

Abstract
DNA microarrays have gained wide use in biomedical research by simultaneously monitoring the expression levels of a large number of genes. The successful implementation of DNA microarray technologies requires the development of methods and techniques for the fabrication of microarrays, the selection of probes to represent genes, the quantification of hybridization, and data analysis. In this paper, we concentrate on probes that are either spotted or synthesized on the glass slides through several aspects: sources of probes, the criteria for selecting probes, tools available for probe selections, and probes used in commercial microarray chips. We then provide a detailed review of one type of DNA microarray: Affymetrix GeneChips, discuss the need to re-annotate probes, review different methods for regrouping probes into probe sets, and compare various redefinitions through public available datasets.
Keywords: Microarray, GeneChips, Probes, Probe sets, Review
2. Introduction
DNA microarray technology has provided an opportunity to simultaneously monitor the expression levels of a large number of genes in response to intentional experiment perturbations such as gene disruptions and drug treatments. The patterns obtained from microarray experiments have helped researchers to understand genetic mechanisms and progress of diseases (1, 2), to predict molecular functions of genes (3, 4), to build functional pathways (5), and to identify novel genes or splice variants (6). The successful implementation of DNA microarray technologies requires the development of methods and techniques for the fabrication of microarrays, the selection of probes to spot, the quantification of hybridization, and data analysis (7-9). Currently, DNA microarrays are manufactured using either cDNA or oligonucletides as gene probes. cDNA microarrays are usually created by spotting amplified cDNA fragments in a high density pattern onto a solid surface such as a glass slide (10, 11). Probes for oligonucletides arrays are either spotted or synthesized directly onto a glass or silicon surface using various technologies including photolithography, ink-jets, and some other technologies (12-14). There are two schemes to detect differently expressed targets when comparing an experimental sample with a reference sample: one- and two-color schemes. In one-color case, images are obtained on a different chip for each sample using a single fluorescent label (for example, phycoerythrin). Different images are then compared to obtain differentially expressed targets. In two-color format, two RNA samples (reference and experimental) are labeled separately with different fluorescent tags (for example, cyanine 3 and cyanine 5 (Cy3, Cy5)), then hybridized to a single microarray and scanned to generate fluorescent images from the two channels. A two-color graphical overlay can then be used to visualize targets that are up-regulated or down-regulated.
Since the emerge of the technology in the mid 1990s, both commercial and academic groups have developed a number of different microarray platforms but the validity of the results remains a subject of concern to the scientific community mainly due to the poor reproducibility among various platforms (15-22). A number of studies have been conducted to compare different platforms but there is no clear consensus. Some claim a significant divergence across platforms, while others believe the level of consensus is acceptable. With extensive attention being devoted to improving the statistical algorithms used to estimate expression levels and detect differential expressed targets, we believe that probe and probe set identity is also an important factor for the poor reproducibility. It is possible that the sequences immobilized to the microarray surface are not the intended ones possibly caused by unavoidable errors introduced during the manufacturing process (23, 24). For example, cDNA probes are usually obtained from cDNA libraries, and the clone misidentification rates within libraries have been estimated as high as 30% (25-27). Additionally, probes are designed to match particular mRNA transcripts, often based on deposited NCBI sequences such as ESTs, cDNAs, or mRNAs. However, those sequences might be incorrect because of sequencing errors such as including foreign vector sequences (28). Furthermore, annotations of probes might also be inaccurate or incomplete due to limited knowledge available at the probe design stage. Usually, probes are selected to represent genes while measures are obtained based on the hybridization with mRNAs. But one gene can have multiple splice variants and it is estimated that the number of genes which can be spliced is between 30% to 99% (29, 30). Accurate quantitation requires knowledge of both the identity of the genes and the splice variants that are expressed. As our knowledge of genomic sequences (particularly for the human genome) increases, annotations for a substantial number of probes for existing microarray platforms need to be corrected. For example, a large portion of the Affymetrix probes (up to 30-40% depending on the actual chip) did not correspond to their intended mRNA reference sequences defined by the highly curated, publicly available RefSeq database (31-33).
A large number of reviews on about DNA microarray technology prior to year 2002 were assembled by Michael Heller (34). Several reviews have been assembled recently mainly focusing on the similarities and differences among different technologies as well as efforts to integrate data from cross-platform comparative studies (9, 19, 21, 35, 36). Here, we address issues and studies related to probe sequence which include probe resources, probe selection during the design stage, and annotation correction by incorporating up-to-date genomic knowledge for data analysis.
3. Microarray Probes and Probe Sets
Table 1 provides an overview of probes or probe sets used in several commercial platforms. Most of these platforms select probes using public resources such as GenBank or RefSeq. Some of them use in-house or commercial resources. For example, both Agilent and CodeLink use a commercial sequence resource, LifeSeq besides public resources.
Table 1.
Probe resources and probe types for some commercial platforms.
| Company | Organisms | Resources | Probe Types |
|---|---|---|---|
| Agilent | Human Mouse Rat |
LifeSeq RefSeq Genbank NIEHS, TRC, PG, Refseq, Ensembl, RIKEN NIA Mouse Gene Index |
60-mer per target |
| Human Mouse Rat |
LifeSeq UniGene |
Spotted cDNA | |
| Affmertix | Human Mouse Rat |
UniGene | 11 to 20 (PM, MM) pairs of 25-mers Per target |
| CodeLink | Human Mouse Rat |
UniGene RefSeq dbEST LifeSeq |
30-mer per target |
| Applied Biosystems genome survey array | Human Mouse Rat |
GenBank Refseq Celera Genomics In-house transcripts Mouse Genome Sequencing Consortium Genome Sequencing and Annotation |
60-mer per target |
| MVG catalog array | Human Mouse Rat |
GenBank RefSeq |
50-mer per target |
| Stanford Functional Genomics Facility Arrays | Human Mouse |
IMAGE CGAP clone set RIKEN full-length cDNA clones NIA 15K Clone set |
Spotted cDNA |
Probes or probe sets need to be chosen to provide sufficient sensitivity (i.e., the ability to detect the rarely expressed transcripts in a complex background), and specificity (i.e., the ability to distinguish measures among transcripts with high sequence similarity), as well as high coverage (i.e., the ability to include all relevant transcripts to the experiment) (37). It is desired to avoid sequences that are ambiguous (i.e., hybridize to multiple transcripts) or highly similar to non-target transcripts (i.e., cross-hybridization). Additionally, redundancy (i.e., several probes or probe sets targeting the same transcripts) can increase the accuracy of measures but it can at the same time reduce the coverage. Furthermore, the successful application also requires correct and up-to-date annotation (i.e., the association of probes with target transcripts) of the probes or probe sets.
3.1. cDNA microarrays
Probes in cDNA microarrays are mostly cDNA clones provided by IMAGE (the Integrated Molecular Analysis of Genomes and their Expression) Consortium. The consortium was initiated in 1993 as a collaborative effort among several academic groups to share high-quality arrayed cDNA libraries and to place sequence, map, and expression data for use in the public domain (38). Researchers can purchase physical clones from authorized distributors, such as Research Genetics/Invitrogen (http://www.resgen.com), the American Type Culture Collection (http://www.atcc.org), and RZPD German Resource Center for Genome Research (http://www.rzpd.de). Most of these clones have the status of expressed sequence tags (ESTs), and their corresponding sequences are collected in the dbEST database (39).
