

Abstract
During bone resorption, abundant factors previously buried in the bone matrix are released into the bone marrow microenvironment, which results in recruitment and differentiation of bone marrow mesenchymal stem cells (MSCs) for subsequent bone formation, temporally and spatially coupling bone remodeling. Parathyroid hormone (PTH) orchestrates the signaling of many pathways that direct MSC fate. The spatiotemporal release and activation of matrix TGF-β during osteoclast bone resorption recruits MSCs to bone-resorptive sites. Dysregulation of TGF-β alters MSC fate, uncoupling bone remodeling and causing skeletal disorders. Modulation of TGF-β or PTH signaling may reestablish coupled bone remodeling and be a potential therapy.
Introduction
In humans, the skeleton undergoes continuous remodeling throughout adulthood. To maintain skeletal integrity, the activity of two cell types must be precisely coordinated. Osteoclasts, which resorb bone, are multinucleated cells derived from macrophages/monocytes in the HSC lineage. Osteoblasts, which deposit calcified bone matrix, are derived from bone marrow mesenchymal stem cells (MSCs; also referred to as bone marrow stromal cells or skeletal stem cells; ref. 1). The bone remodeling cycle is characterized by three distinct phases: (a) initiation, during which osteoclasts are formed and resorb damaged bone; (b) reversal, the transition of osteoclast to osteoblast activity; and (c) formation, when osteoblasts replace the portion of bone that was resorbed (2). Key steps in the reversal period include termination of osteoclast bone resorption and recruitment/differentiation of MSCs (2). Skeletal homeostasis is maintained by precise regulatory mechanisms during the reversal phase.
The bone marrow microenvironment that is created during osteoclast bone resorption directs MSC fate. During osteoclast-mediated bone resorption, multiple factors are released from the bone matrix and/or secreted locally, creating an osteogenic microenvironment that promotes MSC recruitment and osteoblast differentiation for new bone formation (3–6). Parathyroid hormone (PTH), the systemic hormone that regulates calcium homeostasis, plays a major role in orchestrating bone remodeling by modulating the bone marrow microenvironment and regulating osteogenic signaling pathways (7–16). Latent TGF-β1, which is abundant in the bone matrix, is released and activated by osteoclasts to specifically induce migration of MSCs to bone-resorptive sites (17–19). Because these two pathways have specific effects on MSC fate, their coordinated regulation is critical to bone remodeling.
Here, we discuss the current understanding of how the bone marrow microenvironment at resorption sites affects MSCs and contributes to coupled bone remodeling. We specifically focus on how PTH regulates the osteogenic bone marrow microenvironment and how TGF-β promotes MSC recruitment. We also describe specific skeletal diseases that result from disruption of coupled bone remodeling. Finally, we address how modulation of TGF-β or PTH signaling may offer potential therapeutic approaches for bone remodeling disorders.
Bone remodeling dynamically changes the bone marrow microenvironment
The fate of MSCs, including self-renewal, transient amplification, or differentiation, is regulated by the bone marrow microenvironment and systemic factors (20–22). In the reversal phase of bone remodeling, the bone-resorptive microenvironment provides signals that aid in the cessation of bone resorption and the promotion of bone formation by recruitment and differentiation of MSCs. Multiple cytokines, growth factors, and minerals are released from the bone matrix or secreted by local cells. For example, IGF-1 released from the bone matrix or secreted locally during bone remodeling stimulates osteoblast differentiation of MSCs by activation of mammalian target of rapamycin (mTOR) through the PI3K-Akt pathway (3). Semaphorin 4D (SEMA4D), expressed on the cell surface of osteoclasts, binds to its receptor, plexin B1, on osteoblasts to inhibit the RhoA/Rho-associated protein kinase (ROCK) pathway (23). The ROCK pathway normally phosphorylates IRS-1, a key factor in the PI3K-Akt-mTOR pathway (23); therefore, osteoclast expression of SEMA4D can inhibit MSC differentiation by cell-to-cell contact, creating a boundary between bone resorption and formation. Thus, the dynamic changes in the bone marrow microenvironment result in the coordination of the reversal phase during coupled bone remodeling.
