

Abstract
Kinase domain mutations of the epidermal growth factor receptor (EGFR) are common oncogenic events in lung adenocarcinoma. Here we explore the dependency upon asymmetric dimerization of the kinase domain for activation of lung cancer-derived EGFR mutants. We show that while wild-type EGFR and the L858R mutant require dimerization for activation and oncogenic transformation, the exon 19 deletion, exon 20 insertion, and L858R/T790M EGFR mutants do not require dimerization. In addition, treatment with the monoclonal antibody, cetuximab, shrinks mouse lung tumors induced by the dimerization-dependent L858R mutant, but exerts only a modest effect on tumors driven by dimerization-independent EGFR mutants. These data imply that different EGFR mutants show differential requirements for dimerization, and that disruption of dimerization may be among the antitumor mechanisms of cetuximab.
Keywords: epidermal growth factor receptor (EGFR), cetuximab (Erbitux), lung cancer-derived EGFR mutation, receptor dimerization, targeted therapy
INTRODUCTION
Activation of the epidermal growth factor receptor (EGFR) kinase, a member of the ErbB family of receptor tyrosine kinases, plays a central event in cancer pathogenesis (1, 2). Somatic mutations within the kinase domain of the EGFR gene occur in lung adenocarcinoma in about 8% of patients from Europe and North America and 30% of patients from East Asia (3–6) with the L858R mutation in exon 21 and exon 19 in frame-deletions including amino acids 747 to 749 (Ex19Del) accounting for 88% of these mutations (3). These two somatic mutations are highly associated with clinical responses to treatment with the EGFR kinase inhibitors gefitinib and erlotinib (7–9). However, acquisition of a second EGFR mutation, T790M, that most commonly occurs after treatment with gefitinib or erlotinib, renders the L858R and Ex19Del mutants resistant to these drugs (10, 11). In contrast, the exon 20 insertion (Ex20Ins) EGFR mutants, which represents about 6% of the mutations found in lung adenocarcinoma, appear to be inherently resistant to gefitinib and erlotinib (12, 13).
Cetuximab (Erbitux) is a human-mouse chimeric monoclonal antibody that is FDA-approved for treatment of colorectal and head and neck cancer patients (14–17). Although cetuximab is effective against about 10% of colorectal carcinoma, EGFR mutations are found in fewer than 2% of these tumors (18, 19). While the presence of KRAS or BRAF mutation in colorectal cancer is associated with resistance to cetuximab (20, 21), the EGFR characteristics that correlate with colon tumor sensitivity to cetuximab are less well defined. Recently, cetuximab in combination with chemotherapy has been shown to increase survival of non-small cell lung cancer (NSCLC) patients compared to chemotherapy treatment alone (22) but the molecular mechanisms of cetuximab response in lung cancer are likewise undefined.
Binding of cetuximab to the extracellular domain of EGFR may act via immune responses, promoting receptor degradation and antibody-dependent cellular cytotoxicity (ADCC) (23). In addition, structural studies have suggested that cetuximab may prevent receptor activation by directly blocking ligand binding and/or indirectly blocking the extracellular domain rearrangement required for receptor dimerization by interacting with subdomain III of the EGFR extracellular domain (24–26).
Recent three-dimensional structural analyses of EGFR have provided mechanistic insight into the role of extracellular, juxtamembrane and intracellular receptor dimerization in EGFR activation. First, ligand binding to EGFR extracellular domains I and III stabilizes an open receptor structure, enabling dimerization of extracellular domains and juxtamembrane segments (27–29). Subsequently, the EGFR kinase domain undergoes asymmetric dimerization in which the C-lobe of the activator monomer activates the N-lobe of the receiver monomer, similar to cyclin-induced activation of cyclin-dependent kinases, activating EGF receptor signaling (Fig. 1A) (30). Substitution mutation of amino acid residues at the asymmetric dimerization interface, such as L704N (receiver-impairing mutation) in the N-lobe and I941R (activator-impairing mutation) in the C-lobe, disrupt both dimerization and activation (Fig. 1B) (30). Co-expression of receiver-impaired and activator-impaired EGFR mutants can rescue receptor activation through asymmetric dimerization between the intact C-lobe and the intact N-lobe of the respective EGFR mutants (Fig. 1B).
Figure 1. Dimerization disruption has differential effects on the transforming activity of mutant EGFR proteins.

A and B, Proposed model for ligand-mediated EGFR dimerization and activation. EGF induces extracellular dimerization and asymmetric dimerization, resulting in the activation of the receiver monomer. A single mutation at the asymmetric dimerization interface of either the receiver monomer or the activator monomer is sufficient to impair receptor dimerization and activation. Co-expression of receiver-impaired (L704N) and activator-impaired (I941R) mutants can rescue asymmetric dimerization mediated by the intact C-lobe of receiver-impaired and N-lobe of activator-impaired mutants, and thereby activates the activator-impaired receiver mutant. Adapted from Zhang et al., 2006 Cell 115, 1137–1149; Dawson et al. 2005 Mol Cell Biol 25, 7734–7742; and Riese II et al., 2007 BioEssays 6, 558–565. C, D, E and F, L858R mutants are dependent on asymmetric dimerization for their transforming potential whereas Ex19Del, Ex20Ins and L858R/T790M mutants were not. NIH-3T3 cells expressing the indicated EGFR mutants with or without receiver-impairing (L704N) or/and activator-impairing (I941R) mutations were assayed for anchorage-independent growth in soft agar. The bar graph depicts the relative number of colonies in the dimerization-defective mutants normalized to the number of colonies formed by cells expressing the respective parental mutants (n=3, mean +SD).