When dealing with EST or cDNA clones, a common problem is poor specificity caused by unreliable annotations of their sequence data. For example, Taylor et al. found that only 79% of the clones matched to the designated sequences when sequencing 2300 PCR products ordered from a human, sequence-verified cDNA clone library (25). They recommended sequence verification of clones at the final design stage before actually printing them on microarray slides. Halgren et al. documented that only 62.2% of the 1,189 cDNA sequences of clones ordered from the consortium had significant sequence identity to the published data for the ordered clones (26). The IMAGE Consortium is aware of this and does list problematic clones on its web site based on user feedbacks, however there is no consensus as to the actual error rate or the source of the errors.
Redundancy is another problem when using EST or cDNA clones as probes. Highly expressed genes are often represented by multiple clones. There are two potential ways to reduce the redundancy. One is to use clones from a normalized clone library where the number of clones representing each gene has been equalized (40-42). Another way to control the redundancy is the use of clustering data through either pair-wise or genome-based alignment clustering methods. NCBI's UniGene is the most widely used clustering data which was originally generated using pair-wise alignment and currently is based on genome-wide alignment. The TIGR Gene Indices (TGI) is another well known EST clustering data that uses a highly refined protocol to analyze EST sequences, clustered sequences, and identify genes represented by them.
The use of complete cDNA sequence as probes usually imposes the danger of cross-hybridization. A fragment of the cDNA sequence can be used to spot on the array. cDNA fragments are usually chosen to reduce the danger of cross-hybridization caused by either sequence homology or other factors. Kane et al indicated that selected fragments need to be 75% less than similar to non-target transcripts within the 50 mer region to prevent significant cross-hybridization (43). Besides cross-hybridization caused by sequence similarity, there are some unspecific hybridization signals caused by repetitive elements such as Alu-repeats within the cDNA sequence. Utilizing repetitive element databases such as REPBASE (44), one can avoid the complication caused by repetitive elements.
3.2. Oligonucleotide microarrays
The use of oligonucleotides as probes has become popular because they usually have better specificity than cDNAs and also have the capacity to distinguish single-nucleotide polymorphisms (SNPs) and to discern splice variants (37). There are several issues to consider when selecting oligonucleotide probes.
One is the probe length. Currently, probes used in major commercial platforms can be either short (20-30 mers) or long (50-70 mers) oligonucleotides (see Table 1). It was expected that the length of the probes would be associated with sensitivity, signal strength, and specificity (45). For optimal intensity measure, Chou et al. suggested to use long probes (e.g., 150 mer) if no experimental validation is provided (see Figure 1). Accurate gene expression measurements can be achieved with multiple probes per gene, and fewer probes are needed if longer probes rather than shorter probes are used. Comparing to cDNA microarrays, long oligonucleotide microarrays have the advantages of i) distinguishing different transcripts for the same gene or genes from the same gene family, ii) higher specificity, and iii) requiring smaller quantities of mRNA (36, 43).
Figure 1.
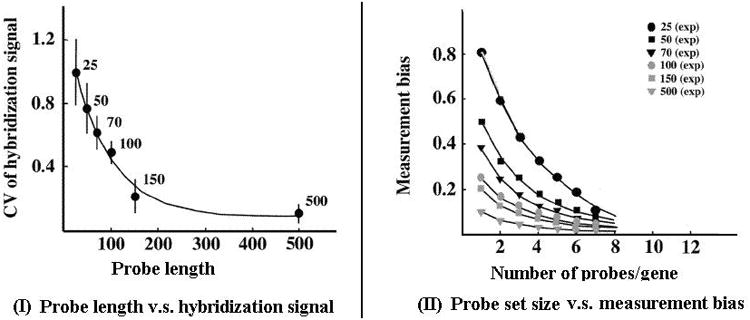
(I) Effect of probe length on the coefficient of variation (CV) in the hybridization signal using different length probes for the same genes. (II) Effect of the number of probes per gene on measurement bias.
The gene region from which a probe is selected can greatly affect specificity and cross hybridization. Coding regions are more conserved and show high degree of similarity with other closely related genes. Hence, probes selected from coding region are the most susceptible to cross-hybridization events. Most probe collections focus on 3’ UTR, in part because of a presumption that oligo dT will be used to prime the RNA populations, and also in part because sequence divergence is typically greater in such regions. However, with more probes distributed in 3’ UTR and less distributed in coding region, it will provide less discrimination among splice variants.
It is difficult to predict whether an oligonucleotide probe will bind efficiently to its target sequence and yield a good hybridization signal on the basis of sequence information alone. It was reported that very high sequence similarity can lead to cross-hybridization even when the sequences have been pre-screened for contiguous perfect match. For example, Hughes et al showed that 18 or more randomly placed mismatches per 60-mer can reduce hybridization to background levels (13). They also suggested that the placement of distinguishing bases at positions relative to the surface has a dramatic impact on the stability of the duplex and therefore can be used to maximize specificity.
3.3. Tools for probe selection
As discussed by Tomiuk and Hofmann, the successful application of each DNA microarray application, depending on the objective of the application, imposes certain criteria for selecting appropriate probes (37). Software tools have been developed to allow users to select appropriate probes or probe sets. Table 2 provides an overview of those tools. Most tools address issues relevant to probe length, cross-hybridization, secondary structure, as well as probe melting temperature.
Table 2.
Features of some probe design tools with respect to oligo length, location, specificity, accessibility, and Tm uniform.
| Probe length | Location | Specificity | Accessibility | Tm Uniform | |
|---|---|---|---|---|---|
| Array Designer | User defined | User can choose 3’ UTR, 5’ UTR, or coding region | BLAST | N/A | N/A |
| Oligo Array 2.0 | Optimal probe length selection within from a user defined range | Backward 3’ UTR end | BLAST | Mfold | Nearest neighbor model |
| Oligodb | User defined | From 5’ UTR to 3’ UTR direction | BLAST | Mfold | Nearest neighbor model based program: melting |
| OligoPicker | User defined within the range between 20 and 100 | Close to 5’ UTR end | BLAST | Self-complementary likelihood | Schildkraut formula |
| PROBEWIZ | Optimal probe length selection within from a user defined range | N/A | BLAST | Unknown | N/A |
| Sarani | Optimal probe length selection within from a user defined range | N/A | BLAST | Unrevealed algorithm | Nearest neighbor model |
| Visual OMP | User defined or Optimal probe length selection | Let user choose visually | BLAST | Shows structure visually | N-stage model |
Most software tools provide users with the freedom to select probe lengths to optimize the performance (46-52). For example, Array Designer (46) allows users to choose specific length for oligonucleotides or PCR primers. The sequence is broken down into small equal-sized fragments according to the size chosen by the user, and then a specific probe is designed for each target. Oligo Array 2.0 (47, 48) allows users to specify oligo length with a range. OligoPicker (49, 50) allows users to choose oligo length from 20 bases to 100 bases long, although it suggests 70 bases as the default. Oligodb (51, 52) treats oligo length one of the required input parameters provided by users. Several tools try to select an optimal probe length given a range (53-56). For example, PROBEWIZ (53, 54), which can design both oligo and PCR primer, lets users input both the minimum and maximum length of the oligonucleotides or PCR primers, and tries to find the optimal length for the best performance. Sarani (55) lets users choose a range of probe length, and automatically make the decision. The Visual OMP (56) gives users flexibilityto either choose a certain oligo length or let the system make decision.
Many oligonucleotide probe design tools take gene regions into consideration. For example, Array Designer (46) allows users to choose their desired oligonucleotide location, such as 3’UTR, 5’UTR, or anywhere else in the sequence. In OligoArray 2.0 (47, 48), normally, the input sequence reads backwards from the 3’ UTR using a moving window according to the oligonucleotide length. The Oligodb (51, 52) lets users choose their desired oligonucleotide probe location from the 5’ UTR to the 3’ UTR. The OligoPicker (49, 50) makes its oligo probes lie as close to the 5’ UTR of the RNA as possible. The Visual OMP (56) can let users choose the oligo probe location visually, and based on the choice, decides the right probe.