Additional properties of the microenvironment also play a role in MSC fate. For example, at fresh resorptive sites, the bone mineral matrix is exposed and lacks a covering of lining cells, providing a stiff elastic microenvironment. The stiff matrix at remodeling sites directly promotes differentiation of MSCs into osteoblasts (24), which may promote the formation of lamellar bone rather than woven bone.
Blood vessels are in close proximity to bone remodeling, and a complex relationship exists between angiogenesis and osteogenesis (25). Factors from the vasculature appear to influence the bone marrow microenvironment. FGF2 is a potent mitogenic factor for several different cell types, including endothelial cells, and can induce angiogenesis by stimulating endothelial cell proliferation, migration, and expression of proteases, growth factors, and integrins involved in angiogenesis (26–29). TGF-β has been shown to enhance production of FGF2 in osteoblasts and positively modulate osteoblast differentiation and bone formation (30–32). In aortic endothelial cells, TGF-β stimulation of a complex containing activin-like kinase receptor 5 (ALK5), TGF-β receptor II (TβRII), and SMAD2 enhances VEGF expression (33), but it is unclear whether there is similar activity at sites of bone remodeling. Hematopoietic stem and progenitor cells (HSPCs) are present in the same niche as MSCs (34). While MSCs and osteoblast precursors can influence the fate of HSPCs (35–37), it is unclear whether HSPCs reciprocally influence MSCs.
PTH modulates the bone marrow microenvironment
The parathyroid gland, the main production site of the calcium homeostasis regulator PTH, evolved in amphibians (38) and represents the transition of aquatic to terrestrial life. Permanent detection of osteoclasts and bone resorption also emerged as vertebrates transitioned to land (39–42), promoting survival by development of lighter cylindrical bones to aid in mobilization and release of calcium from the skeletal matrix. During PTH-mediated osteoclastic bone resorption, growth factors and cytokines are also released from the bone matrix. To protect the integrity of the skeleton in adapting to terrestrial life, PTH regulates bone remodeling by orchestrating signaling of local factors, including TGF-β, Wnts, bone morphogenetic protein (BMP), and IGF-1. Thus, the fate of MSCs and other cells in the microenvironment are indirectly regulated by PTH to integrate systemic control of bone remodeling (Figure 1 and refs. 7–15).
Figure 1. Modulation of the bone marrow microenvironment by PTH-stimulated bone remodeling.

PTH enhances osteoclast bone resorption through direct activation of cells in the osteoblastic cell lineage. During bone remodeling, active TGF-β, IGF-1, and many other bone matrix factors are released to the marrow. PTH orchestrates signaling of local factors, including (but not limited to) TGF-β, Wnts, and BMP. Thus, PTH regulates cellular activities — including those of MSCs, T cells, and other PTH-responsive cells — in the bone marrow to integrate systemic control of bone remodeling. PTH-stimulated bone remodeling expands nestin+ MSC populations, spatially relocates blood vessels closer to sites of new bone formation, and orchestrates the osteogenic bone marrow microenvironment.
PTH modifies many cells that are important in the bone marrow microenvironment. In a lineage tracing study, intermittent PTH treatment increased osteoblast number by converting lining cells to active osteoblasts (43). Lining cells are primarily defined by their location on the bone surface. Further characterization of the nature of lining cells will help to elucidate the relevant cellular mechanisms. PTH stimulates bone marrow CD8+ T cells to produce large amounts of Wnt10b, which activates Wnt signaling in MSCs and osteoblast precursors, thereby increasing osteoblast proliferation and differentiation (44). PTH has also been shown to alter the bone marrow microenvironment by spatially relocating small blood vessels closer to sites of new bone formation, likely secondary to PTH-mediated upregulation of VEGFA and neuropilin 1 and 2 (45). The closer proximity of blood vessels allows more efficient delivery of nutrients to support new bone formation.