Activating mutations in the EGFR kinase domain induce an active conformation of the enzyme that is not dependent on ligand-induced dimerization (12, 31, 32). This observation raises the question of whether EGFR-directed monoclonal antibodies, which can block ligand-induced dimerization, will be effective in treatment of tumors arising from these kinase domain mutations. If the mutants are active even as monomers, antibodies directed at the "upstream" extracellular domain may be ineffective. Alternatively, if asymmetric dimerization is important even when ligand induced dimerization is not (either for trans-autophosphorylation of the receptor itself or for full catalytic activation), then antibody therapy alone or in combination with gefitinib or erlotinib may prove advantageous.
In order to better understand these issues, we have analyzed the requirement for dimerization in tumor-derived EGFR mutants. We have chosen the L858R, Ex19Del, Ex20Ins and T790M mutants for this study because they are common in lung adenocarcinoma. We found that the Ex19Del, Ex20Ins, and T790M mutants are activated and transform cells in a dimerization-independent manner, while L858R mutants, as previously reported (33), are dimerization-dependent. In cellular and animal models, cetuximab inhibits tumorigenesis by the dimerization-dependent L858R EGFR mutant but not by the dimerization-independent mutants.
MATERIALS AND METHODS
Expression Constructs
Wild-type EGFR, L747_E749del, A750P (Ex19Del), D770_N771insNPG (Ex20Ins), L858R/T790M and Ex19Del/T790M mutant EGFR containing vectors were prepared as previously described (12, 37). QuikChange site-directed mutagenesis (Stratagene) was used for generating all mutants described in this study with either wild-type EGFR or the above mutant EGFR in pBabe-puro as a template. C-terminal hemagglutinin (HA) or Myc tagged versions of EGFR were cloned by PCR and ligated into pBabe-puro between SnaBI and SalI. All plasmids were confirmed by sequencing.
Cell culture and generation of cell lines by viral transduction
All NIH-3T3 cell lines stably expressing EGFR mutants were established by retroviral infections and pooled as described previously (12). Cultures were serum-starved for 18 hrs prior to EGF stimulation and harvesting. Epidermal growth factor (EGF, Biosource) stimulations were performed using 25 ng/ml for 5 minutes unless noted in the text. Ba/F3 cells were maintained as previously described (38).
Immunoblotting, immunoprecipitation and antibodies
Cells or homogenized mouse lung tissue were lysed in RIPA buffer supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Calbiochem) and subjected to immunoblotting. For immunoprecipitation, 200–500 µg of protein lysate were incubated with antibody and protein A agarose for 3 hrs at 4 °C. Antibody information is listed in the Supplemental Data.
Anchorage-independent growth assay
Soft agar assays were carried out in triplicate as previously described (12) with minor changes (2 × 105 cells were used per well). Soft agar colonies were photographed after 2–3 weeks and quantified using Image J software (NIH). The data were normalized to number of colonies formed by control cells (see figure legends). The data represent triplicate wells. Each assay was repeated a minimum of two times with comparable results.
Cell growth inhibition assay
For growth inhibition assays, Ba/F3 cells (10,000 cells) were plated in 180 µL media in 96-well flat-bottom plates (Corning). 24hrs after plating, cell culture media were replaced with medium with and without cetuximab. The concentrations of cetuximab used for the assay ranged from 33 ng/mL to 100 mg/mL. The cells were incubated for another 72 hrs and the viable cell numbers were measured using cell counting kit-8 solution (Dojindo, Kumamoto, Japan). Absorbance was measured at 450 nm after 3 hrs. Data are expressed as percentage of growth relative to that of untreated control cells.
Generation of transgenic mice, cetuximab treatment and MRI imaging
L858R, Ex19Del, LTM and DTM bitransgenic mice were previously characterized (44, 53, 54). The generation of Ex20Ins mice is described in Supplemental Data. All mice were housed in a pathogen-free environment at the Harvard School of Public Health and were handled in strict accordance with Good Animal Practice as defined by the Office of Laboratory Animal Welfare, and all animal work was done with Dana-Farber Cancer Institute IACUC approval. Mice were fed a doxycycline diet when they reached 4 weeks of age. For cetuximab experiments, 1 mg per dose was injected intraperitoneally daily and MRI was carried out after 2 weeks of treatment as previously described (44). Tumor burden was documented through MRI imaging after observing panting behavior as described in Supplemental Data.
Immunohistochemistry
Immunochemical analysis was performed as previously described (44). Anti-EGFR and anti-phospho-EGFR (pY1016) were purchased from Cell Signaling Technologies.
