

Abstract
We report a stochastic nanopore sensing method for the detection of Cu2+ ions. By employing a polyhistidine molecule as a chelating agent, and based on the different signatures of the events produced by the translocation of the chelating agent through an α-hemolysin pore in the absence and presence of target analytes, trace amounts of copper ions could be detected with a detection limit of 40 nM. Importantly, although Co2+, Ni2+, and Zn2+ also interacts with the polyhistidine molecule, since the event residence times and/or blockage amplitudes for these metal chelates are significantly different from those of copper chelates, these metal ions do not interfere with Cu2+ detection. This chelating reaction approach should find useful application in the development of nanopore sensors for other metal ions.
Keywords: nanopore sensing, copper, polyhistidine, chelation, biosensor
1. Introduction
Nanopore stochastic sensors have emerged as a label-free and amplification-free technique for various applications, including biosensing (Gao et al., 2009), studying covalent and non-covalent bonding interactions (Luchian et al., 2003; Zhao et al., 2008), investigating biomolecular folding and unfolding (Shim et al., 2009; Talaga et al., 2009), probing enzyme kinetics, sequencing DNA molecules (Kasianowicz et al., 1996; Meller et al., 2000; Howorka et al., 2001; Storm et al., 2005; Sigalov et al., 2008; Jovanovic-Talisman et al., 2009; Derrington et al., 2010), and so on. However, one major hurdle still needs to be overcome before nanopore sensors could be deployed for routine use in the analysis of real-world samples. Thus far, various reported applications of the nanopores were mainly achieved by using protein ion channels. The high sensitivity and selectivity (i.e., molecular recognition) of these biological nanopores are accomplished by modifying the nanopore interior to introduce a variety of new functions (i.e., binding sites) at specific positions (Braha et al., 1997; Braha et al., 2000; Movileanu et al., 2000; Cheley et al., 2002; Guan et al., 2005). However, the protein pore sensors developed by this approach can only be used for sensing a particular substance. To detect different analytes, different nanopores having different binding sites need to be produced, which is time-consuming and inconvenient. Furthermore, the protein pore-based stochastic sensing is not suitable as a deployable tool for extended usage due to the fragility of the biological membrane used in such a sensing platform. It should be noted that development of synthetic nanopores in robust solid-state substrates is currently under intense investigation. Although these stable artificial nanopores are ideal platforms as portable/fieldable stochastic sensors, the current synthetic nanopore technology provides a poor resolution and selectivity due to the difficulty in imparting these nanopores with inner surface functionalities. Therefore, to advance nanopore sensing technology for field-deployable applications, there is a need for the development of other sensor design methodologies, which are fundamentally different from those used in the conventional nanopore sensing.
Recently, highly sensitive and selective nanopore sensors for the detection of Hg2+ were successfully developed, where molecular recognition of the sensors was achieved by using thymine-containing DNA hairpins and based on the interaction between Hg2+ and DNA (Wen et al., 2011; Wang et al., 2013). Nanopore analysis of metal-biomolecule interactions has also been investigated by other researchers (Mereuta et al., 2012; Stefureac et al., 2008; Stefureac et al., 2010). They found that the event signatures of peptides and proteins were significantly different in the absence and presence of metal ions, which may be attributed to the conformational change of the biomolecules induced by the metal ion – biomolecule interaction (Stefureac et al., 2010). In this work, we will demonstrate that a polyhistidine molecule can be utilized as a probe to detect Cu2+ ions based on the chelating reaction between them, providing further evidence that sensitive and selective nanopore sensors could be developed without constructing a binding site in the nanopore interior. It should be noted that copper is an essential trace element that is vital to the human health (Gaggelli et al., 2006). Overload or deficiency of copper is associated with diseases of genetic origin such as Wilson disease (WD) and Menkes disease (MD), respectively. Further, elevated levels of copper have been found in many types of human cancers although mechanisms underlying this phenomenon is not entirely elucidated (Tisato et al., 2010).