PTH-mediated enhancement of MSC transient amplification, differentiation, and function is a part of the integration of the signaling networks of local factors for the spatiotemporal regulation of bone remodeling. PTH can expand nestin+ MSC populations (35, 36, 46), although the precise mechanism of action remains an active area of investigation. PTH stimulates the commitment of MSCs to the osteoblast lineage by enhancing BMP and Wnt signaling. LDL-related protein 6 (LRP6) serves as a coreceptor in the canonical Wnt pathway (47) and interacts with BMP signaling (48). The formation of a ternary complex containing PTH, PTH type 1 receptor (PTH1R), and LRP6 promotes rapid phosphorylation of LRP6, which recruits axin to LRP6 and stabilizes β-catenin (13). Endocytosis of growth factors and GPCRs is known to integrate different signaling pathways (49). PTH binding to PTH1R can induce endocytosis of a PTH1R-LRP6 complex, resulting in enhancement of downstream BMP–phospho-SMAD1 signaling, which ultimately enhances the number of MSCs differentiating into the osteoblast lineage (15). Specifically deleting Lrp6 in mature murine osteoblasts results in a loss of anabolic response to PTH (50). PTH also induces the recruitment of TβRII, a membrane-bound serine/threonine protein kinase receptor, as an endocytic activator. TβRII directly phosphorylates the cytoplasmic domain of PTH1R and facilitates PTH-induced endocytosis of the PTH1R-TβRII complex, which results in downregulation of TGF-β signaling (12).
TGF-β recruits MSCs for bone homeostasis during remodeling
There are more than 40 members in the TGF superfamily, divided into four major subfamilies (51). The TGF-β subfamily, present only in mammals, contains three closely related isoforms, TGF-β1–TGF-β3. TGF-βs are synthesized as large precursor molecules, composed of mature TGF-β and latency-associated protein (LAP). LAP remains noncovalently bound to mature TGF-β as it is secreted, rendering it inactive by masking the ECM of many different tissues, if not all (52–54). TGF-β1 is one of the most abundant cytokines in the bone matrix (200 μg/kg; refs. 55, 56). During tissue injury or remodeling, TGF-β1 in the bone matrix can be activated by cleavage of LAP by osteoclasts (17, 19). A gradient of active TGF-β then signals to transiently recruit perivascular MSCs to the recently resorbed bone surface for osteoblast differentiation and new bone formation (Figure 2 and refs. 18, 57, 58).
Figure 2. Activation of TGF-β recruits MSCs during bone remodeling.
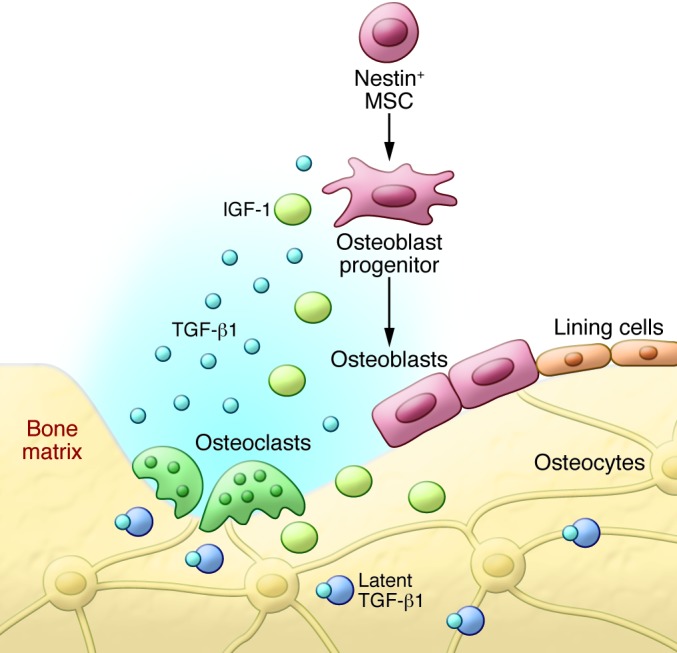
TGF-β1 is released from the bone matrix and activated during osteoclast-mediated bone resorption, creating a gradient. TGF-β1 induces migration of MSCs to the bone remodeling sites to couple bone resorption and formation. The bone-resorptive microenvironment also provides signals that direct the cell lineage–specific differentiation of MSCs.