To avoid cross-hybridization, all probe design tools utilize BLAST to make sure the chosen oligonucleotide probe or probe sets have the lowest similarity to the whole genome comparing to other sequence fragments in the target sequence. For example, OligoPicker (49, 50) uses contiguous base match and at the same time, to reduce the contribution to cross-hybridization by the global similarity, oligonucleotides whose BLAST scores higher than a pre-defined threshold value (around 96%) comparing to all sequences in the same universe are rejected.
Most probe design tools try to avoid secondary structures so that the chosen probes have higher sensitivity. Both OligoArray (47) and Oligodb (51, 52) use program mfold, developed by Zuker et. al. (57), to predict and eliminate secondary structures. The Visual OMP (56) can visually show the structure of each candidate probe so that users can easily reject probes with secondary structures. OligoPicker (49) uses a self-complementary likelihood method to predict secondary structures, and probe candidates are tested for homology to the complementary strand of their cognate sequence using BLAST, but this approach does not take into account the local concentration of the complementary sequence.
To ensure quantitative comparison of gene expressions, microarray hybridization conditions should be similar for all genes in the study, therefore the melting temperature (Tm) of probes should fall in a narrow range. Several tools consider the oligonucleotide melting temperature as an important criteria to choose probes. Oligo Array 2.0 (47, 48) and Sarani (55) apply the Nearest-Neighbor model using DNA parameters develop by SantaLucia et. al.(58) to compute the Tm, and the following formula is used: Tm = (DH°/(DS° + R ln(DNA/4)) -273.15, where R is the gas constant (1.9872 cal/K.mol) and DNA is the DNA concentration. Oligodb (51, 52) uses a program called melting developed by Le Novère et. al. (59), which is also based on nearest neighbor method, to calculate the Tm. The Oligodb (51, 52) does not choose Tm to be an inclusion/exclusion criterion at the Tm computing stage, since the G/C content, which mainly determines Tms, typically varies at scales longer than the transcript length. The user may choose those specific oligos from the output list that fit best the individual respect to Tm and the position in the transcript. OligoPicker (49, 50) first calculates the melting temperature of all sequence using the formula: 64.9 + 41 ×gcCount/oligoLength − 600/oligoLength where gcCount is the number of all Gs and Cs in an oligo and the molar sodium concentration is taken to be 0.1 M (60), and then choose those candidates whose Tm is with 5°C of the median Tm. Visual OMP (56) utilizes a N-Stage model to predict the Tm of a duplex within 2°C on average.
4. Redefinition of Affymetrix Genechips
In order to accomplish high sensitivity and specificity in the presence of a complex background, Affymetrix introduced a system that entails the use of a series of specific and non-specific gene probe sets that are intended to result in a more accurate discrimination between true signal and random hybridization. Each probe set usually consists of 8 to 16 pairs of probes (PM, MM)s where PM probes are perfect matching 25-mer oligos to the target transcripts and MM probes contain sequences with the 13th position of the corresponding PM sequence being modified to the complement nucleotide. Affymetrix claims that probes of approximately 25 nucletoides long provide a very effective balance between signal intensity and related sequence discrimination which allows expression monitoring of thousands of targets. The use of (PM,MM) pairs and multiple pairs for a target transcript allows both absolute and comparative analysis and compensates for variations and noises in the complex background. Affymetrix uses one-color method for obtaining expression measures.
4.1. Issues related to the Affymetrix probes
Probe sets in Affymetrix arrays were either selected based on a set of heuristic rules or on some thermodynamic models (61, 62). For example, candidate probes of the first generation of arrays were chosen from 600 bases at 3’UTR region of each target sequence and rules were used to ensure probes to be unique and have relatively good hybridization performance (61). Mei et al proposed a probe selection method based on the influence of empirical factors on the effective fitting parameters of a thermodynamic model. Probe sets were selected to optimize with respect to probe sensitivity, independence (degree to which probe sequences are non-overlapping), and uniqueness (lack of similarity to sequences in the expressed genomic background) (62).
Table 3 shows examples of the two major problems that necessitate redefining probe sets in the Affymetrix U133A chips for experiments identifying differently expressed transcripts.
Table 3.
Example of ambiguous and mismatched probes and probe sets in the original assignment of Affymetrix HG-U133A chip.
| CLEC2D | NPM1 | |||||
|---|---|---|---|---|---|---|
| Probe set | NM 013269 | NM 001004419 | NM 001004420 | NM 002520 | NM 199185 | NM 001037738 |
| 220132_s_at | 1-11 | 1-11 | 1-11 | - | - | - |
| 221691_x_at | 1,3,4,7,8 | 1,3,4,7,8 | 1,3,4,7,8 | 1-9 | 1-6,9 | 1-11 |
| 200063_s_at | - | - | - | 1-11 | 1-11 | - |
| 221923_s_at | - | - | - | - | - | 1-11 |
A probe set containing some probes that match multiple transcripts - Probes within a probe set do not all target the same set of transcripts. The expression levels measured by those probes will introduce an inconsistency in the quantitation algorithms.
Affymetrix had originally represented the human genes CLEC2D by one probe set 220132_s_at and NPM1 by two probe sets, 221691_x_at and 200063_s_at.
Currently, three RefSeqs represent CLEC2D and three RefSeqs represent NPM1.
The table entries for each probe set (row) identify the probes that match the RefSeqs (columns). For example, all 11 probes in probe set 220132_s_at match NM_013269.
The level of hybridization to probe set 200063_s_at provides a consistent estimate of the composite expression for RefSeqs NM_002520 and NM_199185 of NPM1. The expression of RefSeq NM_001037738 is completely 'transparent' to this probe set. However, the expression of RefSeq NM_001037738 is reflected in the hybridization of probe set 221923_s_at.
In contrast, if we are using probe set 221691_x_at to measure the expression of transcripts of NPM1, the level of hybridization to the probe set could reflect cross-hybridization with RefSeqs of CLEC2D.
Some probes in a probe set do not match the target transcripts – Several probes within a probe set may not match any of the transcripts for the gene that Affymetrix had originally designated for the probe set. The expression levels measured by those probes do not reflect the composite expression of the transcripts of the intended gene and will introduce an inconsistency in the quantitation algorithms.
Probes 7 and 8 of 221691_x_at do not target NM_199185 that represents NPM1, but they do target all three transcripts for CLEC2D.
Therefore, the expression levels measured by 221691_x_at do not consistently reflect the composite expression of the RefSeqs of the intended gene.
4.2. Tools, resources, and studies using Affymetrix probe sequence data
After the probe sequence information was made public by Affymetrix, several recent papers made use of it for improving accuracy and cross-platform consistency (17, 18, 31-33, 63, 64). Table 4 provides an overview of tools, resources, and studies on incorporating probe sequence data into microarray data analysis.
Table 4.
