


Abstract
Homologous recombination is important for the repair of double-strand breaks and daughter strand gaps, and also helps restart stalled and collapsed replication forks. However, sometimes recombination is inappropriate and can have deleterious consequences. To temper recombination, cells have employed DNA helicases that unwind joint DNA molecules and/or dissociate recombinases from DNA. Budding yeast Srs2 is one such helicase. It can act by dissociating Rad51 nucleoprotein filaments, and is required for channelling DNA lesions to the post-replication repair (PRR) pathway. Here we have investigated the role of Srs2 in controlling recombination in fission yeast. Similar to budding yeast, deletion of fission yeast srs2 results in hypersensitivity to a range of DNA damaging agents, rhp51-dependent hyper-recombination and synthetic sickness when combined with rqh1– that is suppressed by deleting rhp51, rhp55 or rhp57. Epistasis analysis indicates that Srs2 and the structure-specific endonuclease Mus81–Eme1 function in a sub-pathway of PRR for the tolerance/repair of UV-induced damage. However, unlike in Saccharomyces cerevisiae, Srs2 is not required for channelling lesions to the PRR pathway in Schizosaccharomyces pombe. In addition to acting as an antirecombinase, we also show that Srs2 can aid the recombinational repair of camptothecin-induced collapsed replication forks, independently of PRR.
INTRODUCTION
Replication forks that encounter bulky lesions in DNA, such as pyrimidine dimers induced by ultraviolet (UV) light, are impeded (1). Repair of UV-induced damage is typically by nucleotide excision repair (NER) (2). However, the presence of a replication fork adjacent to a UV-induced lesion may hinder NER. One potential solution to this is fork regression, which involves the unwinding of nascent DNA strands and re-annealing of parental DNA strands (1). Once the fork is out of the way NER may gain access to the lesion and repair it. The replication fork then has to be reset and the replisome reassembled. Enzymes that drive homologous recombination (HR) can play key roles in both the regression and re-establishment of a replication fork (1). Even if such a mechanism of fork regression coupled to NER operates in vivo, it is clear that cells do not depend solely upon this. Alternative strategies for coping with fork-blocking lesions involve their tolerance rather than their immediate repair. Such mechanisms include: (i) translesion synthesis (TLS), which uses error-prone and error-free DNA polymerases that can replicate past UV photoproducts (reviewed in 3,4); (ii) polymerase ‘skipping’ of the damaged section of the template, which generates a lesion-containing single-strand gap that is later filled in using either HR or TLS (5–7); and (iii) template switching for the polymerase to bypass the lesion, which can be mediated by HR (8). The importance of these mechanisms is underscored by the inability of chicken DT40 cells disabled for HR and TLS to proliferate (9). What factors determine whether a HR-based mechanism is used in preference to TLS are unclear. Certainly there are potential disadvantages with both mechanisms. TLS can be mutagenic, and HR occasionally occurs between allelic or ectopic sequences resulting in loss of heterozygosity and genome rearrangements, respectively.
TLS falls under the general umbrella of the post-replication repair (PRR) pathway (4). In the budding yeast Saccharomyces cerevisiae PRR consists of at least three sub-pathways, two error-free pathways mediated by RAD5 and POL30, respectively, and one error-prone pathway dependent on Polζ (4). Overall control of PRR depends on a ubiquitin-conjugating enzyme Rad6 and a RING finger protein called Rad18 that binds to DNA (4,5). In S.cerevisiae, one factor that is instrumental in directing repair from HR to PRR at stalled forks and/or lesion-containing single-strand gaps is the Srs2 DNA helicase. This has been inferred from three main observations: (i) mutation of SRS2 suppresses the UV sensitivity of rad6, rad18, rad5 and pol30–46 mutants (10–13); (ii) srs2 suppression of PRR mutant UV sensitivity is dependent on the RAD52 epistasis group of recombination genes (10,11,14); and (iii) mutating SRS2 results in hyper-recombination (14).
The RAD52 epistasis group of proteins includes Rad51, Rad52, Rad54, Rad55, Rad57 and Rad59 (reviewed in 15). Rad51 is a homologue of the archetypal recombinase RecA from bacteria. It binds to single-stranded DNA to form a right-handed helical nucleoprotein filament in which pairing and strand exchange between homologous DNA molecules occurs. Filament assembly is variously aided by RPA, Rad52 and the Rad55–Rad57 heterodimer (15). Srs2 seems to direct repair from HR to PRR by dissociating Rad51 nucleoprotein filaments (16,17). This appears to be important for preventing toxic recombination since a diploid srs2 mutant’s sensitivity to UV light is suppressed by semi-dominant mutations in RAD51 (18). Dissociation of Rad51 filaments may also help Srs2 to suppress the formation of crossovers during the repair of DNA double-strand breaks (DSBs) by directing repair to a synthesis-dependent strand annealing (SDSA) mechanism (19). Srs2’s ability to dislodge Rad51 from DNA appears to be especially important when the nucleoprotein filament is aberrant or disabled by mutation. For example, in the absence of Rad54, which promotes Rad51-mediated strand invasion and D-loop formation (20), Srs2 becomes essential for viability (21). Presumably, Rad51 filaments that are rendered useless in the absence of Rad54 block other repair processes if Srs2 does not remove them (16). Srs2 is also important for viability in the absence of the RecQ family DNA helicase Sgs1 (22). Sgs1, like Srs2, has an anti-recombinogenic role, and the poor viability of an srs2 sgs1 double mutant is rescued by deleting rad51 or rad52, suggesting that both helicases process toxic recombination intermediates (22).
Srs2 is structurally related to the bacterial UvrD and Rep helicases, and although no obvious human homologue has yet been found, an orthologue in the fission yeast Schizosaccharomyces pombe has been identified (23,24). The S.pombe srs2 mutant shares a number of phenotypes with the S.cerevisiae srs2 mutant, including hypersensitivity to DNA damaging agents, hyper-recombination and strong genetic interactions with rqh1 and rhp54 (homologues of SGS1 and RAD54, respectively). In this paper, we present further genetic analyses of S.pombe Srs2. Like S.cerevisiae Srs2, we show that S.pombe Srs2 acts as an anti-recombinase, which counters Rhp51-dependent recombination. In addition, we show that, as in S.cerevisiae, Srs2 functions in the PRR pathway for the tolerance/repair of UV damage, which interestingly in S.pombe also appears to involve the structure-specific endonuclease Mus81–Eme1. However, unlike in S.cerevisiae, lesions appear to be channelled efficiently to the PRR pathway without Srs2 in S.pombe. In addition to functioning in the PRR pathway, we also provide evidence that Srs2 functions to promote the recombinational repair of collapsed replication forks. Possible roles for Srs2 in repairing collapsed replication forks are discussed.
MATERIALS AND METHODS
Media and genetic methods
Procedures and media used for the routine growth and maintenance of S.pombe are described by Moreno et al. (25). The complete medium was yeast extract with supplements (YES), the minimal media was Edinburgh minimal medium (EMM) with supplements added where appropriate. Thiamine was used at 4 µM (1.35 µg/ml) where needed. To measure the number of ade+ prototrophs in recombination assays YE medium lacking adenine, and containing 200 µg/ml guanine, to prevent uptake of residual adenine, was used.
Strains and plasmids
The S.pombe strains used in this study are listed in Table 1. The srs2Δura4+ mutant contains a replacement of the entire srs2 open reading frame by a ura4 marker. It was made by first creating an in vitro deletion construct consisting of ∼1 kb regions of genomic DNA that flank the srs2 open-reading frame, which were amplified by PCR, cloned either side of a ura4 marker to make the plasmid pMW454. The cloned genomic DNA in pMW454 was sequenced to confirm that no mutations had been introduced during the PCR. To delete srs2 in vivo, the ura4 marker with flanking genomic fragments was excised from pMW454 and used to transform a diploid wild-type strain to ura+ by one-step gene disruption (26). Stable Ura+ transformants were screened for genuine replacement of srs2 with ura4 by genomic Southern blot analysis. rhp51 was amplified by PCR from a genomic template and cloned as a NdeI–BamHI fragment downstream of the nmt promoter in pREP41. Nucleotide sequencing of the cloned rhp51 gene confirmed that no mutations had been introduced during the PCR.