Signaling by TGF-β in MSCs occurs through the SMAD family of signal transduction proteins. TGF-β binds to two major types of membrane-bound serine/threonine kinase receptors, TβRI and TβRII. TβRII transphosphorylates TβRI (59, 60), and phosphorylated TβRI, in association with either ALK1 or ALK5, phosphorylates receptor-regulated SMADs (R-SMADs; including SMAD1–SMAD3, SMAD5, and SMAD8; refs. 61–64). R-SMADs then rapidly dissociate from the receptor to form complexes with SMAD4 and migrate into the nucleus to regulate transcription of target genes (61, 63–66). Using a series of animal models, inhibitors, siRNAs, and retrovirus-mediated expression, we have found that TGF-β1 MSC migration occurs through the ALK5-SMAD2/3-SMAD4 pathway, with SMAD3 playing a more prominent role and SMAD2 playing a compensatory role (18). Cell type–specific effects of TGF-β signaling are largely determined by the interaction of SMAD2/3 proteins with cell type–specific master transcription factors that specify and maintain specific effects (67). Whereas the master transcription factors in embryonic stem cells, myotubes, and pro-B cells have been determined (Oct4, MyoD1, and PU.1, respectively; ref. 67), MSC master transcription factors have not yet been identified.
Active TGF-β released into the microenvironment can exert specific effects, including proliferation, differentiation, migration, and apoptosis, depending on the cell type and duration of action (51). For example, although TGF-β signaling induces MSC migration, it does not induce osteoblast differentiation (68). The gradient of TGF-β created during osteoclast bone resorption can limit further osteoclast activity. In the short term, low concentrations of active TGF-β can induce macrophage migration via activation of RhoA; however, high concentrations or prolonged exposure of macrophages/monocytes to active TGF-β have been shown to inhibit migration of osteoclast precursors (69). Both high concentrations of and prolonged exposure to TGF-β activate SMAD3-SMAD4 complexes, which in turn activate PKA, resulting in phosphorylation and inactivation of RhoA (69, 70). Thus, the gradient of TGF-β1 generated at the resorption sites likely prohibits further recruitment of osteoclast precursors, protecting it from further resorption during the reversal phase of bone remodeling.
Disorders associated with abnormalities in bone remodeling
High levels of active TGF-β in the bone marrow and abnormalities in bone remodeling are associated with multiple skeletal disorders. Genetic mutations in the TGF-β signaling pathway cause premature activation of matrix latent TGF-β and may manifest with various skeletal defects. There are additional diseases that result in high levels of active TGF-β, which may contribute to the pathology. Here, we discuss how abnormal TGF-β signaling results in uncoupled bone remodeling, mainly by loss of site-directed recruitment of MSCs that causes aberrant bone formation. Direct or indirect inhibition of TGF-β signaling may provide potential therapeutic options for these disorders.
Genetic disorders.
The critical role of TGF-β1 in the reversal phase of bone remodeling is demonstrated by the range of skeletal disorders resulting from mutations in genes involved in TGF-β1 signaling. Camurati-Engelmann disease (CED), characterized by a fusiform thickening of the diaphysis of the long bones and skull, is caused by mutations in TGFB1 that result in premature activation of TGF-β1 (71–74). Approximately 11 different TGFB1 mutations have been identified from families affected by CED (75, 76). All of the mutations are located in the region encoding LAP, either destabilizing LAP disulfide bridging or affecting secretion of the protein, both of which increase TGF-β1 signaling, as confirmed by in vitro cell cultures and mouse models. Bone histology sections from patients with CED show decreased trabecular connectivity despite normal bone histomorphometric parameters with respect to osteoblast and osteoclast numbers (76, 77), suggestive of uncoupled bone remodeling. In vitro, the ratio of active to total TGF-β1 in conditioned medium from cells expressing the CED mutant TGF-β1 is significantly higher and enhances MSC migration (18). Targeted recruitment of MSCs to the bone-remodeling site is likely disrupted, secondary to loss of a TGF-β gradient.
Elevations in TGF-β signaling have also been observed in many genetic connective tissue disorders with craniofacial, skeletal, skin, and cardiovascular manifestations, including Marfan syndrome (MFS), Loeys-Dietz syndrome (LDS), and Shprintzen-Goldberg syndrome (SGS). MFS is caused by mutations in fibrillin and often results in aortic dilation, myopia, bone overgrowth, and joint laxity. Fibrillin is deposited in the ECM and normally binds TGF-β, rendering it inactive. In MFS, the decreased level of fibrillin enhances TGF-β activity (78). LDS is caused by inactivating mutations in genes encoding TβRI and TβRII (79). Physical manifestations include arterial aneurysms, hypertelorism, bifid uvula/cleft palate, and bone overgrowth resulting in arachnodactyly, joint laxity, and scoliosis. Pathologic analyses of affected tissue suggest chronically elevated TGF-β signaling, despite the inactivating mutation (79). The mechanism of enhanced TGF-β signaling remains under investigation. SGS is caused by mutations in the v-ski avian sarcoma viral oncogene homolog (SKI; refs. 80, 81) and causes physical features similar to those of MFS plus craniosynostosis. SKI negatively regulates SMAD-dependent TGF-β signaling by impeding SMAD2 and SMAD3 activation, preventing nuclear translocation of the SMAD4 complex, and inhibiting TGF-β target gene output by competing with p300/CBP for SMAD binding and recruiting transcriptional repressor proteins, such as mSin3A and HDACs (82–84).