An overview of tools, resources, and studies that utilize Affymetrix probe sequence data.
| Purpose | Matching method | Affymetrix chip considered | Software | Resources | |
|---|---|---|---|---|---|
| Gautier | Tools for redefining probe sets | R function: matchprobes (using C library string) | Hgu95Av2, Hgu133A | Altcdfenvs | RefSeq |
| Dai | Resources for redefining probe sets | NA | All human, mouse, and rat GeneChips | NA | UniGene, RegSeq, DoTS, ENSEMNL, Exon |
| Kong | Software for integrating different generations of Affymetrix chips | Blat | Some human and mouse hcips | Crosschip | Human genome assembly was used to filter out absent probes or ambiguous probes |
| Harbig | Sequence-based correction for Hgu133Plus | Blast | Hgu133Plus | NA | Resource for Hgu133Plus |
| Liu | Tools and resources for redefining probe sets | Blat | All GeneChips | AffyProbeMiner | Complete CDS from GenBank Refseq |
The first tool available to use for redefining chip definition files (CDFs) is by Gautier et al. (64) Recognizing the need to incorporate the latest genomic knowledge into microarray data analysis, they developed an open-source tool, an R package “altcdfenvs” which was integrated into the microarray data analysis flow through Bioconductor, an R software system for computational biology and bioinformatics (65). Only sequences in RefSeq were used and the mapping was done using “matchprobes”, a method in altcdfenvs utilizing the standard C library string. The package has been used by DeCook et al. to generate alternative chip definition files (CDFs) to remove unwanted probe pairs (66). Carter et al. (18) also utilized the tool to redefine Affymetrix probe sets by sequence overlap with cDNA microarray probes for the purpose of reducing cross-platform inconsistencies in cancer-associated gene expression measurements. In Carter's study, probes targeting identical transcript sequence regions were shown to give substantially stronger concordance than probes that target identical contiguous transcript molecules at different sequence regions. The study suggests that discrepancies between different platforms are caused by improper cross-platform probe matching. Recently, a web resource, AffyProbeMiner, was developed by Liu et al. to provide pre-computed redefined CDFs as well as software for generating redefinitions (67). Additionally, a web interface is also available. In AffyProbeMiner, probes are grouped into a set if they are mapped to a consistent set of transcripts or genes based on a collection of complete CDSs (CCDSs) obtained from GenBank and RefSeq.
Besides these tools, there are several resources distributing redefined CDFs. One is the work of Dai et al. which provides extensive resources for re-analyzing GeneChip data based on redefining CDFs (33). They reorganized probes on more than a dozen popular GeneChips into gene-, transcript- and exon-specific probe sets utilizing up-to-date genome, cDNA/EST clustering, and single nucleotide polymorphism information. The redefined CDFs were originally available for human, mouse, and rat chips. Recently, several other chips were added. Another resource is by Harbig et al. that used BLAST to match probes with documented and postulated human transcripts and redefined about 37% of the probes on the “U133 plus 2.0” array (31). They found that the original Affymetrix annotation was compromised because of the potential for cross-hybridization with splice variants or transcripts of other genes containing matching sequences. More than 5,000 probe sets were shown to hybridize with multiple transcripts. They proposed a sequence-based identification method and redefined probes to the most closely-related RefSeq sequences. Another resource distributing redefined CDFs is AffyProbeMiner (67), redefined CDFs according to Entrez genes and complete CDSs (CCDSs) are downloadable from its website.
Several other studies aimed to improve the consistency among different generations of GeneChips (17, 63). For example, utilizing the probe sequence information, Elo et al. verified probes according to NCBI mRNA sequences by searching all PM probes against the mRNA sequences using BLAT v. 26 (68). Probes mapped to the same gene according to Entrez GENE were grouped as an alternative probe set. Then they compared a method called probe-level expression change averaging (PECA) to RMA and MAS5 and found that PECA provided better agreement of differentially expressed genes between different generations of GeneChips. Kong et al. used sequence information to increase the compatibility between different generations of GeneChips by filtering probes that were not consistent with their annotations according to the human genome build (17).
4.3. Some statistics of Affymetrix probes
We downloaded all probe sequence information as well as CDFs for each gene expression Affymetrix chip. We obtained the mapping results of several human chips with the current human genome build. We then verified that probes in Affymetrix chips were designed towards 3’UTR end.
Since Affymetrix human arrays were designed using previous version of human genome build, some of the probes may fail to be matched to the current human genome build. Additionally, some of the probes may correspond to multiple locations in the genome. We mapped all sequences in four of the human arrays (U95Av2, U133A, U133B, U133Plus2) to the current human genome build (March, 2006) and then categorized the mapping results into four categories: no exact matching (i.e., 0), unique exact matching (i.e., 1), matching to two locations (i.e., 2), and matching to more than two locations (i.e., >2). Figure 2 shows the results of mapping probes in several Affymetrix human arrays to the current human genome build (March 2006 release). For all chips, the number of probes which can be mapped uniquely to the current genome build is around 80% (March, 2006). However, around 7-10% of the probes failed to be mapped to the current genome and the remaining 7-10% probes were mapped to multiple segments in the genome.
Figure 2.
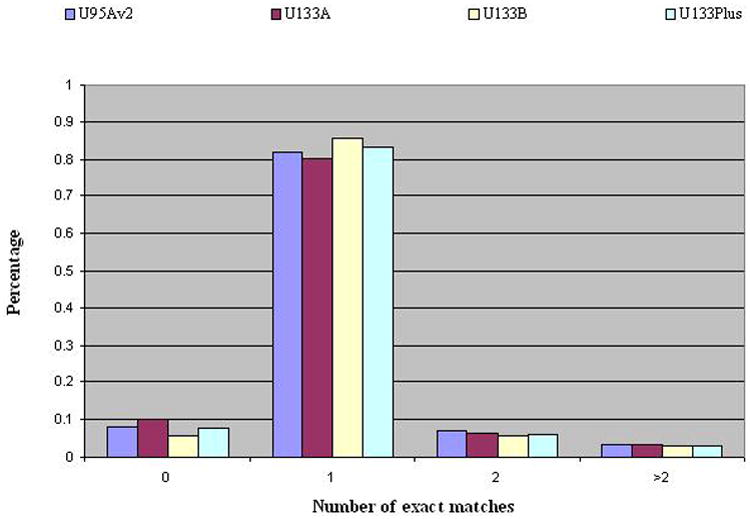
The mapping results of four Affymetrix human chips to the human genome build (March 2006).
Probes in traditional Affymetrix chips are skewed towards the 3’ UTR end. Figure 3 shows the distribution of probes for 51 gene expression Affymetrix chips. The X-axis denotes the distance to the 3’UTR end and the Y-axis denotes the percentage of probes. From Figure 3, we can see that probes in all chips were skewed towards the 3’ UTR end. Such skewed distribution makes it very difficult to disambiguate differential expression of different splice forms of the same gene.
Figure 3.
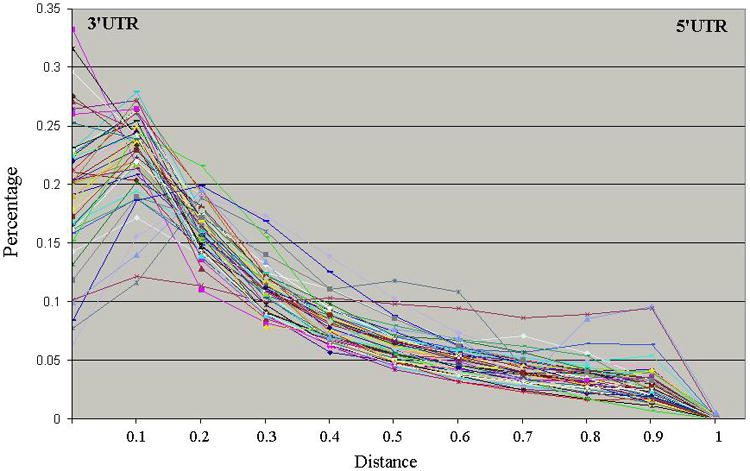
The distribution of probes regarding to the distance to the 3’UTR end for 51 Affymetrix gene expression chips when mapping to complete CDS sequences.