RESULTS
Asymmetric dimerization is required for oncogenic activity of the L858R mutant and ligand-stimulated wild-type EGFR, but is dispensable for Ex19Del, Ex20Ins and L858R/T790M mutants
Given that EGFR becomes activated through formation of asymmetric dimers between the N-lobe and the C-lobe of its kinase domain, we sought to determine whether this dimerization is essential for enzymatic activation of and consequently cellular transformation by EGFR mutants found in lung cancer. To this end, we generated epitope-tagged EGFR expression constructs that combined a receiver-impairing mutation (L704N) and/or an activator-impairing mutation (I941R) with oncogenic Ex19Del, Ex20Ins, L858R, L858R/T790M (LTM) mutants or wild-type EGFR. The single or compound EGFR mutants were expressed in NIH-3T3 cells, which express low to undetectable levels of endogenous ErbB family proteins including EGFR, by retroviral transduction. These EGFR mutant-expressing cells were assayed for their ability to grow in soft agar (12, 34). In this system, the transforming ability of dimerization-dependent mutants is predicted to be abolished by cis mutation of L704 or I941. Furthermore, co-expression of the L704N and I941R mutants, in contrast, is predicted to restore transforming ability that is dimerization-dependent, because the two mutant forms can heterodimerize (Fig. 1B). Therefore, this experiment allows us to test whether specific EGFR kinase domain mutants can induce cellular transformation in a dimerization dependent or independent fashion.
As a control to assess whether cis mutation of L704N or I941R could disrupt biochemical dimerization in various mutant EGFR backgrounds, we performed non-denaturing gel electrophoresis and anti-EGFR immunoblotting of lysates from NIH-3T3 cells expressing the Ex19Del, Ex20Ins and L858R mutants. As predicted, the abundance of a higher molecular weight isoform in the kinase domain mutants was diminished by cis mutation of L704N or I941R but restored by co-expression of both mutants (Supplementary Fig. S1), confirming that the L704N and I941R mutants specifically disrupt dimerization.
Dimerization-impairing cis mutations in EGFR, L704N and I941R, significantly reduced the ability of L858R EGFR mutant to promote colony formation of NIH-3T3 cells upon retroviral transduction (Fig. 1C, L704N or I941R); colony-forming activity was partially or completely restored by co-expression of the L858R/L704N and L858R/I941R mutants in trans (Fig. 1C, L704N&I941R). Consistent with these results, tyrosine-phosphorylation of dimerization-impaired L858R mutants was restored by co-expression of L858R/L704N and L858R/I941R mutants (Supplementary Fig. S2A). Similar findings were obtained for EGF-stimulated NIH-3T3 cells expressing wild-type EGFR (Supplementary Figs. S2B, S2C and S3A).
In contrast, introduction of dimerization-impairing mutations into Ex19Del, Ex20Ins, and LTM mutants had little significant effect on their ability to induce colony formation in NIH-3T3 cells (Fig. 1D, 1E and 1F), suggesting that these mutants can induce cellular transformation without dimerization. EGFR tyrosine-phosphorylation of the L704N compound mutants was not detectable (Supplementary Figs. S4A, S4B and S4C, lane 4), but the I941R compound mutants still display constitutive tyrosine-phosphorylation (Supplementary Figs. S4A, S4B and S4C, lane 45. Furthermore, introduction of additional dimerization-disrupting mutants, the receiver-impairing P699G mutant and activator-impairing M952R mutant, into the Ex20Ins background (30) did not significantly reduce colony formation by Ex20Ins mutants (Supplementary Fig. S3B) but did abolish colony formation by wild-type EGFR with exogenous EGF (Supplementary Fig. S3C). As predicted, constitutive or EGF-induced tyrosine phosphorylation of the dimerization-impaired mutants were significantly diminished (Supplementary Figs. S2C and S4D).
The lung-cancer derived Ex19Del, Ex20Ins, L858R and LTM mutants as well as their cis dimerization-impaired mutants are equivalently expressed on cell surface of NIH-3T3 cells as assayed by immunofluorescence as well as flow cytometry assays (Supplementary Figs. S5A, S5B, S5C and S5D), suggesting that the differential oncogenic activity of dimerization-competent vs. dimerization-impaired mutants is unlikely to be due to alterations in EGFR protein expression in cells.
Taken together, these data suggest that Ex19Del, Ex20Ins and LTM mutants are oncogenic in the absence of asymmetric dimerization, whereas L858R mutants as well as wild-type EGFR acquire their oncogenic potentials following constitutive “inside-out” or ligand-dependent “outside-in” asymmetric dimerization, respectively, which is consistent with recent reports showing that oncogenic activity of L858R mutant may arise by promoting high receptor dimerization (28, 35, 36).
Dimerization-independent EGFR mutants are active in either the receiver or the monomer conformation
As dimerization-dependent activation of EGFR is well-described but dimerization-independent activation has not been reported to our knowledge, we further assessed the ability of EGFR mutants to be activated biochemically without asymmetric dimerization. To do so, we co-expressed dimerization-impaired activating EGFR mutants in which only one heterodimeric partner could be an active kinase, by introducing a kinase-inactivating D837A mutation together with a Myc tag into either a receiver-impaired, obligate activator L704N mutant (Figs. 2A and 2B, lanes 1, 3, and 5) or into an activator-impaired, obligate receiver I941R mutant (Figs. 2A and 2B, lanes 2, 4, and 6). To avoid confounding by receptor autophosphorylation in the analysis, we performed an anti-Myc immunoprecipitation followed by anti-phosphotyrosine immunoblotting, to assess the phosphorylation status of only the kinase-dead monomer (Fig. 2B).
Figure 2. Biochemical analysis of transphosphorylation by obligate activator and obligate receiver monomer forms of EGFR mutants.