The neurocutaneous syndrome neurofibromatosis type 1 (NF1) has been noted to have skeletal features similar to those of CED, MFS, and LDS, including kyphoscoliosis, osteoporosis, and tibial pseudoarthrosis. Hyperactive TGF-β1 signaling has been implicated as the primary factor underlying the pathophysiology of the osseous defects in Nf1fl/–Col2.3Cre mice, a model of NF1 that closely recapitulates the skeletal abnormalities found in human disease (85). The exact mechanisms mediating mutant neurofibromin–associated enhancement of TGF-β production and signaling remain unknown.
Osteoarthritis.
While genetic disorders are rare, they have provided critical insight into the pathophysiology of more common disorders. Uncoupled bone remodeling accompanies the onset of osteoarthritis. TGF-β1 is activated in subchondral bone in response to altered mechanical loading in an anterior cruciate ligament transection (ACLT) mouse model of osteoarthritis (86). High levels of active TGF-β1 induced formation of nestin+ MSC clusters via activation of ALK5-SMAD2/3. MSCs underwent osteoblast differentiation in these clusters, leading to formation of marrow osteoid islets. Transgenic expression of active TGF-β1 in osteoblastic cells alone was sufficient to induce osteoarthritis, whereas direct inhibition of TGF-β activity in subchondral bone attenuated the degeneration of articular cartilage. Knockout of Tgfbr2 in nestin+ MSCs reduced osteoarthritis development after ACLT compared with wild-type mice, which confirmed that MSCs are the target cell population of TGF-β signaling. High levels of active TGF-β1 in subchondral bone likely disrupt the TGF-β gradient and interfere with targeted migration of MSCs. Furthermore, mutations of ECM proteins that bind to latent TGF-βs, such as small leucine-rich proteoglycans (87) and fibrillin (88), or mutations in genes involved in activation of TGF-β, such as in CED (76) and LDS (89), are associated with high osteoarthritis incidence. Osteoblast differentiation of MSCs in aberrant locations appears histologically as subchondral bone osteoid islets and alters the thickness of the subchondral plate and calcified cartilage zone, changes known to be associated with osteoarthritis (90, 91). A computer-simulated model found that a minor increase in the size of the subchondral bone (1%–2%) causes significant changes in the mechanical load properties on articular cartilage, which likely leads to degeneration (86). Importantly, inhibition of the TGF-β signaling pathway delayed the development of osteoarthritis in both mouse and rat models (86).
MSCs in bone loss.
Aging leads to deterioration of tissue and organ function. Skeletal aging is especially dramatic: bone loss in both women and men begins as early as the third decade, immediately after peak bone mass. Aging bone loss occurs when bone formation does not adequately compensate for osteoclast bone resorption during remodeling. Age-associated osteoporosis was previously believed to be due to a decline in survival and function of osteoblasts and osteoprogenitors; however, recent work by Park and colleagues found that mature osteoblasts and osteoprogenitors are actually nonreplicative cells and require constant replenishment from bone marrow MSCs (92). When MSCs fail to migrate to bone-resorptive sites or are unable to commit and differentiate into osteoblasts, new bone formation is impaired. Therefore, insufficient recruitment of MSCs, or their differentiation to osteoblasts, at the bone remodeling surface may contribute to the decline in bone formation in the elderly.
