


Abstract
HIV-1 integrase (IN) catalyzes the integration of the proviral DNA into the cellular genome. The catalytic triad D64, D116 and E152 of HIV-1 IN is involved in the reaction mechanism and the DNA binding. Since the integration and substrate binding processes are not yet exactly known, we studied the role of amino acids localized in the catalytic site. We focused our interest on the V151E152S153 region. We generated random mutations inside this domain and selected mutated active INs by using the IN-induced yeast lethality assay. In vitro analysis of the selected enzymes showed that the IN nuclease activities (specific 3′-processing and non-sequence-specific endonuclease), the integration and disintegration reactions and the binding of the various DNA substrates were affected differently. Our results support the hypothesis that the three reactions may involve different DNA binding sites, enzyme conformations or mechanisms. We also show that the V151E152S153 region involvement in the integration reaction is more important than for the 3′-processing activity and can be involved in the recognition of DNA. The IN mutants may lead to the development of new tools for studying the integration reaction, and could serve as the basis for the discovery of integration-specific inhibitors.
INTRODUCTION
HIV-1 integrase (IN) is a key enzyme of the virus infectious cycle. Integration of the viral DNA into the cell genome is required for retroviral replication and chronic infection (1). Integration of proviral DNA proceeds through a defined set of DNA cutting and joining reactions. One activity of the retroviral integrases is 3′-end processing, in which two nucleotides are removed from the 3′-end of each strand of the viral DNA. A second activity, referred to as DNA strand transfer, is a concerted cleavage–ligation reaction in which the recessed 3′-ends of the viral DNA are covalently joined to host DNA. The final step is the repair of the integration intermediate in which the 5′-ends of viral DNA are joined to host DNA. This process requires the gaps flanking the provirus to be filled in and the two unpaired nucleotides at the 5′-ends of the viral DNA to be removed. In addition to the biologically relevant processing and joining activities operating in the viral proliferation process, IN exhibits two other in vitro endonuclease activities: disintegration, a reversal of the strand transfer step, which is a polynucleotidyl transfer reaction that is relatively independent of viral DNA sequence (2), and a more recently reported non-specific alcoholysis in vitro activity (3–5).
IN displays three independent structural and functional domains as determined by structural, complementation and mutational analyses. The amino-terminal domain (residues 1–50) contains the conserved HHCC motif that binds to one atom of zinc (6). This region is involved in protein–protein interaction and may contribute to the specific recognition of viral DNA ends (7,8). The central catalytic core domain (residues 50–212) contains the D,D(35)E motif which is invariant for retroviral INs, as well as INs of yeast retrotransposons and some bacterial transposases, and is crucial in enzyme activity (9). It can interact with DNA and divalent cations (10–13). The carboxy-terminal domain (residues 213–288) is the least conserved domain. It is involved in non-specific DNA binding and IN oligomerization necessary for the integration process (14–16). Although processing and strand transfer reactions require a full-length IN, disintegration and non-specific alcoholysis can be performed by the isolated catalytic domain. The biological significance of disintegration and non-specific alcoholysis in the viral cycle is unknown.
Little is known about the IN quaternary structure. HIV-1 integrase seems to exist in a dynamic equilibrium of monomers, dimers, tetramers and high-order oligomers (17,18). However, the active conformation has not yet been demonstrated. Mutagenesis, DNA footprinting and structural and molecular dynamic studies have shown that the three domains of IN are involved in DNA binding (19–21). Recent studies support the possible existence of different sites or structures needed in the recognition of the viral and cellular DNA. This suggests that the mechanism involved in the 3′-processing and the strand transfer are not completely identical and could involve different DNA binding sites (19,22,23).
Mutations in any of the D64D116(35)E152 motif were shown to affect the 3′-end processing, strand transfer and disintegration activities, arguing for the existence of a single site directly involved in catalysis of all three reactions (9,24–27). The proximity of these residues in the crystal structure of the avian sarcoma virus (ASV) and HIV-1 integrase core domains supports the notion that they form the catalytic center (28–30). Besides being the most conserved of the three IN domains, the structure of the catalytic core domain presents a striking structural homology with the RNase H domain of HIV-1 reverse transcriptase, Escherichia coli RNase H, Mu transposase and the Holliday junction resolvase RuvC, all enzymes which catalyze substitution reactions on phosphodiester bonds (31).
We have previously described a system in which the expression of HIV-1 IN in haploid RAD52 DNA repair system deficient and AB2 diploid Saccharomyces cerevisiae strains leads to the emergence of a lethal phenotype (23,32,33). This lethal phenotype is abolished when substitutions are introduced into the crucial triad residues. Our results strongly suggest that the lethal phenotype is due to DNA damage produced by the endonuclease-related IN activity in a reaction identical to the IN-associated alcoholysis previously described (3,5). The IN-associated endonuclease activity seems responsible for the lethal effect by inducing DNA breaks in the yeast nuclear genome. Recent results showing that recombinant HIV-1 IN is able to catalyze non-sequence-specific DNA hydrolysis in vitro even in the absence of HIV-1 LTR viral sequences further supports this hypothesis (34). Taken together, these results strongly suggest that IN-induced lethality reflects the cellular activity of the retroviral enzyme.
The IN-induced yeast lethality has already been used for the selection of revertant INs from the inactivated D116A IN (23). In contrast here, we used the yeast lethality assay for selecting mutants of HIV-1 IN generated by random mutagenesis in the V151E152S153 region and selected using the ‘yeast lethality assay’. Here we focus on the E152 residue, a crucial amino acid that has previously been demonstrated as belonging to the catalytic site and involved in all the IN activities (35) as well as the adjacent V151 and S153 residues. Different IN mutants able to induce the yeast lethal phenotype were obtained. Mutated INs were then purified and their in vitro activities were evaluated. Our results demonstrate that IN activities could be affected differently and suggest a precise involvement of the V151 E152S153 region in DNA recognition and 3′-processing.
MATERIALS AND METHODS
Bacterial strains, culture media, transformation procedure, DNA preparation and analysis
The E.coli strain DH5α was used for plasmid amplification and the bacterial transformation was performed as described previously (36). Bacteria were grown on standard LB medium containing 100 µg/ml ampicillin. Plasmids were extracted from bacteria using the boiling method (37). DNA restriction endonucleases and DNA ligase were used under conditions recommended by the supplier (Promega). DNA fragments were analyzed by electrophoresis on 1% agarose gel in the presence of ethidium bromide (0.5 µg/ml) under UV light.
Yeast strains, culture media, growth conditions and yeast transformation
The diploid yeast strain AB2 (MATa/α ura3/ura3 leu2/leu2 ade3/ade3 his3/his3 trp1/trp1) and haploid W839–5C (MATa ura3 leu2 ade3 his3 rad52:TRP1) were used to perform the yeast lethal assay (32). The protein extracts used for IN purification were obtained from the yeast strain JSC310 (MATα ura3-52 leu2 trp1 prb1-1122 pep4-3 prc1-407) transformed with pHIV1-SF2IN or the different IN expressing vectors (32). Culture media: (i) yeast complete medium YEPD (1% yeast extract, 2% bactopeptone, 2% glucose); (ii) yeast selective media: YNB lacking uracil and leucine (0.67% yeast nitrogen base without amino acids, 5.6% glucose); (iii) YCAD lacking uracil (YNB supplemented with 0.5% casamino acids). Amino acids and bases (20–30 mg/l) were added as required. Liquid cultures were performed in Erlenmeyer flasks filled to a fifth of their capacity and then shaken. Solid media were obtained by supplementing liquid media with 2% bacto-agar. Yeast strains were grown at 30°C. Yeast transformation was performed using LiCl as described (38).