5. Comparison Analysis of Different Remapping Methods
Probes in Affymetrix were selected based on the most up-to-date genomic knowledge available at the time of fabrication. As accuracy and completeness in our knowledge of genomic sequences increase, the sequence knowledge used to select those probes may be incorrect now and annotations for them need to be corrected. As we have shown, probes can be regrouped according to different conditions such as genes, transcripts, UniGene clusters, or complete CCDSs. Using two chip types, U95Av2 and U133A, we performed a study to compare different types of redefined CDFs with respect to overlapping among different generations and cross-generation consistency.
5.1. Redefinition used
We downloaded a recent version (version 7) of three types of redefined CDFs of U95Av2 and U133A from the resource website developed by Dai et al.(33), namely UniGene-based, ENTREZ GENE-based, and RefSeq-based. All redefined probe sets in Dai's redefined CDFs contain at least three probe pairs. For UniGene-based redefinition, all PM probes in a probe set must match continuously on the genomic sequence in the same direction with only one perfect match for each probe in the most current genome assembly and all PM probes in the probe set must also correspond to the same UniGene Cluster. Probes with more than one perfect hit on the corresponding genomic sequence were removed. In ENTREZ GENE-based and RefSeq-based redefined CDFs, one probe can appear in multiple probe sets. We also assembled redefined CDFs through AffyProbeMiner web site (August 4, 2006) where probes were grouped based on CCDSs (CCDS) (67). To be consistent, we required all probe sets in the redefined CDFs according to CCDSs contain at least three probe pairs. However, probes mapped to multiple CCDSs were kept in CCDS-based redefinition.
We calculated percentages of probes included in the redefined CDFs as well as percentages of probe sets overlapping between U95Av2 and U133A.
5.2. Data set
For the cross-generation consistency, we used the public data sets from the microarray studies of Yeoh et al. and Ross et al. (69, 70). The data set contained expression data from patients with different leukemia subtypes A total of 360 patient samples were hybridized to U95Av2 arrays and 132 of the same samples were also hybridized to U133A arrays. We selected 40 samples for our analyses, which were hybridized to both array types and and represented two genetically distinct leukemia subtypes: 20 TEL-MEL1 samples and 20 MLL samples.
5.3. Consistency assessment
The comparison study of assessing the consistency across U95Av2 and U133A was conducted in two different ways. One way is to look at the correlation of the gene expression values after redefinition within each pair. A high correlation indicates good consistency between the two platforms. For each of the leukemia subtype, we used RMA to obtain the gene expression values and computed the correlation of the gene expression values for genes that appear in both platforms (U95Av2 and U133A) (71). Another way is to assess the agreement between different platforms when selecting differentially expressed genes between two different subtypes. We computed the proportion of common selected genes among the top K differentially expressed from the two platforms. A high proportion of common genes indicate good agreement between the platforms. We used SAM to select differentially expressed genes (72). We implemented the data analysis using a microarray analysis platform, Bioconductor (http://www.bioconductor.org)(65).
5.4. Comparison outcome
Figure 4 shows the comparison of the four types of redefined CDFs between U95Av2 and U133A according. For each of the three types of Dai, over 95% of probe sets in U95Av2 were overlapped with around 65% of those in U133A. Around 70% of probes were included in the redefined CDFs in both chips of Dai's redefined CDFs. For CCDS-based CDFs, 81.7% in U95Av2 were overlapped with 53.9% in U133A. Around 80% of probes were included in the redefined CDFs.
Figure 4.
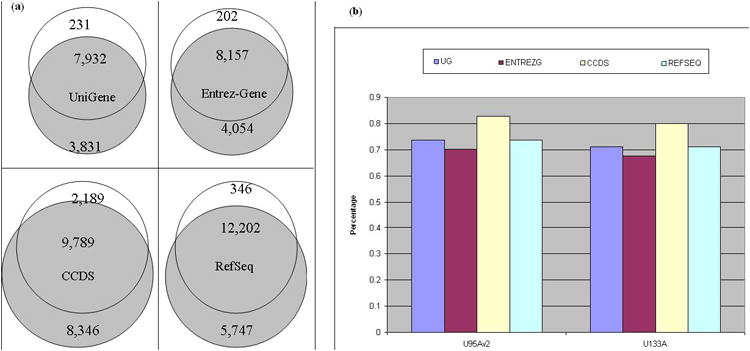
Statitistics of four three redefined CDFs for two array chips: U95Av2 and U133A: UniGene-based redefinition, Entrez-Gene redefinition, CDSs-based, and RefSeq redefinition: (a) Venn-diagram of overlapping between two chips, and (b) percentage of probes included in the redefined probe sets.
The cross-generation consistency results are presented in Figure 5 and Figure 6. Figure 5 shows the boxplot of the correlation. As one can see, using the correlation as a measure of consistency, the REFSEQ and CCDS annotations give better results than ENTREZ Gene and UniGene. From Figure 6a, ENTREZ Gene has better performance if the number of top selected genes is less than 100 when using the proportion of common selected genes among the top K differentially expressed genes as the measure of consistency. However, when the number of top selected genes was over 100, ENTREZG, UniGene, and REFSEQ tended to exhibit similar performance. Comparing to ENTREZG, UniGene, and REFSEQ, the redefinition according to CCDs tends to have poor consistency between different platforms.
Figure 5.
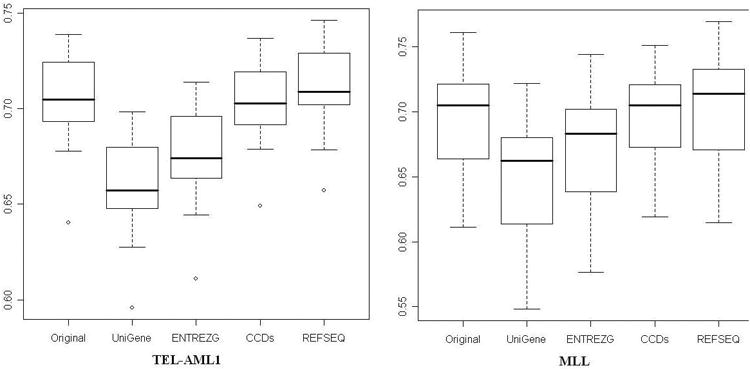
The RMA intensity correlation between technical replicates for two data sets (TEL-AML1 and MLL) on two array generations: U95Av2 and U133A.
Figure 6.
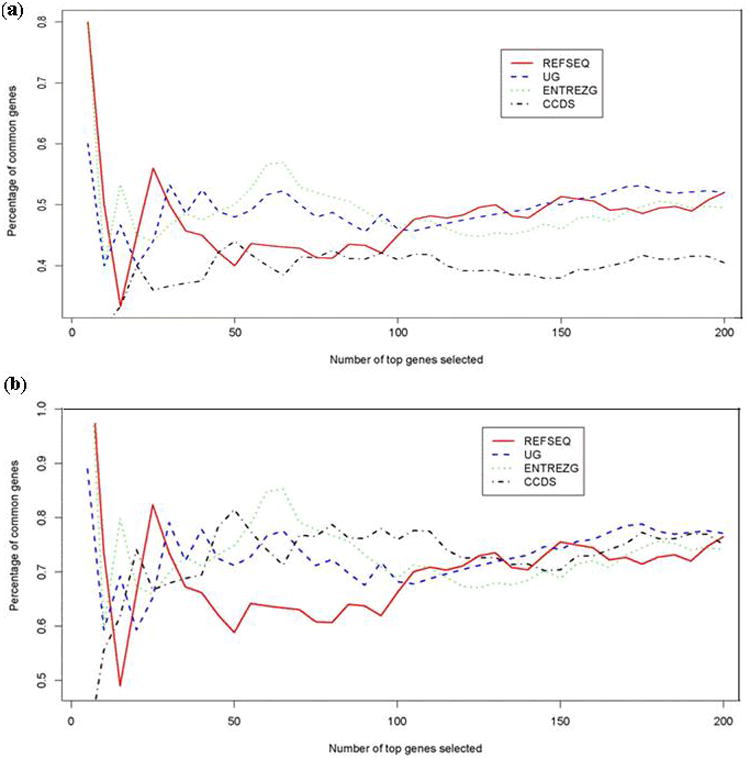
The agreement of U95Av2 and U133A assessed using the proportion of the common top differentially expressed genes between two subtypes (TEL-AML1 and MLL). The bottom figure is the result after removing the difference caused by the different percentages of number of common genes in different redefined CDFs. UG stands for UniGene and ENTREZG represents ENTREZ GENE.
The biology behind DNA microarray suggests that expression levels measured from experiments are on transcript level, not gene level. With the estimation of 30-99% genes exhibiting alternative splicing, DNA microarrays should be designed to permit delineation of differential expression of different transcripts representing alternative splice variants. However, probes in the traditional Affymetrix chips are skewed towards the 3’ UTR end. Such distribution makes it hard to differentiate splice variants. Luckily, the new generation of microarrays has been designed to have such power. For example, the probes in ExonHit microarrays are uniformly distributed along the entire lengths of genes (73). Among the four redefinition methods, UniGene and ENTREZ Gene represent gene-level analysis while REFSEQ and CCDS represent transcript-level analysis. REFSEQ and CCDS have better consistency when using the correlation of common targets between different generation as the consistency measure. CCDS are more comprehensive but less accurate comparing to REFSEQ with respect to splice variants since it contains complete coding sequences from GenBank without expert curation.
Most microarray experiments were conducted to identify differentially expressed transcripts. When using the proportion of common selected targets among the top K differentially expressed targets as the measure of consistency, percentages of common targets in different generations tended to be highly related to the results. For example, according to UniGene, ENTREZG, and REFSEQ, about two thirds of the redefined probe sets in redefined CDFs for U133A are paired with redefined probe sets for U95Av2. They tend to have similar results when K, the number of top selected genes considered, is at least 100. However, only half of the probe sets in CCDS-based CDFs for U133A are paired with those for U95Av2. Consequently, the proportion of common top selected genes tends to be smaller. The correlation between the proportion of common top selected genes and the percentage of common genes for redefined CDFs for U133A is over 95% when K is at least 100. Figure 6b shows the results when taking the percentage of common targets for redefined U133A CDFs into consideration. We can see that different redefinition methods tend to have similar agreement between U95Av2 and U133A when the number of top selected genes considered is at least 100.
6. Conclusion
In this paper, we have reviewed probes and probe sets used in DNA microarrays. Successful microarray applications begin with selecting proper probes that have high specificity and sensitivity. For cDNA spotted microarray, sequence-verification of clones before spotting is also important. Currently, various probe design tools can be used to select high quality probes based on our current genomic knowledge.
Our review and study suggest that the original Affymetrix probe set definition is problematic in many aspects according to the current genomic knowledge. The probe set definition issue is of critical importance, as it can dramatically influence the interpretation and understanding of expression data derived from microarray experiments when using Affymetrix. With several resources available, it is possible to re-analyze microarray data using redefined probe sets and enhance the accuracy of microarray data analysis. Therefore, we recommend to re-interpret existing microarray data with more accurate an dup-to-date genomic knowledge.
Acknowledgments
The authors thank members of the Microarray group in the Department of Biostatistics, Bioinformatics, and Biomathematics at Georgetown University for insightful discussion. The authors also thank Dr. John Weinstein and Dr. Barry Zeeberg from National Cancer Institute (NCI) for their collaboration on AffyProbeMiner.
Abbreviations
- CCDS
complete coding sequence)
- 3’ UTR
3’ untranslated region of mRNA
- 5’ UTR
5’ untranslated region of mRNA 5’
- RMA
Robust Multi-array Average or Robust Multi-chip Average
- SAM
Significant Analysis of Microarrays
- CDF
Chip Definition File
Contributor Information
Hongfang Liu, Email: hl224@georgetown.edu.
Ionut Bebu, Email: ib62@georgetown.edu.
Xin Li, Email: xl35@georgetown.edu.
References
- 1.Miklos GL, Maleszka R. Microarray reality checks in the context of a complex disease. Nat Biotechnol. 2004;22(5):615–21. doi: 10.1038/nbt965. http://dx.doi.org/10.1038/nbt965. [DOI] [PubMed] [Google Scholar]
- 2.Breitling R, Amtmann A, Herzyk P. Iterative Group Analysis (iGA): a simple tool to enhance sensitivity and facilitate interpretation of microarray experiments. BMC Bioinformatics. 2004;5:34. doi: 10.1186/1471-2105-5-34. http://dx.doi.org/10.1186/1471-2105-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park JC, Kim HS, Kim JJ. Bidirectional incremental parsing for automatic pathway identification with combinatory categorial grammar. Pac Symp Biocomput. 2001:396–407. [PubMed] [Google Scholar]
- 4.Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9(12):3273–97. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts CJ, Nelson B, Marton MJ, Stoughton R, Meyer MR, Bennett HA, He YD, Dai H, Walker WL, Hughes TR, Tyers M, Boone C, Friend SH. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science. 2000;287(5454):873–80. doi: 10.1126/science.287.5454.873. http://dx.doi.org/10.1126/science.287.5454.873. [DOI] [PubMed] [Google Scholar]
- 6.Shoemaker DD, Schadt EE, Armour CD, He YD, Garrett-Engele P, McDonagh PD, Loerch PM, Leonardson A, Lum PY, Cavet G. Experimental annotation of the human genome using microarray technology. Nature. 2001;409:922–927. doi: 10.1038/35057141. http://dx.doi.org/10.1038/35057141. [DOI] [PubMed] [Google Scholar]
- 7.Schena M. DNA Microarrays: A Practical Approach. Oxford University Press; 1999. [Google Scholar]
- 8.Mills JC, Roth KA, Cagan RL, Gordon JI. DNA microarrays and beyond: completing the journey from tissue to cell. Nat Cell Biol. 2001;3(8):E175–8. doi: 10.1038/35087108. http://dx.doi.org/10.1038/35087108. [DOI] [PubMed] [Google Scholar]
- 9.Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7(1):55–65. doi: 10.1038/nrg1749. http://dx.doi.org/10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 10.Hegde P, Qi R, Abernathy K, Gay C, Dharap S, Gaspard R, Hughes JE, Snesrud E, Lee N, Quackenbush J. A concise guide to cDNA microarray analysis. Biotechniques. 2000;29(3):548–556. doi: 10.2144/00293bi01. [DOI] [PubMed] [Google Scholar]
- 11.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270(5235):467–70. doi: 10.1126/science.270.5235.467. http://dx.doi.org/10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 12.Pease AC, Solas D, Sullivan EJ, Cronin MT, Holmes CP, Fodor SP. Light-generated oligonucleotide arrays forrapid DNA sequence analysis. Proc Natl Acad Sci U S A. 1994;91(11):5022–6. doi: 10.1073/pnas.91.11.5022. http://dx.doi.org/10.1073/pnas.91.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hughes TR, Mao M, Jones AR, Burchard J, Marton MJ, Shannon KW, Lefkowitz SM, Ziman M, Schelter JM, Meyer MR. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nature Biotechnology. 2001;19:342–347. doi: 10.1038/86730. http://dx.doi.org/10.1038/86730. [DOI] [PubMed] [Google Scholar]
- 14.Nakano YI, Okamoto M, Nishida T. Enriching agent animations with gestures and highlighting effects. Intelligent Media Technology for Communicative Intelligence. 2004;3490:91–98. doi: 10.1007/11558637_10. http://dx.doi.org/10.1007/11558637_10. [DOI] [Google Scholar]
- 15.King HC, Sinha AA. Gene expression profile analysis by DNA microarrays: Promise and pitfalls. JAMA, the journal of the American Medical Association. 2001;286(18):2280–2288. doi: 10.1001/jama.286.18.2280. [DOI] [PubMed] [Google Scholar]
- 16.Jarvinen AK, Hautaniemi S, Edgren H, Auvinen P, Saarela J, Kallioniemi OP, Monni O. Are data from different gene expression microarray platforms comparable. Genomics. 2004;83(6):1164–1168. doi: 10.1016/j.ygeno.2004.01.004. http://dx.doi.org/10.1016/j.ygeno.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Kong SW, Hwang KB, Kim RD, Zhang BT, Greenberg SA, Kohane IS, Park PJ. CrossChip: a system supporting comparative analysis of different generations of Affymetrix arrays. Bioinformatics. 2005;21(9):2116–7. doi: 10.1093/bioinformatics/bti288. http://dx.doi.org/10.1093/bioinformatics/bti288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carter SL, Eklund AC, Mecham BH, Kohane IS, Szallasi Z. Redefinition of Affymetrix probe sets by sequence overlap with cDNA microarray probes reduces cross-platform inconsistencies in cancer-associated gene expression measurements. BMC Bioinformatics. 2005;6(1):107. doi: 10.1186/1471-2105-6-107. http://dx.doi.org/10.1186/1471-2105-6-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardiman G. Microarray platforms- comparisons and contrasts. Pharmacogenomics. 2004;5(5):487–502. doi: 10.1517/14622416.5.5.487. http://dx.doi.org/10.1517/14622416.5.5.487. [DOI] [PubMed] [Google Scholar]
- 20.Petersen D, Chandramouli GVR, Geoghegan J, Hilburn J, Paarlberg J, Kim CH, Munroe D, Gangi L, Han J, Puri R. Three microarray platforms: an analysis of their concordance in profiling gene expression. BMC Genomics. 2005;6(1):63. doi: 10.1186/1471-2164-6-63. http://dx.doi.org/10.1186/1471-2164-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Draghici S, Khatri P, Eklund AC, Szallasi Z. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 2005 doi: 10.1016/j.tig.2005.12.005. http://dx.doi.org/10.1016/j.tig.2005.12.005. [DOI] [PMC free article] [PubMed]
- 22.Yauk CL, Berndt ML, Williams A, Douglas GR. Comprehensive comparison of six microarray technologies. Nucleic Acids Research. 2004;32(15):e124. doi: 10.1093/nar/gnh123. http://dx.doi.org/10.1093/nar/gnh123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knight J. When the chips are down. Nature. 2001;410:860–861. doi: 10.1038/35073680. http://dx.doi.org/10.1038/35073680. [DOI] [PubMed] [Google Scholar]
- 24.Forman-Kay JD. The ‘dynamics’ in the thermodynamics of binding. Nature Structural Biology. 1999;6:1086–1087. doi: 10.1038/70008. http://dx.doi.org/10.1038/70008. [DOI] [PubMed] [Google Scholar]
- 25.Taylor E, Cogdell D, Coombes K, Hu L, Ramdas L, Tabor A, Hamilton S, Zhang W. Sequence verification as quality-control step for production of cDNA microarrays. Biotechniques. 2001;31(1):62–5. doi: 10.2144/01311st01. [DOI] [PubMed] [Google Scholar]
- 26.Halgren RG, Fielden MR, Fong CJ, Zacharewski TR. Assessment of clone identity and sequence fidelity for 1189 IMAGE cDNA clones. Nucleic Acids Research. 2001;29(2):582–588. doi: 10.1093/nar/29.2.582. http://dx.doi.org/10.1093/nar/29.2.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kothapalli R, Yoder SJ, Mane S, Loughran TP., Jr Microarray results: how accurate are they. BMC Bioinformatics. 2002;3(1):22. doi: 10.1186/1471-2105-3-22. http://dx.doi.org/10.1186/1471-2105-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seluja GA, Farmer A, McLeod M, Harger C, Schad PA. Establishing a method of vector contamination identification in database sequences. Bioinformatics. 15:106–110. doi: 10.1093/bioinformatics/15.2.106. http://dx.doi.org/10.1093/bioinformatics/15.2.106. [DOI] [PubMed] [Google Scholar]
- 29.Lee C, Roy M. Analysis of alternative splicing with microarrays: successes and challenges. Genome Biol. 2004;5(7):231. doi: 10.1186/gb-2004-5-7-231. http://dx.doi.org/10.1186/gb-2004-5-7-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boue S, Letunic I, Bork P. Alternative splicing and evolution. Bioessays. 2003;25(11):1031–4. doi: 10.1002/bies.10371. http://dx.doi.org/10.1002/bies.10371. [DOI] [PubMed] [Google Scholar]
- 31.Harbig J, Sprinkle R, Enkemann SA. A sequence-based identification of the genes detected by probesets on the Affymetrix U133 plus 2.0 array. Nucleic Acids Res. 2005;33(3):e31. doi: 10.1093/nar/gni027. http://dx.doi.org/10.1093/nar/gni027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gautier L, Moller M, Friis-Hansen L, Knudsen S. Alternative mapping of probes to genes for Affymetrix chips. BMC Bioinformatics. 2004;5:111. doi: 10.1186/1471-2105-5-111. http://dx.doi.org/10.1186/1471-2105-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33(20):e175. doi: 10.1093/nar/gni179. http://dx.doi.org/10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heller MJ. DNA microarray technology: devices, systems, and applications. Annu Rev Biomed Eng. 2002;4:129–3. doi: 10.1146/annurev.bioeng.4.020702.153438. http://dx.doi.org/10.1146/annurev.bioeng.4.020702.153438. [DOI] [PubMed] [Google Scholar]
- 35.Liu AC, Collins AJ, Zhang ABL, Elliot CM, de Longueville EF, Shippy G, Baker IS, Kawasaki NE, Lee A, Luo Y. [March 1, 2007];Guidance to the MAQC Main Study. http://www.fda.gov/downloads/ScienceResearch/BioinformaticsTools/MicroarrayQualityControlProject/UCM126031.pdf.