There are multiple hypotheses regarding the decreased osteogenic potential of MSCs during aging. For example, during aging, the bone marrow environment has an increased concentration of ROS and lipid oxidation that may decrease osteoblast differentiation, yet increase osteoclast activity (93, 94). MSCs also undergo senescence, which decreases proliferative capacity and contributes to decreased bone formation (95, 96). Cellular senescence involves the secretion of a plethora of factors, including TGF-β, which induces expression of cyclin-dependent kinase inhibitors 2A and 2B (p16INK4A and p15INK4B, respectively; refs. 97).
Microgravity experienced by astronauts during spaceflight causes severe physiological alterations in the human body, including a 1%–2% loss of bone mass every month during spaceflight (98). Several studies have shown decreases in osteoblastic markers of bone formation and increases in bone resorption (99–101). The underlying molecular mechanisms responsible for the apparent concurrent decrease in bone formation and increase in bone resorption remain under investigation. Work by the McDonald group suggests that bone remodeling may become uncoupled under zero-gravity conditions secondary to decreased RhoA activity and resultant changes in actin stress fiber formation (102). In modeled microgravity, cultured human MSCs exhibit disruption of F-actin stress fibers within three hours of initiation of microgravity; the fibers are completely absent after seven days. RhoA activity is significantly reduced, and introduction of an adenoviral construct expressing constitutively active RhoA can reverse the elimination of stress fibers, significantly increasing markers of osteoblast differentiation (102). Under zero-gravity conditions, RhoA is unable to bind to its receptor, and a sufficient number of MSCs may not be able to migrate correctly to the bone-resorptive site for osteoblast differentiation, ultimately leading to bone loss with every cycle of remodeling.
Bone metastases are a frequent complication of cancer and often have both osteolytic and osteoblastic features, indicative of dysregulated bone remodeling. The importance of the bone marrow microenvironment contributing to the spread of cancer was first described in 1889 (103), postulating that tumor cells can grow only if they are in a conducive environment. Activation of matrix TGF-β during bone remodeling plays a central role in the initiation of bone metastases and tumor expansion by regulating osteolytic and prometastatic factors (reviewed in refs. 104–110). For example, TGF-β can induce osteoclastic bone destruction by upregulating tumor cell expression of PTHrP and IL-11. Additionally, upregulation of CXCR4 by TGF-β may home cancer cells to bones.
Potential treatment of uncoupled bone remodeling disorders
Genetically modified mouse models have been used to demonstrate that increased TGF-β signaling leads to diminished mineral concentration and inferior mechanical properties, whereas decreased TGF-β signaling enhances these properties (111). Wild-type mice injected with a relatively low concentration of an antibody against TGF-β show increased bone mineral density, trabecular thickness, and bone volume as a result of elevated osteoblast numbers and decreased osteoclasts (112). In the ACLT rodent model of osteoarthritis, inhibition of TGF-β signaling attenuated articular cartilage damage, delaying osteoarthritis onset (86). Active TGF-β1 concentrations are also high in the subchondral bone in humans with osteoarthritis, suggestive of a similar pathogenesis and a potential role for TGF-β signaling inhibition as a disease-modifying therapy. Several clinical trials have or are being conducted to evaluate the efficacy of inhibiting TGF-β signaling in metastatic cancers, focal segmental glomerulosclerosis, and idiopathic pulmonary fibrosis (112–115). Because abnormal bone remodeling also generates high levels of active TGF-β and leads to skeletal disorders, inhibition of TGF-β signaling could be a potential treatment.
The bone marrow microenvironment may change in disease states and affect TGF-β1–mediated coupled bone remodeling. Improving the osteogenic microenvironment may help restore coupling by enhancing the osteogenic potential of MSCs during the reversal phase of bone remodeling, and therefore may be another potential therapeutic target. PTH modifies the bone marrow microenvironment by orchestrating the signaling of local factors for bone remodeling, reducing ROS, and stimulating Wnt signaling in the bones of old mice (7–16). PTH is an FDA-approved anabolic therapy for osteoporosis, and daily injection of PTH increases bone formation with normal microarchitecture by coupling bone remodeling (7–15). PTH treatment has been shown to attenuate osteoarthritis progression in animal models (116, 117). Translational studies expanding the use of PTH for the treatment of skeletal remodeling disorders are in the beginning phases.