Table 1. Schizosaccharomyces pombe strains.
| Strain | Genotype | Reference |
|---|---|---|
| MCW23 | leu1-32, ura4-D18, his3-D1, ade6-M375 int::pUC8/his3+/ade6-L469, h+ | Laboratory stock |
| MCW42 | leu1-32, ura4-D18, his3-D1, h+ | Laboratory stock |
| MCW121 | rhp54Δura4+, leu1-32, ura4-D18, his3-D1, h+ | Muris et al. (52) |
| MCW149 | rqh1Δura4+, leu1-32, ura4-D18, his3-D1, h+ | Stewart et al. (53) |
| MCW162 | rad8Δura4+, leu1-32, ura4-D18, ade6-704, h+ | Doe et al. (49) |
| MCW431 | rhp55Δarg3+, leu1-32, ura4-D18, his3-D1, arg3-D4, ade6-M375 int::pUC8/his3+/ade6-L469, h+ | Laboratory stock |
| MCW449 | leu1-32, ura4-D18, ade6-704, h+ | Laboratory stock |
| MCW460 | srs2Δura4+, leu1-32, ura4-D18, ade6-704, h+ | This work |
| MCW473 | srs2Δura4+, leu1-32, ura4-D18, his3-D1, h+ | This work |
| MCW547 | srs2Δura4+, leu1-32, ura4-D18, his3-D1, ade6-M375 int::pUC8/his3+/ade6-L469, h+ | This work |
| MCW595 | rhp6Δura4+, leu1-32, ura4-D18, lys1-131, h– | Reynolds et al. (54) |
| MCW680 | srs2Δura4+, rad8Δura4+, leu1-32, ura4-D18, ade6-704, h+ | This work |
| MCW744 | mus81ΔkanR, leu1-32, ura4-D18, ade6-704, h+ | Laboratory stock |
| MCW745 | mus81ΔkanR, leu1-32, ura4-D18, his3-D1, h+ | Laboratory stock |
| MCW747 | srs2Δura4+, mus81ΔkanR, leu1-32, ura4-D18, ade6-704, h+ | This work |
| MCW832 | rqh1Δura4+, rhp55Δarg3+, leu1-32, ura4-D18, his3-D1, arg3-D4, h+ | This work |
| MCW897 | mus81ΔkanR, rhp54Δura4+, leu1-32, ura4-D18, his3-D1, h+ | Laboratory stock |
| MCW928 | rqh1ΔkanR, leu1-32, ura4-D18, his3-D1, h+ | To be described elsewhere |
| MCW1043 | srs2ΔkanR, leu1-32, ura4-D18, his3-D1, h+ | Maftahi et al. (23) |
| MCW1049 | rhp55Δarg3+, leu1-32, ura4-D18, his3-D1, arg3-D4, h+ | Khasanov et al. (32) |
| MCW1086 | srs2Δura4+, rhp55Δarg3+, leu1-32, ura4-D18, his3-D1, arg3-D4, ade6-M375 int::pUC8/his3+/ade6-L469, h+ | This work |
| MCW1088 | rhp51Δarg3+, leu1-32, ura4-D18, his3-D1, arg3-D4, h+ | To be described elsewhere |
| MCW1097 | srs2Δura4+, rqh1ΔkanR, rhp51Darg3+, leu1-32, ura4-D18, his3-D1, arg3-D4 | This work |
| MCW1099 | srs2Δura4+, rhp51Δarg3+, leu1-32, ura4-D18, his3-D1, arg3-D4, h+ | This work |
| MCW1162 | rhp51Δarg3+, leu1-32, ura4-D18, his3-D1, arg3-D4, ade6-M375 int::pUC8/his3+/ade6-L469, h+ | Laboratory stock |
| MCW1137 | mus81ΔkanR, rhp6Δura4+, leu1-32, ura4-D18, his3-D1, lys1-131 | This work |
| MCW1269 | rhp18Δura4+, leu1-32, ura4-D18, ade6-704, h+ | Verkade et al. (48) |
| MCW1270 | srs2Δura4+, rhp18Δura4+, leu1-32, ura4-D18, ade6-704, h+ | This work |
| MCW1271 | mus81ΔkanR, rhp18Δura4+, leu1-32, ura4-D18, ade6-704, h+ | This work |
| MCW1272 | mus81ΔkanR, rad8Δura4+, leu1-32, ura4-D18, ade6-704, h+ | This work |
| MCW1273 | srs2ΔkanR, rhp6Δura4+, leu1-32, ura4-D18, his3-D1 | This work |
| MCW1276 | srs2Δura4+, rhp55Δarg3+, leu1-32, ura4-D18, his3-D1, arg3-D4, h+ | This work |
| MCW1278 | rhp57ΔLEU2+, leu1-32, ura4-D18, his3-D1, ade6-M375 int::pUC8/his3+/ade6-L469, h+ | Laboratory stock |
| MCW1279 | srs2Δura4+, rhp51Δarg3+, leu1-32, ura4-D18, his3-D1, arg3-D4, ade6-M375 int::pUC8/his3+/ade6-L469, h+ | This work |
| MCW1280 | srs2Δura4+, rhp57ΔLEU2+, leu1-32, ura4-D18, his3-D1, arg3-D4, ade6-M375 int::pUC8/his3+/ade6-L469, h+ | This work |
| MCW1282 | srs2Δura4+, rqh1ΔkanR, rhp55Δarg3+, leu1-32, ura4-D18, his3-D1, arg3-D4, h+ | This work |
| MCW1283 | srs2Δura4+, rqh1ΔkanR, rhp57ΔLEU2+, leu1-32, ura4-D18, his3-D1, h+ | This work |
| MCW1284 | srs2Δura4+, rqh1ΔkanR, leu1-32, ura4-D18, his3-D1, arg3-D4, h+ | This work |
Spot assays
Exponentially growing cell cultures were adjusted to a density of 1 × 107 cells/ml, serially diluted by a factor of 10 down to 1 × 104 cells/ml, and 10 µl of each dilution spotted onto YES plates, which contained hydroxyurea (HU), methyl methanesulfonate (MMS) or camptothecin (CPT), or were subsequently UV irradiated using a Stratalinker (Stratagene) as indicated. Plates were incubated at 30°C typically for 3–5 days before being photographed. All spot assays were repeated at least once to ensure that results were reproducible.
Quantitative UV survival assays
Exponentially growing cells were plated to YES in duplicate and UV irradiated. Plates were grown at 30°C for 5–7 days before counting. All data points represent the mean values from at least two independent experiments.
Recombination assays
Mitotic recombination was assayed using strains containing an intrachromosomal recombination substrate consisting of a non-tandem direct repeat of ade6 heteroalleles flanking a functional his3 gene (Fig. 1C). Spontaneous and UV-induced recombinant frequencies were measured as described by Osman et al. (27). Recombination frequencies are mean values from at least three independent assays, and in each assay five independent colonies were tested. Two-sample t-tests were used to determine the statistical significance of differences in recombination frequencies between given strains.
Figure 1.