- 36.Holloway AJ, van Laar RK, Tothill RW, Bowtell DDL. Options available—from start to finish—for obtaining data from DNA microarrays II. Nature Genetics. 2002;32:481–489. doi: 10.1038/ng1030. http://dx.doi.org/10.1038/ng1030. [DOI] [PubMed] [Google Scholar]
- 37.Tomiuk S, Hofmann K. Microarray probe selection strategies. Briefings in Bioinformatics. 2001;2(4):329. doi: 10.1093/bib/2.4.329. http://dx.doi.org/10.1093/bib/2.4.329. [DOI] [PubMed] [Google Scholar]
- 38.Lennon G, Auffray C, Polymeropoulos M, Soares MB. The IMAGE Consortium: an Integrated molecular analysis of genomes and their expression. Genomics. 1996;33(1):151–2. doi: 10.1006/geno.1996.0177. http://dx.doi.org/10.1006/geno.1996.0177. [DOI] [PubMed] [Google Scholar]
- 39.Boguski MS, Lowe TM, Tolstoshev CM. dbEST--database for “expressed sequence tags”. Nat Genet. 1993;4(4):332–3. doi: 10.1038/ng0893-332. http://dx.doi.org/10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- 40.Chen YG. Construction of a normalized cDNA library by mRNA-cDNA hybridization and subtraction. Methods Mol Biol. 2003;221:33–40. doi: 10.1385/1-59259-359-3. [DOI] [PubMed] [Google Scholar]
- 41.Patanjali SR, Parimoo S, Weissman SM. Construction of a Uniform-Abundance (Normalized) cDNA Library. Proceedings of the National Academy of Sciences. 1991;88(5):1943–1947. doi: 10.1073/pnas.88.5.1943. http://dx.doi.org/10.1073/pnas.88.5.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soares MB, Bonaldo MDF, Jelene P, Su L, Lawton L, Efstratiadis A. Construction and Characterization of a Normalized cDNA Library. Proceedings of the National Academy of Sciences. 1994;91(20):9228–9232. doi: 10.1073/pnas.91.20.9228. http://dx.doi.org/10.1073/pnas.91.20.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kane MD, Jatkoe TA, Stumpf CR, Lu J, Thomas JD, Madore SJ. Assessment of the sensitivity and specificity of oligonucleotide (50mer) microarrays. Nucleic Acids Research. 2000;28(22):4552–4557. doi: 10.1093/nar/28.22.4552. http://dx.doi.org/10.1093/nar/28.22.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research. 2005;110(1):462–467. doi: 10.1159/000084979. http://dx.doi.org/10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 45.Chou CC, Chen CH, Lee TT, Peck K. Optimization of probe length and the number of probes per gene for optimal microarray analysis of gene expression. Nucleic Acids Research. 2004;32(12):e99. doi: 10.1093/nar/gnh099. http://dx.doi.org/10.1093/nar/gnh099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. [March 1, 2007];ArrayDesigner. http://www.premierbiosoft.com/dnamicroarray/index.html.
- 47. [March 1, 2007];OligoArray2.0. http://berry.engin.umich.edu/oligoarray2/
- 48.Rouillard JM, Zuker M, Gulari E. OligoArray 2.0: design of oligonucleotide probes for DNA microarrays using a thermodynamic approach. Nucleic Acids Res. 2003;31(12):3057–62. doi: 10.1093/nar/gkg426. http://dx.doi.org/10.1093/nar/gkg426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. [March 1, 2007];OligoPicker. http://pga.mgh.harvard.edu/oligopicker/index.html.
- 50.Wang X, Seed B. Selection of oligonucleotide probes for protein coding sequences. Bioinformatics. 2003;19(7):796–802. doi: 10.1093/bioinformatics/btg086. http://dx.doi.org/10.1093/bioinformatics/btg086. [DOI] [PubMed] [Google Scholar]
- 51. [March 1, 2007];Oligodb. http://oligodb.charite.de/
- 52.Mrowka R, Schuchhardt J, Gille C. Oligodb--interactive design of oligo DNA for transcription profiling of human genes. Bioinformatics. 2002;18(12):1686–7. doi: 10.1093/bioinformatics/18.12.1686. http://dx.doi.org/10.1093/bioinformatics/18.12.1686. [DOI] [PubMed] [Google Scholar]
- 53. [March 1, 2007];ProbeWiz. http://www.cbs.dtu.dk/services/DNAarray/probewiz.php.
- 54.Nielsen HB, Knudsen S. Avoiding cross hybridization by choosing nonredundant targets on cDNA arrays. Bioinformatics. 2002;18(2):321–2. doi: 10.1093/bioinformatics/18.2.321. http://dx.doi.org/10.1093/bioinformatics/18.2.321. [DOI] [PubMed] [Google Scholar]
- 55. [March 1, 2007];Sarani. http://www.strandgenomics.com/saranioverview.html.
- 56. [March 1, 2007];VisualOMP. http://www.dnasoftware.com/Products/VisualOMP/index.htm.
- 57.Zukerman I, Litman D. Natural language processing and user modeling: Synergies and limitations. User Modeling and User-Adapted Interaction. 2001;11(1–2):129–158. doi: 10.1023/A:1011174108613. http://dx.doi.org/10.1023/A:1011174108613. [DOI] [Google Scholar]
- 58.SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci U S A. 1998;95(4):1460–5. doi: 10.1073/pnas.95.4.1460. http://dx.doi.org/10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le Novère N. MELTING, computing the melting temperature of nucleic acid duplex. Bioinformatics. 2001;17(12):1226–7. doi: 10.1093/bioinformatics/17.12.1226. http://dx.doi.org/10.1093/bioinformatics/17.12.1226. [DOI] [PubMed] [Google Scholar]
- 60.Schildkraut C. Dependence of the melting temperature of DNA on salt concentration. Biopolymers. 1965;3(2):195–208. doi: 10.1002/bip.360030207. http://dx.doi.org/10.1002/bip.360030207. [DOI] [PubMed] [Google Scholar]
- 61.Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Norton H. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nature Biotechnology. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. http://dx.doi.org/10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 62.Mei R, Hubbell E, Bekiranov S, Mittmann M, Christians FC, Shen MM, Lu G, Fang J, Liu WM, Ryder T. Probe selection for high-density oligonucleotide arrays. Proceedings of the National Academy of Sciences. 2003;100(20):11237–11242. doi: 10.1073/pnas.1534744100. http://dx.doi.org/10.1073/pnas.1534744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elo LL, Lahti L, Skottman H, Kyläniemi M, Lahesmaa R, Aittokallio T, Journals O. Integrating probe-level expression changes across generations of Affymetrix arrays. Nucleic Acids Research. 2005;33(22):e193. doi: 10.1093/nar/gni193. http://dx.doi.org/10.1093/nar/gni193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gautier L. [March 1, 2007];Alternative CDF environments. 2005 http://www.bioconductor.org/repository/devel/vignette/altcdfenvs.pdfcited.
- 65.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. http://dx.doi.org/10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DeCook R, Lall S, Nettleton D, Howell SH. Genetic Regulation of Gene Expression During Shoot Development in Arabidopsis. Genetics. 2006;172(2):1155–1164. doi: 10.1534/genetics.105.042275. http://dx.doi.org/10.1534/genetics.105.042275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu H, Zeeberg BR, Qu G, Koru AG, Ferrucci A, Kahn A, Ryan C, Nuhanovic A, Munson P, Reinhold WC, Weinstein JN. AffyProbeMiner: a web resource for computing or retrieving accurately redefined Affymetrix probe sets. 2007;23(18):2385–2390. doi: 10.1093/bioinformatics/btm360. http://dx.doi.org/10.1093/bioinformatics/btm360. [DOI] [PubMed] [Google Scholar]
- 68.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002 Apr;12(4):656–664. doi: 10.1101/gr.229202. http://dx.doi.org/10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yeoh EJ, Ross ME, Shurtleff SA, Williams WK, Patel D, Mahfouz R, Behm FG, Raimondi SC, Relling MV, Patel A. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1(2):133–143. doi: 10.1016/S1535-6108(02)00032-6. http://dx.doi.org/10.1016/S1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 70.Ross ME, Mahfouz R, Onciu M, Liu HC, Zhou X, Song G, Shurtleff SA, Pounds S, Cheng C, Ma J. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104(12):3679–3687. doi: 10.1182/blood-2004-03-1154. http://dx.doi.org/10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- 71.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Research. 2003;31(4):e15. doi: 10.1093/nar/gng015. http://dx.doi.org/10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences. 2001;98(9):5116–5121. doi: 10.1073/pnas.091062498. http://dx.doi.org/10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lyddy J. ExonHit Therapeutics. Pharmacogenomics. 2002;3(6):843–846. doi: 10.1517/14622416.3.6.843. http://dx.doi.org/10.1517/14622416.3.6.843. [DOI] [PubMed] [Google Scholar]