Conclusion
In order for bone homeostasis to be maintained through adulthood, bone remodeling must be spatially and temporally regulated. TGF-β is one of the key cytokines responsible for coupling bone resorption with formation, largely by recruitment of MSCs to bone-resorptive sites. Aberrant TGF-β signaling or increased TGF-β activation can result in uncoupled remodeling and cause skeletal diseases/disorders. Therapies that can attenuate TGF-β signaling, either directly by neutralizing TGF-β activity or indirectly by PTH-mediated modulation of the bone marrow microenvironment, may serve as potential therapies for these bone and joint disorders.
Acknowledgments
This work was supported in part by the grants from the NIH, including T32DK007751 (to J.L. Crane) and AR06394 and DK 057501 (to X. Cao). We thank Qin Bian for her help with figure design.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2014;124(2):466–472. doi:10.1172/JCI70050.
References
- 1.Bianco P, et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19(1):35–42. doi: 10.1038/nm.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kular J, Tickner J, Chim SM, Xu J. An overview of the regulation of bone remodelling at the cellular level. Clin Biochem. 2012;45(12):863–873. doi: 10.1016/j.clinbiochem.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 3.Xian L, et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med. 2012;18(7):1095–1101. doi: 10.1038/nm.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crane JL, Cao X. J Mol Med (Berl). Function of matrix IGF-1 in coupling bone resorption and formation. [published online ahead of print September 26, 2013]. doi: 10.1007/s00109-013-1084-3 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pacifici R. Role of T cells in the modulation of PTH action: physiological and clinical significance. Endocrine. 2013;44(3):576–582. doi: 10.1007/s12020-013-9960-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuo C, et al. Osteoblastogenesis regulation signals in bone remodeling. Osteoporos Int. 2012;23(6):1653–1663. doi: 10.1007/s00198-012-1909-x. [DOI] [PubMed] [Google Scholar]
- 7.Bikle DD, et al. Insulin-like growth factor I is required for the anabolic actions of parathyroid hormone on mouse bone. J Bone Miner Res. 2002;17(9):1570–1578. doi: 10.1359/jbmr.2002.17.9.1570. [DOI] [PubMed] [Google Scholar]
- 8.Canalis E, Centrella M, Burch W, McCarthy TL. Insulin-like growth factor I mediates selective anabolic effects of parathyroid hormone in bone cultures. J Clin Invest. 1989;83(1):60–65. doi: 10.1172/JCI113885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lombardi G, Di SC, Vuolo L, Guerra E, Scarano E, Colao A. Role of IGF-I on PTH effects on bone. J Endocrinol Invest. 2010;33(7 suppl):22–26. [PubMed] [Google Scholar]
- 10.Miyakoshi N, Kasukawa Y, Linkhart TA, Baylink DJ, Mohan S. Evidence that anabolic effects of PTH on bone require IGF-I in growing mice. Endocrinology. 2001;142(10):4349–4356. doi: 10.1210/endo.142.10.8436. [DOI] [PubMed] [Google Scholar]
- 11.Pfeilschifter J, Laukhuf F, Muller-Beckmann B, Blum WF, Pfister T, Ziegler R. Parathyroid hormone increases the concentration of insulin-like growth factor-I and transforming growth factor beta 1 in rat bone. J Clin Invest. 1995;96(2):767–774. doi: 10.1172/JCI118121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiu T, Wu X, Zhang F, Clemens TL, Wan M, Cao X. TGF-β type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat Cell Biol. 2010;12(3):224–234. doi: 10.1038/ncb2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wan M, et al. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 2008;22(21):2968–2979. doi: 10.1101/gad.1702708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watson P, Lazowski D, Han V, Fraher L, Steer B, Hodsman A. Parathyroid hormone restores bone mass and enhances osteoblast insulin-like growth factor I gene expression in ovariectomized rats. Bone. 1995;16(3):357–365. doi: 10.1016/8756-3282(94)00051-4. [DOI] [PubMed] [Google Scholar]
