

Abstract
Mechanical resilience of bone tissue decreases with age. The ability to comprehensively probe and understand bone properties could help alleviate this problem. One important aspect of bone quality which has recently been made evident is the presence of dilatational bands formed by osteocalcin (OC) and osteopontin (OPN), which contribute to fracture toughness. However, experimental evidence of the structural role of these two proteins at the organic-mineral interface in bone is still needed. Solid state nuclear magnetic resonance (SSNMR) is emerging as a useful technique in probing molecular level aspects of bone. Here, we present the first SSNMR study of bone tissue from genetically modified mice lacking OC and/or OPN. Probing the mineral phase, the organic matrix and their interface revealed that despite the absence of OC and OPN, the organic matrix and mineral were well preserved, and the overall exposure of collagen to hydroxyapatite (HA) nanoparticles was hardly affected. However, the proximity to HA surface was slightly increased for a number bone components including less abundant amino acids like lysine, suggesting that this is how the tissue compensates for the lack of OC and OPN. Taken together, the NMR data supports the recently proposed model, in which the contribution of OC - OPN to fracture toughness is related to their presence at the extrafibrillar organic-mineral interfaces, where they reinforce the network of mineralized fibrils and form dilatational bands. In effort towards understanding further the structural role of individual amino acids of low abundance in bone, we then explored the possibility of specific 13C enrichment of mouse bone, and report the first SSNMR spectra of 97% 13C lysine-enriched tissues. Results show that such isotopic enrichment allows valuable molecular-level structural information to be extracted, and sheds light on post-translational modifications undergone by specific amino acids in vivo.
Introduction
Bone's ability to resist fracture deteriorates with age,1 which causes substantial economic burden and decreased quality of life.2 Assessing the risk of fracture, followed by appropriate intervention, can reduce this problem.3 Currently in the United States, bone mineral density and assessment of known clinical risk factors are used to estimate the risk of fracture.4 However, a comprehensive understanding of how the organic matrix and its specific components contribute to the structural strength of bone should lead to better assessment of fracture risk and promising therapeutics.
Bone is a complex hierarchical composite, where resistance to crack growth is accomplished by toughening mechanisms across multiple length scales, and where plastic (irreversible) deformation, occurring during fracture, originates at the nanometer scale.5 The properties of the organic matrix (composed mainly of collagen), the mineral phase (composed mainly of carbonated hydroxyapatite, HA) and of their interface at the molecular scale all collectively play important roles in determining the properties of bone at the tissue level.
Bone deformation involves sliding of the mineralized fibrils past each other, which results in shearing of the extrafibrillar matrix.6 Deforming the extrafibrillar matrix leads to energy dissipation via breaking of “sacrificial bonds” and exposing “hidden lengths” of the noncollagenous molecules.7,8 In particular, osteopontin (OPN), one of the most abundant bone noncollagenous proteins (NCPs), has been found to be able to function as “glue” in this extrafibrillar space.9 At lower hierarchal level, it has been shown that the brittle mineral nanoparticles are embedded into the ductile collagen fibers to make them stiffer and to improve the energy dissipation.10 Recently, it was shown that two abundant NCPs, osteocalcin (OC) and OPN, also contribute to whole bone toughness by forming dilatational bands.11 In this study, a model was proposed in which OPN attaches to extrafibrillar mineral aggregates via the linker protein OC. During plastic deformation at yield, OPN and OC stretch to dissipate energy as the mineral surfaces move apart, and thus contribute to arrest the crack growth.11 Consequently, the bone toughness in mice lacking OC and/or OPN (OC-OPN-/-;-/-) was found to decrease as compared to their wildtype littermates (WT).11
Several questions arise regarding the possible origin of this decrease. While increased bone brittleness is clearly associated with absence of the dilatational bands, other structural consequences of OC-OPN removal could have parallel effects. Firstly, OC and OPN are known to regulate biomineralization,12-14 but it is not known whether their removal will affect the structure, composition or hydration of HA platelets. Secondly, the absence of OC and OPN may adversely affect other NCPs which play a role in collagen assembly.15 Lastly, the structure at the organic-mineral interface may become altered, because other noncollagenous components such as glucosaminoglycans,16 citrate,17 as well as various NCPs18 may become exposed to the HA surface when OC and OPN are absent.
Solid state NMR is uniquely suited to address these questions, as it has been shown to be useful in studies of molecular structure of bone mineral19-27 as well as organic matrix.26-29 Particularly, the 13C{31P} REDOR technique (rotational echo double resonance)30 is well adapted to probe the structural arrangement of organic-mineral interfaces at the molecular level, whether in model systems31,32 or directly in biological solids33-37 including powdered bone33,38-40 and, more recently, non-powdered hydrated bone.41,42
Here, we present the first atomic-level study of the structural role of the OC and OPN in intact bone, by performing solid state NMR experiments on bone tissue from mice genetically modified to lack OC and/or OPN. We describe NMR analyses of the mineral phase, organic matrix and their mutual interface, and draw conclusions on the role played by these NCPs on molecular level structure of bone tissue, especially at the organic-mineral interface. As an extension of this study, in the last part of the manuscript, we demonstrate how specific 13C isotope enrichment of some bone amino acids in-vivo can provide more detailed insight into structural role of individual organic matrix components.
Materials and Methods
Sample preparation
All studies were carried out in compliance with NIH and IACUC guidelines for animal use.
Osteocalcin and osteopontin knock-out mice (OC-/-, OPN-/-) were provided by Gerard Karsenty and Susan Rittling, respectively.43,44 Double knock out mice (OC-OPN-/-;-/-) were made by crossing the compound heterozygotes. All animals were transferred to a C57 Bl6 background through 10 generations, and wildtype littermates (WT) were used as a control.
Hind limbs were wrapped in saline soaked gauze stored at -80°C.45 Femora were extracted and epiphyses with soft tissue were removed. The diaphyseal shafts were separated by razor blade into large longitudinal sections. The marrow and trabecular tissue were removed from the intramedullary canal and the sections were washed twice by 60 s agitation by vortex mixer in 1 ml of saline. The clean cortical sections were inserted in the 3.2 mm Bruker MAS rotor (Figure 1). The rotor chamber contained saline (to maintain the hydration of the tissue and act as ballast during the rotation),42 and was sealed with a PTFE membrane (to avoid any change in hydration of the tissue during the experiment). The MRI image in Figure 1 shows the arrangement of the bone sections (dark) in the saline (grey) inside the rotor chamber. The femora of 16 different mice were analyzed by NMR: 5 WT, 4 OC-/-, 3 OPN-/-, and 4 OC-OPN-/-;-/-.
Figure 1.
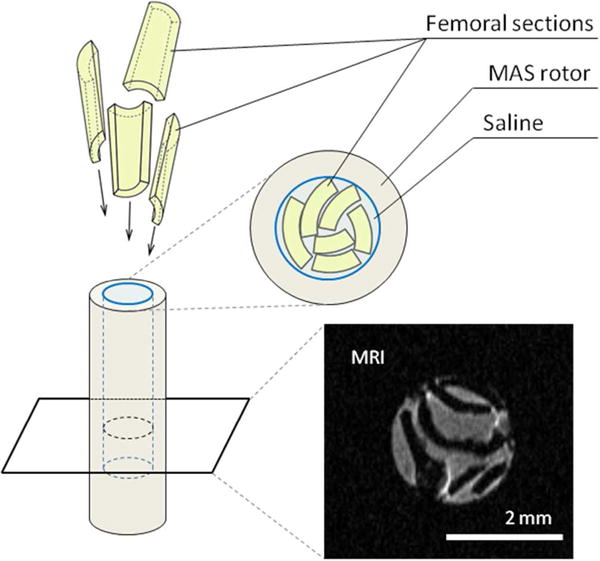
Schematic representation of the packing of femoral mid-diaphyseal sections into the NMR rotor. A representative 1H MRI image of a transverse cross-section of the rotor shows the actual arrangement of the bone sections (black) surrounded by saline (grey). Images confirm that the bone fragments remained intact. No powdering, chemical treatment or drying was required to probe the molecular structure of the mouse femora.
Mouse femora enriched with 13C-6 lysine were obtained as a gift from the Matthias Mann group at the Max Plank Institute of Biochemistry (Martinsried, Germany). Femora from 13C-lysine enriched C57Bl/6 mice were harvested, wrapped in saline soaked gauze and stored at -80°C prior to NMR analysis.
Solid state NMR experiments
All solid state NMR experiments were carried out on a Bruker 600 MHz wide bore NMR spectrometer, equipped with an E-Free triple resonance 3.2 mm magic angle spinning (MAS) probe (Bruker BioSpin, Karlsruhe, Germany) tuned to 1H-13C-31P resonance. The temperature was controlled at 4°C and MAS frequency was 12.5 kHz in all experiments. 100 kHz Spinal 64 1H decoupling during acquisition was used in all experiments. The 31P chemical shifts were referenced to synthetic hydroxyapatite (Ca10(PO4)6(OH)2), used as a secondary reference (at 2.8 ppm with respect to an 85% H3PO4 solution). The 13C chemical shifts were referenced to external adamantane (high frequency signal at 38.5 ppm with respect to tetramethylsilane (TMS)). All 13C NMR signals were assigned as proposed by Aliev46 based on previous literature, with the exception of some of the lysine peaks, as further detailed at the end of this manuscript.
1H- 31P HETCOR
Two-dimensional (2D) 1H→ 31P cross-polarization magic angle spinning (CPMAS) experiments, referred to as heteronuclear correlation (HETCOR) experiments, were acquired on intact bone samples in order to resolve and individually characterize the resonances from various 31P environments. Spectra were acquired with contact times of 0.3, 0.6, 1.5, 3, 5 and 8 ms, following an initiating 2.5 μs 1H 90° pulse. A 2 s relaxation delay was used, and States-TPPI was employed for quadrature detection with 64 transients acquired for each increment, yielding experimental time ∼4 hours per 2D spectrum.
Sine apodization was applied to the time domain of the direct 31P dimension. In the indirect (1H) dimension, the time domain data was zero-filled to 256 points and sine apodization function was applied prior to Fourier transformation. Experimentally derived 1D 31P spectra for individual HPO42-, H2O- PO43- and OH- PO43- components were calculated as sums of F2 spectral rows over the respective 1H regions. The 31P signal was characterized in terms of peak intensity, width, and chemical shift by deconvolution using the DMfit software package.47 The CP buildup rates were determined by fitting bi-exponential function25 using Origin (OriginLab, Northampton, MA). Experiments were carried out for samples from all four experimental groups (WT, OC-/-, OPN-/- and OC-OPN-/-;-/-), but only the comparison between the WT and OC-OPN-/-;-/- specimens is discussed here. In order to assess the reproducibility of the spectral measurements, duplicate data sets were acquired for one WT sample and one OC-OPN-/-;-/-.
1H- 13C CPMAS
Variable-amplitude 1H→13C CPMAS experiments were carried out in order to compare the organic matrix of the samples in terms of the 13C signals. A 2.5 μs 1H 90° pulse and 2.5 ms contact time were used, and the acquisition window was 30.5 ms. The relaxation delay was set to 2 s and a minimum of 8192 scans were acquired for each sample.
Prior to Fourier transform, 40 Hz line broadening was applied to the FID (Free Induction Decay). In order to overcome ambiguity arising from overlap of certain signals, each spectrum was deconvoluted independently four times and the resulting parameters were averaged. The experimental groups included distinct bone samples from 5, 4, 3, and 4 separate WT, OC-/-, OPN-/- and OC-OPN-/-;-/- mice, respectively. To compare the effects of OC and OPN removal, the parameters from individual animals from each group were averaged.
13C {31P} REDOR
In order to probe the organic-mineral interface, the 13C {31P} REDOR experiment was carried out on samples from WT and genetically modified mice. The 13C {31P} REDOR used the same CP parameters and relaxation delay as in the 1H→ 13C CPMAS experiments described above. During the REDOR period, an 8 μs 180° pulse on 13C was applied at the midpoint to refocus chemical shift evolution, and a train of rotor-synchronized 180° dephasing pulses was applied on the 31P over a dephasing period of 10.2 ms, in order to acquire the “dephased” spectra. Reference spectra were acquired with no power in the 31P dephasing pulses. Scans were recorded with the dephased and reference spectra interleaved at the scan level in order to minimize the effect of possible hardware drift and/or temperature differences from RF heating over long acquisition times, relative to serial acquisitions. For each spectrum, 47,000 to 100,000 scans were time-averaged. The data were processed as described for the 1H-31C CPMAS spectra. Each sample studied with CP MAS experiments was probed also by the 13C{31P} REDOR, and the measured parameters were averaged within experimental groups.
Results
Bone mineral
In the 1D 31P MAS NMR spectra of all mouse groups, a single highly overlapped signal was observed centered at ∼3 ppm. The comparison of the 1D CPMAS 31P NMR spectra of the WT and OC-OPN-/-;-/- samples did not reveal any clear difference, suggesting that overall the mineral phase remains structurally similar in the absence of OC and OPN. More detailed information on the specific entities within mineral phase was extracted from the 2D 1H-31P HETCOR spectra (Figure 2A). Three main 31P environments of bone HA were resolved and as a result, the corresponding peak widths and chemical shifts (Table S1 in the supporting information), as well as 1H-31P CP buildup rates, could be determined for the resonances in each environment (Figure 2B, Table 1).
Figure 2.
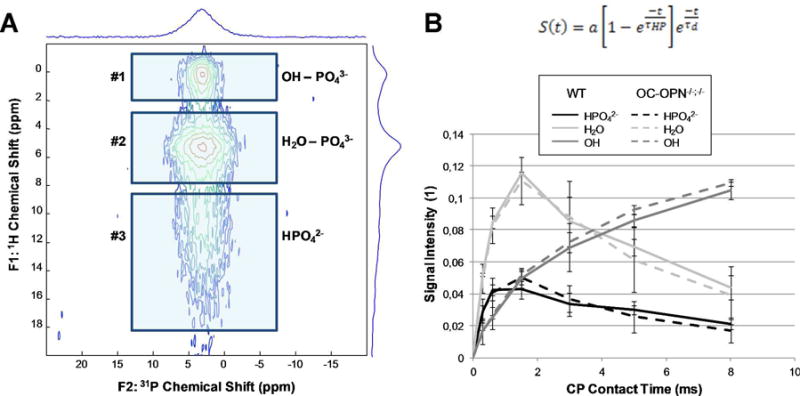
(A) 1H-31P HETCOR spectrum of WT bone at 1.5 ms contact time, where three main 31P environments of bone hydroxyapatite are resolved. (B) Bi-exponential function (top) used to determine CP buildup and decay constants τD and τHP from the CP buildup curves of each of the three 31P environments (bottom). For each specimen, the intensity axis is normalized by the sum of areas under the 3 related buildup curves. Solid lines correspond to the measurements on the WT samples, and dashed lines to the OC-OPN-/-;-/- samples. The error bars correspond to difference between 2 measurements on the same sample.