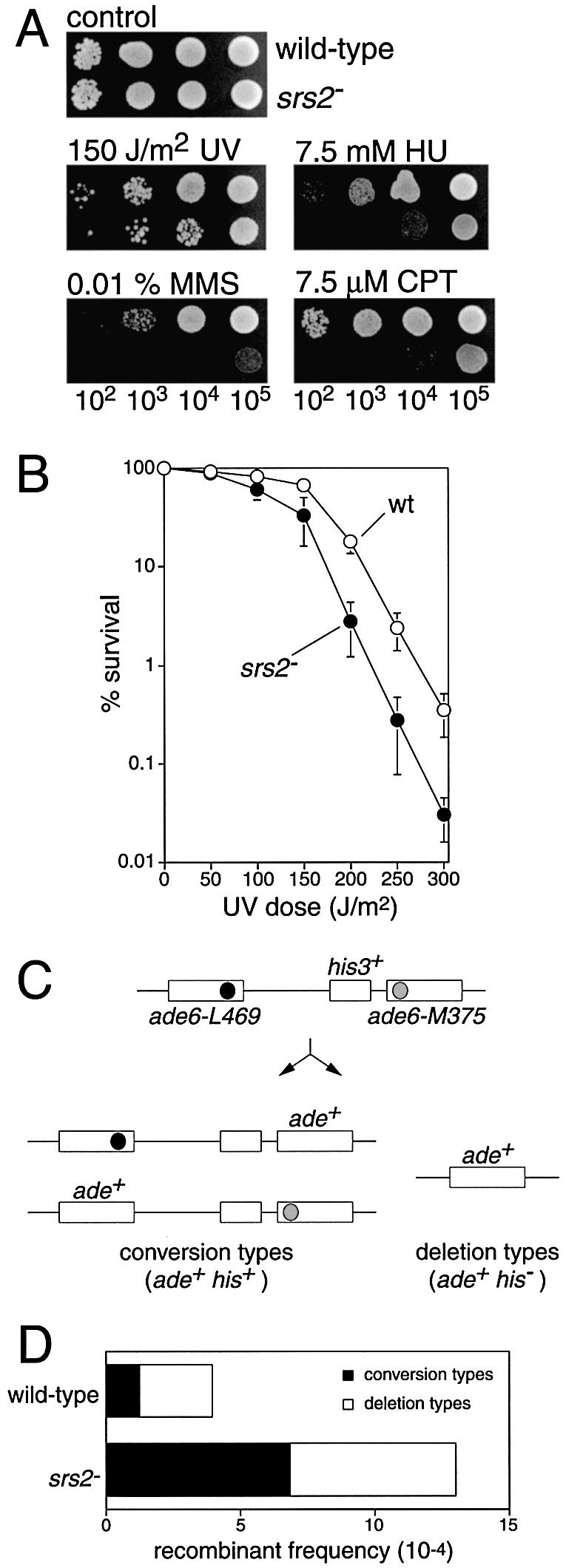
Phenotypes of the srs2 deletion mutation strain. (A) Spot assay comparing wild-type (MCW42) and srs2Δ (MCW473) strains for sensitivity to UV, HU, MMS and CPT. Plates were incubated at 30°C for 3 days before being photographed. (B) UV survival curves of wild-type (MCW42) and srs2Δ (MCW473) strains. (C) Schematic of the recombination substrate and recombinant products. Solid and grey circles represent the ade6-L469 and ade6-M375 mutations, respectively. (D) Comparison of the spontaneous recombinant frequencies of wild-type (MCW23) and srs2Δ (MCW547) strains.
Viability testing
Cultures of exponentially growing cells were counted and a known number of cells were plated to YES plates in duplicate. Plates were counted after growth at 30°C for 5 days for control strains and 10 days for the mus81 rhp54 double mutant strain. Data are mean values from three independent experiments.
RESULTS
DNA damage sensitivity and hyper-recombination of srs2Δ
The identification of the homologue of the S.cerevisiae Srs2 DNA helicase in S.pombe has previously been reported by two groups (23,24). We had independently identified the same candidate Srs2 homologue by a BLAST search of the S.pombe database, and had made a null mutant strain by deleting the entire srs2 open reading frame and replacing it with a ura4+ marker. Consistent with the published data we found that the srs2Δ strain is viable and grows normally as a haploid indicating that srs2 is not an essential gene.
There are conflicting reports concerning the sensitivity of S.pombe srs2 mutant strains to DNA damage. In one report, srs2Δ is shown to have a mild hypersensitivity to UV light, the ribonucleotide reductase inhibitor, HU, the alkylating agent, MMS, and the radiomimetic, bleomycin (24). However, in a second report, srs2Δ was shown to have the same sensitivity as a wild-type strain to UV and HU (23). To assess the sensitivity of our srs2Δ mutant strain we spotted dilutions of wild-type and srs2Δ cells onto nutrient agar plates that were either subsequently exposed to UV or contained HU, MMS or the topoisomerase I (Top1) poison CPT (Fig. 1A). The srs2Δ strain is much more sensitive to HU, MMS and CPT than the wild-type strain, and slightly more sensitive to UV. The increase in UV sensitivity was confirmed by a quantitative UV survival experiment (Fig. 1B). These results corroborate the data from Wang et al. (24) and show that Srs2 is required for aiding survival following treatment with agents that damage DNA and cause replication fork arrest. Moreover, the hypersensitivity to CPT indicates that Srs2 plays a role in the repair of collapsed replication forks. CPT collapses forks by inhibiting the religation step during Top1’s reaction cycle. This leads to the formation of persistent single-strand nicks at which DNA polymerase run-off can occur, which creates single-end breaks (SEBs) (28,29).
srs2 mutant strains of both S.cerevisiae and S.pombe exhibit hyper-recombination (14,24). In S.pombe this has been assessed previously by measuring inter-homologue recombination in an srs2Δ diploid strain (24). To see whether srs2Δ affects inter/intra-chromatid recombination we used strains containing a direct repeat of ade6– heteralleles and measured the frequency of Ade+ recombinants (Fig. 1C and D). A His+ marker between the repeats enabled us to distinguish recombinants that had lost (deletion types) or retained (conversion types) the intervening DNA between the repeats (Fig. 1C). In wild-type cells the average frequency of Ade+ recombinants is ∼3.5 × 10–4 of which ∼70% are deletion types and ∼30% are conversion types. In the srs2Δ strain the frequency of Ade+ recombinants is ∼3-fold higher than in the wild type and ∼50% of these recombinants are conversion types. These data show that Srs2 acts to limit inter/intra-chromatid recombination, particularly that which gives rise to conversion types.
Genetic interactions between srs2 and rhp51, rhp54, rhp55 and rhp57
Both genetic and biochemical analyses indicate that a prime function of Srs2 in S.cerevisiae is to dissociate Rad51 nucleoprotein filaments, thereby channelling potential recombination substrates down other pathways of repair. In the absence of Srs2, Rad54, which promotes Rad51-mediated strand invasion, becomes essential (21). Presumably, unproductive Rad51 nucleoprotein filaments fail to be dislodged and therefore block other means of lesion repair/tolerance, resulting in loss of viability (16). This idea is consistent with the fact that the lethality of a sgs1 rad54 double mutant is suppressed by mutations in RAD51, RAD52, RAD55 and RAD57 (30,31). In S.pombe the homologue of Rad54 is called Rhp54 and is likewise essential in the absence of Srs2 (23). The unviability of an srs2 rhp54 strain is suppressed by deleting rhp51 (S.pombe RAD51), consistent with the idea that Srs2 is required to process Rhp51 nucleoprotein filaments (23). During our studies of srs2 we have independently confirmed these findings (data not shown). We also looked to see what affect srs2Δ would have on the sensitivity of a rhp51 mutant strain to UV, HU, MMS and CPT (Fig. 2). The sensitivities of the srs2Δ rhp51Δ strain are indistinguishable from those of a rhp51Δ strain (Fig. 2A and B). This is consistent with the idea that Srs2 is only required to process Rhp51 nucleoprotein filaments. However, it should be noted that a rhp51 single mutant is considerably more sensitive to UV, HU, MMS and CPT than an srs2 single mutant, so any additive increase in sensitivity could go unnoticed in our assays. In S.cerevisiae the Rad55–Rad57 heterodimer is believed to promote the formation and stability of the Rad51 nucleoprotein filament (15). In the absence of the homologues of these proteins in S.pombe (Rhp55 and Rhp57, respectively) Rhp51-dependent recombination is ablated (our unpublished data). However, a rhp51Δ mutant is more sensitive to DNA damaging agents than either rhp55Δ or rhp57Δ mutants, which indicates that Rhp51 can still promote repair/tolerance in the absence of Rhp55–Rhp57 (32,33). Based on this we wondered whether deleting srs2 would improve the stability of the Rhp51 nucleoprotein filament in the absence of Rhp55–Rhp57 enabling it to more efficiently promote DNA repair. This would be reminiscent of the way that srs2 suppresses certain RAD51 and RAD52 non-null mutant alleles in S.cerevisiae (34). However, we found that an srs2Δ rhp55Δ strain is in fact more sensitive to UV, MMS and CPT, although not HU, than its component single mutant strains (Fig. 2). For UV the increase in sensitivity is approximately additive (Fig. 2A and B), whereas for MMS and CPT it appears to be more than additive (Fig. 2A). These data suggest that there may be a greater need for Srs2 to dislodge Rhp51 in the absence of Rhp55–Rhp57. Perhaps without Rhp55–Rhp57 Rhp51 forms more non-functional or aberrant nucleoprotein filaments that need to be processed by Srs2.