Plasmid vectors
The IN expression vectors were derived from the yeast/E.coli shuttle plasmid pBS24.1 previously described (32). All the other vectors expressing the mutated INs were constructed as described in the mutagenesis section below. The pUC19 vector substrate of the endonucleolytic IN activity was purchased (Gibco) and maintained in E.coli DH5α bacterial strain. The pUC19 IN used for the generation of mutated IN-encoding genes was constructed in the laboratory by inserting the IN gene between SacI SalI cloning sites into the vector.
Random mutagenesis and lethality test
The protocol used for random mutagenesis of HIV-1 IN is schematically described in Figure 1. The specific mutagenic PCR in the V151E152S153 region was performed using the following primers. Primer 9298: (5′-TGC TGT CTT AAG GTG TTC GAG CAT GAT GTC TTA CCT GTC CTA TAA TTT TCT TTA ATT-3′). Primer 9299: (5′-GGC ATT CCC TAC AAT CCC CAA AGT CAA CGA GTA NNN NNN NNN ATG AAT AAT GAA TTA AAG AAA AT-3′). (The bold characters indicate the randomly mutated sequence corresponding to the DNA sequence encoding amino acids V151, E152 and S153 in the integrase gene.) The oligomer 9299 contains a random sequence of 9 nt in place of the nucleotides encoding the V151E152 S153 region of HIV-1 IN. The PCR was performed in two steps. First, the two primers (10 ng each) were mixed in 50 µl of 200 µM dNTP, 1× PCR Goldstar buffer, 1.5 mM MgCl2 and 1.5 U Taq DNA polymerase (Goldstar Eurogentec) following one cycle of annealing: 1 min at 94°C, 1 min at 37°C and 2 min at 72°C. Secondly, the double-stranded oligonucleotides were PCR amplified by adding 1 µM of oligonucleotides 9301 (5′-TGCTGTCTTAA GGT-3′) and 9300 (5′-ATTTGGCATTCCC-3′) for 20 cycles of 30 s at 94°C, 1 min at 55°C and 2 min at 72°C.
Figure 1.

Random mutagenesis of HIV-1 IN and mutant selection strategy. The construction of a library of HIV-1 IN variants containing random nucleotide substitutions in the V151E152S153 region is described in Materials and Methods. The different mutated IN encoding genes were introduced into the expression vector pHIVSF2IN to express the corresponding mutated INs in the IN-sensitive yeast strains AB2 and W839–5C. The yeast lethal assay was performed under the conditions described in Materials and Methods and the clones presenting lethality were selected.
The amplified DNA was digested with BsmI and AflI (Gibco BRL), adding 50 mM NaCl and 10 mM MgCl2, and purified on the QIAquick PCR purification kit (Qiagen). The pUC19IN plasmid was also digested by these two enzymes and ligated with a double-stranded DNA insert containing the single site NotI, which allows a positive selection of correctly cloned mutants. This new plasmid pUC19IN-NotI was transformed in the bacterial strain DH5α. After extraction it was digested with BsmI and AflII. The vector was then purified with the QIAquick gel extraction kit (Qiagen), ligated to the 95 bp randomly mutated inserts using T4 DNA ligase (Promega) and transformed into competent DH5α. A plasmid library was prepared from a culture of transformed cells in LB Amp medium containing 100 µg/ml ampicillin with Miniprep Express Matrix kit (BIO101-Ozyme). The plasmids were pooled and digested with SacI, SalI and NotI in order to eliminate inserts without random sequences. The 1491 bp inserts were eluted from a 1% agarose gel and purified with QIAquick gel extraction kit. The plasmid pHIVSF2IN was digested with SacI and SalI, purified on QIAquick gel extraction kit and ligated with the inserts. The ligation mixture was used for transformation of competent bacteria DH5α, the clones obtained by selection on agar plates of LB Amp were pooled and plasmids were prepared from an overnight culture in LB Amp medium. The pHIVMUTIN plasmids containing randomly mutated sequences were transformed in the two IN-sensitive yeast strains: the diploid AB2 and the haploid W839–5C. The selection of mutants was performed using the lethality assay on agar plates of YNB 5.6% glucose without leucine.
Sequencing
Yeast cells were grown overnight on YCAD–URA medium and DNA was extracted using the protocol described by Robzyk and Kassir (39). The DNA was used for transformation of competent bacteria DH5α and plasmid purification was performed from clones selected on LB Amp medium using the BIO101 (Promega) protocol of purification. The whole IN encoding gene of all mutated HIV-1 IN proteins, including the randomized segment, was checked by PCR-based sequencing (ABI Prism Big Dye terminator cycle sequencing ready reaction kit; Applied Biosystems). The primer was an 18mer (5′-TAG CAG GAA GAT GGC CAG-3′) corresponding to the position encoding amino acid residues 104–110 of HIV-1 IN in the gene. The amount of DNA used was 0.5 µg.
Yeast lethal assay
After generation of the mutant library, the effect of these mutations on IN related to the lethality in yeast was observed using the ‘drop test’ (32). In this test, 3 µl droplets of plasmid-containing yeast standard suspension (∼20 000 URA+ yeast cells) were deposited on YNB solid medium lacking leucine and containing 5.6% glucose to allow IN expression. After incubation for 3 days at 28°C, the effect of IN expression on yeast was determined either by visual observation or by measuring the survival rates of the cells as described previously (32). Yeast cell survival rates were evaluated by a comparison of the colony-forming unit number with the cell number as determined with a hemacytometer.
Purification of integrase
Obtaining yeast extracts containing recombinant integrase. JSC310 (IN) yeast cells were grown for 3 days on a YCAD selective medium, precultured in YNB liquid medium and then incubated in YPDA complete medium for 3 days. Cells were harvested for 10 min at 4 000 g and broken with glass beads in 5 mM HEPES pH 7.6, 5 mM EDTA and 1 mM DTT. The lysate was centrifuged at 12 000 g for 20 min and the pellet was solubilized in 1 M NaCl and 10 mM CHAPS. Final NaCl concentration of the extracts was attained by addition of salt or after dialysis, according to the affinity column elution procedure used in the subsequent purification steps.
Integrase purification procedure. Purification was performed as previously described (40) except that only one heparin column was used during the procedure. Purified IN solutions were kept at –80°C. Proteins were analyzed by electrophoresis in a 12% SDS–PAGE and western blot using monoclonal mAb 44 anti-IN (a generous gift from Dr A. Leavitt, University of California at San Francisco, USA).
In vitro integrase assays
All assays were performed in 20 mM HEPES pH 7.5, 10 mM DTT, 7.5 mM MnCl2 and 0.05% NP40. For the 3′-processing, integration and disintegration activities, IN (5 pmol) and radiolabeled oligonucleotide (1 pmol) in a total volume of 20 µl were used. The reaction mixture was incubated at 37°C for 1 h and then stopped by adding 10 µl of loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue) and heating at 90°C for 5 min. The reaction products were analyzed by electrophoresis on 12% or 15% polyacrylamide gels with 7 M urea in Tris–borate–EDTA (TBE) pH 7.6 and autoradiographed. The sequence of the oligonucleotides used in the 3′-processing and strand transfer assays was as follows. ODN 70 (21mer): 5′-GTG TGG AAA ATC TCT AGC AGT-3′; ODN 71 (19mer): 5′-GTG TGG AAA ATC TCT AGC A-3′; ODN 72 (21mer): 5′-ACT GCT AGA GAT TTT CCA CAC-3′.