A, Legend for schematic diagrams used in panel B. B, The Ex19Del and Ex20Ins mutants were constitutively active irrespective of monomeric position in the asymmetric dimer complex, whereas the activator-impaired monomer, not the receiver-impaired monomer, was specifically active in the L858R mutant and wild-type EGFR. Levels of tyrosine phosphorylation in the receiver-impaired monomer and activator-impaired monomer were compared among different EGFR mutants as well as wild-type EGFR by immunoprecipitation under denaturing conditions followed by immunoblotting with 4G10 antibody (top panel). The same blot was stripped and reprobed with anti-Myc antibody to detect total EGFR (middle panel). The relative levels of phosphorylation on the activator-impaired monomer were normalized to that of the receiver-impaired monomer and graphed (bottom panel, n=3, mean +SD). In the schematic diagrams, the possible asymmetric dimer formation of co-expressed dimerization-impaired (L704N and I941R) EGFR mutants, the activation status of each monomer, and the direction of phosphorylation are indicated.
We found that the Ex19Del and Ex20Ins mutant forms of EGFR are active kinases either in the obligate activator or obligate receiver forms, while the L858R mutant is only an active kinase in the obligate receiver form. Tyrosine phosphorylation was specifically detected on the immunoprecipitated kinase-dead obligate activator L858R/L704N/D837A-Myc EGFR monomer, but not on the kinase-dead obligate receiver L858R/I941R/D837A-Myc EGFR monomer (Fig. 2B, lanes 1 and 2, respectively), indicating that the enzymatic activity of the obligate receiver L858R monomer, but not the obligate activator monomer, is specifically activated through receptor dimerization (Fig. 2B, L858R schematics); similar results were obtained for wild-type EGFR with ligand stimulation (Supplementary Fig. S6, lanes 2 and 4 and WT schematics). In contrast, tyrosine phosphorylation was observed on Myc-tagged L704N/D837A and I941R/D837A forms of both the Ex19Del and Ex20Ins mutants (Fig. 2B, lanes 3, 4, 5 and 6). We conclude that either the obligate receiver or the obligate activator form of the Ex19Del and Ex20Ins mutants could trans-phosphorylate their kinase-dead heterodimeric partners (Fig. 2B, Ex19Del and Ex20 Ins schematics).
More broadly, we conclude that this ability of the Ex19Del and Ex20Ins mutants to be active in either the receiver or the activator conformation explains their ability to induce cellular transformation in the absence of canonical asymmetric dimerization, in contrast to the L858R mutant and wild-type EGFR which are active only in the dimerization-activated receiver conformation.
Differential effects of cetuximab on Ba/F3 cells that express dimerization-dependent L858R mutant EGFR compared to dimerization-independent mutants including L858R/T790M
The identification of dimerization-dependent and independent EGFR mutants raises the question whether the potency of cetuximab against distinct EGFR mutants is correlated with dimerization, given that inhibition of dimerization is one proposed mechanism of action for cetuximab (24). Ba/F3 cells can be engineered so that their survival without exogenous growth factors depends on different EGFR mutants (37, 38). Consistent with our previous results (37, 38), gefitinib inhibited the growth of Ba/F3 cells expressing either the L858R or the Ex19Del mutants, but did not show any effect on the cells transformed by either L858R/T790M (LTM), Ex19Del/T790M (DTM) or Ex20Ins mutants (Supplementary Figs. S8A). Because Ba/F3 cells expressing wild-type EGFR does not transform the cells, we did not include these cells for the assay.
Interestingly, cetuximab was able to suppress the growth of Ba/F3 cells expressing the L858R mutant in a dose-dependent manner, with an IC50 of 0.31 µg/ml, but had no effect on Ba/F3 cells expressing Ex19Del, Ex20Ins, LTM or DTM mutants (Fig. 3A). The lack of response to cetuximab was not due to the impaired cell surface expression of the mutant EGFR, as each mutant was expressed at high levels (Supplementary Fig. S7). However, in Ba/F3 cells, the Ex20Ins and T790M mutants are expressed at somewhat reduced levels compared to the L858R or Ex19Del mutants. To further characterize the biochemical effects of cetuximab on mutant EGFR, we performed immunoblotting of EGFR-expressing Ba/F3 cell lysates with either anti-EGFR or anti-phosphotyrosine antibodies before or after cetuximab treatment. Consistent with previous reports (39, 40), we observed that cetuximab induced EGF receptor degradation and down-regulation of phosphorylation in a dose-dependent manner (Fig. 3B). This effect occurred not only in the cetuximab-sensitive L858R mutant, but also in the cetuximab-resistant Ex19Del and Ex20Ins mutants, indicating that receptor degradation may not be sufficient for an anti-proliferative effect in EGFR-expressing Ba/F3 cells. Our results suggest that dimerization dependency and cetuximab sensitivity in Ba/F3 cells are correlated: the dimerization-dependent L858R mutant is cetuximab-sensitive while the dimerization-independent Ex19Del, Ex20Ins and LTM mutants are not. However, we could not assess directly whether cetuximab blocks dimerization, because this antibody binds directly to the EGF receptor, thereby confounding cross-linking analysis.
Figure 3. Differential pharmacological effects of cetuximab against oncogenic mutant EGFR in vitro and in vivo.

A, Cetuximab suppresses the growth of Ba/F3 cells dependent upon the L858R mutant, but not other tested oncogenic EGFR mutants. Ba/F3 cells transformed with the indicated EGFR mutants were treated with either cetuximab at the concentrations indicated and assayed for viability after 72 hours of drug treatment. The results are indicated as mean +/− SD of sextuplicate wells and are representative of three independent experiments. B, Cetuximab-induced receptor degradation and down-regulation of receptor phosphorylation occurred with similar kinetics in L858R, Ex19Del and Ex20Ins mutants. Cell lysates prepared from Ba/F3 cells expressing L858R or Ex19Del or Ex20Ins mutants following cetuximab treatment with concentrations indicated were blotted with phospho-tyrosine (4G10) or EGFR antibodies. C and D, While the growth of H3255 cells was partially or completely suppressed by cetuximab or gefitinib, respectively, these drugs had no effect on the growth of H3255GR cells. H3255 and H3255GR cells harboring L858R, or L858R/T790M mutations, respectively, were treated with gefitinib for 3 days (C) or cetuximab for 7 days (D) and cells were assayed for cell viability using MTS reagents. The results are indicated as mean +/- SD of sextuplicate wells and are representative of three independent experiments.
To determine whether similar patterns of cetuximab response are observed in lung cancer-derived cell lines, recognizing of course that these cell lines have complex and divergent genetic backgrounds, we tested the effects of cetuximab on NCI-H3255, NCI-H3255GR and PC9 cells, harboring L858R, LTM or Ex19Del EGFR mutations, respectively. Consistent with previous reports (41–43), the growth of NCI-H3255 and PC9 cells, but not NCI-H3255GR cells, was inhibited by gefitinib in a dose-dependent manner (Fig. 3C and Supplementary Fig. S8B). In contrast, cetuximab had different effects on the growth of these cell lines. The proliferation of NCI-H3255 cells bearing L858R mutant EGFR, but not of NCI-H3255GR nor of PC9 cells, was suppressed by cetuximab although the inhibition of NCI-H3255 cell proliferation is not as potent as for Ba/F3 cells transformed with L858R mutant EGFR (Fig. 3D and Supplementary Fig. S8C).
Dimerization-independent EGFR mutants promote lung tumorigenesis in vivo despite cetuximab treatment that blocks autophosphorylation
To further investigate the relationship between dimerization dependency and cetuximab response among oncogenic mutants of EGFR, we expanded our studies to include transgenic mouse models utilizing tetracycline-inducible expression of L858R, LTM, Ex19Del, DTM or Ex20Ins mutant EGFR, which develop poorly differentiated lung adenocarcinomas with bronchioloalveolar carcinoma (BAC) features after induction with doxycycline for 6–8 weeks as previously described (44)(Supplementary Fig. S9). Cetuximab was administered to these tumor-bearing mice by daily intraperitoneal (I.P.) injection. After 2 weeks, changes in tumor burden in the mice were documented with magnetic resonance imaging and compared to images taken prior to cetuximab treatment. The findings were consistent with previous experiments (44) showing that L858R driven lung cancers responded to cetuximab treatment, with approximately 15% of the tumor remaining after the two week treatment (Fig. 4A, L858R). A partial response to cetuximab was also observed in the transgenic mice expressing the Ex19Del mutant, which is somewhat different from what we observed in vitro with this mutant (Fig. 4A, Ex19Del). In contrast, the size of the overall tumors driven by either the LTM, the DTM, or the Ex20Ins mutations were only slightly decreased or even increased after cetuximab treatment, showing that cetuximab is no longer effective against these three mutants in vivo (Fig. 4A, LTM, DTM, and Ex20Ins). Taken together, these data demonstrate that cetuximab has differential efficacy against mutant EGFR in vivo as well in vitro.
Figure 4. Differential response to cetuximab of EGFR-driven tumors in transgenic mice.

A, Cetuximab is effective against EGFR L858R mutant-induced tumors but not many other EGFR mutant induced-tumors in mice. Bitransgenic mice expressing the indicated tetracycline-inducible EGFR mutants were treated with doxycycline from 4 weeks of age. Initial tumor burden was documented through MRI imaging following an observed panting (top panel of MRI images). Cetuximab was administered for 2 weeks followed by reimaging by MRI (bottom panel). The volume of tumor in each mouse was measured and the relative change upon cetuximab treatment is graphed (n=3, *P < 0.05, **P < 0.005, mean +SD). B, Cell lysates prepared from the tumor nodules of L858R, Ex19Del, L858R/T790M, Ex19Del/T790M and Ex20Ins transgenic mice before or after 2 weeks cetuximab treatment were subjected to immunoblotting with anti-phospho-Y1092, anti-phospho-Y1197 and anti-EGFR antibodies. Tubulin served as a loading control. Due to the lack of remaining tumor nodules in L858R mice after cetuximab treatment, we prepared the lysates using normal lung tissue in lane 2. C, Sections of lung tumors from cetuximab-untreated and cetuximab-treated mice were analyzed by immunohistochemistry using antibodies against total EGFR and phospho-Y1197 EGFR.
Given that several lung cancer-derived mutants of EGFR are resistant to cetuximab, we asked whether their cetuximab resistance is due to the inability of cetuximab to block receptor autophosphorylation. However, immunoblotting and immunohistochemistry demonstrate that cetuximab treatment of lung cancers driven by cetuximab-resistant Ex20Ins and T790M double mutants of EGFR leads to vastly reduced EGFR autophosphorylation (Figs. 4B, 4C and Supplementary Fig. S10). This reduced phosphorylation phenotype cannot be measured with the cetuximab-sensitive L858R mutant as tumors are completely ablated (Fig. 4B, lane 2). Taken together, these results suggest that cetuximab can block receptor autophosphorylation of dimerization-independent EGFR mutants, and that these mutants are cetuximab resistant because they are still active despite such blockade.
DISCUSSION
We have shown that wild-type EGFR and the L858R mutant require asymmetric dimerization for activation and oncogenic transformation, but the Ex19Del, Ex20Ins and T790M mutants are dimerization-independent, although we cannot formally exclude the formation of transient dimers that are not detected in steady-state cross-linking experiments.
Based on our findings, we propose a model of wild-type and mutant EGFR activation (Fig. 5). Wild-type EGFR is activated by ligand binding, which induces extracellular dimerization of the receptor, which in turn triggers “outside-in” asymmetric dimerization of the N- and C- lobes in the kinase domain, resulting in enzymatic activation of the receiver monomer and subsequent autophosphorylation on the C-terminal tail of the activator monomer (Fig. 5A). Thus the wild-type receptor requires ligand binding, dimerization, and autophosphorylation for its activity.
Figure 5. Proposed model of cellular transformation by wild-type EGFR and oncogenic mutants.
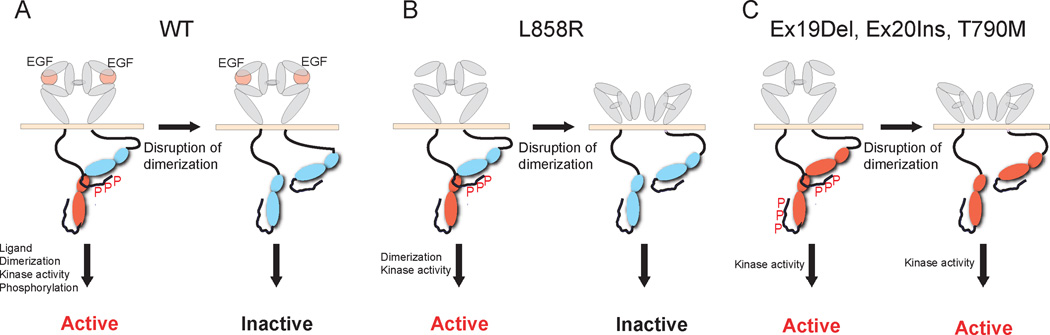
The L858R EGFR mutant is able to undergo constitutive asymmetric dimerization in the absence of ligand which in turn promotes “inside-out” extracellular dimerization (Fig. 5B); ligand increases dimerization but is not required. Prevention of dimerization impairs the oncogenic activity of the L858R mutant because the receiver monomer is no longer active. The Ex19Del and Ex20Ins mutants and the LTM double mutants similarly dimerize in the absence of ligand, but do not require dimerization for their activity (Fig. 5C). These mutant forms of EGFR are dimerization-independent.
Because several of the lung cancer-derived EGFR mutants do not require asymmetric dimerization for activation, disruption of dimerization is not effective in blocking their transforming activity. The therapeutic monoclonal antibody, cetuximab, potently inhibited the oncogenic activity of the dimerization-dependent L858R mutant, but had no effect on the dimerization-independent oncogenic activity of the Ex20Ins mutant or T790M double mutants, and a partial effect on the Ex19Del mutant. Thus, dependency of cellular transformation on receptor dimerization is correlated with cetuximab sensitivity to the mutant EGFR, consistent with a previously proposed model suggesting that inhibition of receptor dimerization is the primary mechanism for cetuximab activity (24). However, we were not able to demonstrate the disruption of EGFR dimerization by cetuximab directly, as direct binding of cetuximab to the EGFR renders cross-linking experiments uninformative. One alternative hypothesis to explain why dimerization-independent oncogenic mutants are cetuximab insensitive is that these EGFR mutants are not significantly expressed on the cell surface due to differential cellular localization and therefore inaccessible to cetuximab. However, several lines of evidence argue against the cell surface expression hypothesis, most notably the fact that autophosphorylation of the Ex20Ins mutant in mouse tumors is completely blocked by cetuximab treatment (Figure 4B and 4C) with no apparent physiological consequence given that autophosphorylation is not required for signaling, which is consistent with recent reports (45–47). In addition, the levels of total and phosphorylated mutant EGFR in Ba/F3 cells are downregulated by cetuximab in a dose-dependent manner irrespective of cetuximab-sensitivity, suggesting that mutant EGFR expressed on the cell surface are accessible to cetuximab (Figure 3B). Interestingly, the decreased levels of Ex19Del and Ex20Ins mutants induced by high dose of cetuximab treatment do not show any effect on the viability of these cell lines (Figure 3B). This finding may suggest that unlike dimerization-dependent L858R mutant, Ex19Del and Ex20Ins mutants are able to induce oncogenic activation even at low levels. Because some residual expression and phosphorylation of Ex19Del and Ex20Ins mutants at high concentrations of cetuximab was observed in Ba/F3 cells, we cannot exclude the possibility that diminished degradation effect of cetuximab on these mutant EGFR may be partly responsible for the cetuximab resistance. Another alternative hypothesis is that cetuximab insensitive oncogenic mutants act by heterodimerization, however, deletion experiments in mice provide evidence that the most plausible heterodimeric partner, ERBB3, is not required for EGFR-driven lung tumorigenesis (K. Politi, personal communication). Thus, we believe that dimerization independence remains the most parsimonious explanation for cetuximab insensitivity in the model systems studied here. Nevertheless, more studies are needed to be done on an epithelial background model system to investigate whether heterodimerization with any RTK proteins also contribute to the cetuximab insensitivity of dimerization-independent mutant EGFR.
While we observed a correlation of cetuximab sensitivity with the requirement of EGFR mutants for dimerization in cellular and animal model systems, the picture in lung cancer-derived cell lines is more complex. In many ways, this is reminiscent of gefitinib sensitivity, for which there are cells and tumors with gefitinib-sensitive EGFR mutations that are nevertheless resistant to the drug, due to additional genomic events such as the T790M mutation (10, 11), PTEN deletion (48) and MET amplification (49). In addition, the experimental conditions utilized for the various assays using NSCLC cell lines may likely account for the different results in regards to sensitivity to cetuximab (39, 42). Indeed, we observed a partial cell growth inhibition in this study (Fig. 3D) only when NCI-H3255 cells were treated with cetuximab for longer periods of time than those reported by Mukohara et al. Moreover, as previously proposed in mouse xenograft studies with NSCLC cell lines, receptor degradation induced by cetuximab may contribute to the antitumor effect of the drug against mutant EGFR (39, 40, 50).
Given that tumor models of the cetuximab-resistant, erlotinib-resistant, and dimerization-independent L858R/T790M compound EGFR mutant can be treated effectively with the simultaneous administration of cetuximab and an irreversible EGFR inhibitor, BIBW 2992 or afatinib (51, 52), a next step in research is to test the hypothesis that afatinib may render EGFR mutants dimerization-dependent.
Supplementary Material
Acknowledgements
We thank Hideo Watanabe, Amit Dutt, Derek Chiang, Barbara Weir and Zhang Li for helpful discussion. We also thank Dyane Bailey, Chris Fiore and Massimo Loda of The Center for Molecular Oncologic Pathology, DFCI for their technical assistance and professional advice, respectively.
Grant Support
The research was supported in part by NCI R01 CA116020 (MM and MJE).
Footnotes
Disclosure of Potential Conflicts of Interest
No conflicts of interest were disclosed.
REFERENCES
- 1.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 2.Ritter CA, Arteaga CL. The epidermal growth factor receptor-tyrosine kinase: a promising therapeutic target in solid tumors. Semin Oncol. 2003;30:3–11. doi: 10.1053/sonc.2003.50027. [DOI] [PubMed] [Google Scholar]
- 3.Chan SK, Gullick WJ, Hill ME. Mutations of the epidermal growth factor receptor in non-small cell lung cancer -- search and destroy. Eur J Cancer. 2006;42:17–23. doi: 10.1016/j.ejca.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England journal of medicine. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 5.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 7.Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Janne PA, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26:2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 8.Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, et al. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:839–844. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida K, Yatabe Y, Park JY, Shimizu J, Horio Y, Matsuo K, et al. Prospective validation for prediction of gefitinib sensitivity by epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer. J Thorac Oncol. 2007;2:22–28. [PubMed] [Google Scholar]
- 10.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 11.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasaki H, Endo K, Takada M, Kawahara M, Kitahara N, Tanaka H, et al. EGFR exon 20 insertion mutation in Japanese lung cancer. Lung Cancer. 2007;58:324–328. doi: 10.1016/j.lungcan.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 14.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 15.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 16.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 17.Mendelsohn J. Antibody-mediated EGF receptor blockade as an anticancer therapy: from the laboratory to the clinic. Cancer Immunol Immunother. 2003;52:342–346. doi: 10.1007/s00262-002-0354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barber TD, Vogelstein B, Kinzler KW, Velculescu VE. Somatic mutations of EGFR in colorectal cancers and glioblastomas. N Engl J Med. 2004;351:2883. doi: 10.1056/NEJM200412303512724. [DOI] [PubMed] [Google Scholar]
- 19.Tsuchihashi Z, Khambata-Ford S, Hanna N, Janne PA. Responsiveness to cetuximab without mutations in EGFR. The New England journal of medicine. 2005;353:208–209. doi: 10.1056/NEJM200507143530218. [DOI] [PubMed] [Google Scholar]
- 20.Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 21.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 22.Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009;373:1525–1531. doi: 10.1016/S0140-6736(09)60569-9. [DOI] [PubMed] [Google Scholar]
- 23.Harding J, Burtness B. Cetuximab: an epidermal growth factor receptor chemeric human-murine monoclonal antibody. Drugs Today (Barc) 2005;41:107–127. doi: 10.1358/dot.2005.41.2.882662. [DOI] [PubMed] [Google Scholar]