2. Materials and methods
2.1. Chemicals and Reagents
Peptide H6 (Hexa-His, Sequence: HHHHHH) was purchased from Genscript Corporation (Piscataway, NJ), while peptides H10 (Sequence: HHHHHHHHHH) and H3 (Sequence: GHHPHG) were chemically synthesized by Biomatik Corparation (Wilmington, DE). Other chemicals such as Cu(NO3)2 (99.999%), Co(NO3)2 (99.999%), Ni(NO3)2 (99.999%), Zn(NO3)2 (99.999%), NaCl (99.999%), HCl (ACS reagent, ≤1 ppm heavy metals), and Trizma base (BioXtra grade, ≥99.9%) were bought from Sigma (St. Louis, MO). All of the peptide samples and chemicals were dissolved in HPLC-grade water (ChromAR, Mallinckrodt Baker). The stock solutions of metal salts were prepared at concentrations of 1.0 mM each, while those of peptides were prepared at 5.0 mM each, and were kept at −20°C before and after use. The electrolyte solution used in this work contained 1.0 M NaCl and 10 mM tris, with the pH of the solutions adjusted to pH 6.5, pH 7.5, or pH 8.5 using HCl. Lipid 1,2-diphytanoylphosphatidylcholine was obtained from Avanti Polar Lipids (Alabaster, AL). Teflon film was purchased from Goodfellow (Malvern, PA). αHL protein pores were produced using a procedure as described by our previous work (Wang et al., 2009).
2.2. Electrical Recording
A bilayer of 1,2-diphytanoylphosphatidylcholine was formed on an aperture (150 μm) in a Teflon septum (25 μm thick) that divided a planar bilayer chamber into two compartments, cis and trans. The formation of the bilayer was achieved by using the Montal-Mueller method (Montal et al., 1972). The experiments were performed under a series of symmetrical buffer conditions with a 2.0 mL solution comprising 1.0 M NaCl, and 10 mM Tris·HCl at 22 ± 1°C. Unless otherwise noted, the αHL proteins were added to the cis compartment, which was connected to “ground”, while peptides and metal salts were added to the trans compartment. The final concentration of the αHL proteins used for the single channel insertion was 0.2–2.0 ng·mL−1. Currents were recorded with a patch clamp amplifier (Axopatch 200B, Molecular Devices, Sunnyvale, CA, USA). They were low-pass filtered with a built-in four-pole Bessel filter at 10 kHz and sampled at 50 kHz by a computer equipped with a Digidata 1322 A/D converter (Molecular Devices).
2.3. Data Analysis
Data were analyzed with the following software: pClamp 10.2 (Molecular Devices), Origin 8.0 (Microcal, Northampton, MA), and SigmaPlot 12.0 (Systat Software Inc., San Jose, CA). Conductance values were obtained from the amplitude histograms after the peaks were fit to Gaussian functions. Mean residence times (τoff values) for peptides and Cu(II)-peptide complexes were obtained from the residence time histograms by fitting the distributions to single exponential functions by the Levenberg-Marquardt procedure (Movileanu, et al., 2005). Each single-channel current trace was recorded for at least 2 minutes. At least three separate experiments were carried out for each sample.
3. Results and discussion
3.1. Effect of peptide probes on Cu2+ detection
The principle for nanopore detection of Cu2+ ions is based on the interaction between a copper-chelating agent and Cu2+. As shown in Scheme 1, without Cu2+ ions, the interactions between the copper-chelating agents and the protein pore produce only one type of events. In contrast, after addition of Cu2+ ions to the solution, they will interact with the copper-chelating agent molecules to form copper chelates. The interactions between these copper chelates and the protein pore result in events having significantly different signatures (e.g., residence times or blockage amplitudes) from those in the absence of Cu2+ ions, which permits the copper chelates to be readily recognized.
Scheme 1.

Nanopore detection of Cu2+ ions using a chelating agent probe. The interactions between the chelating agent and the nanopore in the absence and presence of Cu2+ ions produce events having significantly different signatures, thus permitting the free chelating agents and Cu2+-chelating agent complexes to be readily differentiated.
To demonstrate this concept, our initial experiments were carried out at pH 7.5 with three short peptides in the mutant α-hemolysin (M113F)7 pore in the absence and presence of Cu2+ ions. It has been shown that the (M113F)7 protein could provide an enhanced resolution for biomolecule recognition compared with that observed with the wild-type α-hemolysin pore. The peptides used included H3, H6, and H10, which contains 3, 6, and 10 histidines, respectively. It should be noted that peptides possess a range of potential donor atoms, and are very effective ligands for a variety of metal ions with high specificities (Gaggelli et al., 2006). The complexes formed exist in various conformations. Taking histidine-containing peptides for example, the histidine residue possesses a very efficient nitrogen donor in its side chain imidazole ring, and provides two nitrogen donors and a six-membered chelate ring for coordination. Terdentate binding makes histidine a primary low molecular weight chelator in living systems. It is worth mentioning that the administration of Cu-histidine, when initiated early in life, is the most effective treatment for Menkes disease, a genetic neurodegenerative disorder due to the impaired copper metabolism (Tümer et al., 1996).