- 15.Yu B, et al. Parathyroid hormone induces differentiation of mesenchymal stromal/stem cells by enhancing bone morphogenetic protein signaling. J Bone Miner Res. 2012;27(9):2001–2014. doi: 10.1002/jbmr.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jilka RL, et al. Decreased oxidative stress and greater bone anabolism in the aged, when compared to the young, murine skeleton with parathyroid hormone administration. Aging Cell. 2010;9(5):851–867. doi: 10.1111/j.1474-9726.2010.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dallas SL, Rosser JL, Mundy GR, Bonewald LF. Proteolysis of latent transforming growth factor-beta (TGF-β)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-β from bone matrix. J Biol Chem. 2002;277(24):21352–21360. doi: 10.1074/jbc.M111663200. [DOI] [PubMed] [Google Scholar]
- 18.Tang Y, et al. TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009;15(7):757–765. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oreffo RO, Mundy GR, Seyedin SM, Bonewald LF. Activation of the bone-derived latent TGF β complex by isolated osteoclasts. Biochem Biophys Res Commun. 1989;158(3):817–823. doi: 10.1016/0006-291X(89)92795-2. [DOI] [PubMed] [Google Scholar]
- 20.Owen M, Friedenstein AJ. Stromal stem cells: marrow-derived osteogenic precursors. Ciba Found Symp. 1988;136:42–60. doi: 10.1002/9780470513637.ch4. [DOI] [PubMed] [Google Scholar]
- 21.Sacchetti B, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131(2):324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 22.Woodbury D, Schwarz EJ, Prockop DJ, Black IB. Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res. 2000;61(19):364–370. doi: 10.1002/1097-4547(20000815)61:4<364::AID-JNR2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 23.Negishi-Koga T, et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat Med. 2011;17(11):1473–1480. doi: 10.1038/nm.2489. [DOI] [PubMed] [Google Scholar]
- 24.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 25.Chim SM, et al. Angiogenic factors in bone local environment. Cytokine Growth Factor Rev. 2013;24(3):297–310. doi: 10.1016/j.cytogfr.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 26.Seghezzi G, et al. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol. 1998;141(7):1659–1673. doi: 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beck L, Beck L, Jr, D’Amore PA. Vascular development: cellular and molecular regulation. FASEB J. 1997;11(5):365–373. [PubMed] [Google Scholar]
- 28.Klein S, Giancotti FG, Presta M, Albelda SM, Buck CA, Rifkin DB. Basic fibroblast growth factor modulates integrin expression in microvascular endothelial cells. Mol Biol Cell. 1993;4(10):973–982. doi: 10.1091/mbc.4.10.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein S, Bikfalvi A, Birkenmeier TM, Giancotti FG, Rifkin DB. Integrin regulation by endogenous expression of 18-kDa fibroblast growth factor-2. J Biol Chem. 1996;271(37):22583–22590. doi: 10.1074/jbc.271.37.22583. [DOI] [PubMed] [Google Scholar]
- 30.Sabbieti MG, et al. Prostaglandins differently regulate FGF-2 and FGF receptor expression and induce nuclear translocation in osteoblasts via MAPK kinase. Cell Tissue Res. 2005;319(2):267–278. doi: 10.1007/s00441-004-0981-8. [DOI] [PubMed] [Google Scholar]
- 31.Sobue T, et al. Regulation of fibroblast growth factor 2 and fibroblast growth factor receptors by transforming growth factor β in human osteoblastic MG-63 cells. J Bone Miner Res. 2002;17:502–512. doi: 10.1359/jbmr.2002.17.3.502. [DOI] [PubMed] [Google Scholar]
- 32.Sabbieti MG, et al. Prostaglandins regulate the expression of fibroblast growth factor-2 in bone. Endocrinology. 1999;140(1):434–444. doi: 10.1210/endo.140.1.6442. [DOI] [PubMed] [Google Scholar]
- 33.Shao ES, Lin L, Yao Y, Bostrom KI. Expression of vascular endothelial growth factor is coordinately regulated by the activin-like kinase receptors 1 and 5 in endothelial cells. Blood. 2009;114(10):2197–2206. doi: 10.1182/blood-2009-01-199166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park D, Sykes DB, Scadden DT. The hematopoietic stem cell niche. Front Biosci (Landmark Ed). 2012;17:30–39. doi: 10.2741/3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calvi LM, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 36.Mendez-Ferrer S, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krause DS, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med. 2013;19(11):1513–1517. doi: 10.1038/nm.3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okabe M, Graham A. The origin of the parathyroid gland. Proc Natl Acad Sci U S A. 2004;101(51):17716–17719. doi: 10.1073/pnas.0406116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiss RE, Watabe N. Studies on the biology of fish bone. III. