

Abstract
Iron is an essential but potentially harmful nutrient, poorly soluble in aerobic conditions, and not-freely available in the human host. To acquire iron, bacteria have evolved high affinity iron acquisition systems that are expressed under iron limitation often in conjunction with virulence determinants. Because excess iron can be dangerous, intracellular iron must be tightly controlled. In mycobacteria, IdeR functions as a global iron dependent transcriptional regulator, but because inactivation of ideR is lethal for Mycobacterium tuberculosis, it has not been possible to use genetics to fully characterize this protein’s function or examine the requirement of iron regulation during tuberculosis infection. In this work, a conditional M. tuberculosis ideR mutant was generated and used to study the basis of IdeR’s essentiality. This investigation uncovered positive regulation of iron storage as a critical aspect of IdeR’s function in regular culture and a prominent factor for survival under stresses associated with life in macrophages. Moreover, this study demonstrates that IdeR is indispensable in the mouse model of tuberculosis, thereby linking iron homeostasis to virulence in M. tuberculosis.
Introduction
Most living organisms, including Mycobacterium tuberculosis (Mtb) the causative agent of tuberculosis, are strictly dependent on iron. As a cofactor of enzymes involved in basic cellular functions ranging from respiration to DNA replication, iron is essential for life. Due to the poor aqueous solubility of ferric ion (Fe3+) in the presence of oxygen and at neutral pH, free iron is not found in the mammalian host, but is sequestered in complexes with iron binding proteins such as transferrin, lactoferrin and ferritin or bound to protoporphyrins in heme and hemoproteins (Weinberg, 1984). Therefore, to establish a productive infection, pathogens must be able to actively compete for iron in the host. To obtain iron, Mtb synthesizes and secretes siderophores (mycobactins), small Fe3+ chelators, that sequester iron and deliver it to the bacterium, via specialized Fe3+-siderophore transporters (Snow, 1970; Gobin et al., 1995; Rodriguez and Smith, 2006). Although essential, iron can also be harmful, because it readily catalyzes the generation of toxic reactive oxygen species (ROS), from normal products of aerobic respiration in the Fenton reaction (Keyer, 1996). To set the boundaries between iron sufficiency, deficiency and toxicity, bacteria have evolved iron sensors that tightly control intracellular iron. This control is implemented at the level of transcription. Transcriptional regulators of the Fur (ferric uptake regulator) or the DtxR (diphtheria toxin regulator) families govern iron metabolism in bacteria (Waldron and Robinson, 2009). The mycobacterial iron dependent regulator IdeR is a metal binding transcriptional regulator of the DtxR family. Functional and structural studies support a model for IdeR-mediated transcriptional repression in which iron ligation stabilizes the dimeric form of the protein, and generates conformational changes that optimally orient the DNA-binding helix in each monomer, for DNA binding. Two dimers bind to promoters containing a 19 bp conserved sequence (Gold et al., 2001), forming a dimer-dimer DNA complex (Feese et al., 2001; Chou et al., 2004; Semavina et al., 2006). Recently, a structural binding site for Zn(II) was unveiled in IdeR in addition to the regulatory Fe(II) binding site. Zn(II) binding was found to increase the promoter binding affinity of IdeR-Fe(II) possibly by increasing the efficiency of dimer formation (Stapleton et al., 2013).
Disruption of ideR is possible in the saprophytic mycobacterium, Mycobacterium smegmatis, but is lethal for Mtb, implying an essential role for IdeR in this pathogen (Rodriguez et al., 2002). Previously, we obtained a rare Mtb ideR mutant (ST22), rescued by secondary mutation(s) that seem to reduce iron import, since as a result of this mutation(s) ST22 grew very poorly in low iron medium (Rodriguez et al., 2002). Nonetheless, transcriptomic analyses of ST22, compared to its ideR complemented derivative and the parental strain, grown in high iron, revealed the requirement of IdeR for iron dependent repression of more than 50 genes involved in diverse functions including iron metabolism, transcriptional regulation, stress response, central metabolism and secretion (Rodriguez et al., 2002). Even though, studies with ST22 provided information about the IdeR regulon, it is not possible to use this mutant to fully understand the consequences of IdeR’s depletion in Mtb, due to the complicated effects of the suppressor mutation(s), whose identification has proven very difficult. In this work, we generated a conditional ideR mutant of Mtb and utilized it to determine the direct effects of IdeR depletion in culture and also in macrophages and mice. The results revealed novel aspects of IdeR essentiality and demonstrate the requirement of this regulator for intracellular multiplication and mice colonization.
Results
Generation of an ideR conditional mutant in Mtb
An ideR conditional mutant was generated using the TetR/Pip OFF system (Boldrin et al., 2010). The native promoter of ideR was replaced by a Pip-repressible promoter by homologous recombination. This was done in an H37Rv-derived strain (TB38.2) in which a copy of the Pip and TetR repressors had been integrated in the chromosome at the attB site (Boldrin et al., 2010) (Fig. S1). In this strain, pip expression is driven by a TetR-dependent promoter. ATc induces the Pip-encoding gene and allows tight repression of ideR. Replacement of the native ideR promoter by the Pip repressible promoter was confirmed by PCR amplification and sequencing of a fragment encompassing the Pip dependent promoter and ideR. The resulting strain was named ST217. ATc dependent IdeR synthesis was examined by Immunoblot of cell lysates prepared from the ideR conditional mutant (ideRcm) and parental strain treated or not with sub-inhibitory concentrations of ATc and cultured in standard 7H9 (an iron rich medium) or in minimal medium (MM) containing a low (~2μM) (LIMM) or high (50 μM) concentration of FeCl3 (HIMM). Without ATc, IdeR levels in the ideRcm and the wild type were similar (Fig S2). In contrast, addition of ATc repressed synthesis of IdeR in the ideRcm independently of the medium’s composition and iron concentration (Fig. 1). Culture in the presence of 600 ng/ml ATc for 48 hr resulted in reduction in IdeR to a level below the limit of detection of Immunoblot. ATc exposure did not affect ideR expression in the parental strain under any condition tested (data not shown). Based on these results, it was concluded that the ideRcm strain was ideal to study the direct effects of IdeR’s depletion.
Figure 1. IdeR synthesis is repressed by ATc in the ideR conditional mutant.

Cells were grown in 7H9 (top panel), MM low iron (middle panel) and MM high iron (bottom panel) without or with ATc (ng.ml−1) for one or two days. Protein extracts were obtained as described in Materials and Methods. Levels of IdeR were detected by Immunoblotting using anti-IdeR antibodies. SirR was used as control for an unregulated protein and was detected using anti-SirR antibodies.
IdeR and iron sensing
To adapt to iron limitation, Mtb up-regulates iron uptake genes such as those involved in siderophore synthesis and transport. When iron is available, iron uptake genes are repressed and iron storage genes are induced (Rodriguez et al., 2002). IdeR-binding sites are found in the promoters of iron acquisition and storage genes, and the expression of those genes is no longer iron responsive in the suppressor-rescued-ideR mutant strain (ST22) indicating that they are regulated by IdeR (Gold et al., 2001; Rodriguez et al., 2002). To validate the effect of ideR repression on iron dependent gene regulation in the conditional mutant, RNA was extracted from the ideRcm cultured under permissive (no ATc) and non-permissive (with ATc) conditions as well as from the parental strain. In each case the strains were cultured in iron sufficient medium, since iron ligation is necessary for IdeR’s promoter binding activity. Transcripts of the following iron regulated representative genes were measured by qRT-PCR: mbtB, the first gene in the siderophore synthesis cluster mbtB-H; (Quadri et al., 1998), irtA encoding a subunit of the iron-siderophore importer IrtAB (Rodriguez and Smith, 2006) and the ferritin encoding gene bfrB (Cole et al., 1998). mbtB and irtA are normally repressed by iron, whereas bfrB is induced by iron (Rodriguez et al., 2002). Expression of mbtB and irtA was repressed while that of bfrB was induced in the ideRcm not treated with ATc just as in the parental strain. The opposite was observed in the presence of ATc: mbtB and irtA were derepressed, (30 and 10 fold respectively), while bfrB was down-regulated (10 fold), in the ideRcm in comparison to the parental strain (Fig. 2). These results demonstrated that Mtb’s normal transcriptional response to iron is abolished when ideR expression is repressed.
Figure 2. Effect of ideR repression on the expression of iron regulated genes.

mRNA transcripts of, (A) mbtB, (B) irtAB and (C) bfrB obtained from cells grown in high iron medium with or without ATc were analyzed by quantitative RT-PCR with SYBR green. The data are expressed as the relative quantity of the respective mRNA normalized to 16S ribosomal RNA and presented as the arithmetic means ± standard deviations from three biological replicates. * p < 0.01
Constitutive expression of siderophore synthesis genes was linked to uncontrolled mycobactin production. Approximately 100 times more mycobactin was extracted from the ideRcm cultured with ATc than without ATc or from the parental strain (Fig. 3). Taken together, the results of qRT-PCR and the quantification of mycobactin confirmed that ideR repression leads to iron unresponsiveness in Mtb.
Figure 3. Constitutive synthesis of siderophores in IdeR deficient cells.
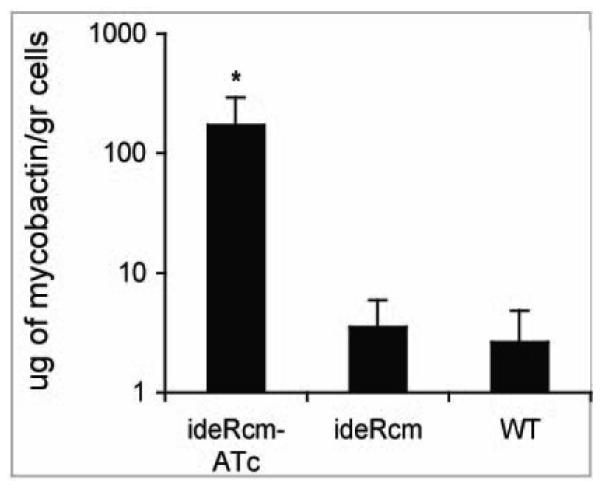
Cell associated mycobactin was extracted from (WT) and the ideR conditional mutant (ideRcm) strain cultured with or without ATc in high iron medium and quantified as described in materials and methods. The data shows the mean±standard deviations of three biological replicates. * p < 0.01
IdeR is required for growth in standard culture conditions
Growth analyses confirmed that ideR repression prevented normal growth of Mtb in Middlebrook 7H9 broth (Fig. 4A). Thus, explaining the failure of previous studies in obtaining an ideR mutant in that medium. Middlebrook 7H9 and 7H10 agar are complex media and contain high and variable levels of iron (~80-100 μM). To determine whether the requirement of IdeR in standard culture was related to the nutrient composition or to the content of iron in the medium, we examined how ideR repression affected the capacity of Mtb to multiply in a defined minimal medium (MM) containing limited or sufficient concentrations of iron. Cells that had been grown in 7H10 without ATc were transferred to a low (LIMM) or high iron minimal medium (HIMM) in the presence or absence of ATc. Repression of ideR did not affect Mtb’s replication in MM when iron was limited (Fig. 4B). However, when iron was abundant Mtb’s growth was greatly diminished (Fig. 4C). Thus, IdeR is indispensable in vitro under conditions of high availability of iron. It is important to note that a direct link between iron and IdeR essentiality could not be established with the original ideR mutant ST22, since that strain showed just the opposite phenotype in culture that is, normal growth in high iron and defective replication in low iron (Rodriguez, et al 2002), probably because the secondary mutation conferred iron deficiency, thereby, confounding the effects of high iron. This, highlights once more the relevance of using the conditional mutant to study IdeR’s function.
Figure 4. Effect of ideR repression on adaptation to iron changes in vitro.

Wild type parental strain (squares) and the ideR conditional mutant (circles) were grown in 7H10 and then transferred to 7H9 (panel A), MM containing low (2 μM) (panel B), or high (50 μM) FeCl3 (panel C). Without ATc (open symbols) and with ATc (filled symbols). Growth was monitored by the increase in the optical density at 540 nm. Data are expressed as the mean ± standard deviations from three biological replicates.
IdeR is required to control intracellular iron
So far, the results show that without IdeR, Mtb is unable to sense iron, repress the iron acquisition machinery and multiply under conditions of iron sufficiency. Given the known toxicity of free iron, unrestricted iron uptake and enhanced free iron in the cell might be the cause for the poor growth of IdeR deficient cells in high iron culture. Measurement of total and free iron content showed highly increased iron levels in IdeR depleted cells (Fig. 5). Enhanced intracellular iron in the absence of IdeR correlated with increased sensitivity to streptonigrin, an antibiotic that requires iron for its bactericidal activity and therefore is often used to indirectly assess free-iron levels in the cell (Fig. S3). Collectively, the results indicate that IdeR repression results in unrestrained uptake and harmful accumulation of iron in the cell.
Figure 5. Iron content.
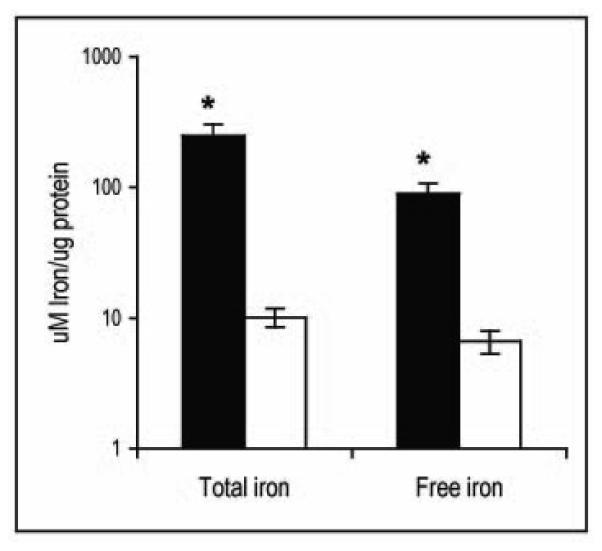
Total and free iron in the wild type parental strain (open bars) and in the ideR conditional mutant (filled bars) cultured in 7H9 in the presence of ATc was measured as described in materials and methods. Data are expressed as the mean ± standard deviations from three biological replicates. * p < 0.01
IdeR and iron storage
The Mtb genome encodes a ferritin (BfrB) and a bacterioferitin (BfrA), two proteins related to the ferritin superfamily of iron storage proteins. Expression of bfrB is highly induced in high iron and this induction requires IdeR (Rodriguez et al., 2002). bfrA on the other hand is modestly induced in high iron but this induction is also IdeR dependent (Gold et al., 2001). Previously, we showed that BfrB functions as the primary iron storage protein. On the other hand obvious phenotypes were not observed in a bfrA mutant, so the role of this protein is unknown (Pandey and Rodriguez, 2012). The increased sensitivity to iron and the enlarged intracellular iron pool exhibited by the ideR conditional mutant under non-permissive conditions are features observed also in a bfrB mutant (Pandey and Rodriguez, 2012) which suggests that compromised iron storage could contribute to the iron hypersensitivity of IdeR deficient cells. To determine whether restoring iron storage alleviates the effects of deregulated iron uptake in the absence of IdeR, the ideRcm was transformed with a low copy plasmid carrying bfrA or bfrB transcribed from an IdeR independent promoter. Expression of bfrB, but not bfrA greatly restored growth of the ideRcm cultured with ATc in high iron medium (Fig.6). These results indicate that reinstating iron storage suppress the effects of deregulated iron uptake, consistent with iron toxicity underlying the phenotype of poor growth in high iron. From the results of these experiments we conclude that IdeR mediated upregulation of ferritin is critical for Mtb adaptation to conditions of iron sufficiency.
Figure 6. Effect of restoring iron storage on the sensitivity of IdeR deficient Mtb to high iron.

Growth of the ideR conditional mutant in iron rich medium in the absence of ATc (◆), in the presence of ATc (■), and in the presence of ATc when expressing bfrA
 or bfrB (●) constitutively.
or bfrB (●) constitutively.
IdeR, iron and oxidative stress
Iron plays a determinant role in resistance to oxidative stress due to its ability to catalyze the generation of hydroxyl radicals (OH.) from H2O2 in the Fenton reaction (Imlay et al, 1988). The results shown above indicate that IdeR deficiency ultimately leads to increased levels of intracellular iron which may accelerate the Fenton reaction. To test this hypothesis, we examined the ability of the ideR conditional mutant to withstand exposure to H2O2 under iron sufficiency. The ideRcm and the parental strain treated or not with ATc were subjected to low concentrations of hydrogen peroxide and the proportion of survivors was determined by plating for CFUs. Repression of ideR rendered Mtb highly sensitive to hydrogen peroxide (Fig. 7A). However, addition of the cell permeable iron chelator 2,2’ dipyridyl (DPI) or constitutive expression of bfrB protected the ideRcm from peroxide, indicating the involvement of iron in potentiating peroxide toxicity (Fig. 7B). These results support the interpretation that enhanced free iron exacerbates oxidative stress in IdeR deficient cells. This is in agreement with the increased sensitivity to oxidative stress exhibited by a bfrB mutant of Mtb, reported previously (Pandey and Rodriguez, 2012). Constitutive expression of bfrA modestly enhanced resistance to oxidative stress suggesting that BfrA may function under oxidative stress. Thus, the data indicates that through its role in maintaining iron homeostasis, IdeR is required for Mtb’s ability to overcome oxidative stress.
Figure 7. Sensitivity of Mtb strains to hydrogen peroxide.

(A). Wild type parental strain (WT) and the ideR conditional mutant (ideRcm) cultured with or without ATc were treated with hydrogen peroxide for 48 hrs. The percentage of survivors was determined after plating for CFUs and comparing the number of colonies obtained to that of a non treated culture. (B). Shown is the pecentage of survival of the ideRcm without or with DPI, ideRcm expressing bfrB constitutively (ideRcm-bfrB) and ideRcm expressing bfrA constitutively (ideRcm-bfrA) in the presence of ATc and hydrogen peroxide. Data are the mean ± standard deviations from three biological replicates. * p < 0.01.
IdeR is required for resistance to reactive nitrogen intermediates (RNI)
Together with hydrogen peroxide, nitric oxide (NO) produced by the macrophage nitric oxide synthase is mycobacteriocidal and as such has an important contribution to host defense against Mtb (Nathan and J. B. Hibbs, 1991; Chan et al., 1995). Mtb, however, has evolved various mechanisms to resist NO toxicity (Lee et al., 2009). To determine whether IdeR contributes to NO resistance, we evaluated the sensitivity of the ideR conditional mutant to acidified sodium nitrite. At an acidic pH, nitrite is partially protonated to nitrous acid which dismutates to generate NO and NO2 (Conner and Grisham, 1995). As depicted in figure 8A, ideR repression drastically potentiates killing by nitrite. To determine if this effect was dependent on iron, we examined whether iron chelation or constitutive expression of bfrB or bfrA suppressed nitrite toxicity in IdeR deficient cells. Although, DPI addition had no significant effect, BfrB and to a less extent BfrA protected from killing by nitrite, suggesting that iron potentiates NO toxicity (figure 8B). Consistent with a role of BfrB and BfrA in protection against NO toxicity was the strong and moderate sensitivity to nitrite observed in the bfrB and bfrA mutants of H37Rv, respectively (Fig. S4). These results unveiled a previously unknown role of ferritin and bacterioferritin in resistance of Mtb to NO. They also indicate that as activator of bfrB and bfrA, IdeR, is vital for Mtb’s resistance to RNI.
Figure 8. Sensitivity of Mtb strains to Nitric Oxide.

(A) Wild type parental strain and the ideR conditional mutant (ideRcm) cultured with or without ATc were treated with acidified Sodium Nitrite for 48 hrs. The percentage of survivors was determined after plating for CFUs and comparing the number of colonies obtained with that of a non treated culture. (B) Shown is the pecentage of survival of ideRcm without or with DPI, ideRcm expressing bfrB constitutively (ideRcm-bfrB) and ideRcm expressing bfrA constitutively (ideRcm-bfrA) in the presence of ATc and Sodium Nitrite. Data are the mean ± standard deviations from three biological replicates. * p < 0.01
IdeR is necessary for proliferation of M. tuberculosis in human macrophages
Induction of ideR expression has been detected in Mtb isolated from macrophages (Hobson et al., 2002) suggesting that IdeR may be active intracellularly. However, the IdeR requirement in macrophages is unknown. Based on the results obtained in this study, IdeR functionality in the macrophage will likely depend on the availability of iron in the phagosome. The requirement of siderophores for growth in macrophages (De Voss et al., 2000) may be interpreted as the phagosome being a low iron environment. However, induction of bfrB in intracellular bacteria (Homolka et al., 2010) and direct measurement of iron in the phagosome (Wagner et al., 2005) suggest that the resting macrophage-phagosome is not iron limiting. To determine whether IdeR is necessary for Mtb to multiply in macrophages, we employed the human promyelomonocytic cell line THP-1 after PMA induced differentiation into macrophages. To control ideR expression, ATc was added to the macrophage culture medium and maintained during the experiment. Control infections without ATc and with the parental strain under identical conditions were conducted simultaneously. The ideRcm replicated in macrophages similarly to the wild type in the absence of ATc. Addition of ATc had no effect on the replication of the wild type strain (Fig. 9A), however, it impaired proliferation of the ideRcm. Furthermore, the decline in colony forming units (CFUs) recovered from macrophages infected with the ideRcm in the presence of ATc suggested that absence of IdeR leads to death of Mtb in these cells (Fig. 9A). Considering the increased sensitivity of IdeR-depleted bacilli to oxidative stress in vitro, and since inactivated THP-1 cells produce little or no NO (Sato and Tomioka, 1999), we considered whether the oxidative burst may be responsible for the death of the ideRcm in these cells. To investigate that possibility, THP-1 cells were treated with DPHI, an inhibitor of the NADPH oxidase responsible for the phagocyte defensive-oxidative burst. We also examined whether the ideRcm constitutively expressing bfrA and bfrB had improved survival in macrophages. Inhibiting the oxidative burst and restoring iron storage alone or in combination rescued IdeR-depleted cells and allowed some growth (Fig, 9B) however, they were not sufficient to restore normal bacterial proliferation. These results demonstrate that IdeR deficiency prevents Mtb from adapting to the phagosome environment and proliferate. The inability of IdeR lacking Mtb to multiply in THP-1 is probably due to altered expression of genes essential for intracellular replication other than ferritins since, in contrast to the observations in culture, restoring iron storage under oxidative stress did not allow normal replication in macrophages.
Figure 9. Replication of Mtb strains in THP-1 cells.

THP-1 cells were induced to differentiate into macrophages and infected with (A) Wild type parental strain (triangles) or the ideR conditional mutant (circles) plus ATc (filled symbol) or minus ATc (open symbols) as described in Materials and Methods. Data shows the number of CFUs recovered from infected macrophages at increasing time intervals. (B) Shown is the number of CFUs recovered from macrophages infected with the ideR conditional mutant in the absence (○) or presence of ATc (●), with ATc and DPHI (◇) ideRcm expressing bfrB constitutively (□) with ATc, ideRcm expressing bfrB constitutively with ATc plus DPHI (△) and ideRcm expressing bfrA constitutively (□) with ATc. Each day for seven days after infection, cells were lysed and released mycobacteria were plated onto 7H10 to determine number of CFUs. Data are the mean ± standard deviations from three biological replicates.
IdeR is indispensable during mouse infection
To assess whether Mtb requires IdeR to establish a successful infection, we used the mouse model. Female C57BL-6 mice were aerosol infected with the ideRcm or the wild type parental strain (TB38.2) and fed a diet containing or not the ATc analog doxycycline (doxy). At the indicated time intervals post infection, 3 mice were sacrificed. Lung homogenates were plated for CFUs in 7H10 agar without ATc. Similar numbers of CFUs were obtained from the lungs of mice infected with the parental strain fed the regular or the doxy containing diet, indicating that doxy had no effect on the course of the infection with the wild type strain (Fig 10). Animals infected with the ideRcm showed deficient lung colonization as bacteria were present at numbers bellow the detection limit of the assay (12 bacteria/lung) at early time points (1 day and 2 weeks post infection) whether they were fed a regular diet or a doxy containing diet (data not shown). This was despite the ideRcm and the wild type strain inocula having the same number of viable cells. This indicated that both repression of ideR and also expression driven by a heterologus promoter decreased Mtb’s resistance to innate host defense. At later time points bacilli were recovered from the lungs of mice infected with the ideRcm and fed a regular diet (although, at a lower frequency than the parental strain). At nine weeks post infection a low number of colonies (2.5 log less than the wild type) were recovered from the lungs of one of the three mice fed the doxy diet whereas the lungs from the other two mice had no colonies (Fig 10). In a replicate experiment we decided to examine ideR expression in colonies recovered at 9 weeks from animals infected with the ideRcm and fed the doxy containing diet. Protein extracts from twenty colonies grown in vitro, with and without ATc were analyzed by Western Blot for ATc dependent IdeR synthesis as done before (Fig. 1). All of the colonies had lost repression of ideR. Subsequent PCR amplification and sequence analysis of the Pip repressible promoter driving the expression of ideR, identified different point mutations in the Pip operator which most likely are responsible for the loss of ideR repression. This indicates that there is a strong selection pressure on IdeR deficient bacteria and further that IdeR is absolutely essential during infection.
Figure 10. Replication of Mtb strains in mouse lungs.

Mice were aerosol infected with the same number of viable cells of the ideR conditional mutant (ideRcm) and the parental wild type strain (WT). A group of mice infected with each strain was fed doxy 2000 ppm in the food for the duration of the experiment while another group was fed a regular diet. At the indicated time points 3 mice were sacrificed, the lungs homogenized and dilutions of the homogenate plated on 7H10 without ATc to enumerate CFUs. WT regular diet (open bars), WT doxy containing diet (striped bars), ideRcm regular diet (dark bars), ideRcm doxy diet (grey bars). Although no colonies were recovered from mice infected with the ideRcm fed doxy at 5 weeks, the detection limit of the assay (12 colonies/lungs) was plotted to introduce a visible bar. Data shows the mean and standard deviations of the CFUs recovered from the lungs of 3 infected animals. The experiment was repeated twice with similar results.
Discussion
This investigation was conducted to understand the basis of IdeR essentiality using a genetic approach. An Mtb conditional mutant was created in which ideR expression is down-regulated by addition of ATc to the medium. The combined effects of ideR repression and the natural turn-over of the protein resulted in decrease of cellular IdeR to undetectable levels, enabling us to examine IdeR’s role in culture and during infection. IdeR deficiency led Mtb into an iron unresponsive state. Despite abundance of iron, genes like the siderophore synthesis and transport genes that should be repressed to limit iron uptake were derepressed, whereas the ferritin encoding gene bfrB, which is required in high iron was not induced. As a consequence, siderophore synthesis was no longer iron repressed, mycobactin accumulated and intracellular iron increased. Consequently, iron availability in the medium was found to be the main determinant of Mtb’s requirement for IdeR in culture. IdeR-deficient Mtb supplied with abundant iron exhibited a marked growth defect, whereas limiting the amount of iron available in the medium or restoring the capacity to store surplus iron by expressing bfrB partially restored growth of the mutant in high iron medium. Considering these results we can now understand how the ideR mutant was originally obtained (Rodrigez et al 2002). The selection for this mutant was done under iron sufficiency. It is possible that in those conditions the suppressor mutation(s) may have restricted iron assimilation rescuing the ideR mutant while leading to iron deficiency. This is consistent with the normal growth of that strain in high iron medium and the deficient growth under iron limitation (Rodriguez et al 2002).
Surplus free iron can cause oxidative stress due to the propensity of iron to interact with hydrogen peroxide and superoxide (species that inevitably rise from partial reduction of molecular oxygen during aerobic respiration), generating hydroxyl radicals, which rapidly diffuse and damage all biomolecules, including DNA (Imlay et al., 1988). IdeR deficient cells were exquisitely sensitivity to peroxide and this effect could be suppressed by iron chelation or restoring iron storage, indicating that increased intracellular iron in IdeR deficient cells is fueling the Fenton reaction (Fig. 11). IdeR depletion resulted also in increased sensitivity to NO and this effect was suppressed by bfrB, and to a less extend by bfrA. This finding combined with the increased sensitivity to NO of the individual H37Rv derived bfrB and bfrA mutants (Fig S4) indicated that ferritin and bacterioferitin are determinants of NO resistance and that as a positive regulator of those genes, IdeR is required for NO resistance. Although, it is beyond the scope of this study to define the precise events underlining the enhanced sensitivity of IdeR deficient cells to NO, it is worth noting that it has been long known from studies in Escherichia coli that NO potentiates peroxide toxicity in an iron dependent manner (Pacelli et al., 1995). Various mechanisms have been postulated to explain this effect. On one hand peroxynitrous acid generated by the reaction of NO with superoxide modifies iron sulfur clusters releasing iron, thereby increasing the levels of intracellular iron and accelerating the Fenton reaction (Pacelli et al., 1995). In addition, by binding to heme in the quinol oxidases, NO blocks respiration leading to accumulation of NADH, and diverting the electron flow towards reduction of ferric iron thereby, replenishing ferrous iron needed for the Fenton reaction (Woodmansee and Imlay, 2003). Protection by BfrB and BfrA from NO toxicity suggested surplus iron as the potentiator of NO toxicity, however, iron chelation with DPI was not protective. It is possible that BfrA and BfrB protect from NO toxicity in other ways in addition to their role in iron sequestration. For instance, being a heme containing protein, BfrA could have a protective effect by scavenging NO. Additional studies are needed to clearly establish the mechanism by which ferritin and bacterioferritin protect Mtb from NO toxicity.
Figure 11. IdeR is responsible for maintaining iron homeostasis in Mtb.

When iron is available (left panel) and the cell has satisfied its iron needs, IdeR binds Fe+2 and is active as repressor of siderophore synthesis, export and import genes and as activator of iron storage genes. IdeR mediated response to iron is required to prevent iron overload and iron mediated toxicity. In the absence of IdeR, (right panel), Mtb has no control over the amount of iron taken-in and has no storage capability. In consequence, free iron increases in the cell, accelerating the Fenton chemistry that produces toxic oxygen radicals which damage all biomolecules, including DNA.
The use of the conditional ideR mutant allowed us to demonstrate the requirement of IdeR for intracellular growth. IdeR deficient mycobacteria were killed in non-activated THP-1 cells. This was at least in part due to iron mediated oxidative damage since inhibiting the macrophage oxidase as well as restoring iron storage protected Mtb from killing. These results clearly support the Fenton reaction in the phagosome of THP-1 cells as an effective defense against Mtb. However, reducing oxidative stress was not sufficient to completely restore normal bacterial replication, suggesting that IdeR’s role in macrophages extends beyond protection against oxidative damage. Considering that IdeR regulates the expression of many more genes besides those involved in iron metabolism (Rodriguez, et al 2002), it is likely that regulation of some of those genes is essential for Mtb to manipulate the macrophage environment and create a niche that supports proliferation. Additional studies using various types of macrophages are needed to identify the regulatory pathways controlled by IdeR that are required for intracellular growth. Since IdeR activity is dependent on iron, the requirement for IdeR in THP-1 implies that there is sufficient iron available for IdeR activity including upregulation of bfrB. This finding supports the phagosome in naive macrophages as a compartment where Mtb can achieve iron sufficiency, as previously suggested by the direct assessment of the mycophagosome metal content (Wagner et al., 2005). Mtb’s ability to manipulate endosomal trafficking and maintain selective fusion of the phagosome with vesicles containing iron loaded transferrin, which Mtb can efficiently use as iron source (Clemens and Horwitz, 1996; Olakanmi et al., 2002; Wagner et al., 2005), is likely critical for Mtb to achieve iron sufficiency. Furthermore, satisfying its iron requirement may be a strong driving force behind Mtb’s manipulation of the macrophage environment. One of the antimicrobial effects of macrophage activation may indeed be preventing the bacterium from easily accessing transferring-bound iron by overcoming the phagosome-lysosome fusion arrest. This idea is consistent with the decreased iron detected in activated macrophages’ phagosomes (Wagner et al., 2005).
Given the role of IdeR in resistance to oxidative and NO stress and the deficient replication of IdeR depleted cells in macrophages, it was not unexpected that the absence of IdeR will attenuate Mtb growth in mice. However, the colonization defect of the ideR conditional mutant under permissive conditions was surprising. We wandered whether the strain had loss phthiocerol dimycocerosates (PDIMs) production as observed in some attenuated variants of H37Rv (Kirksey et al 2011). However, no difference in PDIMs production in the ideR conditional mutant and the parental strain was detected (data not shown). Thus, we concluded that expression of ideR driven from an heterologous promoter decreased the ability of Mtb to colonize the lungs, and ideR depletion impeded normal replication. IdeR deficiency posed such a strong selective pressure that in mice fed doxy, bacteria that survive were also the ones able to restore ideR expression. Although we did not analyze all the survivors, a large proportion of them had mutations that allowed them to synthesize ideR even in the presence of ATc. This could be due to instability of the Tet/Pip repression system. Alternatively, it is conceivable that increased levels of intracellular iron result in high mutation rates due to enhanced ROS mediated DNA damage as previously observed in E. coil (Keyer, 1996). Consequently, mutations that restored IdeR expression in the ideR conditional mutant as well as the suppressor mutations in ST22 may be reflecting a high mutagenic rate in iron overloaded cells. This is a possibility that is currently under investigation.
According to the results of this report, lack of IdeR leads Mtb into self-inflicted, lethal “iron intoxication” that can be alleviated by restoring the normal capacity to store iron. However, during infection IdeR seems to have a more pleotropic, indispensable role. We previously reported that enhanced intracellular free iron as a result of defective iron-storage in a bfrB mutant, dramatically potentiated the bacteriocidal effects of several clinically relevant antibiotics (Pandey and Rodriguez, 2012). Since IdeR is necessary for induction of bfrB, inhibition of IdeR might have a similar effect. This is relevant in the context of developing new strategies to eliminate Mtb.
Experimental Procedures
Bacterial strains, media and chemicals
Escherichia coli strains JM109 and XL-10 (Stratagene) used for cloning were grown in Luria-Bertani (LB) broth. Mtb strains were maintained in 7H10 agar or 7H9 broth (Difco) supplemented with 0.2% glycerol, 0.05% Tween 80, 0.5% bovine serum albumin (BSA), 0.2% dextrose and 0.085% NaCl (ADN). Minimal medium (MM) was used to grow Mtb under iron defined conditions. MM contains 0.5 % w/v asparagine, 0.5% w/v KH2P04, 0.2% glycerol, 0.05% Tween-80 and 10% ADN. The pH was adjusted to 6.8. To lower the trace metal contamination, the medium was treated with Chelex-100 (Bio-Rad) according to the manufacturer’s instructions. Chelex was removed by filtration and then the medium was supplemented with 0.5 mg.L−1 ZnCl2, 0.1 mg.L−1 MnS04, and 40 mg.L−1 MgS04. This medium contains less than 2 μM residual iron as determined by atomic absorption spectroscopy.
Where indicated, antibiotics were used at the following concentrations: Hygromycin (Hyg) 100 μg.ml−1, Streptomycin (Strep) 20 μg.ml−1, Spectinomycin (Spec) 75 μg.ml−1 and Anhydrotetracycline (ATc) as indicated.
Generation of the ideR conditional mutant
The TetR/Pip OFF system (Boldrin et al., 2010) was used to generate a conditional mutant of ideR. The native ideR promoter was replaced by the Streptomyces coelicolor pristinamycin responsive protein (Pip) dependent promoter Pptr by homologous recombination. Briefly 500 bp from the 5’ region of ideR were amplified by PCR using the primers IdeR-Rev524 and IdeR-Fw4 (Table S1) including restriction sites for NsiI and NheI in the forward and reverse primers respectively, and inserted into the suicide vector pFRA50 (Boldrin et al., 2010) in front of the repressible promoter Pptr, generating plasmid pSM811. pSM811 was electroporated into the H37Rv derivative strain,TB38.2, which harbors the Tet/Pip system (Boldrin et al., 2010) in the integrative plasmid pFRA42B (Boldrin et al., 2010). Transformants were selected on 7H10 agar containing Hyg, the selection marker in pFRA50. Transformants were screened by colony PCR using a forward primer specific for Pptr (PptrIdeRFw7) and a reverse primer from the 3’ end of ideR (PptrIdeRRev900) to identify clones in which the ideR promoter has been replaced by Pptr by homologous recombination. PCR products were sequenced using the same primers used for amplification. Analysis of DNA sequences was performed using Vector NTI (Invitrogen) and the correct strain was named ST217.
Complementation with bfrA or bfrB
The Mtb bfrA and bfrB were PCR amplified using primers (BfrAFwpMV261, BfrARevpMV261 for bfrA and BfrBFwpMV261, BfrBRevpMV261 for bfrB (Table S1) and the PCR products were cloned into the BamHI site of pMV261 by In-fusion cloning (Clontech). In this plasmid, bfrA or bfrB expression is driven by the hsp60 promoter. The resulting plasmids pSM866 and pSM865 (for those containing bfrA and bfrB respectively) were then electroporated into the ideR conditional mutant strain ST217 and transformants were selected in Kan containing medium. The resulting strains were named ST236 and ST248 (ST217 constitutively expressing bfrB and bfrA, respectively) (Table 1).
Table 1.
Plasmids and strains
| Name | Specification | Source |
|---|---|---|
| pFRA50 | Suicide vector containing the Pptr
promoter. |
(Boldrin et al., 2010) |
| pMV261 | Expression vector | (Stover et al., 1991) |
| pSM811 | pFRA50 containing 500 bp of the 5 end of ideR cloned in front of the Pptr promoter. |
This work |
| pSM865 | Mtb bfrB in pMV261 | This work |
| pSM866 | Mtb bfrA in pMV261 | This work |
| Mtb Strains | This work | |
| TB38.2 | H37Rv with tetR and pip integrated in the chromosome |
(Boldrin et al., 2010) |
| ST214 | Δ bfrB | (Pandey, 2012) |
| ST215 | Δ bfrA | (Pandey, 2012) |
| ST217 | ideR conditional mutant (ideRcm) | This work |
| ST236 | ST217 carrying pSM865 | This work |
| ST248 | ST217 carrying pSM866 | This work |
Immunodetection
Protein extracts were prepared from bacteria grown in 7H9 and MM with or without 50 μM FeCl3 in the presence or absence of ATc. Cultures were harvested by centrifugation at time 0, 24 and 48 hours after inoculation. The bacterial pellet was washed twice with PBS to remove ADC and resuspended in PBS containing “Complete” protease inhibitor cocktail (Roche Diagnostics). Cells were broken by three 30 sec pulses in a BeadBeater with 200 μm zirconia silica beads (Sigma Aldrich). Unbroken cells and debris were removed by centrifugation (20.8 × g for 15 min). Protein concentration in the cell lysate was determined using the Bio-Rad DC protein assay (Bio-Rad). Protein extracts containing 10 μg of protein were separated by SDS-PAGE and processed for Western blotting. The blot was probed with a polyclonal antiserum against IdeR pre-absorbed with a protein extract prepared from a M. smegmatis ideR mutant. A polyclonal antibody that detects the SirR protein was used as a loading control. Western blot signals were detected with ECL-Prime Western Blot Detection Reagent (GE Healthcare).
Mycobactin determination: Mycobacterial strains were grown in 7H9 to mid logarithmic phase in the presence or absence of ATc (600 μg.ml−1). Bacterial cells were collected by centrifugation and Fe+3-mycobactin was extracted in ethanol and chloroform and quantified as previously described (Rodriguez et al., 2002).
Quantification of intracellular iron
The ideR conditional mutant and the wild type parental strain were cultured in 50 ml of 7H9 with or without ATc (600 ng/ml). Extraction and quantification of iron was done as previously described (Riemer, et al 2004). Briefly, cells were harvested by centrifugation, washed twice with cold PBS and the pellets resuspended in 5 ml NaOH (50mM) and zirconia beads (0.1-mm diameter; Biospec Products Inc., OH). Cells were disrupted by two 1 min pulses in a BeadBeater. To quantify bound iron, the lysate (0.1 ml) was mixed with 10 mM HCL (0.1 ml) and 0.1 ml of iron releasing reagent (1.4 M HCl plus 4.5% (w/v) aqueous solution of KMnO4; 1:1) and incubated at 60°C for two hours. After cooling, 30 μl iron detection reagent (6.5 mM ferrozine, 6.5 mM neocuproine, 2.5 M ammonium acetate, 1M ascorbic acid in water) were added and absorbance of the sample at 550nm was measured. The total free iron concentration was determined as above without addition of ascorbic acid in the iron-detection reagent. The iron concentration in the samples was determined based on a standard curve obtained with increasing concentrations of ferric chloride and normalized to protein content which was determined using the BioRad Dc protein assay (BioRad).
Streptonigrin sensitivity
The microplate version of the Alamar Blue Assay (MABA) (Franzblau et al., 1998) was used to assess the susceptibility of mycobacterial strains to streptonigrin when cultured in the presence or absence of 600 ng.ml−1 of ATc. Briefly, 2×105 bacteria in microtiter plates were incubated in 7H9 containing 0.6 μg.ml×1 of streptonigrin, a concentration previously determined not to affect a wild type strain. 20 μl of Alamar Blue were added and 24 hours later the fluorescence emitted by cell-reduced Alamar blue was measured in a GloMax-Multi-detection System (Promega).
Sensitivity to hydrogen peroxide and Nitric oxide
Mtb strains were cultured in 7H9 to logarithmic phase in the absence of ATc, diluted to OD540 0.05 and incubated with or without 5mM H2O2 in the presence or absence of ATc (600 ng.ml−1). To test the sensitivity to nitric oxide, cells were incubated in acidified 7H9 (pH 5.5) with or without 3 mM Sodium Nitrite in the presence or absence of ATc (600 ng.ml−1). Each day an aliquot of all the cultures was taken for serial dilutions and plated for CFUs to determine the number of survivors.
RNA extraction
Mtb strains were grown to early log phase in 7H9 medium in the presence or absence of 600 ng.ml−1 ATc. Cells were collected by centrifugation and the pellets resuspended in 1 ml of TRI reagent (Molecular Research Center, OH) and immediately transferred to a tube containing 0.5 ml zirconia beads (0.1-mm diameter; Biospec Products Inc., OH). Cells were disrupted by two 1 min pulses in a BeadBeater. RNA was purified as described previously using RNeasy columns (Qiagen) (Rodriguez et al., 2002).
Quantitative RT and real-time PCR with SYBR green
For reverse transcription, the ThermoScript™ RT-PCR System (Invitrogene) with random hexamer primers was used according to the manufacturer instructions. 200 ng of RNA was used for cDNA synthesis. Control reactions not treated with ThermoScript™ were also prepared to exclude significant DNA contamination. After cDNA amplification, the samples were diluted 50 fold for PCR amplification. Quantitative PCR was performed in a Bio Rad i-cycler using SYBR green qPCR superMix Universal (Invitrogen). PCR conditions were identical for all reactions. The 25 μl reaction mixture contained 6 ng of cDNA template, 12.5 μl of SYBR Green Master Mix 2 μl mix containing primers (10 pM) and 7.5 μl water. After 10 min at 94° to activate the DNA polymerase, a set of 40 cycles of 30 sec at 95 °C, annealing at 55°C for 30 sec and elongation at 72 °C for 30 sec was run. Primers used for qRT-PCR are listed in Table S2. Fluorescence was measured during the annealing step and plotted automatically for each sample. The results were normalized to the amount of 16S ribosomal RNA. In order to obtain a standard curve for the RT-PCR, PCR was performed with each primer set by using calibrated amounts of chromosomal DNA and these reactions were performed at the same time as the RT-PCR. These standard curves were used to calculate the amount of cDNA for each gene present in the test samples.
Macrophage infection
Infection of THP-1 human monocytic cells was performed as previously described (Rodriguez and Smith, 2006). Briefly, 1×105 THP-1 cells per well were differentiated with 50 nM phorbol myristate acetate in 96 well plates and infected with Mtb strains, pre-grown in 7H9 medium without ATc, at a multiplicity of infection of 0.05 bacterium per macrophage. After 4 hours of incubation at 37°C in 5% CO2 atmosphere, the medium was removed and the cells were washed with warm PBS to remove extracellular bacteria. Finally, 100 μl of warm RPMI with or without ATc (1000 ng.ml−1) was added to each well and the plate was incubated at 37°C in 5% CO2 atmosphere. RPMI with or without ATc was replaced every 48 hrs. At indicated time points after infection, triplicate wells for each Mtb strain infection were treated with 0.05% sodium dodecyl sulfate (SDS) to lyse the macrophages and numbers of CFU were determined by plating appropriate serial dilutions on 7H10 plates. When indicated the Nox-2 inhibitor diphenyleneiodonium chloride (DPHI) (Sigma Aldrich) was included at a concentration of 10 μM. Cells were pre-incubated in RPMI with DPHI for 2 hours before infection and then DPHI was maintained throughout the experiment.
Mouse aerosol infection
C57BL/6 female mice were infected via aerosol as described previously (Rodriguez and Smith, 2006). Briefly, for each strain tested, a 10 ml bacterial suspension of 1×106 bacilli.ml−1 in saline containing 0.04% Tween 80 was used. Aerosols were generated with a Lovelace nebulizer (In-tox Products, Alburquerque, NM) and animals were exposed to the aerosol for 30 minutes. Upon infection, a group of mice infected with wild type or the conditional ideR mutant were fed a diet containing the tetracycline analog doxycycline at 2000 ppm (BioServe), while a second group of infected mice were fed a control diet lacking doxycycline. At the indicated time points after infection, 3 mice were sacrificed and their lungs were removed and homogenized in PBS-Tween 80. Dilutions of the homogenates were plated onto 7H10 agar to determine CFUs.
Supplementary Material
Acknowledgments
We thank Issar Smith for helpful discussions and critical reading of the manuscript. Krishna Kurthkoti and Blas Peixoto for their assistance, and Riccardo Manganelli for providing us with the Tet/Pip system constructs. This work was supported by NIH grant AI044856 (GMR).
References
- Boldrin F, Casonato S, Dainese E, Sala C, Dhar N, Palu G, Riccardi G, Manganelli R. Development of a repressible mycobacterial promoter system based on two transcriptional repressors. Nucleic Acids Res. 2010;38:e134. doi: 10.1093/nar/gkq235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J, Tanaka K, Carroll D, Flynn J, Bloom BR. Effects of Nitric Oxide Synthase Inhibitors on Murine Infection with Mycobacterium tuberculosis. Infect Immun. 1995;63:736–740. doi: 10.1128/iai.63.2.736-740.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CJ, Wisedchaisri G, Monfeli RR, Oram DM, Holmes RK, Hol WG, Beeson C. Functional studies of the Mycobacterium tuberculosis iron-dependent regulator. J Biol Chem. 2004;279:53554–53561. doi: 10.1074/jbc.M407385200. [DOI] [PubMed] [Google Scholar]
- Clemens DL, Horwitz MA. The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J Exp Med. 1996;184:1349–1355. doi: 10.1084/jem.184.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. (3rd) 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Conner EM, Grisham MB. Nitric Oxide: Biochemistry, Physiology and Pathophysiology. Methods. Acompanion to Meth in Enzymol. 1995;7:3–13. [Google Scholar]
- De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y., III. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc Natl Acad Sci. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. B.C.E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feese MD, Ingason BP, Goranson-Siekierke J, Holmes RK, Hol WG. Crystal structure of the iron-dependent regulator from Mycobacterium tuberculosis at 2.0-A resolution reveals the Src homology domain 3-like fold and metal binding function of the third domain. J Biol Chem. 2001;276:5959–5966. doi: 10.1074/jbc.M007531200. [DOI] [PubMed] [Google Scholar]
- Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, Gilman RH. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J Clin Microbiol. 1998;36:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobin J, Moore CH, Reeve JR, Jr, Wong DK, Gibson BW, Horwitz MA. Iron acquisition by Mycobacterium tuberculosis : Isolation and characterization of a family of iron-binding exochelins. Proc Natl Acad Sci USA. 1995;92:5189–5193. doi: 10.1073/pnas.92.11.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B, Rodriguez GM, Marras MP, Pentecost M, Smith I. The Mycobacterium tuberculosis IdeR is a dual functional regulator that controls transcription of genes involved in iron acquisition, iron storage and survival in macrophages. Mol Microbiol. 2001;42:851–865. doi: 10.1046/j.1365-2958.2001.02684.x. [DOI] [PubMed] [Google Scholar]
- Hobson RJ, McBride AJA, Kempsell KE, Dale JW. Use of an arrayed promoter-probe library for the identification of macrophage-regulated genes in Mycobacterium tuberculosis. Microbiology. 2002;148:1571–1579. doi: 10.1099/00221287-148-5-1571. [DOI] [PubMed] [Google Scholar]
- Homolka S, Niemann S, Russell DG, Rohde KH. Functional genetic diversity among Mycobacterium tuberculosis Complex clinical Isolates:Delineation of Conserved core and Lineage-Specific Transcriptomes during Intracellular survival. PlosPathogens. 2010;6:e1000988. doi: 10.1371/journal.ppat.1000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA, Chin SM, Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988;240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- Keyer K, Imlay. JA. Superoxide accelerates DNA damage by elevating free iron levels. Pro Natl Acad Sci USA. 1996;193:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirksey MA, Tischler AD, Simeone R, Hisert KB, Uplekar S, Guilhot C, McKinney JD. Spontaneous Phthiocerol Dimycocerosate-Deficient variants of Mycobacterium tuberculosis are susceptible to gamma Interferon-mediated immunity. Infect Immun. 2011;79:2829–2838. doi: 10.1128/IAI.00097-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WL, Gold B, Darby C, Brot N, Jiang X, Carvalho LPS, Jacobs WR, Jr, Nathan C. Mycobacterium tubeculosis expresses methionine sulpoxide reductases A and B that protect from killing by nitrite and hypochlorite. Mol Microbiol. 2009;71:583–593. doi: 10.1111/j.1365-2958.2008.06548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madigan CA, Cheng TY, Layre E, Young DC, McConnell MJ, Dbono CA, Murry JP, Wey JR, Barry C.E.r., Rodriguez GM, Matsunaga I, Rubin EJ, Moody DB. Lipidomic discovery of deoxysiderophores reveals a revised mycobactin biosynthesis pathway in Mycobacterium tuberculosis. Pro Natl Acad Sci USA. 2012;109:1257–1262. doi: 10.1073/pnas.1109958109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan CF, J. B. Hibbs J. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr Opin Immunol. 1991;3:65. doi: 10.1016/0952-7915(91)90079-g. [DOI] [PubMed] [Google Scholar]
- Olakanmi O, Schlesinger LS, Ahmed A, Britigan BE. Intraphagosomal Mycobacterium tuberculosis acquires iron from both extracellular transferrin and intracellular iron pools: Impact of interferon-gamma and hemochromatosis. J Biol Chem. 2002;23:23. doi: 10.1074/jbc.M209768200. [DOI] [PubMed] [Google Scholar]
- Pacelli R, Wink DA, Cook JA, Krishna MC, DeGraff W, Friedman N. Nitric oxide potentiates hydrogen peroxide-induced killing of Escherichia coli. J Exp Med. 1995;182:1469–1479. doi: 10.1084/jem.182.5.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R, Rodriguez GM. A Ferritin Mutant of Mycobacterium tuberculosis is Highly suceptible to Killing by Antibiotics and Is Unable to Establish a chronic Infection in Mice. Infect and Immun. 2012;80:3650–3659. doi: 10.1128/IAI.00229-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadri LEN, Sello J, Keating TA, Weinreb PH, Walsh CT. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem and Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Drigen R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 2004;331:370–375. doi: 10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- Rodriguez GM, Smith I. Identification of an ABC Transporter Required for Iron Acquisition and Virulence in Mycobacterium tuberculosis. J Bacteriol. 2006;188:424–430. doi: 10.1128/JB.188.2.424-430.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK, Smith I. ideR, An essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect Immun. 2002;70:3371–3381. doi: 10.1128/IAI.70.7.3371-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryndak MB, Wang S, Smith I, Rodriguez GM. The Mycobacterium tuberculosis high-affinity iron importer, IrtA, contains an FAD-binding domain. J Bacteriol. 2012;192:861–869. doi: 10.1128/JB.00223-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Tomioka H. Antimicrobial activities of benzoxazinorifamycin (KRM-1648) and clarithromycin aagainst Mycobacterium Avium-intracellular complex within murine peritoneal macrophages, human macrophage-like cells and human alveolar epithelial cells. J Antimicrob Chemother. 1999;43:351–357. doi: 10.1093/jac/43.3.351. [DOI] [PubMed] [Google Scholar]
- Semavina M, Beckett D, Logan TM. Metal-linked dimerization in the iron dependent regulator from Mycobacterium tuberculosis. Biochemistry. 2006;45:12480–12490. doi: 10.1021/bi060797s. [DOI] [PubMed] [Google Scholar]
- Shiloh MU, MacMicking Nicholson S., Brause J.e., Potter S, Marino M, Fang F, Dinaeur M, Nathan C. Phenotype of mice and macrophage deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 1999;10:29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- Snow GA. Mycobactins: iron chelating growth factors from mycobacteria. Bacteriol Rev. 1970;34:99–125. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton B, Walker L, Logan TM. Z(II) stimulation of Fe(II)-activated repression in the iron-dependent repressor from Mycobacterium tuberculosis. Biochemistry. 2013;19:1927–1938. doi: 10.1021/bi301608p. [DOI] [PubMed] [Google Scholar]
- Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, et al. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- Wagner D, Maser J, Lai B, Cai Z, Barry CE, Honer Zu Bentrup K, Russell DG, Bermudez LE. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell's endosomal system. J Immunol. (3rd) 2005;174:1491–1500. doi: 10.4049/jimmunol.174.3.1491. [DOI] [PubMed] [Google Scholar]
- Waldron K, Robinson N. How do bacterial cells ensure that metalloproteins get the correct metal? Nat Rev. 2009;6:25–36. doi: 10.1038/nrmicro2057. [DOI] [PubMed] [Google Scholar]
- Weinberg ED. Iron withholding: A defense against infection and neoplasia. Physiolo Rev. 1984;64:65–102. doi: 10.1152/physrev.1984.64.1.65. [DOI] [PubMed] [Google Scholar]
- Woodmansee AN, Imlay JA. A mechanism by which nitric oxide accelerates the rate of oxidative DNA damge in Excherichia coli. Mol Microbiol. 2003;49:11–22. doi: 10.1046/j.1365-2958.2003.03530.x. [DOI] [PubMed] [Google Scholar]
- Yeowell HN, White JR. Iron requirement in the bactericidal mechanism of streptonigrin. Antimicrob Agents Chemother. 1982;22:961–968. doi: 10.1128/aac.22.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.