Our experimental results (Fig. 1) showed that, at +120 mV, the event signatures (residence time and blockage amplitude) of peptides H3 and H6 didn’t change much in the absence and presence of Cu2+ ions. However, when peptide H10 was used as the chelating agent to detect copper ions, significant difference in its event signature was observed before and after addition of Cu2+ ions. Specifically, in the absence of Cu2+ ions, H10 produced a single population of short-lived translocation events with a mean residual current of ~ 15 pA and a mean residence time of ~ 3 ms, which is typical for a short peptide of 10 amino acids (Movileanu et al., 2005). In sharp contrast, upon addition of Cu2+ ions to the electrolyte solution, in addition to the short-lived events, a new type of long-lived events having a mean residual current of ~6.0 pA and a mean residence time of ~ 40 ms was observed (Fig. 2). Furthermore, with an increase in the concentration of added Cu2+, the frequency of the long-lived events increased, while that of the short-lived ones decreased. It is apparent that these long-lived events were attributed to the Cu2+-peptide complexes. In addition to the peptide conformational change (Mereuta et al., 2012, and Stefureac et al., 2008, Stefureac et al., 2010), another possible reason why the event signatures of H10 were significantly different before and after addition of Cu2+ ions is that free peptides and Cu2+-peptide chelates have different charge states. It is well known that at pH 7.5, histidine is almost at its isoelectric point, and hence peptide H10 would be neutral molecules. After H10 molecules are complexed with Cu2+ ions, the Cu2+-peptide chelates are positively charged. The positively charged nature of the copper chelates are supported by the voltage dependence experiment, where the mean residence time of the long-lived events first increased and then decreased significantly as the applied potential bias increased (Supporting Information, Fig. S1a). This voltage dependent phenomenon is well documented for charged species in nanopores (Jayawardhana et al., 2009). Since the largest residence time of H10 was observed at +100 mV (Fig. S1), this voltage bias was used throughout the remaining experiments unless otherwise noted.
Figure 1.
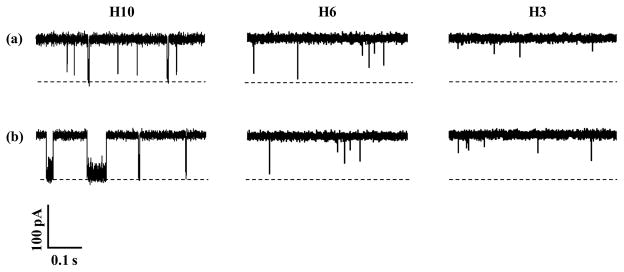
Typical single-channel current recording traces, showing the effect of peptide probes on Cu2+ detection. The experiments were performed at +120 mV with the (M113F)7 α-hemolysin pore and three peptides H10, H6 and H3 in the (a) absence and (b) presence of Cu2+ ions in a buffer solution comprising 1.0 M NaCl and 10 mM Tris•HCl (pH 7.5). Dashed lines represent the levels of zero current. The concentration of Cu2+ was 10 μM, while those of H10, H6, and H3 were 50 μM, 500 μM, and 10 μM, respectively.
Figure 2.

Event signatures of peptide H10 before and after addition of Cu2+ ions. (a) Scatter plots of event residence time versus amplitude; (b) Amplitude histograms; (c) Residence time histograms of short-lived events; and (d) Residence time histograms of long-lived events. The experiments were performed with the (M113F)7 α-hemolysin pore in a buffer solution comprising 1.0 M NaCl and 10 mM Tris•HCl (pH 7.5) at +100 mV (cis at ground) in the presence of 40 μM H10 peptide samples.
3.2. pH effect on the detection of Cu2+ ions
The protein ion channel properties such as conductance and ion selectivity are sensitive to the pH of electrolyte solution (Gu et al., 2000; Gu et al., 2001; Merzlyak et al., 2005). For example, Bayley and coworkers reported that when the pH of the electrolyte decreased from 11.0 to 7.5 and then to 5.0, the charge selectivity of the wild-type αHL pore changed from cation selective to weakly anion selective, and then to anion selective (Gu et al., 2001). Our previous studies demonstrated that a reduction in the pH of the electrolyte solution could result in an increase in the event frequency and residence time of biomolecules and terrorist agents in the nanopore, and/or improve the event signature contrast between the target analyte and other matrix components, thus offering an enhanced resolution and better sensitivity for the nanopore sensor (de Zoysa et al., 2011; Gupta et al., 2013). The improved nanopore resolution and sensitivity might be attributed to several factors, such as the enhanced electro-osmotic flow, the pH effect on the charge state of the target analyte, and the analyte/protein ion channel structure or conformational change.
To examine whether the pH of the electrolyte solution will affect Cu2+ detection, the interactions between peptide H10 and the (M113F)7 pore in the absence and presence of Cu2+ were investigated in 1 M NaCl solutions at three different pH values (i.e., pH 6.5, pH 7.5, and pH 8.5). Our experimental results (Fig. 3, and Supporting Information, Fig. S2) showed that, in the absence of Cu2+ ions, H10 produced events having a larger frequency but a smaller mean residence time as the pH of the solution decreased. Although the conductivities of these three different pH electrolytes were quite stable (~79 mS/cm), the results are reasonable considering the charge state of the peptide H10 at different pH values. As mentioned previously, the pI (isoelectric point) of histidine is around 7.5, and hence, H10 is positively charged at pH 6.5 while negatively charged at pH 8.5. Therefore, under an applied voltage bias of +100 mV, the electrophoretic effect will facilitate the positively charged H10 (at pH 6.5) and retard the negatively charged H10 (at pH 8.5) translocating through the nano-channel, respectively. However, in the presence of Cu2+, although the pH effect on the event frequency showed a same trend as that without Cu2+, the effect of the solution pH on the event residence time was quite different from that in the absence of Cu2+ ions (Fig. 3, and Supporting Information, Fig. S3). Specifically, when the solution pH decreased from pH 8.5 to pH 6.5, the frequency of the Cu2+-peptide complex events increased by 4.8 folds (from ~ 0.84 s−1 to ~ 4.0 s−1 at 10 μM concentration of Cu2+). Among the three pH values, Cu2+-peptide complexes showed the largest mean residence time of ~186.6 ms at pH 6.5, while the corresponding event residence times at pH 7.5 and pH 8.5 were 40.0 ms, and 80.1 ms, respectively. As discussed previously, as the solution pH decreased from pH 8.5 to pH 7.5 and to pH 6.5, peptide H10 was negatively charged, neutral, and positively charged, respectively. After complexation with Cu2+, the charge state of the Cu2+-peptide chelates should be more positive than the free peptides. If the electrophoretic effect was the dominant factor affecting the event residence time, Cu2+-peptide complexes should produce the smallest residence time at pH 6.5, which is not in agreement with our experimental result. Therefore, it is more likely that the event mean residence time of Cu2+-peptide chelates is mainly affected by their structures or conformations. For example, the solution pH might affect the stoichiometric ratio of the Cu2+-peptide complexes or the positions in the peptide chain at which histidines bind to Cu2+ ions. This interpretation is supported by both literature and our dose-response experiment. It is well known that the coordination properties of a histidine residue within a peptide sequence depend enormously on its position in a peptide chain, further, the metal-peptide complexes formed can exist in a variety of conformations that are dependent not only on the concentrations of both the peptide and metal ion but also on the pH of the reaction medium (Daniele et al., 1991; Sjöberg, 1997; Gaggelli et al., 2003; Belosi et al., 2004). It was also reported that the solution pH would affect the binding affinity of the Cu2+-peptide interaction significantly (Viles et al., 1999). Our experimental results (Fig. 3) showed that, at pH 7.5, the event residence time was quite stable with an increase in the concentration of added Cu2+, suggesting the formation of a fixed ratio of Cu2+-H10 complexes. In contrast, at pH 6.5 or pH 8.5, the event residence time increased as the Cu2+ concentration increased, indicating that at these pH values, the formed metal-peptide complexes might have different stoichiometric ratios or different conformations, which is dependent on the concentrations of peptides and metal ions. Since the event mean residence time of Cu2+-peptide complexes was quite stable at different copper concentrations at pH 7.5, it could serve as a signature to differentiate Cu2+ from other metal ions, and hence this pH was used in the remaining experiments.
Figure 3.
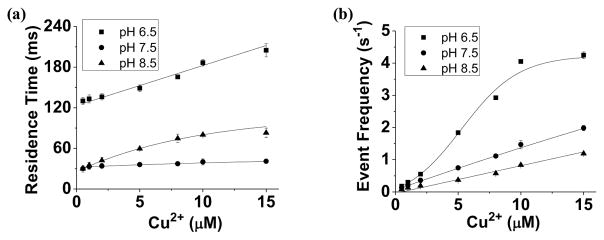
Effect of solution pH on nanopore detection of Cu2+. Dependence of the (a) event mean residence time and (b) frequency of Cu2+-peptide complexes on the solution pH and the concentration of added Cu2+ ions. The experiments were performed with the (M113F)7 α-hemolysin pore in a buffer solution comprising 1.0 M NaCl and 10 mM Tris•HCl at +100 mV in the presence of 40 μM H10.
3.3. Selectivity of the Cu2+ Nanopore Sensor
His-tagging is one of the most widespread strategies used to purify recombinant proteins for biochemical and structural studies because the histidine residues of the proteins could bind to several types of immobilized metal ions, including nickel, cobalt and copper, under specific buffer conditions. To test the selectivity of this nanopore sensor, Co2+, Ni2+, and Zn2+ which have very similar ligand-binding characteristics to Cu2+ were examined. Early studies with other detection techniques have shown that these metal ions, especially Ni2+, could potentially interfere with the Cu2+ detection (Liu et al., 1999; Yang et al., 2001; Dong et al., 2012). The Ni2+ interference could be suppressed with the addition of other chelating agents such as dimethylglyoxime (Liu et al., 1999). To improve the selectivity for Cu2+ detection, other non-chelating reaction approaches are currently under intensive investigation. For example, highly sensitive and selective detection of Cu2+ has been achieved based on Cu2+-controlled enzymatic/Cu2+-catalyzed chemical reactions (Yin et al., 2009; Lou et al, 2011; Yao et al., 2013). Our single-channel recording experiments (Supporting Information, Fig. S4) showed that indeed all these three metal ions could interact with H10 to form metal-peptide complexes. Fortunately, the event residual currents of Zn2+-peptide and Cu2+-peptide complexes were quite different (10.9 ± 0.1 pA vs. 6.1 ± 0.1 pA), which allows these two complexes easily to be differentiated and even simultaneously detected. On the other hand, although the events of Ni2+-peptide and Co2+-peptide complexes had similar blockage amplitudes to those of Cu2+-peptide chelates, their event residence times were much smaller than that of Cu2+-peptide complexes (5.9 ± 0.6 ms, 13.7 ± 0.8 ms, and 40.0 ± 1.2 ms for Co2+, Ni2+, and Cu2+, respectively). By using an appropriate residence time cut-off value (e.g., 15 ms), we can separate the events of Cu2+-peptide complex from those of others, thus successfully analyzing Cu2+ in the presence of Ni2+ (Fig. 4) and/or Co2+. In the case of analyzing a solution mixture consisting of Cu2+, Ni2+, Co2+, and Zn2+ ions, selective detection of Cu2+ could still be achieved if only the events having the residence times larger than 15 ms, and with residual currents within the range from 4 pA to 10 pA were included in the data analysis. Under this condition, the nanopore sensor responded selectively toward Cu2+ ions, by at least 6-fold or more, relative to the other metal ions (Fig. 5). If equal concentrations of Cu2+, Ni2+, Co2+, and Zn2+ ions were present in the solution mixture, ~77% of the total events were contributed by the Cu2+-peptide complexes, while 3.2%, 7.4%, and 12.6% of the events were due to Ni2+-, Co2+-, and Zn2+-peptide complexes, respectively (Supporting Information, Fig. S5). Further improvement in the sensor selectivity was possible by analyzing events within a narrower amplitude range. However, this approach would result in a smaller event frequency, thus requiring a longer single-channel recording time to collect enough Cu2+-peptide complex events. In addition, it is worth mentioning that, by modifying the data analysis condition, e.g., only including events with residual currents larger than 10 pA, and having the residence times larger than 20 ms, our nanopore sensor can selectively detect Zn2+ ions in the presence of other metal ions.