Figure 2.

Epistasis analysis of srs2, rhp51 and rhp55 mutants. (A) Spot assays comparing wild-type (MCW42), srs2Δ (MCW473), rhp51Δ (MCW1088), rhp55Δ (MCW1049), srs2Δ rhp51Δ (MCW1099) and srs2Δ rhp55Δ (MCW1276) strains for sensitivity to UV, HU, MMS and CPT. Plates were incubated at 30°C for 4 days before being photographed. (B) UV survival curves of the same strains as in (A). Colonies were grown at 30°C for 5–7 days before being counted. Error bars represent standard deviations about the mean.
In S.pombe the generation of conversion-type recombinants depends on Rhp51 together with its accessory proteins Rhp55 and Rhp57 (27) (Fig. 3). If the hyper-recombination of an srs2 mutant is due to unconstrained Rhp51-dependent recombination then conversion types in an srs2Δ strain should be abolished by deleting either rhp55 or rhp57. To test this we constructed srs2Δ rhp55Δ and srs2Δ rhp57Δ strains containing the direct repeat substrate shown in Figure 1C, and measured their Ade+ recombinant frequencies with and without exposure to UV light (Fig. 3). In accord with our prediction, conversion types are largely abolished in srs2Δ rhp55Δ and srs2Δ rhp57Δ strains. However, the residual levels of conversion types in these double mutant strains are slightly higher than their respective rhp55Δ and rhp57Δ single mutant strains. This difference is statistically significant (P ≤ 0.03) and may be due to the residual activity of ill-formed Rhp51 nucleoprotein filaments that would be dissociated in srs2+ cells. rhp55Δ and rhp57Δ mutants, although deficient in conversion type recombination, exhibit ∼4- to 5-fold higher frequencies of deletion types compared with wild type. Interestingly the srs2Δ rhp55Δ and srs2Δ rhp57Δ strains exhibit statistically significant increased frequencies of deletion type recombinants compared with their respective rhp55Δ and rhp57Δ single mutant strains (P ≤ 0.003). Furthermore, in both cases, the frequency of these deletion types is induced by UV. In contrast, recombination is not induced by UV in the rhp55Δ and rhp57Δ single mutant strains. These data indicate that Srs2 acts to block the formation of some spontaneous and all UV-induced deletion types in rhp55Δ and rhp57Δ mutants. To see whether this reflects Srs2’s potential for processing partially active Rhp51 nucleoprotein filaments, we next compared the frequency of recombination of a rhp51Δ strain with that of an srs2Δ rhp51Δ strain (Fig. 3). Like the rhp55 and rhp57 single mutant strains, the rhp51Δ strain produces hardly any conversion type recombinants, is hyper-recombinant for deletion types and shows no significant increase in recombination following UV irradiation. The srs2Δ rhp51Δ strain exhibits no significant difference from the rhp51Δ strain for the frequency and type of recombinants produced with or without UV. These data indicate that the recombination induced by deletion of srs2, in rhp55Δ and rhp57Δ backgrounds, depends on rhp51, and is consistent with the idea that Srs2 dissociates partially active Rhp51 nucleoprotein filaments.
Figure 3.

Bar chart showing the effect of rhp55, rhp57 and rhp51 mutations on the hyper-recombination of an srs2Δ mutant strain. Spontaneous and UV-induced Ade+ recombinant frequencies were measured using strains containing the recombination substrate shown in Figure 1C. The strains used were MCW547 (srs2Δ), MCW431 (rhp55Δ), MCW1278 (rhp57Δ), MCW1086 (srs2Δ rhp55Δ), MCW1280 (srs2Δ rhp57Δ), MCW1162 (rhp51Δ) and MCW1279 (srs2Δ rhp51Δ). The mean frequencies (×10–4) of conversion types (ct) and deletion types (dt) (± standard deviation) are as follows: 1.2 ± 0.5 (ct) and 2.6 ± 0.8 (dt) (wild-type, no UV); 4.3 ± 2.1 (ct) and 4.5 ± 2.2 (dt) (wild-type, 25 J/m2 UV); 10.2 ± 4.0 (ct) and 11.1 ± 4.7 (dt) (srs2Δ, no UV); 17.5 ± 4.6 (ct) and 17.3 ± 8.5 (dt) (srs2Δ, 25 J/m2 UV); 0.27 ± 0.19 (ct) and 10.7 ± 3.5 (dt) (rhp55Δ, no UV); 0.48 ± 0.43 (ct) and 11.7 ± 3.5 (dt) (rhp55Δ, 25 J/m2 UV); 0.16 ± 0.10 (ct) and 13.8 ± 5.5 (dt) (rhp57Δ, no UV); 0.19 ± 0.15 (ct) and 13.2 ± 6.6 (dt) (rhp57Δ, 25 J/m2 UV); 0.63 ± 0.50 (ct) and 16.7 ± 7.8 (dt) (srs2Δ rhp55Δ, no UV); 1.2 ± 1.3 (ct) and 27.4 ± 13.6 (dt) (srs2Δ rhp55Δ, 25 J/m2 UV); 0.38 ± 0.30 (ct) and 26.9 ± 10.4 (dt) (srs2Δ rhp57Δ, no UV); 0.68 ± 0.86 (ct) and 53.2 ± 23.8 (dt) (srs2Δ rhp57Δ, 25 J/m2 UV); 0.05 ± 0.05 (ct) and 17.5 ± 5.8 (dt) (rhp51Δ, no UV); 0.06 ± 0.11 (ct) and 17.1 ± 7.9 (dt) (rhp51Δ, 25 J/m2 UV); 0.10 ± 0.20 (ct) and 20.1 ± 8.1 (dt) (srs2Δ rhp51Δ, no UV); 0.06 ± 0.10 (ct) and 24.9 ± 10.5 (dt) (srs2Δ rhp51Δ, 25 J/m2 UV).
Deleting rhp51, rhp55 or rhp57 suppresses the poor growth of an srs2 rqh1 double mutant
In S.cerevisiae an srs2 sgs1 double mutant is poorly viable (22). This phenotype is suppressed by deleting members of the RAD52 epistasis group including RAD51, RAD55 and RAD57 (22). Similar poor growth/viability is seen with an srs2 rqh1 double mutant of S. pombe (Fig. 4A) (23,24). However, there are conflicting data concerning whether the poor growth of an srs2 rqh1 strain can be suppressed by blocking recombination. Maftahi et al. (23) have shown suppression with deletion of either rhp51 or rhp57, whereas Wang et al. (24) concluded that viability is made worse by deleting rhp51 in an srs2 rqh1 mutant background. To provide a further independent test of this we constructed three triple mutant strains each containing a deletion of srs2 and rqh1 combined with a deletion in either rhp51, rhp55 or rhp57. These were then compared with an srs2Δ rqh1Δ double mutant for growth (Fig. 4B). Each triple mutant grew better than the srs2Δ rqh1Δ double mutant. In the case of the srs2Δ rqh1Δ rhp51Δ triple mutant we confirmed that suppression was due to the deletion of rhp51, rather than by some other spontaneous mutation, by complementing the improved growth with a plasmid-borne copy of rhp51 expressed from a thiamine repressible nmt promoter (Fig. 4C). These data corroborate the results reported by Maftahi et al. (23). The failure of Wang et al. (24) to observe suppression of srs2Δ rqh1Δ poor growth by deletion of rhp51 may relate to the particular rhp51Δ mutant that they used in their studies (23).