- 24.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJW, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 25.Ferguson KM. Structure-based view of epidermal growth factor receptor regulation. Annual review of biophysics. 2008;37:353–373. doi: 10.1146/annurev.biophys.37.032807.125829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaborit N, Larbouret C, Vallaghe J, Peyrusson F, Bascoul-Mollevi C, Crapez E, et al. Time-resolved fluorescence resonance energy transfer (TR-FRET) to analyze the disruption of EGFR/HER2 dimers: a new method to evaluate the efficiency of targeted therapy using monoclonal antibodies. J Biol Chem. 2011;286:11337–11345. doi: 10.1074/jbc.M111.223503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol Cell Biol. 2005;25:7734–7742. doi: 10.1128/MCB.25.17.7734-7742.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arkhipov A, Shan Y, Das R, Endres NF, Eastwood MP, Wemmer DE, et al. Architecture and membrane interactions of the EGF receptor. Cell. 2013;152:557–569. doi: 10.1016/j.cell.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, Huang Y, et al. Conformational Coupling across the Plasma Membrane in Activation of the EGF Receptor. Cell. 2013;152:543–556. doi: 10.1016/j.cell.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 31.Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papakyriakou A, Vourloumis D, Tzortzatou-Stathopoulou F, Karpusas M. Conformational dynamics of the EGFR kinase domain reveals structural features involved in activation. Proteins. 2009;76:375–386. doi: 10.1002/prot.22353. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007;450:741–744. doi: 10.1038/nature05998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bishayee A, Beguinot L, Bishayee S. Phosphorylation of tyrosine 992, 1068, and 1086 is required for conformational change of the human epidermal growth factor receptor c-terminal tail. Mol Biol Cell. 1999;10:525–536. doi: 10.1091/mbc.10.3.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shan Y, Eastwood MP, Zhang X, Kim ET, Arkhipov A, Dror RO, et al. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell. 2012;149:860–870. doi: 10.1016/j.cell.2012.02.063. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Longo PA, Tarrant MK, Kim K, Head S, Leahy DJ, et al. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nature structural & molecular biology. 2011;18:1388–1393. doi: 10.1038/nsmb.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuza Y, Glatt KA, Jiang J, Greulich H, Minami Y, Woo MS, et al. Allele-dependent variation in the relative cellular potency of distinct EGFR inhibitors. Cancer Biol Ther. 2007;6:661–667. doi: 10.4161/cbt.6.5.4003. [DOI] [PubMed] [Google Scholar]
- 38.Jiang J, Greulich H, Janne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005;65:8968–8974. doi: 10.1158/0008-5472.CAN-05-1829. [DOI] [PubMed] [Google Scholar]
- 39.Perez-Torres M, Guix M, Gonzalez A, Arteaga CL. Epidermal growth factor receptor (EGFR) antibody down-regulates mutant receptors and inhibits tumors expressing EGFR mutations. J Biol Chem. 2006;281:40183–40192. doi: 10.1074/jbc.M607958200. [DOI] [PubMed] [Google Scholar]
- 40.Doody JF, Wang Y, Patel SN, Joynes C, Lee SP, Gerlak J, et al. Inhibitory activity of cetuximab on epidermal growth factor receptor mutations in non small cell lung cancers. Mol Cancer Ther. 2007;6:2642–2651. doi: 10.1158/1535-7163.MCT-06-0506. [DOI] [PubMed] [Google Scholar]
- 41.Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, et al. Differential Effects of Gefitinib and Cetuximab on Non-small-cell Lung Cancers Bearing Epidermal Growth Factor Receptor Mutations. J Natl Cancer Inst. 2005;97:1185–1194. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- 43.Ono M, Hirata A, Kometani T, Miyagawa M, Ueda S, Kinoshita H, et al. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther. 2004;3:465–472. [PubMed] [Google Scholar]
- 44.Ji H, Li D, Chen L, Shimamura T, Kobayashi S, McNamara K, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–495. doi: 10.1016/j.ccr.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 45.Rothenberg SM, Engelman JA, Le S, Riese DJ, 2nd, Haber DA, Settleman J. Modeling oncogene addiction using RNA interference. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12480–12484. doi: 10.1073/pnas.0803217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maegawa M, Arao T, Yokote H, Matsumoto K, Kudo K, Tanaka K, et al. Epidermal growth factor receptor lacking C-terminal autophosphorylation sites retains signal transduction and high sensitivity to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer science. 2009;100:552–557. doi: 10.1111/j.1349-7006.2008.01071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cho J, Pastorino S, Zeng Q, Xu X, Johnson W, Vandenberg S, et al. Glioblastoma-derived epidermal growth factor receptor carboxyl-terminal deletion mutants are transforming and are sensitive to EGFR-directed therapies. Cancer research. 2011;71:7587–7596. doi: 10.1158/0008-5472.CAN-11-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69:3256–3261. doi: 10.1158/0008-5472.CAN-08-4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 50.Steiner P, Joynes C, Bassi R, Wang S, Tonra JR, Hadari YR, et al. Tumor growth inhibition with cetuximab and chemotherapy in non-small cell lung cancer xenografts expressing wild-type and mutated epidermal growth factor receptor. Clin Cancer Res. 2007;13:1540–1551. doi: 10.1158/1078-0432.CCR-06-1887. [DOI] [PubMed] [Google Scholar]
- 51.Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. The Journal of clinical investigation. 2009;119:3000–3010. doi: 10.1172/JCI38746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Janjigian YY, Groen HJ, Horn L, Smit EF, Fu Y, Wang F, et al. Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J Clin Oncol. 2011;29(Suppl) abstr 7525. [Google Scholar]
- 53.Meyerhardt JA, Clark JW, Supko JG, Eder JP, Ogino S, Stewart CF, et al. Phase I study of gefitinib, irinotecan, 5-fluorouracil and leucovorin in patients with metastatic colorectal cancer. Cancer chemotherapy and pharmacology. 2007;60:661–670. doi: 10.1007/s00280-006-0411-6. [DOI] [PubMed] [Google Scholar]
- 54.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.