Figure 4.

Nanopore detection of Cu2+ in the presence of Ni2+ ions. The experiments were performed at +100 mV in the presence of 40 μM H10.
Figure 5.
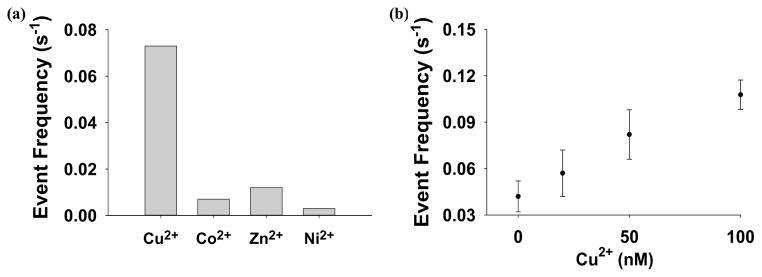
(a) Selectivity and (b) dose-response curve for the Cu2+ ion nanopore sensor system. Only the events having the residence times larger than 15 ms, and with residual currents within the range from 4 pA to 10 pA were included in the data analysis.
3.4. Improving the Sensitivity of the Cu2+ Nanopore Sensor
Under the current experimental conditions (i.e., peptide H10, α-hemolysin (M113)7 pore, pH 7.5, +100 mV applied voltage bias), Cu2+ ions could be detected with a detection limit (defined as the concentration corresponding to three times the standard deviation of a blank signal) of ~500 nM in a 1 min second single-channel recording. The relatively poor detection limit was mainly due to the low event frequency (0.2 s−1•μM−1 Cu2+) of this sensing system. It should be mentioned that this Cu2+ sensor had an extremely low background, and hence increasing the event frequency offers the potential to greatly lower its detection limit. Since the frequency of the Cu2+-H10 complex events increased significantly with an increase in the applied potential (Supporting Information, Fig. S1), the feasibility of utilizing the voltage-dependence to improve sensor sensitivity was then investigated at +160 mV. Note that at this voltage, the mean residence time of the Cu2+-H10 complex events was ~20 folds larger than that of free H10 peptides, and hence could still be readily recognized (Supporting Information, Fig. S7). Although a further increase in the applied potential may lead to more events, the lipid bilayer used in the experiments would become less stable. With this experimental condition, the detection limit for Cu2+ detection could be improved to ~40 nM (Fig. 5). In addition, our experiments with the pH effect on Cu2+ detection demonstrated that the event frequency increased by ~3 folds when the solution pH was reduced from pH 7.5 to pH 6.5 (Fig. 3). Although lowering the pH of the electrolyte solution provides a feasible approach to further improve the sensitivity for Cu2+ detection, a disadvantage of this approach is that the sensor selectivity will possibly be worse since the event residence time of the Cu2+-peptide complexes varied with the Cu2+ concentration at pH 6.5, so that the events of Cu2+-peptide and other metal-peptide complexes might overlap to some extent, thus interfering with Cu2+ detection.
4. Conclusions
A sensitive and selective nanopore sensor for the detection of Cu2+ ions was successfully developed by using a polyhistidine molecule as a chelating agent. In our nanopore sensor design, the chelating agent provides the required molecular recognition and plays a major role in the sensitivity and selectivity of the nanopore sensor. Hence, modification of the inner surface of the nanopore to construct recognition sites for the target analyte is not necessary, which simplifies the fabrication of nanopore sensors (refer to Supporting information, Fig. S5, for Cu2+ detection with the wild-type α-hemolysin pore). This nanopore sensor design strategy should find useful application in the development of stochastic sensors for other substances, especially in situations where construction of binding sites in the nanopore interior is difficult.
Supplementary Material
Highlights.
We developed a sensitive stochastic nanopore sensor for the detection of Cu2+ using a polyhistidine probe as the chelating agent.
The sensor was highly selective to Cu2+ ions.
Experimental conditions such as the polyhistidine probe, solution pH, and the applied voltage bias significantly affected the sensor sensitivity.
The chelating reaction approach simplified the nanopore sensor design.
Acknowledgments
This work was financially supported by the Defense Advanced Research Projects Agency (HR0011-10-C-0226), the National Institutes of Health (1R011HG005095), and Department of Homeland Security (HSHQDC-09-C-00091).
Appendix A. Supporting Information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Belosi B, Gaggelli E, Guerrini R, Kozlowski H, Euczkowski M, Mancini FM, Remelli M, Valensin D, Valensin G. ChemBioChem. 2004;5:349–359. doi: 10.1002/cbic.200300786. [DOI] [PubMed] [Google Scholar]
- 2.Braha O, Walker B, Cheley S, Kasianowicz JJ, Song L, Gouaux JE, Bayley H. Chem Biol. 1997;4:497–505. doi: 10.1016/s1074-5521(97)90321-5. [DOI] [PubMed] [Google Scholar]
- 3.Braha O, Gu LQ, Zhou L, Lu X, Cheley S, Bayley H. Nat Biotechnol. 2000;17:1005–1007. doi: 10.1038/79275. [DOI] [PubMed] [Google Scholar]
- 4.Cheley S, Gu LQ, Bayley H. Chem Biol. 2002;9:829–838. doi: 10.1016/s1074-5521(02)00172-2. [DOI] [PubMed] [Google Scholar]
- 5.Daniele PG, Zerbinati O, Zelano V, Ostacoli G. J Chem Soc, Dalton Trans. 1991:2711–2715. [Google Scholar]
- 6.Derrington IM, Butler TZ, Collins MD, Manrao E, Pavlenok M, Niederweis M, Gundlach JH. Proc Natl Acad Sci USA. 2010;107:16060–16065. doi: 10.1073/pnas.1001831107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Zoysa RSS, Krishantha DMM, Zhao Q, Gupta J, Guan X. Electrophoresis. 2011;32:3034–3041. doi: 10.1002/elps.201100216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong Y, Wang R, Li G, Chen C, Chi Y, Chen G. Anal Chem. 2012;84:6220–6224. doi: 10.1021/ac3012126. [DOI] [PubMed] [Google Scholar]
- 9.Gaggelli E, D’Amelio N, Valensin D, Valensin G. Magn Reson Chem. 2003;41:877–883. [Google Scholar]
- 10.Gaggelli E, Kozlowski H, Valensin D, Valensin G. Chem Rev. 2006;106:1995–2044. doi: 10.1021/cr040410w. [DOI] [PubMed] [Google Scholar]
- 11.Gao C, Ding S, Tan Q, Gu L. Anal Chem. 2009;81:80–86. doi: 10.1021/ac802348r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu L, Bayley H. Biophys J. 2000;79:1967–1975. doi: 10.1016/S0006-3495(00)76445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu L, Cheley S, Bayley H. J Gen Physiol. 2001;118:481–493. doi: 10.1085/jgp.118.5.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guan X, Gu LQ, Cheley S, Braha O, Bayley H. ChemBioChem. 2005;6:1875–1881. doi: 10.1002/cbic.200500064. [DOI] [PubMed] [Google Scholar]
- 15.Gupta J, Zhao Q, Wang G, Kang X, Guan X. Sensor Actuat B. 2013;176:625–631. [Google Scholar]
- 16.Howorka S, Cheley S, Bayley H. Nat Biotechnol. 2001;19:636–639. doi: 10.1038/90236. [DOI] [PubMed] [Google Scholar]
- 17.Jayawardhana DA, Crank JA, Zhao Q, Armstrong DW, Guan X. Anal Chem. 2009;81:460–464. doi: 10.1021/ac801877g. [DOI] [PubMed] [Google Scholar]
- 18.Jovanovic-Talisman T, Tetenbaum-Novatt J, McKenney AS, Zilman A, Peters R, Rout MP, Chait BT. Nature. 2009;457:1023–1027. doi: 10.1038/nature07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasianowicz JJ, Brandin E, Branton D, Deamer D. Proc Natl Acad Sci USA. 1996;93:13770–13773. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu A, Chen D, Lin C, Chou H, Chen C. Anal Chem. 1999;71:1549–1552. [Google Scholar]
- 21.Lou T, Chen L, Chen Z, Wang Y, Chen L, Li J. ACS Appl Mater Interfaces. 2011;3:4215–4220. doi: 10.1021/am2008486. [DOI] [PubMed] [Google Scholar]
- 22.Luchian T, Shin SH, Bayley H. Angew Chem Int Ed. 2003;42:3766–3771. doi: 10.1002/anie.200351313. [DOI] [PubMed] [Google Scholar]