Figure 4.

The poor viability of an srs2Δ rqh1Δ double mutant strain is rescued by deletion of rhp51, rhp55 or rhp57. (A) Tetrad analysis of spores from a cross between srs2Δ (MCW473) and rqh1Δ (MCW149) mutant strains. Spores were micro-dissected as grids onto YES agar plates and incubated for 5 days at 30°C before being photographed. The growth of spores from four separate asci is shown. The genotype of each spore is shown on the right of the panel. wt, wild-type; s, srs2Δ single mutant; q, rqh1Δ single mutant; qs, rqh1Δ srs2Δ double mutant. (B) Streak plate showing the relative growth on complete medium of the srs2Δ rqh1Δ double mutant strain (MCW1284) and the srs2Δ rqh1Δ rhp51Δ (MCW1097), srs2Δ rqh1Δ rhp55Δ (MCW1282) and srs2Δ rqh1Δ rhp57Δ (MCW1283) triple mutant strains. (C) Complementing the improved growth of an srs2Δ rqh1Δ rhp51Δ triple mutant strain by expression of rhp51 from a plasmid. The schematic at the top of the panel shows the order in which strains were streaked onto selective media. The strains carry either the plasmid pREP41 or a derivative of it expressing rhp51 from a thiamine repressible nmt promoter. Strains were initially streaked onto EMM containing thiamine followed by two successive rounds of replica plating onto EMM lacking thiamine. By the second replica plating the nmt promoter is fully derepressed and the expression of rhp51 is therefore switched on. The strains used were MCW42 (wild-type), MCW473 (srs2Δ), MCW928 (rqh1Δ), MCW1088 (rhp51Δ) and MCW1097 (srs2Δ rqh1Δ rhp51Δ). (D) Spot assay showing the relative sensitivities of wild-type (MCW42), srs2Δ (MCW473), rqh1Δ (MCW928), rhp55Δ (MCW1049), rqh1Δ rhp55Δ (MCW832) and srs2Δ rqh1Δ rhp55Δ (MCW1282) strains to UV, HU and MMS.
Suppression of rqh1Δ DNA damage sensitivity by rhp55Δ is independent of Srs2
The hypersensitivity of a rqh1Δ mutant to certain DNA damaging agents is suppressed by deleting rhp51, rhp55 or rhp57 (Fig. 4D, and our unpublished data) (35). As Srs2 appears to control residual Rhp51 activity in rhp55Δ and rhp57Δ mutants, we looked to see whether it is required for rhp55Δ’s suppression of rqh1Δ’s sensitivity to UV, HU and MMS (Fig. 4D). The srs2Δ rqh1Δ rhp55Δ strain behaves like a rqh1Δ rhp55Δ strain and is noticeably less sensitive to UV, HU and MMS than a rqh1Δ single mutant (Fig. 4D). These data suggest that residual Rhp51 activity in the absence of Rhp55 and Srs2 does not necessitate the action of Rqh1.
Srs2’s relationship to the PRR pathway in S.pombe
To investigate whether Srs2 is involved in the PRR pathway in S.pombe we first compared the UV sensitivities of srs2Δ and rhp6Δ (homologue of RAD6) single mutant strains with that of an srs2Δ rhp6Δ double mutant (Fig. 5A). The double mutant is only slightly more sensitive to UV than the most sensitive single mutant (rhp6) indicating that srs2 is more or less epistatic with rhp6 for UV sensitivity. The same analysis was performed with two further PRR genes [rhp18 (RAD18 homologue) and rad8 (RAD5 homologue)]. In both cases srs2 is epistatic with the PRR gene for UV sensitivity (Fig. 5B and C). These data indicate that Srs2 functions in the PRR pathway for the tolerance/repair of UV-induced DNA damage. The slight increase in UV sensitivity of the srs2Δ rhp6Δ double mutant compared with a rhp6Δ single mutant may be due to the fact that Rhp6 is likely to have roles separate from PRR (36). Possibly, defects in these non-PRR functions enable some UV-induced damage to be processed by a mechanism that involves Srs2.
Figure 5.

Epistasis analysis of srs2, rhp6, rhp18 and rad8 mutants. (A–C) Survival curves showing the relative sensitivity to UV of wild-type (MCW449), srs2Δ (MCW460), rhp6Δ (MCW595), rhp18Δ (MCW1269), rad8Δ (MCW162), srs2Δ rhp6Δ (MCW1273), srs2Δ rhp18Δ (MCW1270) and srs2Δ rad8Δ (MCW680) strains. (D) Spot assay showing the relative sensitivities to UV and CPT of wild-type (MCW449), srs2Δ (MCW460), rhp18Δ (MCW1269), rad8Δ (MCW162), srs2Δ rhp18Δ (MCW1270) and srs2Δ rad8Δ (MCW680) strains.
We also compared the srs2 and PRR mutant strains for sensitivity to CPT (Fig. 5D and data not shown). Whereas an srs2Δ mutant is hypersensitive to CPT, rhp6Δ, rhp18Δ and rad8Δ mutants are about as sensitive as a wild-type strain. Furthermore, the srs2Δ rhp6Δ, srs2Δ rhp18Δ and srs2Δ rad8Δ double mutant strains exhibit the same sensitivity as an srs2Δ single mutant. Together with the data in Figure 2B, these results suggest that Srs2 and Rhp51 function together, independently of the PRR pathway, for the repair of SEBs.
Genetic interactions between srs2 and mus81
Mus81–Eme1 is a heterodimeric endonuclease that appears to be required for the cleavage of a range of different DNA junctions that are formed during HR and the regression of stalled replication forks (37–41). Similar to srs2, mus81 has strong genetic interactions with both rqh1and rhp54, with the mus81 rqh1 double mutant being unviable (38,42), and the mus81 rhp54 double mutant exhibiting severely reduced viability (Fig. 6A and B). To investigate the genetic interaction between srs2 and mus81 we constructed a double mutant strain and compared it with wild-type, and srs2 and mus81 single mutant strains for growth and sensitivity to HU, UV, MMS and CPT. The srs2Δ mus81Δ double mutant grows just as well as a mus81Δ single mutant (data not shown). It is also indistinguishable from a mus81Δ strain for sensitivity to HU, UV and MMS (Fig. 6C and D). In the case of HU and MMS, the mus81 mutant is considerably more sensitive than the srs2 mutant, and this makes it difficult to tell whether deletion of srs2 adds to the sensitivity of the mus81Δ strain or not. However, in the case of UV the difference in sensitivity between the single mutants is not so marked, enabling us to conclude that srs2 and mus81 are epistatic for UV sensitivity. This suggests that they function in the same pathway for the repair/tolerance of UV-induced damage. In contrast, the srs2Δ mus81Δ double mutant is noticeably more sensitive to CPT than either of the single mutants (Fig. 6C). The increase in sensitivity appears to be synergistic, suggesting that Mus81 and Srs2 promote the repair of SEBs by separate but overlapping pathways.
Figure 6.
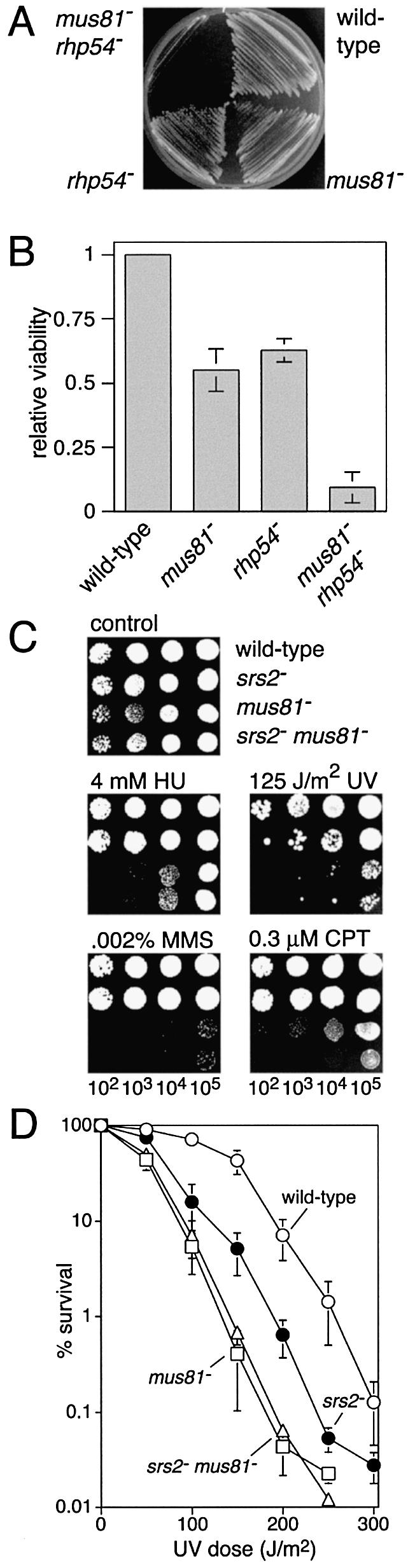
Genetic interactions between mus81 and rhp54, and mus81 and srs2. (A) Streak plate showing the relative growth on complete medium of the mus81Δ rhp54Δ double mutant strain (MCW897) compared with wild-type (MCW42), mus81Δ (MCW745) and rhp54Δ (MCW121) control strains. The plate was photographed after 4 days growth at 30°C. (B) Histogram showing the relative viability compared with wild-type (MCW42) of mus81Δ (MCW745), rhp54Δ (MCW121) and mus81Δ rhp54Δ (MCW897) strains on complete medium. (C) Spot assay showing the relative sensitivities of wild-type (MCW449), srs2Δ (MCW460), mus81Δ (MCW744) and srs2Δ mus81Δ (MCW747) strains to UV, HU, MMS and CPT. (D) Survival curves showing the relative sensitivity to UV of wild-type (MCW449), srs2Δ (MCW460), mus81Δ (MCW744) and srs2Δ mus81Δ (MCW747) strains.
Mus81 and the PRR pathway
From the data in Figures 5 and 6 it is evident that srs2 is epistatic with both mus81 and the PRR pathway genes for UV sensitivity. It was therefore important to establish the genetic relationship between mus81 and the PRR pathway. Double mutants of mus81Δ with rhp6Δ, rhp18Δ and rad8Δ were made and compared with their respective single mutant strains for sensitivity to UV (Fig. 7). Each double mutant is more sensitive than either of its relative single mutant strains, and the increase is no more than additive. These data show that both Mus81 and the PRR pathway can function independently of each other to promote the repair/tolerance of UV-induced damage. However, the fact that they share in common an epistatic interaction with srs2 suggests that there is a sub-pathway of PRR that depends on both Srs2 and Mus81.
Figure 7.

Epistasis analysis of the mus81 mutant with rhp6, rhp18 and rad8 mutants for UV sensitivity. The strains used were MCW449 (wild-type), MCW744 (mus81Δ), MCW595 (rhp6Δ), MCW1269 (rhp18Δ), MCW162 (rad8Δ), MCW1137 (mus81Δ rhp6Δ), MCW1271 (mus81Δ rhp18Δ) and MCW1272 (mus81Δ rad8Δ).
DISCUSSION
In S.cerevisiae a key function of the Srs2 DNA helicase is to prevent or limit potentially deleterious recombination. This is a function that Srs2 shares with the RecQ family DNA helicase Sgs1, as judged by the severe growth defects of an srs2 sgs1 double mutant, which can be suppressed by blocking recombination at a presynaptic stage. These features of Srs2 behaviour are conserved in S.pombe as reported here and by Maftahi et al. (23). However, as discussed below, Srs2 is less critical for the PRR pathway in S.pombe than it is in S.cerevisiae. We also highlight below the genetic interactions between srs2 and mus81, and Srs2’s role in promoting the recombinational repair of SEBs.
The involvement of Srs2 and Mus81 in a sub-pathway of PRR
In S.cerevisiae, deletion of srs2 suppresses the UV sensitivity of rad6, rad18 and rad5 mutants (10–13). This has been taken as evidence that Srs2 is required to channel DNA lesions into the PRR pathway. In S.pombe, mutation of srs2 exerts no suppression on the UV sensitivity of rhp6–, rhp18– or rad8– strains. Therefore, in contrast to S.cerevisiae, Srs2 does not appear to be essential for channelling DNA lesions to the PRR pathway in S.pombe. This may be because PRR proteins are more able to compete with HR proteins for access to lesion-containing DNAs. In this regard it is worth noting that in S.pombe, PRR proteins can work upstream of HR proteins, at least in some situations. Rhp18, for example, is active throughout the cell cycle in response to UV-induced damage, and in G2, Rhp51 foci formed in response to UV are partially dependent on Rhp18 (35). Another possibility for why Srs2 appears to be less critical for PRR in S.pombe than in S.cerevisiae is redundancy of function with Rqh1. In S.cerevisiae, over-expression of Sgs1 can partially suppress the need for Srs2 (43). Perhaps in S.pombe endogenous levels of Rqh1 are more able to compensate for the lack of Srs2 than they are in S.cerevisiae. In this regard it is interesting to contemplate the possibility that in humans, where a homologue of Srs2 has not been identified, the role of Srs2 has been taken on by one of five known RecQ DNA helicases (44).
Even though Srs2 is not required to channel lesions to PRR in S.pombe, it still plays some role in the PRR response to UV-induced damage, as judged by its epistatic relationship to rhp6, rhp18 and rad8 for UV sensitivity. Interestingly, this sub-pathway of PRR also appears to depend on Mus81, based on the epistatic relationship between srs2 and mus81 for UV sensitivity. In S.cerevisiae it is thought that Srs2 channels lesions to the PRR pathway by dissociating Rad51 nucleoprotein filaments. If the same is true in S.pombe, what possible role could Mus81–Eme1 have in this pathway? Mus81–Eme1 is a structure-specific endonuclease, which can potentially cleave a variety of DNA junctions in vivo (37–41). These include the aberrant structures that can form when replication forks are blocked, the D-loops that are formed by strand invasion reactions, and 3′ flaps that can form during the repair of DSBs and single-strand gaps by SDSA. It is difficult to see how any of these known activities of Mus81–Eme1 could collude with Srs2 to dislodge Rhp51 from DNA and direct repair to the PRR pathway. Possibly Srs2 only acts after strand invasion, and although in vitro it is unable to dissociate an intact D-loop (17), perhaps it can do so if the base of the D-loop is first nicked by Mus81–Eme1 (40). Alternatively, as suggested by Fabre and co-workers, Srs2 and Mus81–Eme1 could work together in a SDSA-type mechanism for the repair of lesion-containing gaps (45). In this model, Rhp51 catalyses strand invasion from the damaged DNA into an intact sister chromatid to form a D-loop. The 3′ end of the invading strand primes DNA synthesis enabling recovery of the missing genetic information. Srs2 then dissociates the D-loop so that the invading strand re-anneals to the lesion-containing strand. Over-replication will result in the formation of a 3′ flap, which could be removed by Mus81–Eme1. It is unclear what role PRR proteins could play in this mechanism. One possibility is that when Mus81–Eme1 removes the 3′ flap it generates a small single-stranded gap (39,40). Conceivably, PRR proteins could be involved in the repair of this gap.
Do the shared genetic interactions of srs2 and mus81 point to a common pathway?
Srs2 and Mus81 appear to work in a common pathway responding to UV-induced damage. However, the synergistic increase in CPT sensitivity of a mus81 srs2 double mutant, compared with mus81 and srs2 single mutants, indicates that Mus81 and Srs2 also function in independent pathways for the repair of SEBs. Interestingly, both mus81 and srs2 have strong genetic interactions with rqh1 and rhp54. However, these common interactions are not necessarily indicative of Mus81 and Srs2 functioning in the same pathway. As shown here and elsewhere, the poor growth of an srs2 rqh1 double mutant is suppressed by mutation of rhp51 (23). In contrast, the unviability of a mus81 rqh1 double mutant is not rescued by rhp51 mutation (C. L. Doe, F. Osman, J. Dixon and M. C. Whitby, unpublished data). These data suggest that Rqh1 is needed in an srs2– background to process recombination intermediates, whereas in a mus81– background it is needed to process other kinds of intermediates that are formed independently of Rhp51. As mentioned earlier, the unviability of an srs2 rhp54 double mutant may reflect a need for Srs2 to dissociate Rhp51 nucleoprotein filaments that are incompetent for normal strand invasion in the absence of Rhp54 (16). What underlies the poor growth of a mus81 rhp54 double mutant is not certain. In fact, this interaction was a surprise to us, since in S.cerevisiae, MUS81 and RAD54 exhibit a purely epistatic relationship and their proteins physically interact (46). One possible explanation for the strong genetic interaction between mus81 and rhp54 in S.pombe is that they both contribute to the stability of joint molecules formed by Rhp51-mediated strand invasion. Based on the activities of S.cerevisiae and human Rad54 in vitro, Rhp54 could do this in a number of ways, from dissociating proteins that might impede Rhp51-mediated strand invasion, to altering the supercoiling of the target DNA so that its strands can be readily unpaired for D-loop formation (20). We have recently shown that Mus81–Eme1 can cleave D-loops in vitro, and this activity could provide an additional way of helping to stabilize the strand invasion (40). It is worth noting here that Srs2 may also aid joint molecule stability by dissociating Rhp51 from the displaced single strand at a D-loop, thereby preventing potential reversal of strand invasion (17).
Srs2 and the repair of collapsed replication forks
The hypersensitivity of an srs2 mutant to CPT indicates that Srs2 promotes the repair of SEBs formed when replication forks collapse at strand nicks in the template DNA. This role for Srs2 must be distinct from PRR because rhp6, rhp18 and rad8 mutants show no hypersensitivity to CPT. Interestingly, S.pombe PRR mutants are hypersensitive to ionizing radiation (47–49). So, although they might not be important for SEB repair, PRR proteins do play a role in the repair of DSBs. This provides evidence for mechanistic differences between SEB and DSB repair.
Srs2 seems to function in the Rhp51-dependent pathway for SEB repair. We suspect that this pathway proceeds via strand invasion from the broken arm of the replication fork back into the intact arm. This generates a D-loop, which may act as a site for replisome reassembly (1). What might Srs2’s involvement be here? One possibility, which is mentioned above, is that it could help to stabilize the D-loop by dissociating any Rhp51 that nucleates onto the displaced strand (17). Promoting the recycling of a finite resource of Rhp51 may also aid in the efficient repair of multiple SEBs. Alternatively, Srs2’s requirement for SEB repair could reflect its potential involvement in DNA damage checkpoint responses. Srs2 in S.cerevisiae is phosphorylated as part of the intra-S checkpoint response to DNA damage, and is required for ‘switching off’ the checkpoint response after the damage has been repaired (50,51). This might be achieved by dissociating Rad51 and/or checkpoint proteins from the DNA (16). It will be interesting to see whether Srs2 behaves similarly in S.pombe, and whether this, or any of the above potential activities, underlies Srs2’s involvement in repairing collapsed replication forks.
Acknowledgments
ACKNOWLEDGEMENTS
We thank David Sherratt for his encouragement and Julie Dixon for excellent technical support. We would also like to thank Greg Freyer, Matthew O’Connell, Albert Pastink and Hideo Shinagawa for the gift of strains. This work was supported by a project grant (065278/Z/01/Z) and a Senior Research Fellowship from the Wellcome Trust awarded to M.C.W.
REFERENCES
- 1.McGlynn P. and Lloyd,R.G. (2002) Recombinational repair and restart of damaged replication forks. Nature Rev. Mol. Cell Biol., 3, 859–870. [DOI] [PubMed] [Google Scholar]
- 2.Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem., 65, 43–81. [DOI] [PubMed] [Google Scholar]
- 3.Prakash S. and Prakash,L. (2002) Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev., 16, 1872–1883. [DOI] [PubMed] [Google Scholar]
- 4.Lehmann A.R. (2000) Replication of UV-damaged DNA: new insights into links between DNA polymerases, mutagenesis and human disease. Gene, 253, 1–12. [DOI] [PubMed] [Google Scholar]
- 5.Prakash L. (1981) Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations. Mol. Gen. Genet., 184, 471–478. [DOI] [PubMed] [Google Scholar]
- 6.Rupp W.D. and Howard-Flanders,P. (1968) Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J. Mol. Biol., 31, 291–304. [DOI] [PubMed] [Google Scholar]
- 7.Rupp W.D., Wilde,C.E., 3rd, Reno,D.L. and Howard-Flanders,P. (1971) Exchanges between DNA strands in ultraviolet-irradiated Escherichia coli. J. Mol. Biol., 61, 25–44. [DOI] [PubMed] [Google Scholar]
- 8.Higgins N.P., Kato,K. and Strauss,B. (1976) A model for replication repair in mammalian cells. J. Mol. Biol., 101, 417–425. [DOI] [PubMed] [Google Scholar]
- 9.Yamashita Y.M., Okada,T., Matsusaka,T., Sonoda,E., Zhao,G.Y., Araki,K., Tateishi,S., Yamaizumi,M. and Takeda,S. (2002) RAD18 and RAD54 cooperatively contribute to maintenance of genomic stability in vertebrate cells. EMBO J., 21, 5558–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aboussekhra A., Chanet,R., Zgaga,Z., Cassier-Chauvat,C., Heude,M. and Fabre,F. (1989) RADH, a gene of Saccharomyces cerevisiae encoding a putative DNA helicase involved in DNA repair. Characteristics of radH mutants and sequence of the gene. Nucleic Acids Res., 17, 7211–7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schiestl R.H., Prakash,S. and Prakash,L. (1990) The SRS2 suppressor of rad6 mutations of Saccharomyces cerevisiae acts by channeling DNA lesions into the RAD52 DNA repair pathway. Genetics, 124, 817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lawrence C.W. and Christensen,R.B. (1979) Metabolic suppressors of trimethoprim and ultraviolet light sensitivities of Saccharomyces cerevisiae rad6 mutants. J. Bacteriol., 139, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broomfield S. and Xiao,W. (2002) Suppression of genetic defects within the RAD6 pathway by srs2 is specific for error-free post-replication repair but not for damage-induced mutagenesis. Nucleic Acids Res., 30, 732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rong L., Palladino,F., Aguilera,A. and Klein,H.L. (1991) The hyper-gene conversion hpr5-1 mutation of Saccharomyces cerevisiae is an allele of the SRS2/RADH gene. Genetics, 127, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Symington L.S. (2002) Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev., 66, 630–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veaute X., Jeusset,J., Soustelle,C., Kowalczykowski,S.C., Le Cam,E. and Fabre,F. (2003) The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature, 423, 309–312. [DOI] [PubMed] [Google Scholar]
- 17.Krejci L., Van Komen,S., Li,Y., Villemain,J., Reddy,M.S., Klein,H., Ellenberger,T. and Sung,P. (2003) DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature, 423, 305–309. [DOI] [PubMed] [Google Scholar]
- 18.Aboussekhra A., Chanet,R., Adjiri,A. and Fabre,F. (1992) Semidominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to procaryotic RecA proteins. Mol. Cell. Biol., 12, 3224–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ira G., Malkova,A., Liberi,G., Foiani,M. and Haber,J. (2003) Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell, 115, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan T.L., Kanaar,R., Wyman,C., Hsiang,Y.H., Lihou,M.G. and Liu,L.F. (2003) Rad54, a Jack of all trades in homologous recombination. DNA Repair (Amst.), 2, 787–794. [DOI] [PubMed] [Google Scholar]
- 21.Palladino F. and Klein,H.L. (1992) Analysis of mitotic and meiotic defects in Saccharomyces cerevisiae SRS2 DNA helicase mutants. Genetics, 132, 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gangloff S., Soustelle,C. and Fabre,F. (2000) Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nature Genet., 25, 192–194. [DOI] [PubMed] [Google Scholar]
- 23.Maftahi M., Hope,J.C., Delgado-Cruzata,L., Han,C.S. and Freyer,G.A. (2002) The severe slow growth of Δsrs2 Δrqh1 in Schizosaccharomyces pombe is suppressed by loss of recombination and checkpoint genes. Nucleic Acids Res., 30, 4781–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang S.W., Goodwin,A., Hickson,I.D. and Norbury,C.J. (2001) Involvement of Schizosaccharomyces pombe Srs2 in cellular responses to DNA damage. Nucleic Acids Res., 29, 2963–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno S., Klar,A. and Nurse,P. (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol., 194, 795–823. [DOI] [PubMed] [Google Scholar]
- 26.Rothstein R. (1991) Targeting, disruption, replacement and allele rescue: integrative DNA transformation in yeast. Methods Enzymol., 194, 281–301. [DOI] [PubMed] [Google Scholar]
- 27.Osman F., Adriance,M. and McCready,S. (2000) The genetic control of spontaneous and UV-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Curr. Genet., 38, 113–125. [DOI] [PubMed] [Google Scholar]
- 28.Liu L.F., Duann,P., Lin,C.T., D’Arpa,P. and Wu,J. (1996) Mechanism of action of camptothecin. Ann. N. Y. Acad. Sci., 803, 44–49. [DOI] [PubMed] [Google Scholar]
- 29.Nitiss J.L. and Wang,J.C. (1996) Mechanisms of cell killing by drugs that trap covalent complexes between DNA topoisomerases and DNA. Mol. Pharmacol., 50, 1095–1102. [PubMed] [Google Scholar]
- 30.Schild D. (1995) Suppression of a new allele of the yeast RAD52 gene by overexpression of RAD51, mutations in srs2 and ccr4, or mating-type heterozygosity. Genetics, 140, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein H.L. (2001) Mutations in recombinational repair and in checkpoint control genes suppress the lethal combination of srs2Δ with other DNA repair genes in Saccharomyces cerevisiae. Genetics, 157, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khasanov F.K., Savchenko,G.V., Bashkirova,E.V., Korolev,V.G., Heyer,W.D. and Bashkirov,V.I. (1999) A new recombinational DNA repair gene from Schizosaccharomyces pombe with homology to Escherichia coli RecA. Genetics, 152, 1557–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsutsui Y., Morishita,T., Iwasaki,H., Toh,H. and Shinagawa,H. (2000) A recombination repair gene of Schizosaccharomyces pombe, rhp57, is a functional homolog of the Saccharomyces cerevisiae RAD57 gene and is phylogenetically related to the human XRCC3 gene. Genetics, 154, 1451–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milne G.T., Ho,T. and Weaver,D.T. (1995) Modulation of Saccharomyces cerevisiae DNA double-strand break repair by SRS2 and RAD51. Genetics, 139, 1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laursen L.V., Ampatzidou,E., Andersen,A.H. and Murray,J.M. (2003) Role for the fission yeast RecQ helicase in DNA repair in G2. Mol. Cell. Biol., 23, 3692–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang H., Kahana,A., Gottschling,D.E., Prakash,L. and Liebman,S.W. (1997) The ubiquitin-conjugating enzyme Rad6 (Ubc2) is required for silencing in Saccharomyces cerevisiae. Mol. Cell. Biol., 17, 6693–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boddy M.N., Gaillard,P.-H.L., McDonald,W.H., Shanahan,P., Yates 3rd,J.R. and Russell,P. (2001) Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell, 107, 537–548. [DOI] [PubMed] [Google Scholar]
- 38.Doe C.L., Ahn,J.S., Dixon,J. and Whitby,M.C. (2002) Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. J. Biol. Chem., 277, 32753–32759. [DOI] [PubMed] [Google Scholar]
- 39.Whitby M.C., Osman,F. and Dixon,J. (2003) Cleavage of model replication forks by fission yeast Mus81-Eme1 and budding yeast Mus81-Mms4. J. Biol. Chem., 278, 6928–6935. [DOI] [PubMed] [Google Scholar]
- 40.Osman F., Dixon,J., Doe,C.L. and Whitby,M.C. (2003) Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis. Mol. Cell, 12, 761–774. [DOI] [PubMed] [Google Scholar]
- 41.Gaillard P.H., Noguchi,E., Shanahan,P. and Russell,P. (2003) The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Mol. Cell, 12, 747–759. [DOI] [PubMed] [Google Scholar]
- 42.Boddy M.N., Lopez-Girona,A., Shanahan,P., Interthal,H., Heyer,W.D. and Russell,P. (2000) Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol. Cell. Biol., 20, 8758–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mankouri H.W., Craig,T.J. and Morgan,A. (2002) SGS1 is a multicopy suppressor of srs2: functional overlap between DNA helicases. Nucleic Acids Res., 30, 1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hickson I.D. (2003) RecQ helicases: caretakers of the genome. Nature Rev. Cancer, 3, 169–178. [DOI] [PubMed] [Google Scholar]
- 45.Fabre F., Chan,A., Heyer,W.D. and Gangloff,S. (2002) Alternate pathways involving Sgs1/Top3, Mus81/ Mms4 and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl Acad. Sci. USA, 99, 16887–16892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Interthal H. and Heyer,W.D. (2000) MUS81 encodes a novel helix-hairpin-helix protein involved in the response to UV- and methylation-induced DNA damage in Saccharomyces cerevisiae. Mol. Gen. Genet., 263, 812–827. [DOI] [PubMed] [Google Scholar]
- 47.Rowley R. and Zhang,J. (1999) Caffeine-mediated override of checkpoint controls. A requirement for rhp6 (Schizosaccharomyces pombe). Genetics, 152, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verkade H.M., Teli,T., Laursen,L.V., Murray,J.M. and O’Connell,M.J. (2001) A homologue of the Rad18 postreplication repair gene is required for DNA damage responses throughout the fission yeast cell cycle. Mol. Genet. Genomics, 265, 993–1003. [DOI] [PubMed] [Google Scholar]
- 49.Doe C.L., Murray,J.M., Shayeghi,M., Hoskins,M., Lehmann,A.R., Carr,A.M. and Watts,F.Z. (1993) Cloning and characterisation of the Schizosaccharomyces pombe rad8 gene, a member of the SNF2 helicase family. Nucleic Acids Res., 21, 5964–5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vaze M.B., Pellicioli,A., Lee,S.E., Ira,G., Liberi,G., Arbel-Eden,A., Foiani,M. and Haber,J.E. (2002) Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol. Cell, 10, 373–385. [DOI] [PubMed] [Google Scholar]
- 51.Liberi G., Chiolo,I., Pellicioli,A., Lopes,M., Plevani,P., Muzi-Falconi,M. and Foiani,M. (2000) Srs2 DNA helicase is involved in checkpoint response and its regulation requires a functional Mec1-dependent pathway and cdk1 activity. EMBO J., 19, 5027–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muris D.F., Vreeken,K., Carr,A.M., Murray,J.M., Smit,C., Lohman,P.H. and Pastink,A. (1996) Isolation of the Schizosaccharomyces pombe RAD54 homologue, rhp54+, a gene involved in the repair of radiation damage and replication fidelity. J. Cell Sci., 109, 73–81. [DOI] [PubMed] [Google Scholar]
- 53.Stewart E., Chapman,C.R., Al-Khodairy,F., Carr,A.M. and Enoch,T. (1997) rqh1+, a fission yeast gene related to the Bloom’s and Werner’s syndrome genes, is required for reversible S phase arrest. EMBO J., 16, 2682–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reynolds P., Koken,M.H., Hoeijmakers,J.H., Prakash,S. and Prakash,L. (1990) The rhp6+ gene of Schizosaccharomyces pombe: a structural and functional homolog of the RAD6 gene from the distantly related yeast Saccharomyces cerevisiae. EMBO J., 9, 1423–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]