The 3′-processing reaction was performed in the presence of a double-stranded DNA substrate formed by the ODN 70 hybridized to ODN 72. Substrates were either labeled on the 5′-end (5′-radiolabeled ODN 70 hybridized to ODN 72) or on the 3′-end. The latter was prepared as follows: the ODN 71 (lacking the GT in the 3′-end) was annealed with the 21mer ODN 72 by heating for 3 min at 90°C and slow cooling. This double-stranded DNA was labeled at the 3′-end with [α-32P]dGTP and [α-32P]TTP in the presence of the exonuclease-free Klenow fragment of E.coli DNA polymerase. For the strand transfer assays, the substrate was formed by the 5′-end radiolabeled ODN 71 hybridized with ODN 72. Disintegration assays were performed using a ‘Y-like’ 5′-end radiolabeled dumbbell substrate as described previously: TGC TAG TTC TAG CAG GCC CTT GGG CCG GCG CTT GCG CC (41). The activities were evaluated by scanning the product bands using the NIH software.
The endonucleolytic reaction was performed using 5 pmol of IN incubated with 200 ng of pUC19 plasmid for 2 h at 37°C in the reaction mix described above. The reactions were stopped by addition of 2 µl of 95% formamide, 20 mM EDTA and 0.05% bromophenol blue stop solution. Samples were analyzed on a 1% agarose minigel containing ethidium bromide (0.5 µg/ml). Electrophoresis was carried out for 30 min at 100 V at room temperature. DNA was detected by fluorescence upon exposure to UV light. Activity was evaluated by quantification of the bands corresponding to the different topological forms of the plasmid using the NIH software after scanning.
DNA binding assays
Nitrocellulose filters (0.45 µm) (Whatman) were washed in wash buffer (200 mM HEPES pH 7.5, 10 mM DTT, 0.05% NP-40, 7.5 mM MnCl2, 0.01% BSA) for 10 min. Integrase was mixed with the different radiolabeled substrates used for processing, strand transfer or disintegration, with ratios of 5 pmol of IN for 0.2 pmol of substrate in 200 µl of activity buffer (200 mM HEPES pH 7.5, 10 mM DTT, 0.05% NP-40, 7.5 mM MnCl2) at 4°C and filtrated with the multifilter Millipore system at a constant flow. The filters were washed three times with 300 µl of wash buffer and then dried and counted in a scintillation counter (Wallac 1409). All the experiments were reproduced independently three times and the final results were given as the means of the three values ± SD.
RESULTS
Screening of integrases mutated in the V151E152S153 region
To determine the importance of the V151E152S153 residues on the activity of HIV-1 IN, we generated random sequence substitutions in the wild-type gene using the protocol described in Figure 1. After mutagenesis, cloning in the pHIV1SF2IN expression vector and transformation of the E.coli DH5α strain, a library of 326 bacterial clones was obtained encoding the corresponding number of IN proteins. To identify active mutants of IN among this library, we used a genetic selection system in which the expression of active HIV-1 IN in yeast leads to a lethal phenotype as described previously (32).
The plasmid DNA extracted from the 326 pooled bacterial colonies was used to transform in parallel both the IN-sensitive diploid AB2 and haploid W839–5C yeast strains previously described. After yeast transformation, 1923 clones from both strains were obtained. The ratio between the initial library size (326) and the clones obtained (1923) corresponds to a good level of representativity from the initial mutant library. The lethality test was then performed. Among the total 1923 clones obtained, 1868 did not show any modification of their growth under lethal IN expression conditions and thus were eliminated. In contrast, 55 clones presenting both a decrease in yeast growth and survival rate when the retroviral enzyme was expressed were used for further studies, since they should correspond to IN enzymatically active mutants.
Identification of the mutations
The 55 selected clones were sequenced in the whole IN encoding gene. Twelve IN mutants carried mutations outside the V151E152S153 interest site and were eliminated. The 43 remaining clones were distributed in 15 IN families carrying different amino acid substitutions in the V151E152S153 region (Table 1). After random mutagenesis, one of the selected clones carried the wild-type V151E152S153 sequence. Since the latter was generated by the same random mutagenesis protocol, we used it as a wild-type positive control in our selection system after checking the absence of mutation in the other parts of the protein.
Table 1. Sequence of the V151 E152S153 region of the selected integrases.
| IN | No. of selected clones | V151E152S153 sequence | Nucleotide sequence |
|---|---|---|---|
| WT IN | 5 | VES | GTA GAA TCT |
| d1 | 3 | TEQ | ACT GAG CAA |
| d46 | 2 | LEL | CTG GAA CTG |
| d34 | 8 | LVF | TTG GTA TTT |
| d37 | 3 | AMA | GCT ATG GCG |
| d30 | 2 | YVP | TAC GTT CCA |
| r14 | 2 | HST | CAT TCT ACC |
| r24 | 1 | ALT | GCT TTA ACT |
| r1 | 1 | KLT | AAA CTA ACT |
| r9 | 3 | SSP | TCG AGC CCG |
| r17 | 2 | HGL | CAC GGC CTT |
| r21 | 1 | VNR | GTG AAC AGA |
| r22 | 1 | ESN | GAA TCA AAT |
| r34 | 6 | TSM | ACT AGC ATG |
| r36 | 3 | TFC | ACT TTT TGC |
Sequence selection was carried out as described in Materials and Methods. The 43 selected mutants were distributed in the 15 families presented here. The names of the IN mutants were chosen arbitrarily: d means that they were selected in the diploid yeast strain and r in the haploid RAD52 deficient strain. The number of clones in each family and both nucleotide and amino acid sequences corresponding to the V151E152S153 region are indicated.
Yeast lethal phenotype analysis of the selected mutants
It is possible to have several plasmids inside yeast cells. Thus we isolated each vector expressing the corresponding mutants from a single bacterial clone and reintroduced it into both diploid AB2 and haploid rad52– yeast strains. Next we checked the lethality under the conditions described previously. Similar results were obtained with both strains. The phenotype observed for the 15 different types of mutants in the AB2 diploid cells are shown in Figure 2.
Figure 2.
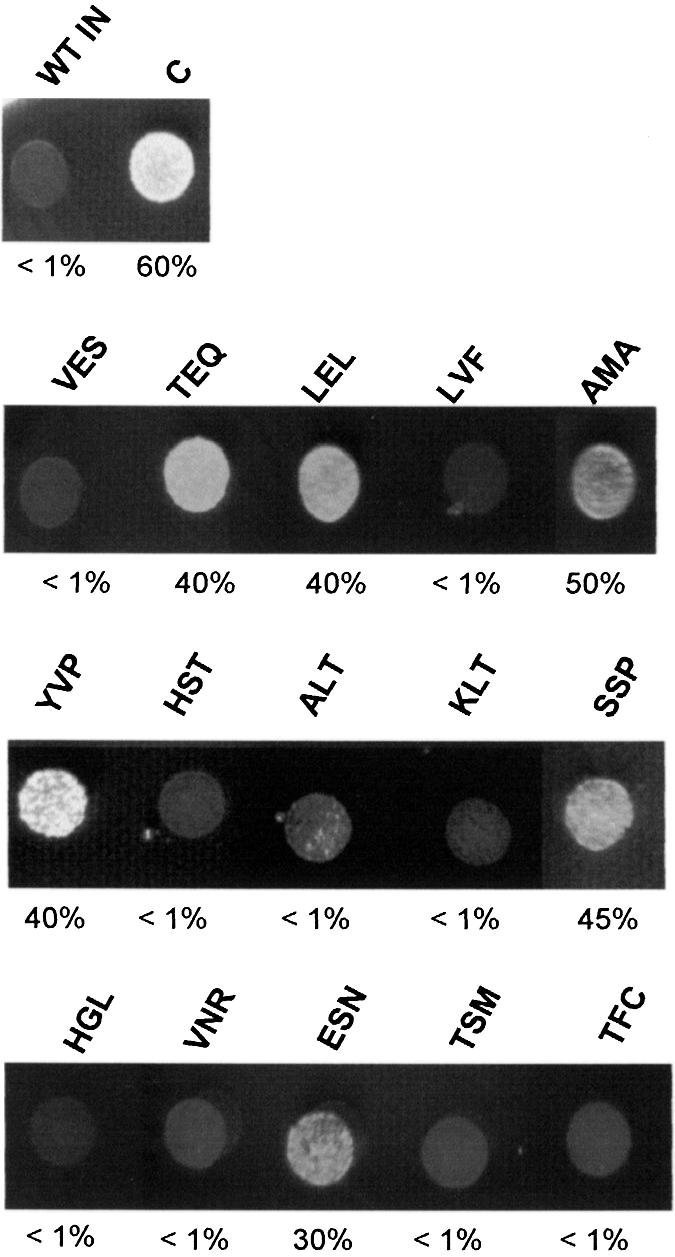
Yeast lethal phenotype induced by the selected integrase mutants. Drop tests: the INs described in Table 1 were expressed in the diploid AB2 yeast strain under minimal IN expression (5.6% glucose) on solid YNB media lacking leucine in order to perform the lethality test. Photographs were taken after incubating yeast cells for 3 days at 28°C. AB2 (WT IN) expressing the wild-type integrase and AB2 (C) expressing no integrase were used as controls. Percentages given for the ‘drop test’ experiments correspond to the yeast cell survival rates in the absence of integrase (C) or in the presence of wild-type integrase (WT IN) or the mutated integrase as indicated. The yeast cell survival rates were evaluated as described in Materials and Methods. The survival rates, measured as described in Materials and Methods, are shown in the figure.
All yeast cells expressing HIV-1 IN mutants presented a decrease in both growth and survival rate when compared with yeast cells expressing no IN. We also observed different degrees of lethality compared with the one induced by the wild-type IN, suggesting that the selected mutants may show different levels of enzymatic activities. As expected, the wild-type control (VES) presented a lethal phenotype. The most efficient INs able to produce the yeast lethal phenotype were the proteins carrying the following sequences in the V151E152S153 region: LVF, HST, ALT, KLT, HGL, VNR, TSM and TFC. The other selected mutants presented survival rates from 30 to 50% and corresponded to enzymes carrying the following sequences: TEQ, LEL, AMA, YVP, SSP and ESN.
Purification of the mutated integrases
In order to study the effect of amino acid substitutions on the in vitro activity of each retroviral mutated IN recombinant enzyme, we expressed it in the JSC310 yeast strain under conditions allowing a high level of IN expression as described previously (32). After expression, the mutated enzymes were purified as the wild-type IN. In all cases a protein of 32 kDa apparent size was detected in a Coomassie Blue stained SDS–PAGE. These 32 kDa purified mutated proteins were positive in a western blot in the presence of monoclonal anti-IN antibodies (Fig. 3). Differences in yields in the induction and purification steps observed between the mutants were not significantly different from those observed in different preparations of the wild-type IN.
Figure 3.

Western blot analysis of the purified integrase fractions. All INs were purified according to the procedure described in Materials and Methods. Each purified fraction (2.5 µl) was loaded on SDS–PAGE gel and subjected to western blot analysis using the monoclonal mAb 44 anti-IN.
In vitro disintegration, strand transfer and processing activities of the mutated integrases
The in vitro disintegration, strand transfer and 3′-processing reactions were performed under the optimal conditions previously established. In all cases the same amount of IN protein was used.
Disintegration. The fact that the HIV-1 IN catalytic core is sufficient to catalyze the disintegration reaction suggests that this catalytic step is less stringent than in the case of the 3′-end processing and strand transfer reactions. This latter characteristic allows many genetic variants of IN to catalyze the disintegration step while lacking the other activities. Regardless of its relevance in vivo, the disintegration reaction has been very useful in the characterization of several mutants, particularly those encoding substitutions in the core domain (41).
We therefore assayed the disintegration activity of all mutated purified proteins in the presence of the ‘Y-oligomer’ disintegration substrate (Fig. 4). Only the wild-type control IN (VES) and the TEQ mutant were able to catalyze the standard disintegration reaction. Mutant LEL showed an activity with a different specificity than that expected for wild-type IN, producing products of greater size than the wild-type disintegration 14mer product. Comparison of overexposed gels obtained with the other IN mutants showed that these bands were observed for all proteins at different intensities. Different intensities were also observed for different preparations of the same variant of IN.
Figure 4.

Disintegration activity of the integrases. (A) Reactions were performed as described under Materials and Methods. The different INs (5 pmol) were incubated with the DNA disintegration substrate (1 pmol) for 1 h at 37°C. (B) Products were separated on denaturing 12% polyacrylamide gel and autoradiographed. S corresponds to the dumbbell substrate and P to the disintegrated product. Integrases are named by the amino acid sequence present in the V151E152S153 region.
Strand transfer. Each purified protein was tested for strand transfer activity on a duplex substrate formed by ODN 71/ODN 72. Strand transfer activity was detected only with the control IN enzyme carrying the wild-type sequence (Fig. 5). No activity with this assay was observed with the other mutated proteins, suggesting that the wild-type sequence V151E152S153 region is absolutely necessary for this activity.
Figure 5.

Strand transfer activity of the integrases. (A) Reactions were performed as described under Materials and Methods. The different INs (5 pmol) were incubated with the DNA strand transfer substrate (1 pmol) for 1 h at 37°C. (B) Products were separated on denaturing 12% polyacrylamide gel and autoradiographed. S corresponds to the preprocessed 19mer substrate and IP to the integration products. Integrases are named according to the amino acid sequence present in the V151E152S153 region.
3′-processing. The 3′-processing reaction was assayed in the presence of the duplex 5′-end radiolabeled ODN 70 hybridized to ODN 72. As reported in Figure 6A, wild-type IN showed a strong 3′-processing activity with production of the –2 nt processed product. An interesting result was obtained with the mutated integrases: all proteins exhibited the 3′-processing activity, although at different levels. The LEL, YVP, HST and HGL mutants presented a processing activity close to that of the wild type, in contrast with other mutants which presented reduced levels of 3′-processing activity.
Figure 6.

3′-processing activity of the integrases. Reactions were performed as described under Materials and Methods. The different INs (5 pmol) were incubated with the DNA substrate (1 pmol) for 1 h at 37°C. Products were separated on denaturing (A) 12% or (B) 15% polyacrylamide gel and autoradiographed. Integrases are named according to the amino acid sequence present in the V151E152S153 region. The 3′-processing reaction was performed in the presence of (A) a 5′-end radiolabeled ODN 70 hybridized to ODN 72 or (B) a 3′-end GT-radiolabeled substrate. S corresponds to the substrate and P to the processed products. The arrowhead indicates the position of the GT cyclic dinucleotide.
With all proteins, wild-type and mutated integrases, we observed, besides the 19 nt product, other low-molecular-weight oligonucleotides. This was not the case with a D116A integrase, a protein purified in the same way that is completely inactive for all IN activities. To address the question of the origin of these low-molecular-weight bands, we studied the non-sequence-specific endonuclease activity that has been associated with IN (5,34).
Since in the assay with the 5′-end-labeled ODN it is difficult to differentiate between the sequence-specific 3′-processing and the non-sequence-specific endonuclease activity of IN, we performed a 3′-processing test, but this time in the presence of a substrate containing the labeled GT in the 3′-end. In this case we should observe only the specific dinucleotide product of the 3′-processing activity. As shown in Figure 6B, the IN-dependent removal of the [32P](GT) dinucleotide was observed once again for all proteins, except the D116A mutated IN. We therefore concluded that the mutated (VES region) INs were able to perform the sequence-specific 3′-processing reaction.
Non-sequence-specific endonuclease activity of the mutated integrases
In addition to the sequence-specific 3′-processing reaction, HIV-1 IN carries a second non-sequence-specific endonuclease activity (5), which seems to be involved in the lethal effect observed in yeast (34). Thus we assayed this latter reaction in vitro under the conditions described in Materials and Methods.
The non-sequence-specific activity of IN was monitored by observing nicking of close circular DNA by an assay described previously (34). We therefore tested the capacity of the different enzymes to induce DNA breaks inside the pUC19 plasmid. As shown in Figure 7, wild-type VES IN and mutants AMA, YVP and HST were the most active enzymes in the non-sequence-specific endonuclease activity assay.
Figure 7.

Non-sequence-specific endonucleolytic activity of the integrases. Supercoiled (CC) pUC19 DNA (200 ng) was incubated as described in Materials and Methods with the different INs (5 pmol) for 2 h at 37°C. The products were separated on 1% agarose gel in the presence of ethidium bromide (50 µg/ml) and visualized under UV light. CC, native closed circular form of pUC19; OC, open circular form; L, linear form. Integrases are named according to the amino acid sequence present in the V151E152S153 region.
Taken together, these results indicate that mutations in the E152 region seem to affect the activities of the mutated enzymes differently. To address whether this could be due to changes in the binding affinities for the different in vitro DNA substrates, we determined the DNA binding capacity of the selected INs.
DNA binding of the mutated integrases
All mutants were analyzed for their capacity to bind DNA molecules corresponding to the processing, strand transfer and disintegration of ODN substrates. The percentages of substrate retained by mutated IN proteins on nitrocellulose filters were compared with that obtained with the wild-type IN. This approach makes it possible to measure rapidly the general ability of IN to bind DNA.
As shown in Figure 8, the binding of the substrates to IN was differently affected by the mutations depending on the type and the position of the amino acid substitutions. Both decrease and increase effects on DNA binding were detected compared with the wild-type level of DNA binding. The fact that the DNA binding capacity of mutated INs was partially impaired or affected by mutations in the V151E152S153 region means that this region is involved in the binding of the physiological substrates. Furthermore, some mutants conserving the E152 catalytic amino acid but carrying mutations in the adjacent amino acids present affected DNA binding (see mutants TEQ and LEL). This suggests that the catalytic E152 is not involved alone in that process but rather the whole region, or that the structure of the region is important and that the adjacent V151 and S153 could be critical for the binding of both viral and cellular DNA.
Figure 8.

DNA binding properties of integrases. Equal concentrations of WT control IN or mutated integrases (5 pmol) were incubated with the radiolabeled olignucleotide (0.2 pmol) corresponding to (A) 3′-processing, (B) strand transfer or (C) disintegration reaction substrates. Following incubation the mixtures were filtered through nitrocellulose filters and extensively washed. The extent of DNA bound to each enzyme was quantified by scintillation counting. Bars represent the means ± SD of triplicate independent experiments. Integrases are named according to the amino acid sequence present in the V151E152S153 region.
The binding of the different substrates can also be affected independently by the mutations (see mutants TEQ, LEL, HST or KLT). This result suggests that the positioning and/or the binding mechanism of the different substrates is not the same, supporting the hypothesis of different binding sites and binding processes for corresponding viral and host DNA.
DISCUSSION
We have previously shown that HIV-1 IN causes a lethal effect due to its activity when expressed in yeast (32,34). Here we used the lethality test to screen only the active mutated INs from a population of variants containing random nucleotide substitutions in the V151E152S153 region. The acidic D,D(35)E triad located in the catalytic core of IN has been shown to be crucial for the reaction catalyzed by retroviral integrases and other related enzymes. The V151E152S153 region was targeted since our aim was to determine the importance of E152 and its neighboring amino acids on the different enzyme activities of HIV-1 IN. Forty-three INs mutated in the region of interest were selected as being able to induce the yeast lethal phenotype, meaning that IN remained active despite lacking the glutamic acid in position 152. Mutated IN proteins were distributed in 15 families of INs carrying the following residues in this region: VES (wild type), TEQ, LEL, LVF, AMA, YVP, HST, KLT, ALT, SSP, HGL, VNR, ESN, TSM and TFC. Their in vitro biochemical properties were studied to gain further information about the involvement of the V151E152S153 region in the reactions catalyzed by the retroviral enzyme and its ability to bind a DNA substrate.
Integrase activities
Recombinant HIV-1 INs carrying mutations introduced into the V151E152S153 site were assayed for the different activities. As summarized in Table 2, the effect of mutagenesis on the various activities was markedly different. The nuclease activities of the retroviral integrase (3′-processing and non-sequence-specific nuclease) were less affected by the mutations than the strand transfer or the disintegration reactions. These results suggest that the V151E152S153 region is less involved in the nuclease activities of IN than in the strand transfer and disintegration. This may be explained by the fact that the crucial amino acid triad D,D(35)E is not involved simultaneously in the same enzyme activities performed by IN in vivo. This hypothesis is supported by other reports based on the effect of IN inhibitors or mutagenesis studies showing that processing and strand transfer may involve different mechanisms, structures or/and DNA binding abilities.
Table 2. In vitro characteristics of the different integrases.
| Mutant | VES region sequence | Survival rate (%) | In vitro IN activities | DNA binding | |||||
|---|---|---|---|---|---|---|---|---|---|
| Processing (%) | Integration (%) | Disintegration (%) | Non-sequence-specific nuclease (%) | P (%) | ST (%) | D (%) | |||
| WT IN | VES | <1 | 30 ± 2 | 30 ± 1 | 50 ± 2 | 10 ± 3 | 100 | 100 | 100 |
| d1 | TEQ | 40 | 15 ± 1 | – | 40 ± 1 | 5 ± 1 | 60 | 140 | 90 |
| d46 | LEL | 40 | 25 ± 1 | – | – | 10 ± 3 | 110 | 50 | 80 |
| d34 | LVF | <1 | 15 ± 2 | – | – | 10 ± 2 | 60 | 50 | 50 |
| d37 | AMA | 50 | 15 ± 2 | – | – | 15 ± 3 | 50 | 75 | 50 |
| d30 | YVP | 40 | 25 ± 3 | – | – | 15 ± 3 | 40 | 90 | 110 |
| r14 | HST | <1 | 30 ± 1 | – | – | 20 ± 4 | 10 | 25 | 80 |
| r24 | ALT | <1 | 25 ± 1 | – | – | 10 ± 3 | 70 | 95 | 60 |
| r1 | KLT | <1 | 15 ± 2 | – | – | 10 ± 1 | 50 | 65 | 120 |
| r9 | SSP | 45 | 15 ± 1 | – | – | 5 ± 1 | 70 | 50 | 110 |
| r17 | HGL | <1 | 25 ± 1 | – | – | 10 ± 3 | 60 | 110 | 150 |
| r21 | VNR | <1 | 20 ± 1 | – | – | 5 ± 2 | 100 | 160 | 110 |
| r22 | ESN | 30 | 20 ± 2 | – | – | 5 ± 1 | 50 | 110 | 70 |
| r34 | TSM | <1 | 25 ± 1 | – | – | 5 ± 2 | 70 | 125 | 120 |
| r36 | TFC | <1 | 25 ± 4 | – | – | 5 ± 3 | 140 | 275 | 150 |
All the characteristics of the mutated and wild-type integrases are listed. The survival rates of the diploid AB2 yeast strain expressing the corresponding mutant are shown. For INs activities, the results are expressed as the percentage means of substrate consumed ± SD of triplicate independent experiments; – corresponds to no detected activity. Data for the DNA binding represent the percentage of DNA binding capacity of the INs compared with the binding observed with wild-type IN (corresponding to 100% binding capacity).
Studies using IN inhibitors have already indicated that both activities could be affected differently (22). In their report, Hazuda et al. (22) showed that the antiviral activity of diketo acids is due exclusively to inhibition of the strand transfer activity of HIV-1 IN, while the processing reaction is not affected. We found similar results with a peptide isolated by using the phage display technique which is able to inhibit the strand transfer activity of IN without affecting processing (C. Desjobert, V. Richard de Soultrait, V. Parissi, M. Fournier and S. Litvak, unpublished data).
Crystallographic studies using the IN catalytic core domain have recently shown that the inhibitor 5CITEP binds centrally in the active site between Asp 64, Asp 116 and Glu 152 (42). In the 5CITEP structure, there was a change in the orientation of the Glu152 side chain. IN crystal structures can be strikingly different in the positioning of the active site residues, particularly Glu152. The conformational flexibility exhibited by Glu152 is consistent with the notion that the active site itself is flexible and may not adopt a well-defined conformation until IN has assembled on the viral end.
In addition to inhibition, mutagenesis studies have also helped in distinguishing between processing and strand transfer. It has been demonstrated that processing is almost completely abolished by mutagenesis of the zinc-coordinating residues of the N-terminal part of IN, whereas the strand transfer activity is much less affected (25).
Furthermore, we have already reported that mutants can behave differently concerning processing and strand transfer activities. Search for D116A mutated IN revertants showed that both activities can be restored at different levels by a second site mutation in other regions of the enzyme (23).
DNA binding
The core domain of IN interacts with DNA and is probably involved in the recognition of the viral DNA (23,43–45). It therefore seemed interesting to determine whether the degree of IN activities due to mutations of the E152 region was related to DNA binding. DNA substrates for processing, strand transfer and disintegration were used in assaying DNA binding to IN. Our results indicated that mutations in the E152 region can affect the binding of IN to DNA, thus confirming the importance of this domain in that process. Other groups have previously reported that the central region of IN plays a role in the selection of the target site in the DNA strand transfer reaction, confirming that the core domain of the enzyme is bound to DNA (4,46). Taken together, these results suggest that residues in the E152 region of IN are involved in DNA binding leading to target DNA selection.
The more pronounced effect of these mutations on strand transfer suggests that the strand transfer substrate may be positioned in the vicinity of the active E152, or that IN adopts a structure involving the E152 immediate region and recognizing the strand transfer substrate. However, when the E152 residue is conserved and the adjacent amino acids are changed, the binding of the two substrates is affected differently (mutants TEQ and LEL; see Fig. 8 and Table 2). The latter result suggests that the binding site for the target DNA could be structurally distinct from that of the donor substrate. In brief, our observations suggest that the E152 amino acid could be involved in DNA binding by IN. Moreover, those results support the hypothesis that the recognition of the two types of DNA (cellular and viral) could also involve the amino acids at positions 151 and 153, indicating that the whole V151E152S153 region and not only the catalytic amino acid E152 must be important for DNA binding and enzyme activity.
The HST mutant is a paradigm of the differential effect of mutations. This enzyme was strongly affected in the binding of the processing and strand transfer substrates but not for the disintegration DNA substrate corresponding to the complex formed between the connected viral and cellular DNA. This result suggests that this DNA complex could bind to a structure or site in the enzyme that could be different from the two other probable sites interacting independently with the viral and the target DNA.
Our results are supported by previous data from other laboratories. Other studies showed the importance of the E152 region of HIV-1 IN for DNA binding and especially the amino acids K156 and K159 (47). Molecular modeling studies indicated that residues 49–69 and 139–152 are in close proximity to target DNA (8,19). V151E152S153 residues belong to the α3 helix, which has been suggested to correspond to the DNA-binding α2 helix of EcoRI (48). Chow’s group (49) showed that residue S153 was positioned to bind target DNA but was not involved in target site selection. This residue, in addition to the adjacent V151E152 amino acids, could be important for the recognition of viral and cellular DNA. The site selection process may involve a larger region motif in the proximity of the catalytic site comprising the α2 helix and/or multiple regions of IN.
The analysis of the D116A revertants previously described (23) led us to conclude that this residue was involved in the selection of the DNA target. Our data indicate that at least the D116- and E152-including regions must act in concert for the selection of the DNA target, i.e. consistent with the hypothesis of the involvement of different parts of the protein, or different structures, in the DNA interaction. In previously proposed models, the C-terminal domain, which presents a non-specific DNA binding property, seems to interact first with the viral DNA before recognizing the central catalytic domain. The central catalytic core of IN may therefore provide the required specificity to recognize the viral DNA ends allowing the 3′-processing reaction. After accomplishing the reaction, the viral DNA and the host cellular DNA should be moved to another position or induce structure changes in the IN protein, leading to the positioning of the two partners in a structural environment allowing strand transfer. In a final step, the complex formed by the inserted viral DNA into cellular host genome could be moved to a new site in the protein before being released. Based on the data from the literature and on our own results, we propose a model of the integration process involving three potential DNA binding sites in the IN protein (Fig. 9). This model, consistent with one proposed previously based on different approaches (7), does not take into account the oligomerization of the enzyme and its involvement in DNA binding property of IN.
Figure 9.

Model for the integration reactions and IN–DNA complex formation. This model does not take into account the oligomerization state of the integrase and is based on our own results and data from other groups reported in the text. The viral DNA could bind to a first DNA binding site in the catalytic center of the enzyme (A) and be positioned for the 3′-processing reaction (B). Then the cellular DNA could be bound to the protein (C) and structure changes might modify the positioning of the two substrates to allow the strand transfer reaction in a secondary site/structure (D). After the integration of the viral DNA into the cellular DNA, a final change of position of the complex could lead it to a new site or structure (E) from which it could be released (F). The positioning of the substrates for the strand transfer reaction could involve the V151E152S153 region. In all cases, the three domains of the protein (core, C-terminal and N-terminal) should be required for DNA binding and the reaction process.
As multimerization, activity and DNA binding are closely related, however, the mutation introduced in the catalytic core could affect the oligomerization state of the selected mutants. When we compare our purification procedure with the data reported by Leh et al. (50) about the relationship between IN solution concentration and multimerization state we would expected to obtain an equilibrium between monomers and dimers of the enzyme. This has been confirmed by gel filtration of our purified wild-type IN (manuscript in preparation) and will be used as a basis for further studies in our laboratory of the mutants’ oligomerization process.
Cation binding
As the catalytic triad D64D116E152 has been shown to bind divalent cations, the altered activity observed for the selected mutants could be explained by modification of the enzyme affinity for those cations. The selection of mutated INs in the E152 catalytic region able to catalyze the 3′-processing was a surprising result since interaction with the cations in this domain, crucial for the activity, should be expected to be affected by the mutations. But according to published structures of the IN core domain the E152 amino acid is different from the two other catalytic residues by the fact that, even if it points in the general direction of D64 and D116, it does not seem to participate in the magnesium binding (51). These data could explain that mutations in this region could have a less drastic effect than those in the vicinity of the two other catalytic carboxylic residues.
We performed all our IN assays in the presence of Mn2+. In vitro studies have suggested that the Mg2+- and Mn2+-dependent activities are not functionally equivalent in terms of reaction specificity. IN displays more non-sequence-specific nuclease activity in the presence of Mn2+ than in the presence of Mg2+ (50). In contrast, the enzyme is more rigid in the presence of Mg2+. The increased rigidity of the protein in the presence of Mg2+ may also give rise to more specificity in binding DNA.
Moreover, it has been reported that IN mutations that confer virologic resistance reduce the affinity and activity of DKAs in Mg2+ but not Mn2+ (52). The mutations may shift the active site residues, altering the affinity or position of the metals. Mn2+ is compatible with a wider range of reactive group geometries than Mg2+, and thus Mn2+-catalyzed IN reactions and inhibitions may be less sensitive to changes in reactive group geometries. Modelization of our mutated INs is under way and should give us new data about the structural effect of the introduced mutations.
Integrase activities and yeast lethal effect
The screening system used for the selection of mutants of IN is based on the DNA nuclease activity of IN, which induces a lethal phenotype when expressed in yeast cells (32). We previously used this system to select revertants from the inactivated D116A IN and we postulated that a non-sequence-specific activity catalyzed by IN was responsible for the lethal effect (23,34). All mutants selected in the present work showed both processing and non-sequence-specific activities in vitro, which again supports the involvement of the nuclease activity of HIV-1 IN in the yeast lethal effect. The correlation between in vitro IN activity and lethal phenotype in yeast confirms the potentiality of the yeast lethal system in the study of the structure–function relationship of IN. Previous data from our laboratory also indicated that the IN effect in this yeast eukaryotic model could mimic the in vivo integration mechanism observed in human infected cells (33,34,53).
In conclusion, mutation of the V151E152S153 IN region affects differently the enzyme activities of HIV-1 IN and its DNA binding properties. Our results also indicate that this region is probably less important in the nuclease IN activities than for the integration and disintegration processes, and suggest strongly that the V151E152S153 region could be involved in the selection of the integration target site.
Targeting the V151E152S153 region with model inhibitors could impair both DNA binding and target site selection, as well as catalysis. Thus the mutants described in this work constitute new tools for studying the reaction mechanism and the definition of such inhibitors. Furthermore, the present data support the use of the yeast lethal system in studying the role of different enzyme domains in the integration process. They allow the fast selection of recombinant forms and lead to the discovery of new targets in other IN domains.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are very grateful to Dr A. Leavitt (University of California at San Francisco, USA) for the generous gift of anti-HIV-1 IN antibodies, Dr M. Castroviejo (CNRS-University Bordeaux 2, UMR 5097) for useful discussions and Professor Ray Cooke (English Department, University Bordeaux 2) for editing the manuscript. This work was supported by the French National Research Agency against AIDS (ANRS). V.P. and V.R.-S. benefited from postdoctoral fellowships from Ensemble contre le SIDA (SIDACTION) and C.D. has a MNERT PhD fellowship.
REFERENCES
- 1.Asante-Appiah E. and Skalka,A.M. (1999) HIV-1 integrase: structural organization, conformational changes and catalysis. Adv. Virus Res., 52, 351–369. [DOI] [PubMed] [Google Scholar]
- 2.Chow S.A. (1992) Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science, 255, 723–726. [DOI] [PubMed] [Google Scholar]
- 3.Katzman M. and Sudol,M. (1996) Nonspecific alcoholysis, a novel endonuclease activity of human immunodeficiency virus type 1 and other retroviral integrases. J. Virol., 70, 2598–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katzman M. and Sudol,M. (1998) Mapping viral DNA specificity to the central region of integrase by using functional human immunodeficiency virus type 1/visna virus chimeric proteins. J. Virol., 72, 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katzman M., Sudol,M., Pufnock,J.S., Zeto,S. and Skinner,L.M. (2000) Mapping target site selection for the non-specific nuclease activities of retroviral integrase. Adv. Virus Res., 66, 87–100. [DOI] [PubMed] [Google Scholar]
- 6.Zheng R., Jenkins,T.M. and Craigie,R. (1996) Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization and enhances catalytic activity. Proc. Natl Acad. Sci. USA, 93, 13659–136429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heuer T.S. and Brown,P.O. (1997) Mapping features of HIV-1 integrase near selected sites on viral and target DNA molecules in an active enzyme–DNA complex by photo-cross-linking. Biochemistry, 36, 10655–10665. [DOI] [PubMed] [Google Scholar]
- 8.Heuer T.S. and Brown,P.O. (1998) Photo-cross-linking studies suggest a model for the architecture of an active human immunodeficiency virus type 1 integrase–DNA complex. Biochemistry, 37, 6667–6678. [DOI] [PubMed] [Google Scholar]
- 9.Kulkosky J., Jones,K.S., Katz,R.A., Mack,J.P. and Skalka,A.M. (1992) Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol., 12, 2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plasterk R.H. (1995) The HIV integrase catalytic core. Nat. Struct. Biol., 2, 87–90. [DOI] [PubMed] [Google Scholar]
- 11.Gerton J.L. and Brown,P.O. (1997) The core domain of HIV-1 integrase recognizes key features of its DNA substrates. J. Biol. Chem., 272, 25809–25815. [DOI] [PubMed] [Google Scholar]
- 12.Shibagaki Y. and Chow,S.A. (1997) Central core domain of retroviral integrase is responsible for target site selection. J. Biol. Chem., 272, 8361–8369. [DOI] [PubMed] [Google Scholar]
- 13.Wlodawer A. (1999) Crystal structures of catalytic core domains of retroviral integrases and role of divalent cations in enzymatic activity. Adv. Virus. Res., 52, 335–350. [DOI] [PubMed] [Google Scholar]
- 14.Eijkelenboom A.P., Lutzke,R.A., Boelens,R., Plasterk,R.H., Kaptein,R. and Hard,K. (1995) The DNA-binding domain of HIV-1 integrase has an SH3-like fold. Nat. Struct. Biol., 2, 807–810. [DOI] [PubMed] [Google Scholar]
- 15.Lutzke R.A. and Plasterk,R.H. (1998) Structure-based mutational analysis of the C-terminal DNA-binding domain of human immunodeficiency virus type 1 integrase: critical residues for protein oligomerization and DNA binding. J. Virol., 72, 4841–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eijkelenboom A.P., Sprangers,R., Hard,K., Puras Lutzke,R.A., Plasterk,R.H., Boelens,R. and Kaptein,R. (1999) Refined solution structure of the C-terminal DNA-binding domain of human immunovirus-1 integrase. Proteins, 36, 556–564. [DOI] [PubMed] [Google Scholar]
- 17.Hickman A.B., Palmer,I., Engelman,A., Craigie,R. and Wingfield,P. (1994) Biophysical and enzymatic properties of the catalytic domain of HIV-1 integrase. J. Biol. Chem., 269, 29279–29287. [PubMed] [Google Scholar]
- 18.Deprez E., Tauc,P., Leh,H., Mouscadet,J.F., Auclair,C. and Brochon,J.C. (2000) Oligomeric states of the HIV-1 integrase as measured by time-resolved fluorescence anisotropy. Biochemistry, 39, 9275–9284. [DOI] [PubMed] [Google Scholar]
- 19.Chen J.C., Krucinski,J., Miercke,L.J., Finer-Moore,J.S., Tang,A.H., Leavitt,A.D. and Stroud,R.M. (2000) Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc. Natl Acad. Sci. USA, 97, 8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lins R.D., Adesokan,A., Soares,T.A. and Briggs,J.M. (2000) Investigations on human immunodeficiency virus type 1 integrase/DNA binding interactions via molecular dynamics and electrostatics calculations. Pharmacol. Ther., 85, 123–131. [DOI] [PubMed] [Google Scholar]
- 21.Dirac A.M. and Kjems,J. (2001) Mapping DNA-binding sites of HIV-1 integrase by protein footprinting. Eur. J. Biochem., 268, 743–751. [DOI] [PubMed] [Google Scholar]
- 22.Hazuda D.J., Felock,P., Witmer,M., Wolfe,A., Stillmock,K., Grobler,J.A., Espeseth,A., Gabryelski,L., Schleif,W., Blau,C. et al. (2000) Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science, 287, 646–650. [DOI] [PubMed] [Google Scholar]
- 23.Parissi V., Caumont,A.B., de Soultrait,V.R., Calmels,C., Pichuantes,S., Litvak,S. and Dupont,C.H. (2000) Selection of amino acid substitutions restoring activity of HIV-1 integrase mutated in its catalytic site using the yeast Saccharomyces cerevisiae. J. Mol. Biol., 295, 755–765. [DOI] [PubMed] [Google Scholar]
- 24.Drelich M., Wilhelm,R. and Mous,J. (1992) Identification of amino acid residues critical for endonuclease and integration activities of HIV-1 IN protein in vitro. Virology, 188, 459–468. [DOI] [PubMed] [Google Scholar]
- 25.Engelman A. and Craigie,R. (1992) Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol., 66, 6361–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.vanGent D.C., Groeneger,A.A. and Plasterk,R.H. (1992) Mutational analysis of the integrase protein of human immunodeficiency virus type 2. Proc. Natl Acad. Sci. USA, 89, 9598–9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leavitt A.D., Shiue,L. and Varmus,H.E. (1993) Site-directed mutagenesis of HIV-1 integrase demonstrates differential effects on integrase functions in vitro. J. Biol. Chem., 268, 2113–2119. [PubMed] [Google Scholar]
- 28.Dyda F., Hickman,A.B., Jenkins,T.M., Engelman,A., Craigie,R. and Davies,D.R. (1994) Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science, 266, 1981–1986. [DOI] [PubMed] [Google Scholar]
- 29.Bujacz G., Jaskolski,M., Alexandratos,J., Wlodawer,A., Merkel,G., Katz,R.A. and Skalka,A.M. (1995) High-resolution structure of the catalytic domain of avian sarcoma virus integrase. J. Mol. Biol., 253, 333–346. [DOI] [PubMed] [Google Scholar]
- 30.Bujacz G., Jaskolski,M., Alexandratos,J., Wlodawer,A., Merkel,G., Katz,R.A. and Skalka,A.M. (1996) The catalytic domain of avian sarcoma virus integrase: conformation of the active-site residues in the presence of divalent cations. Structure, 4, 89–96. [DOI] [PubMed] [Google Scholar]
- 31.Yang W. and Steitz,T.A. (1995) Recombining the structures of HIV integrase, RuvC and RNase H. Structure, 3, 131–132. [DOI] [PubMed] [Google Scholar]
- 32.Caumont A.B., Jamieson,G.A., Pichuantes,S., Nguyen,A.T., Litvak,S. and Dupont,C.H. (1996) Expression of functional HIV-1 integrase in the yeast Saccharomyces cerevisiae leads to the emergence of a lethal phenotype: potential use for inhibitor screening. Curr. Genet., 29, 503–510. [DOI] [PubMed] [Google Scholar]
- 33.Parissi V., Caumont,A., Richard de Soultrait,V., Dupont,C.H., Pichuantes,S. and Litvak,S. (2000) Inactivation of the SNF5 transcription factor gene abolishes the lethal phenotype induced by the expression of HIV-1 integrase in yeast. Gene, 247, 129–136. [DOI] [PubMed] [Google Scholar]
- 34.Parissi V., Caumont,A., Richard de Soultrait,V., Desjobert,C., Calmels,C., Fournier,M., Gourgue,G., Bonneu,M., Tarrago-Litvak,L. and Litvak.,S. (2003) The lethal phenotype observed after HIV-1 integrase in yeast cells is related to DNA repair and recombination events. Gene, 322, 157–168. [DOI] [PubMed] [Google Scholar]
- 35.Bujacz G., Alexandratos,J., Qing,Z.L., Clement-Mella,C. and Wlodawer,A. (1996) The catalytic domain of human immunodeficiency virus integrase: ordered active site in the F185H mutant. FEBS Lett., 398, 175–178. [DOI] [PubMed] [Google Scholar]
- 36.Hanahan D. (1983) Studies on transformation of E.coli with plasmids. J. Mol. Biol., 166, 557–580. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular cloning: a laboratory manual (2nd edn). Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 38.Chen D.C., Yang,B.C. and Kuo,T.T. (1992) One-step transformation of yeast in stationary phase. Curr. Genet., 21, 83–84. [DOI] [PubMed] [Google Scholar]
- 39.Robzyk K. and Kassir,Y. (1992) A simple and highly efficient procedure for rescuing autonomous plasmids from yeast. Nucleic Acids Res., 20, 3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caumont A., Jamieson,G., Richard de Soultrait,V., Parissi,V., Fournier,M., Zakharova,O.D., Bayandin,R., Litvak,S., Tarrago-Litvak,L. and Nevinsky,G.A. (1999) High affinity interaction of HIV-1 integrase with specific and non-specific single-stranded short oligonucleotides. FEBS Lett., 455, 154–158. [DOI] [PubMed] [Google Scholar]
- 41.Chow S.A. (1997) In vitro assays for activities of retroviral integrase. Methods, 12, 306–317. [DOI] [PubMed] [Google Scholar]
- 42.Goldgur Y., Craigie,R., Cohen,G.H., Fujiwara,T., Yoshinaga,T., Fujishita,T., Sugimoto,H., Endo,T., Murai,H. and Davies,D.R. (1999) Proc. Natl Acad. Sci. USA, 96, 13040–13043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kulkosky J., Katz,R.A., Merkel,G. and Skalka,A.M. (1995) Activities and substrate specificity of the evolutionarily conserved central domain of retroviral integrase. Virology, 206, 448–456. [DOI] [PubMed] [Google Scholar]
- 44.Pahl A. and Flugel,R.M. (1995) Characterization of the human spuma retrovirus integrase by site-directed mutagenesis, by complementation analysis and by swapping the zinc finger domain of HIV-1. J. Biol. Chem., 270, 2957–2966. [DOI] [PubMed] [Google Scholar]
- 45.Goulaouic H. and Chow,S.A. (1996) Directed integration of viral DNA mediated by fusion proteins consisting of human immunodeficiency virus type 1 integrase and Escherichia coli LexA protein. J. Virol., 70, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerton J.L. and Brown,P.O. (1997) The core domain of HIV-1 integrase recognizes key features of its DNA substrates. J. Biol. Chem., 272, 25809–25815. [DOI] [PubMed] [Google Scholar]
- 47.Jenkins T.M., Esposito,D., Engelman,A. and Craigie,R. (1997) Critical contacts between HIV-1 integrase and viral DNA identified by structure-based analysis and photo-crosslinking. EMBO J., 16, 6849–6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venclovas C. and Siksnys,V. (1995) Different enzymes with similar structures involved in Mg(2+)-mediated polynucleotidyl transfer. Nat. Struct. Biol., 2, 838–841. [DOI] [PubMed] [Google Scholar]
- 49.Appa R.S., Shin,C.G., Lee,P. and Chow,S.A. (2001) Role of the non-binding region and α helices within the core domain of retroviral integrase in selecting target sites for integration. J. Biol. Chem., 246, 45848–45855. [DOI] [PubMed] [Google Scholar]
- 50.Leh H., Brodin,P., Bischerour,J., Deprez,E., Tauc,P., Brochon,J.C., LeCam,E., Coulaud,D., Auclair,C. and Mouscadet,J.F. (2000) Determinants of Mg2+-dependent activities of recombinant human immunodeficiency virus type 1 integrase. Biochemistry, 39, 9285–9294. [DOI] [PubMed] [Google Scholar]
- 51.Goldgur Y., Dyda,F., Hickman,A.B., Jenkins,T.M., Craigie,R. and Davies,D.R. (1998) Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc. Natl Acad. Sci. USA, 95, 9150–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grobler J.A., Stillmock,K., Hu,B., Witmer,M., Felock,P., Espeseth,A.S., Wolfe,A., Egbertson,M., Bourgeois,M., Melamed,J. et al. (2002) Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl Acad. Sci. USA, 99, 6661–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Richard de Soultrait V., Caumont,A., Durrens,P., Calmels,C., Parissi,V., Recordon,P., Bon,E., Desjobert,C., Tarrago-Litvak,L. and Fournier,M. (2002) HIV-1 integrase interacts with yeast microtubule-associated proteins. Biochim. Biophys. Acta, 1575, 40–48. [DOI] [PubMed] [Google Scholar]