- 23.Meller A, Nivon L, Brandin E, Golovchenko J, Branton D. Proc Natl Acad Sci USA. 2000;97:1079–1084. doi: 10.1073/pnas.97.3.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mereuta L, Schiopu I, Asandei A, Park Y, Hahm K, Luchian T. Langmuir. 2012;28:17079–17091. doi: 10.1021/la303782d. [DOI] [PubMed] [Google Scholar]
- 25.Merzlyak PG, Capistrano MF, Valeva A, Kasianowicz JJ, Krasilnikow OW. Biophys J. 2005;89:3059–3070. doi: 10.1529/biophysj.105.066472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montal M, Mueller P. Proc Natl Acad Sci USA. 1972;69:3561–3566. doi: 10.1073/pnas.69.12.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Movileanu L, Howorka S, Braha O, Bayley H. Nat Biotechnol. 2000;18:1091–1095. doi: 10.1038/80295. [DOI] [PubMed] [Google Scholar]
- 28.Movileanu L, Schmittschmitt JP, Scholtz JM, Bayley H. Biophys J. 2005;89:1031–1045. doi: 10.1529/biophysj.104.057406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shim JW, Tan Q, Gu L. Nucleic Acids Res. 2009;37:972–982. doi: 10.1093/nar/gkn968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sigalov G, Comer J, Timp G, Aksimentiev A. Nano Lett. 2008;8:56–63. doi: 10.1021/nl071890k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sjöberg S. Pure Appl Chem. 1997;69:1549–1570. [Google Scholar]
- 32.Stefureac RI, Lee JS. Small. 2008;4:1646–1650. doi: 10.1002/smll.200800585. [DOI] [PubMed] [Google Scholar]
- 33.Stefureac RI, Madampage CA, Andrievskaia O, Lee JS. Biochem Cell Biol. 2010;88:347–358. doi: 10.1139/o09-176. [DOI] [PubMed] [Google Scholar]
- 34.Storm AJ, Storm C, Chen J, Zandbergen H, Joanny JF, Dekker C. Nano Lett. 2005;5:1193–1197. doi: 10.1021/nl048030d. [DOI] [PubMed] [Google Scholar]
- 35.Talaga DS, Li J. J Am Chem Soc. 2009;131:9287–9297. doi: 10.1021/ja901088b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tisato F, Marzano C, Porchia M, Pellei M, Santini C. Med Res Rev. 2010;30:708–749. doi: 10.1002/med.20174. [DOI] [PubMed] [Google Scholar]
- 37.Tümer Z, Horn N, Tønnesen T, Christodoulou J, Clarke JT, Sarkar B. Nat Genet. 1996;12:11–3. doi: 10.1038/ng0196-11. [DOI] [PubMed] [Google Scholar]
- 38.Viles JH, Cohen FE, Prusiner SB, Goodin DB, Wright PE, Dyson HJ. Proc Natl Acad Sci USA. 1999;96:2042–2047. doi: 10.1073/pnas.96.5.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D, Zhao Q, de Zoysa RSS, Guan X. Sensor Actuat B. 2009;139:440–446. [Google Scholar]
- 40.Wang G, Zhao Q, Kang X, Guan X. J Phys Chem B. 2013;117:4763–4769. doi: 10.1021/jp309541h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wen S, Zeng T, Liu L, Zhao K, Zhao Y, Liu X, Wu HC. J Am Chem Soc. 2011;133:18312–18317. doi: 10.1021/ja206983z. [DOI] [PubMed] [Google Scholar]
- 42.Yang W, Jaramillo D, Gooding JJ, Hibbert DB, Zhang R, Willett GD, Fisher KJ. Chem Commun. 2001;19:1982–1983. doi: 10.1039/b106730n. [DOI] [PubMed] [Google Scholar]
- 43.Yao Z, Yang Y, Chen X, Hu X, Zhang L, Liu L, Zhao Y, Wu HC. Anal Chem. 2013;85:5650–5653. doi: 10.1021/ac401386v. [DOI] [PubMed] [Google Scholar]
- 44.Yin BC, Ye BC, Tan W, Wang H, Xie CC. J Am Chem Soc. 2009;131:14624–14625. doi: 10.1021/ja9062426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Q, Jayawardhana DA, Guan X. Biophys J. 2008;94:1267–1275. doi: 10.1529/biophysj.107.117598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.