Table 1.
CP dynamics for the three 31P environments of bone mineral in OC-OPN-/-;-/- mutants and their WT littermates, and in equine subchondral bone (ESB) (Sample 27 in ref. 25). The errors correspond here to errors of fitting and do not reflect the experimental error.
| WT | OC-OPN-/-;-/- | ESB | ||
|---|---|---|---|---|
|
| ||||
| HPO42- | τHP (ms) | 0.3 ± 0.1 | 0.5 ± 0.1 | 0.4 ± 0.2 |
| τd (ms) | 8.9 ± 1.2 | 5.5 ± 0.5 | ||
|
| ||||
| H2O-PO43- | τHP (ms) | 0.6 ± 0.1 | 0.7 ± 0.1 | 0.6 ± 0.1 |
| τd (ms) | 6.2 ± 0.9 | 5.3 ± 0.5 | ||
|
| ||||
| OH-PO43- | τHP (ms) | 2.5 ± 0.4 | 2.6 ± 0.3 | 2.9 ± 0.5 |
| τd (ms) | >105 | >105 | ||
|
| ||||
| Total | τHP (ms) | 0.4 ± 0.1 | 0.5 ± 0.1 | |
| τd (ms) | 17.5 ± 3.4 | 15.9 ± 3.3 | ||
The first 31P environment corresponds to the phosphates present in close proximity to the HA hydroxyl groups (i.e. within the crystalline domains of the mineral platelets). As shown in Figure 2, these phosphates exhibit a relatively slow monotonous CP buildup, with relatively slow decay. The second and third environments are phosphates which are found in vicinity of H2O and which correspond to HPO42- anions, respectively. These two signals build up and also decay faster in CP. No significant differences in the buildup rate constants τHP and τd were detected here for the three environments between the WT and OC-OPN-/-;-/- bone specimens (apart from the τd for HPO42- in OC-OPN-/-;-/-). Interestingly, the τHP constants are also very similar to those reported previously for equine subchondral bone (Table 1).25
The isotropic chemical shifts of each of the three 31P environments hardly vary with contact time. The resonances for the H2O-associated phosphate and the HPO42- anion are shifted upfield relative to the peak of OH-PO43-, and for HPO42- this shift is on average 0.2 ± 0.1 ppm (Figure S1 A, Table S1). When this shift is plotted against the contact time, it appears that the HPO42- may be shifted slightly more in OC-OPN-/-;-/- than in WT (Figure S1 B). This is, however, a very small difference.
For all three 31P environments, the peak widths at half-maximum decrease with contact time, but are very similar between WT and OC-OPN-/-;-/- samples at any given contact time (Table S1, Figure S2). The linewidths of the 1H resonances in the F1 dimension are also very similar (Table S1). The F1 dimension projections actually suggested the presence of two overlapping 1H signals for the OH-PO43- peak (Figure S3), but the parameters of these signals were similar between WT and OC-OPN-/-;-/- as well (Table S1).
From all the comparisons shown above, we conclude that the global chemical structure of the calcium phosphate phase in “normal” bone of 6 month old mice is very similar to that of bone which formed in the absence of OC and OPN.
Bone organic matrix
The organic matrix and its changes related to the removal of OC and OPN were studied by 1H→13C CPMAS NMR. Such spectra were acquired for each animal, and one example from each group is presented in Figure 3. The spectra are dominated by the signals of collagen, the most abundant bone protein. In addition, signals from carbonate substituents in the HA mineral phase are detected in the 168-171 ppm range,48 and from GAG/citrate species at ∼76 ppm.39,40,49 At first inspection, there are no large differences between the 1H-13C CP spectra of the WT and mutant groups.
Figure 3.

Comparison of 1H-13C CPMAS NMR spectra of individual bones from mice without OC, OPN or both OC-OPN, and their WT control. The asterisk marks spinning sidebands.
The signal intensity, peak width and chemical shifts were examined closely for each specimen, and are listed in supplementary information for the WT and OC-OPN-/-;-/- mice (Table S2). Due to differences in CP dynamics, absolute signal intensities in CP spectra are not reliable for quantifying the relative 13C concentrations of particular components. Thus, although some small changes in signal intensity are detected (for example for the peaks assigned mainly to GAG/citrate and glycine-C2), quantitative interpretation was not attempted here. In contrast, the peak width contains information on the structural order of the given species. In materials like bone and cartilage, narrower peaks have been associated with more order.25,50 Comparing the widths measured in the 1H→13C CP spectra of all samples reveals that the differences are small and all within error. Finally, we also compared relative chemical shifts between major well-resolved peaks in WT and OC-OPN-/-;-/-. Changes in chemical shift were within error for most signals. Indeed, the largest change was measured between peaks assigned mainly to glycine C-2 and proline C-4 (Figure 3), which appeared slightly closer to each other in WT (25.0 ± 0.1 ppm) than in OC-OPN-/-;-/- (25.1 ± 0.1 ppm).
Organic-mineral interface
In order to probe the organic-mineral interface, 13C{31P} REDOR spectra were acquired (Figure 4A). In this NMR technique, a 1D 13C CP-MAS spectrum is acquired, in which 13C-31P dipolar couplings are re-introduced (red color), leading to decreased signal intensity of the 13C atoms in close proximity to 31P (i.e. in close proximity to the HA surface), in comparison to a reference spectrum (black color) which is acquired in the absence of the 31P recoupling pulses. The relative intensity decrease between both spectra (ΔS/S0) is referred to as “dephasing”. The magnitude of dephasing depends on the number and arrangement of the 13C and 31P nuclei and increases with their spatial proximity.30,51 The effective spatial range probed is controlled by the length of the dephasing period (i.e. the number of recoupling 31P π pulses). Here, a single dephasing period of 10.2 ms was used in all samples, since this dephasing period was shown to provide a well suited balance in the sensitive detection of the desired 13C signals and in the ability to discriminate the 13C signals of bone according to their proximity to 31P in the HA surface.39,42
Figure 4.

(A) 13C {31P} REDOR NMR dephased (red) and reference spectra (black) of WT and OC-OPN-/-;-/- mice bones. (B) Peak dephasing values ΔS/S0 and (C) assignment to amino acids,52 for WT, OC-/-, OPN-/- and OC-OPN-/-;-/- groups. The asterisks on the spectra in (A) mark spinning sidebands. All dephasing values within a given group in (B) were averaged, and the error bars represent the standard deviation. The “G/C” notation in (B) stands for GAG/citrate.
The dephasing values for carbon signals of WT, OC-/-, OPN-/- and OC-OPN-/-;-/- groups are presented in Figure 4. The goal of these analyses was to see whether progressive trends in ΔS/S0 emerge, as bone tissue becomes impaired by one and both proteins (i.e. a progressive increase or decrease in ΔS/S0). Some heavily overlapped spectral regions cannot be deconvoluted into a unique combination of model peaks. In such regions, multiple combinations of model peaks can “fit” the measured spectra, resulting in higher variation of the peak parameters. Such overlap is in particular observed in the region between 165 and 185 ppm, corresponding to carboxyl/carbonyl/carbonate signals (Figure 4A). To address this variation, multiple deconvolution solutions were determined and averaged for each spectrum, and multiple samples from each experimental group were measured (Figure 4B).
The signal assigned to GAG/citrate is weak relative to the dominant signals, but shows the largest dephasing, in agreement with previous reports.39,40 This weak signal is more difficult to model and the dephasing consequently carries relatively larger error bars (Figure 4B). A trend towards increased dephasing in the GAG/citrate peak upon removal of OC and OPN is noted: the dephasing is lower in WT (59 ± 17 %) than in OC-OPN-/-;-/- (69 ± 24 %). However, these dephasing means are not significantly different (p > 0.05).
All signals in the amino acid region from 10 to 75 ppm (Figure 4A) undergo 31P dependent dephasing to some degree. The amino acids which are not among the main collagen components (glycine, proline, hydroxyproline and alanine) are much less abundant and induce weaker signals. Nevertheless, due to the high resolution and sensitivity of the spectra acquired at 14 T, and the use of multiple samples in each experimental group, it was possible to determine the extent of dephasing in number of individual signals (Figure 4B). Although quantifying the minor signals inherently carries larger errors, the mean dephasing values can still be compared between the experimental groups. The dephasing in this amino-acid region ranged from ∼7 % for the signal assigned mainly to glycine C-2 (Peak #1, Figure 4), this carbon being most often buried in the core of the collagen helices, to ∼19% for carbon signals from arginine/lysine/leucine (Peak #11, Figure 4). Furthermore, we observed that two spectral regions assigned to overlapping resonances of arginine/lysine/leucine (Peak #11 Figure 4), and resonances of glutamic acid/lysine/isoleucine (Peak #12, Figure 4), become slightly more sensitive to dephasing in bones lacking the OC and/or OPN, with the maximum dephasing occurring in absence of both proteins. Statistical analyses show that the signal of arginine/lysine/leucine (peak #11) is significantly more dephased in OC-OPN-/-;-/- than in WT (p < 0.018, t-test).
Finally, concerning the high frequency region of the spectrum, the dephasing of various carboxylate (COO-) groups was not quantified here due to the weakness and overlap of the signals. However, in agreement with published reports,33,39 this dephasing is strong and consistent with the view that many of these charged functions can be found associated with HA surfaces. In contrast, backbone carbonyl (C=O) groups of proteins tend to be buried in the protein structure further away from the HA surface, and indeed dephase to a much lesser degree than carbons of pending amino acid sidechains. Finally, the signal of carbonates (CO32-), which are found substituted into the lattice of the HA crystallites and hence close to phosphates, present a substantial dephasing. However, no significant changes in dephasing associated with removal of OC and OPN could be detected between 165 and 185 ppm.
Overall, the analysis of the dephasing values shows that changes upon removal of OC-OPN are weak, but occur in signals of some amino acids (including the charged sidechain-carrying lysine and arginine) and possibly also of GAG/citrate. Taken together, our results show that the organic-mineral interface arrangement is slightly altered in the OC-OPN-/-;-/- mice, as further discussed below.
Discussion
To our knowledge, this article reports the first multinuclear solid state NMR study on genetically modified and intact mice bones, namely OC and/or OPN knock-out mice bones. The novel method of rotor packing employed here allows stable spinning of bone specimens that have not been treated in any way and that remain fully hydrated throughout the experiment. This is crucial, in particular, to maintain the structure at the organic-mineral interface. The reproducibility of the spinning of these intact samples is particularly noteworthy, as they are even more inhomogeneous than osteoporotic human bone.42 After analysis, the samples are re-useable for other measurements or analyses, which is very important considering the cost of (double) knock-out mice bone specimens. Using this packing method, we carried out a comprehensive SSNMR study to investigate the effect of genetic removal of OC and OPN on bone structure in terms of the mineral phase, organic matrix and their mutual interface.
Bone mineral
It is conceivable that the absence of both OC and OPN would affect the structure and/or hydration of the HA platelets in bone. From the 1D 31P and 2D 1H-31P NMR experiments carried out here on the WT and mutant mice bones, no substantial difference in chemical shift, peak width, and CP build-up rates were observed for the different environments of the 31P in the mineral (Tables 1 and S1, Figures S1 and S2). These atomic-scale studies of bone mineral thus yielded comprehensive evidence that, despite the removal of OC and OPN, the HA component of the tissue is overall remarkably preserved in terms of structural order and composition. It should be noted that previous studies had shown that the CP dynamics in osteoarthritic and healthy equine subchondral bones are also very similar, even in terms of 1H→31P CP dynamics.25
Although the overall chemical composition of the mineral component seems identical between OC-OPN-/-;-/- and WT samples, we note that the 1H-31P HETCOR technique would neither be sensitive to the changes in the geometrical proportions of the platelets, nor to the changes in concentration of (ultra)trace amounts of substitutions in the HA lattice, involving cations such as Mg2+ or Na+.
Bone organic matrix
To obtain information on how the organic matrix changes in response to the removal of OC and OPN, we have examined the 1H→13C CPMAS NMR spectra. Variations in concentration of specific organic molecules or amino acids would be seen as changes in signal intensity. Collagen hydration alterations would be revealed through broadening of peaks of hydroxyproline and alanine,28 and changes of conformation or binding could slightly affect the chemical shift of the peaks. However, no substantial differences between WT and OC-OPN-/-;-/- mice were observed in any of these parameters. The very slight difference in relative chemical shift between proline-C4 and glycine-C2 signals may perhaps be associated with conformational change in collagen, where glycine and proline play important role in maintaining its helicity. However, measurements on a larger number of samples, in conjunction with computational modeling, would be needed to confirm this hypothesis.
Taken together, our results indicate that the removal of OC and OPN bone is not associated with any major difference in the overall composition and structure of the organic matrix. No concentration change associated with up- or down-regulation of particular amino-acids or of GAGs/citrate could be detected. No collagen disordering was observed upon OC and OPN removal. Collagen is thus remarkably well preserved at the molecular scale in the absence of OC and OPN.
Organic-mineral interface
The OC-OPN-/-;-/- mice bones are known to have inferior fracture toughness compared to their wildtype littermates.11 Here, we have provided comprehensive evidence that both collagen order and structure of HA are globally preserved at the molecular scale in OC-OPN-/-;-/- bone. It is thus unlikely that the inferior toughness would be caused by inferior quality of collagen or mineral. Instead, the inferior toughness could be linked to changes in structure of the different interfaces where OC and OPN are absent, and notably the organic-mineral interface.
To investigate how the organic-mineral interface changes in the absence of the OC and OPN, we employed an established 13C{31P} REDOR NMR technique on a series samples from WT and OC/OPN null mice. All amino acid signals undergo some degree of 31P-induced dephasing. The dephasing of the strongest 13C signals of the collagen-abundant amino acids fell within the range of 5-15% (Peaks #1-#10, Figure 4), but a pattern that would correspond to the removal of either OC or OPN did not emerge. Collagen interactions with HA thus do not seem to be strongly affected by the removal of OC and OPN. This is in agreement with the observation that the full volume of fibrillar gap regions is occupied by HA,53 while OC-OPN form dilatational bands in the extrafibrillar matrix.11
The dephasing in REDOR spectra, however, increased slightly in signals assigned to C atoms of (i) glutamic acid, arginine, lysine, leucine and isoleucine (Figure 4, peaks #11 and #12), and (ii) GAG/citrate39,40 (peak at centered at ∼76 ppm, Figure 4), despite their overall concentration being apparently unchanged between WT and mutant specimens (Table S2). Although more samples would need to be analyzed to confirm the significance of the difference in the GAG/citrate signal dephasing, statistically significant results were obtained for other peaks, clearly showing that some organic fragments are employed differently in the OC-OPN-/-;-/- bone, such that they are more likely to interface with HA. Given that some of these species carry charged pending groups (guadinidium in the case of arginine, ammonium in the case of lysine, carboxylate or sulfate in the case of GAGs/citrate), increased exposure to HA surface would be a means for the mutant bone to compensate for the absence of OC and OPN at the mineral surface.
Despite such compensation, published fracture toughness measurements show that OC-OPN-/-;-/- bone has lower fracture toughness than WT bone.11 The changes in the nature of the ionic functions interacting at the HA surface could be the cause of the fracture toughness decrease in mutant bones. Ionic interactions were indeed proposed to play an important role in the function of the dilatational bands11 by linking the OC to HA.54 Thus, we speculate that bone fails in attempt to compensate for the missing OC-OPN dilatational bands (known to interact with the HA surface) by employing alternative charged organic molecules like proteins, that would bind closely to the HA surface via amino acids including glutamic acid, arginine and/or lysine.
All in all, the NMR results presented here, which provide molecular-level information on OC-OPN-/-;-/- bone specimens, support the previously proposed model,11 in which the OC-OPN dilatational bands are located in the extrafibrillar space, and act as a dedicated and effective mechanism that improves toughness of the network of mineralized collagen fibrils.
Perspective: study of low-abundance amino acids in bone
The two 13C peaks in the amino-acid region whose dephasing responds most to the removal of OC and OPN (Peaks #11 and #12 in Figure 4) can belong to several amino acids. Lysine is common for both signals and deserves a closer examination, because it is site of crosslinking55,56 and thus essential to collagen structure. Lysine has very low abundance in bone and its overlap with other signals of similar abundance makes its individual contribution difficult to quantify. However, enrichment of bone by 13C labeled lysine could conceivably help overcome these difficulties. To assess the feasibility and benefits of such enrichment, we probed bone tissue of 13C lysine-enriched mice.
The first benefit of the 13C enrichment is the obvious signal enhancement (Figure 5). Similar signal to noise ratio can be obtained in only a fraction of the experimental time needed for natural abundance samples. More importantly, the 13C lysine signals in these specifically labeled mouse bones are now fully resolved, unlike the corresponding natural abundance signals which would remain highly overlapped and uninterpretable regardless of much longer acquisition times. Indeed, all lysine carbons from C-2 to C-6 can be sensitively detected. A direct benefit of this specific labeling of a single amino acid is the possibility to better assign its 13C resonances in bone, as these can differ from the resonances observed in solution NMR. Indeed, we show here unambiguously that Lys C-2 resonates at ∼54.2 ppm and Lys C-6 is at ∼40.5, rather than ∼59.6 ppm and ∼38.6 ppm as suggested previously for assignments of parchment collagen.46 It should be noted that thanks to the labeling, other signals that would otherwise remain undetected also become apparent in just a few scans (e.g. signals at ∼45 and 68 ppm, Figure 5), as further discussed below.
Figure 5.
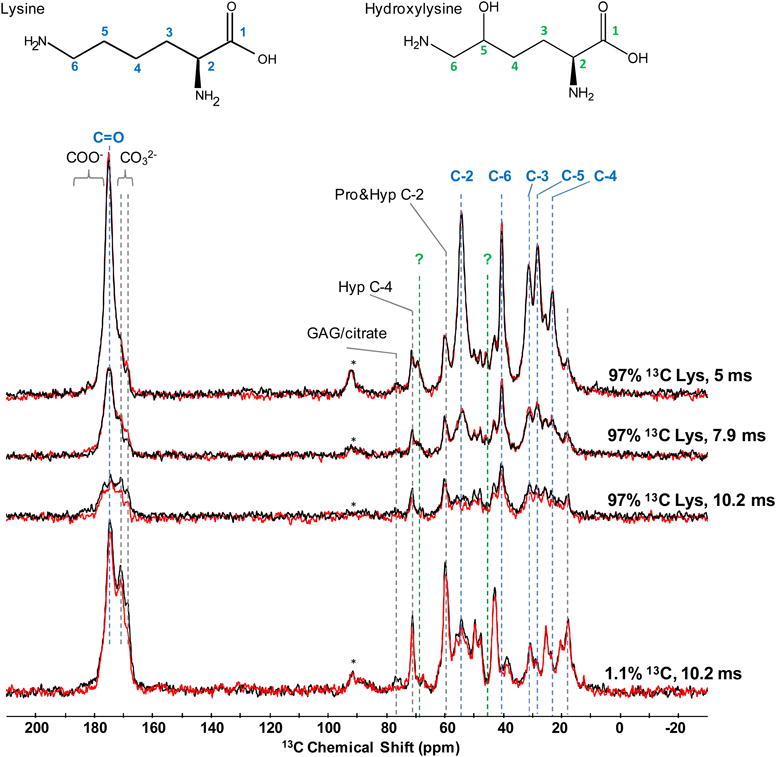
13C{31P} REDOR MAS NMR spectra of mouse bone containing 13C at its natural abundance (1.1%) and mouse fed diet containing 97% 13C enriched lysine. Approximately 200000 scans were accumulated during ∼50 hours acquisition of pair of the natural abundance spectra. Vertical axis of the natural abundance spectra is scaled by factor of 0.25 for clarity. Three dephasing periods (5.0, 7.9 and 10.2 ms) for 13C lysine enriched bone were each acquired by accumulating 20000 -25000 scans over 5.7 – 6.8 hours.
We note here that uniform 13C enrichment would provide exciting new opportunities to investigate the structure of bone tissues with atomic resolution. Although uniform 13C enrichment would reintroduce the spectral complexity and overlap as observed in the 1D 13C spectrum at natural abundance, enrichment would enable the arsenal of multidimensional heteronuclear CP-MAS experiments that efficiently use 13C-13C couplings and chemical shift correlations to provide resolved crosspeaks in highly complex and uniformly 13C-enriched systems.57-59 For example, distances between distinct 13C atoms could be probed within the organic phase in enriched bone using standard DARR (Dipolar Assisted Rotational Resonance) and/or PDSD experiments (Proton Driven Spin Diffusion),60,61 whereas distances between the organic and mineral components could potentially be measured using derivatives of the REDOR and TEDOR (Transferred Echo Double Resonance) experiments that can detect weak couplings in the presence of strong couplings, and can even use the strong couplings for enhanced spectral resolution.62,63 Even at ∼20% 13C enrichment, a ∼20 fold increase in NMR signal intensity would be realized in a 1D 13C spectrum (leading to ∼400 fold decrease in experimental time for equivalent signal to noise ratio) relative to a sample at natural abundance, and provide adequate spin densities to enable most advanced multidimensional experimental techniques.
For the 13C-lysine enriched sample studied here, the REDOR dephasing is weak, and at ∼10 ms, it is in the order of magnitude of the dephasing observed for the WT mice studied at natural abundance (Figure 4). However, in order to fully conclude on the role of lysine in the organic mineral interface with respect to the OC and OPN and their removal, it would be necessary to implement this 13C enrichment in the genetically modified mice as well, which would require in itself a whole new investigation.
Interestingly, the study of the 13C NMR spectrum from the 13C enriched mouse shows that the intensity of the signals at ∼45 and 68 ppm are enhanced beyond their normal natural abundance. The resonance at ∼68 ppm has a similar chemical shift as threonine C-3, which resonates at ∼67.6 ppm. However, isotopic enrichment of threonine due to catabolic scrambling of label from 13C-lysine is not evident, based on the lack of enhanced C-2 and C-4 signals of threonine at 62.5 and 23 ppm, respectively. Alternatively, the enhanced signal at 68 ppm more likely corresponds to C-5 of hydroxylysine. Indeed, this assignment is consistent with the fact that (i) all other C resonances hydroxylysine are expected to fall at similar positions as those of lysine, with the exception of C-6, which should resonate at ∼45 ppm (and thus correspond to the other newly detected signal peak), and (ii) lysine is known to undergo post-translational modifications, including the formation of hydroxylysine.56 This observation thus demonstrates that enrichment of specific compounds in small animal diet by 13C can be used to investigate how compounds become integrated in the organic matrix in bone or other tissues in vivo. This capability can yield interesting and novel insights into critical processes involving the metabolism and post-translational modifications of specific amino acids, where these processes occur, and how they impact the structure and functional properties of intact tissues. Hurdles towards fully exploiting the potential of this specific amino acid labeling for SSNMR include developing number of specifically-enriched comprehensive diets, and implementing these diets in the existing animal models.
Conclusion
In this article, the first SSNMR study of genetically modified intact mouse bone tissue was carried out, in order to investigate the structural role of two specific non-collagenous bone proteins OC and OPN, who are known to improve bone toughness. The mice lacking OC and OPN and their wild-type littermates were probed by a comprehensive set of NMR techniques focusing on i) the mineral, ii) the organic matrix and iii) their interface. We present evidence that the mineral and the organic matrix undergo no major chemical or structural changes. In contrast, a small change takes place in the organic-mineral interface, where amino acids like lysine appear to have increased HA surface exposure in absence of OC and OPN. NMR evidence presented here supports the claim that the contribution of OC and OPN to toughness is related to their presence in the extrafibrillar organic-mineral interfaces, where they reinforce the network of mineralized fibrils by forming dilatational bands.
For the first time, we also report solid state NMR study of specifically and highly 13C enriched mouse bone. We show number of benefits of such 13C enrichment in SSNMR studies of intact bone, which include providing information on amino acid post-translational modifications. Overall, SSNMR shows promise in extracting highly detailed structural and biological information at the molecular-level from intact hydrated tissues of animal models.
Supplementary Material
Acknowledgments
Authors acknowledge the following funding: Partner University Fund between Université Montpellier 2 and Rensselaer Polytechnic Institute; NIAMS AR49635, Ajit Prabhu Fellowship. Authors wish to acknowledge the kind gift of the 13C lysine-enriched bone tissue received from the group of Prof Matthias Mann.
Footnotes
Supporting Information: Additional spectral parameters extracted from the fitting of the 1H-31P HETCOR spectra (Figures S1 to S3 and Table S1), and of 1H-13C CPMAS NMR spectra (Table S2) of WT and OC-OPN-/-;-/- bone specimens. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Martin B. Aging and strength of bone as a structural material. Calcif Tissue Int. 1993;53:S34–9. doi: 10.1007/BF01673400. [DOI] [PubMed] [Google Scholar]
- 2.Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465–75. doi: 10.1359/jbmr.061113. [DOI] [PubMed] [Google Scholar]
- 3.Dempster DW. Osteoporosis and the burden of osteoporosis-related fractures. Am J Managed Care. 2011;17:S164–69. [PubMed] [Google Scholar]
- 4.Kanis JA, Oden A, Johansson H, Borgstrom F, Strom O, McCloskey E. FRAX and its applications to clinical practice. Bone. 2009;44:734–43. doi: 10.1016/j.bone.2009.01.373. [DOI] [PubMed] [Google Scholar]
- 5.Launey ME, Buehler MJ, Ritchie RO. On the mechanistic origins of toughness in bone. Annu Rev Mater Res. 2010;40:25–53. [Google Scholar]
- 6.Gupta HS, Seto J, Wagermaier W, Zaslansky P, Boesecke P, Fratzl P. Cooperative deformation of mineral and collagen in bone at the nanoscale. Proc Natl Acad Sci USA. 2006;103:17741–6. doi: 10.1073/pnas.0604237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fantner GE, Hassenkam T, Kindt JH, Weaver JC, Birkedal H, Pechenik L, Cutroni JA, Cidade GAG, Stucky GD, Morse DE, Hansma PK. Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture. Nat Mater. 2005;4:612–6. doi: 10.1038/nmat1428. [DOI] [PubMed] [Google Scholar]
- 8.Gupta HS, Fratzl P, Kerschnitzki M, Benecke G, Wagermaier W, Kirchner HOK. Evidence for an elementary process in bone plasticity with an activation enthalpy of 1 eV. J R Soc Interface. 2007;4:277–82. doi: 10.1098/rsif.2006.0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fantner GE, Adams J, Turner P, Thurner PJ, Fisher LW, Hansma PK. Nanoscale ion mediated networks in bone: osteopontin can repeatedly dissipate large amounts of energy. Nano Lett. 2007;7:2491–8. doi: 10.1021/nl0712769. [DOI] [PubMed] [Google Scholar]
- 10.Buehler MJ. Molecular nanomechanics of nascent bone: fibrillar toughening by mineralization. Nanotechnology. 2007;18:295102/1–295102/9. [Google Scholar]
- 11.Poundarik AA, Diab T, Sroga GE, Ural A, Boskey AL, Gundberg CM, Vashishth D. Dilatational band formation in bone. Proc Natl Acad Sci U S A. 2012;109:19178–83. doi: 10.1073/pnas.1201513109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romberg RW, Werness PG, Riggs BL, Mann KG. Inhibition of hydroxyapatite-crystal growth by bone-specific and other calcium-binding proteins. Biochemistry. 1986;25:1176–80. doi: 10.1021/bi00353a035. [DOI] [PubMed] [Google Scholar]
- 13.Hunter GK, Kyle CL, Goldberg HA. Modulation of crystal formation by bone phosphoproteins: structural specificity of the osteopontin-mediated inhibition of hydroxyapatite formation. Biochem J. 1994;300:723–8. doi: 10.1042/bj3000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter GK, Haushka PV, Poole AR, Rosenberg LC, Goldberg HA. Nucleation and inhibition of hydroxyapatite formation by mineralized tissue proteins. Biochem J. 1996;317:59–64. doi: 10.1042/bj3170059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinek N, Shahab J, Sodek J, Ringuette M. Is SPARC an evolutionarily conserved collagen chaperone? J Dent Res. 2007;86:296–305. doi: 10.1177/154405910708600402. [DOI] [PubMed] [Google Scholar]
- 16.Vejlens L. Glycosaminoglycans of human bone tissue. I. Pattern of compact bone in relation to age. Calcif Tissue Res. 1971;7:175–90. doi: 10.1007/BF02062605. [DOI] [PubMed] [Google Scholar]
- 17.Dickens F. The citric acid content of animal tissues, with reference to its occurrence in bone and tumour. Biochem J. 1941;35:1011–23. doi: 10.1042/bj0351011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sroga GE, Vashishth D. Effects of bone matrix proteins on fracture and fragility in osteoporosis. Curr Osteoporos Rep. 2012;10:141–50. doi: 10.1007/s11914-012-0103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santos RA, Wind RA, Bronnimann CE. 1H CRAMPS and 1H-31P HetCor experiments on bone, bone mineral, and model calcium phosphate phases. J Magn Reson Ser B. 1994;105:183–7. doi: 10.1006/jmrb.1994.1120. [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, Ackerman JL, Kim HM, Rey C, Barroug A, Glimcher MJ. Nuclear magnetic resonance spin-spin relaxation of the crystals of bone, dental enamel, and synthetic hydroxyapatites. J Bone Miner Res. 2002;17:472–80. doi: 10.1359/jbmr.2002.17.3.472. [DOI] [PubMed] [Google Scholar]
- 21.Wu Y, Ackerman JL, Strawich ES, Rey C, Kim HM, Glimcher MJ. Phosphate ions in bone: identification of a calcium-organic phosphate complex by 31P solid-state NMR spectroscopy at early stages of mineralization. Calcif Tissue Int. 2003;72:610–26. doi: 10.1007/s00223-002-1068-8. [DOI] [PubMed] [Google Scholar]
- 22.Pourpoint F, Diogo CC, Gervais C, Bonhomme C, Fayon F, Dalicieux SL, Gennero I, Salles JP, Howes AP, Dupree R, Hanna JV, Smith ME, Mauri F, Guerrero G, Mutin PH, Laurencin D. High-resolution solid state NMR experiments for the characterization of calcium phosphate biomaterials and biominerals. J Mater Res. 2011;26:2355–68. [Google Scholar]
- 23.Kaflak-Hachulska A, Samoson A, Kolodziejski W. 1H MAS and 1H→31P CP/MAS NMR study of human bone mineral. Calcif Tissue Int. 2003;73:476–86. doi: 10.1007/s00223-002-2111-5. [DOI] [PubMed] [Google Scholar]
- 24.Cho G, Wu Y, Ackerman JL. Spectroscopy detection of hydroxyl ions in bone mineral by solid-state NMR. Science. 2003;300:1123–7. doi: 10.1126/science.1078470. [DOI] [PubMed] [Google Scholar]
- 25.Maltsev S, Duer MJ, Murray RC, Jaeger C. A solid-state NMR comparison of the mineral structure in bone from diseased joints in the horse. J Mater Sci. 2007;42:8804–10. [Google Scholar]
- 26.Kolodziejski W. Solid-State NMR Studies of Bone. Top Curr Chem. 2005;246:235–70. doi: 10.1007/b98652. [DOI] [PubMed] [Google Scholar]
- 27.Singh C, Rai RK, Sinha N. Experimental aspect of solid-state nuclear magnetic resonance studies of biomaterials such as bones. Solid State Nucl Magn Reson. 2013 doi: 10.1016/j.ssnmr.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 28.Zhu P, Xu J, Sahar N, Morris MD, Kohn DH, Ramamoorthy A. Time-resolved dehydration-induced structural changes in an intact bovine cortical bone revealed by solid-state NMR spectroscopy. J Am Chem Soc. 2009;131:17064–5. doi: 10.1021/ja9081028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber F, Boehme J, Scheidt HA, Gruender W, Rammelt S, Hacker M, Schulz-Siegmund M, Huster D. 31P and 13C solid-state NMR spectroscopy to study collagen synthesis and biomineralization in polymer-based bone implants. NMR Biomed. 2012;25:464–75. doi: 10.1002/nbm.1649. [DOI] [PubMed] [Google Scholar]
- 30.Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J Magn Reson. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Ndao M, Ash JT, Breen NF, Goobes G, Stayton PS, Drobny GP. A 13C{31P} REDOR NMR investigation of the role of glutamic acid residues in statherin-hydroxyapatite recognition. Langmuir. 2009;25:12136–43. doi: 10.1021/la901647n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ndao M, Ash JT, Stayton PS, Drobny GP. The role of basic amino acids in the molecular recognition of hydroxyapatite by statherin using solid state NMR. Surf Sci. 2010;604:L39–42. doi: 10.1016/j.susc.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reid DG, Duer MJ, Murray RC, Wise ER. The organic-mineral interface in teeth is like that in bone and dominated by polysaccharides: universal mediators of normal calcium phosphate biomineralization in vertebrates? Chem Mater. 2008;20:3549–50. [Google Scholar]
- 34.Reid DG, Jackson GJ, Duer MJ, Rodgers AL. Apatite in kidney stones is a molecular composite with glycosaminoglycans and proteins: evidence from nuclear magnetic resonance spectroscopy, and relevance to Randall's plaque, pathogenesis and prophylaxis. J Urol. 2011;185:725–30. doi: 10.1016/j.juro.2010.09.075. [DOI] [PubMed] [Google Scholar]
- 35.Duer MJ, Friscic T, Murray RC, Reid DG, Wise ER. The mineral phase of calcified cartilage: its molecular structure and interface with the organic matrix. Biophys J. 2009;96:3372–8. doi: 10.1016/j.bpj.2008.12.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duer MJ, Friscic T, Proudfoot D, Reid DG, Schoppet M, Shanahan CM, Skepper JN, Wise ER. Mineral surface in calcified plaque is like that of bone: further evidence for regulated mineralization. Arterioscler Thromb Vasc Biol. 2008;28:2030–4. doi: 10.1161/ATVBAHA.108.172387. [DOI] [PubMed] [Google Scholar]
- 37.Akiva-Tal A, Kababya S, Balazs YS, Glazer L, Berman A, Sagi A, Schmidt A. In situ molecular NMR picture of bioavailable calcium stabilized as amorphous CaCO3 biomineral in crayfish gastroliths. Proc Nat Acad Sci USA. 2011;108:14763–8. doi: 10.1073/pnas.1102608108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaeger C, Groom NS, Bowe EA, Horner A, Davies ME, Murray RC, Duer MJ. Investigation of the nature of the protein-mineral interface in bone by solid-state NMR. Chem Mater. 2005;17:3059–61. [Google Scholar]
- 39.Wise ER, Maltsev S, Davies ME, Duer MJ, Jaeger C, Loveridge N, Murray RC, Reid DG. The organic-mineral interface in bone is predominantly polysaccharide. Chem Mater. 2007;19:5055–7. [Google Scholar]
- 40.Hu YY, Rawal A, Schmidt-Rohr K. Strongly bound citrate stabilizes the apatite nanocrystals in bone. Proc Natl Acad Sci U S A. 2010;107:22425–9. doi: 10.1073/pnas.1009219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rai RK, Sinha N. Dehydration-induced structural changes in the collagenhydroxyapatite interface in bone by high-resolution solid-state NMR spectroscopy. J Phys Chem C. 2011;115:14219–27. [Google Scholar]
- 42.Nikel O, Laurencin D, Bonhomme C, Sroga GE, Besdo S, Lorenz A, Vashishth D. Solid state NMR investigation of intact human bone quality: balancing issues and insight into the structure at the organic-mineral interface. J Phys Chem C. 2012;116:6320–31. doi: 10.1021/jp2125312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg CM, Bradley A, Karsenty G. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382:448–52. doi: 10.1038/382448a0. [DOI] [PubMed] [Google Scholar]
- 44.Rittling SR, Matsumoto HN, McKee MD, Nanci A, An XR, Novick KE, Kowalski AJ, Noda M, Denhardt DT. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Miner Res. 1998;13:1101–11. doi: 10.1359/jbmr.1998.13.7.1101. [DOI] [PubMed] [Google Scholar]
- 45.Although a recent study suggests that the best preservation conditions for bone tissue on the long term consists in keeping it at -20°C and in 70% ethanol solution (see ref. 27), we have found that the use of ethanol is unsuitable for our mice bone samples, as this strongly alters their macroscopic mechanical properties, and hence some aspects of their structure.
- 46.Aliev AE. Solid-state NMR studies of collagen-based parchments and gelatin. Biopolymers. 2005;77:230–45. doi: 10.1002/bip.20217. [DOI] [PubMed] [Google Scholar]
- 47.Massiot D, Fayon F, Capron M, King I, Le Calve S, Alonso B, Durand JO, Bujoli B, Gan Z, Hoatson G. Modelling one- and two-dimensional solid-state NMR spectra. Magn Reson Chem. 2002;40:70–6. [Google Scholar]
- 48.Mason HE, Kozlowski A, Phillips BL. Solid-state NMR study of the role of H and Na in AB-type carbonate hydroxylapatite. Chem Mater. 2008;20:294–302. [Google Scholar]
- 49.Reid DG, Duer MJ, Jackson GE, Murray RC, Rodgers AL, Shanahan CM. Citrate occurs widely in healthy and pathological apatitic biomineral: mineralized articular cartilage, and intimal atherosclerotic plaque and apatitic kidney stones. Calcif Tissue Int. 2013;93:253–260. doi: 10.1007/s00223-013-9751-5. [DOI] [PubMed] [Google Scholar]
- 50.Chow WY, Taylor AM, Reid DG, Gallagher JA, Duer MJ. Collagen atomic scale molecular disorder in ochronotic cartilage from an alkaptonuria patient, observed by solid state NMR. J Inherited Metab Dis. 2011;34:1137–40. doi: 10.1007/s10545-011-9373-x. [DOI] [PubMed] [Google Scholar]
- 51.Duer MJ. Solid State NMR Spectroscopy: Principles and Applications. Blackwell Science. 2002 [Google Scholar]
- 52.Assignments shown were based on ref. 42 (except for lysine – as detailed later in this article), and include even some of the least abundant amino acids of bone (which had not been represented in ref. 42). For a complete assignment of all C resonances in the amino acid region see ref. 46
- 53.Alexander B, Daulton TL, Genin GM, Lipner J, Pasteris JD, Wopenka B, Thomopoulos S. The nanometre-scale physiology of bone: steric modelling and scanning transmission electron microscopy of collagen-mineral structure. J R Soc Interface. 2012;9:1774–86. doi: 10.1098/rsif.2011.0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dowd TL, Rosen JF, Li L, Gundberg CM. The three-dimensional structure of bovine calcium ion-bound osteocalcin using 1H NMR spectroscopy. Biochemistry. 2003;42:7769–79. doi: 10.1021/bi034470s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knott L, Bailey AJ. Collagen cross-links in mineralizing tissues: a review of their chemistry, function, and clinical relevance. Bone. 1998;22:181–7. doi: 10.1016/s8756-3282(97)00279-2. [DOI] [PubMed] [Google Scholar]
- 56.Yamauchi M, Shiiba M. Lysine hydroxylation and cross-linking of collagen. Methods Mol Biol. 2008;446:95–108. doi: 10.1007/978-1-60327-084-7_7. [DOI] [PubMed] [Google Scholar]
- 57.McDermott A. Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Annu Rev Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 58.Higman VA, Flinders J, Hiller M, Jehle S, Markovic S, Fiedler S, Rossum BJ, Oschkinat H. Assigning large proteins in the solid state: a MAS NMR resonance assignment strategy using selectively and extensively 13C-labelled proteins. J Biomol NMR. 2009;44:245–60. doi: 10.1007/s10858-009-9338-7. [DOI] [PubMed] [Google Scholar]
- 59.Comellas G, Rienstra CM. Protein structure determination by Magic-Angle Spinning solid-state NMR, and insights into the formation, structure, and stability of amyloid fibrils. Annu Rev Biophys. 2013;42:515–36. doi: 10.1146/annurev-biophys-083012-130356. [DOI] [PubMed] [Google Scholar]
- 60.Rienstra CM, Tucker-Kellogg L, Jaroniec CP, Hohwy M, Reif B, McMahon MT, Tidor B, Lozano-Perez T, Griffin RG. De novo determination of peptide structure with solid-state magic-angle spinning NMR spectroscopy. Proc Natl Acad Sci U S A. 2002;99:10260–5. doi: 10.1073/pnas.152346599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zech SG, Wand AJ, McDermott AE. Protein structure determination by high-resolution solid-state NMR spectroscopy: Application to microcrystalline ubiquitin. J Am Chem Soc. 2005;127:8618–26. doi: 10.1021/ja0503128. [DOI] [PubMed] [Google Scholar]
- 62.Jaroniec CP, Filip C, Griffin RG. 3D TEDOR NMR experiments for the simultaneous measurement of multiple carbon-nitrogen distances in uniformly 13C,15N-labeled solids. J Am Chem Soc. 2002;124:10728–42. doi: 10.1021/ja026385y. [DOI] [PubMed] [Google Scholar]
- 63.Graesser DT, Wylie BJ, Nieuwkoop AJ, Franks WT, Rienstra CM. Long-range 19F-15N distance measurements in highly-13C,15N-enriched solid proteins with 19F-dephased REDOR shift (FRESH) spectroscopy. Magn Reson Chem. 2007;45:S129–34. doi: 10.1002/mrc.2126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.