of protein therapeutics. Drug Discov. Today 13, 695–701 (2008). [DOI] [PubMed] [Google Scholar]
- Nygren P.-A. Alternative binding proteins: affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 275, 2668–76 (2008). [DOI] [PubMed] [Google Scholar]
- Walker R. a. & Dearing S. J. Expression of epidermal growth factor receptor mRNA and protein in primary breast carcinomas. Breast Cancer Res. Treat. 53, 167–76 (1999). [DOI] [PubMed] [Google Scholar]
- Hirsch F. R. et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol. 21, 3798–807 (2003). [DOI] [PubMed] [Google Scholar]
- Colquhoun a. J. & Mellon J. K. Epidermal growth factor receptor and bladder cancer. Postgrad. Med. J. 78, 584–9 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löfblom J. et al. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 584, 2670–80 (2010). [DOI] [PubMed] [Google Scholar]
- Zahnd C. et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res. 70, 1595–605 (2010). [DOI] [PubMed] [Google Scholar]
- Summerford C. & Samulski R. J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 72, 1438–45 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiburg iGEM team 2010. Tumor Targeting by N-terminal Fusion to Virus Capsid. igem.org. 2013-12-17. URL:http://2010.igem.org/wiki/index.php?title=Team:Freiburg_Bioware/Project/Targeting_Fusion&oldid=208826. Accessed: 2013-12-17. (Archived by WebCite® at http://www.webcitation.org/6Lw2xwyyD).
- Grieger J. C., Johnson J. S., Gurda-Whitaker B., Agbandje-McKenna M. & Samulski R. J. Surface-exposed adeno-associated virus Vp1-NLS capsid fusion protein rescues infectivity of noninfectious wild-type Vp2/Vp3 and Vp3-only capsids but not that of fivefold pore mutant virions. J. Virol. 81, 7833–43 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux K. et al. Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J. Virol. 79, 11776–87 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S. J. et al. Targeted gene delivery to vascular tissue in vivo by tropism-modified adeno-associated virus vectors. Circulation 109, 513–9 (2004). [DOI] [PubMed] [Google Scholar]
- Girod a. et al. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 5, 1438 (1999). [DOI] [PubMed] [Google Scholar]
- Wobus C. E. et al. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 74, 9281–93 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington K. H. et al. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J. Virol. 78, 6595–609 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. H., Richert N., Ito S., Merlino G. T. & Pastan I. Characterization of epidermal growth factor receptor gene expression in malignant and normal human cell lines. Proc. Natl. Acad. Sci. U. S. A. 81, 7308–12 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath D., Rao V. A. & Plunkett W. Mechanisms of apoptosis induction by nucleoside analogs. Oncogene 22, 9063–74 (2003). [DOI] [PubMed] [Google Scholar]
- Vermes I., Haanen C., Steffens-Nakken H. & Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 184, 39–51 (1995). [DOI] [PubMed] [Google Scholar]
- Zawilska J. B., Wojcieszak J. & Olejniczak A. B. Prodrugs: A challenge for the drug development. Pharmacol. Rep. 65, 1–14 (2013). [DOI] [PubMed] [Google Scholar]
- Paez J. G. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–500 (2004). [DOI] [PubMed] [Google Scholar]
- Perez R., Crombet T., de Leon J. & Moreno E. A view on EGFR-targeted therapies from the oncogene-addiction perspective. Front. Pharmacol. 4, 53 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigout L. et al. Altering AAV tropism with mosaic viral capsids. Mol. Ther. 11, 856–65 (2005). [DOI] [PubMed] [Google Scholar]
- Morrison J. et al. Cetuximab retargeting of adenovirus via the epidermal growth factor receptor for treatment of intraperitoneal ovarian cancer. Hum. Gene Ther. 20, 239–51 (2009). [DOI] [PubMed] [Google Scholar]
- Ardiani a., Sanchez-Bonilla M. & Black M. E. Fusion enzymes containing HSV-1 thymidine kinase mutants and guanylate kinase enhance prodrug sensitivity in vitro and in vivo. Cancer Gene Ther. 17, 86–96 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginn S. L., Alexander I. E., Edelstein M. L., Abedi M. R. & Wixon J. Gene therapy clinical trials worldwide to 2012 - an update. J. Gene Med. 15, 65–77 (2013). [DOI] [PubMed] [Google Scholar]
- Moullier P. & Snyder R. O. International efforts for recombinant adeno-associated viral vector reference standards. Mol. Ther. 16, 1185–8 (2008). [DOI] [PubMed] [Google Scholar]
- Che Part:pSB1C3. Registry of Standard Biological Parts. 2013-12-17. URL:http://parts.igem.org/Part:pSB1C3?title=Part:pSB1C3. Accessed: 2013-12-17. (Archived by WebCite® at http://www.webcitation.org/6Lw3Vr2iw).
- Mullen C. a., Kilstrup M. & Blaese R. M. Transfer of the bacterial gene for cytosine deaminase to mammalian cells confers lethal sensitivity to 5-fluorocytosine: a negative selection system. Proc. Natl. Acad. Sci. U. S. A. 89, 33–7 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt C. et al. Polyethylenimine, a cost-effective transfection reagent. Signal Transduct. 6, 179–184 (2006). [Google Scholar]
- Rohr U.-P. et al. Fast and reliable titration of recombinant adeno-associated virus type-2 using quantitative real-time PCR. J. Virol. Methods 106, 81–8 (2002). [DOI] [PubMed] [Google Scholar]
- Rohr U.-P. et al. Quantitative real-time PCR for titration of infectious recombinant AAV-2 particles. J. Virol. Methods 127, 40–5 (2005). [DOI] [PubMed] [Google Scholar]
- Nakai H., Storm T. A. & Kay M. A. Recruitment of single-stranded recombinant adeno-associated virus vector genomes and intermolecular recombination are responsible for stable transduction of liver in vivo. J. Virol. 74, 9451–63 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N., Kondo I., Gamou S., Behzadian M. a. & Shimizu Y. Genetic analysis of hyperproduction of epidermal growth factor receptors in human epidermoid carcinoma A431 cells. Somat. Cell Mol. Genet. 10, 45–53 (1984). [DOI] [PubMed] [Google Scholar]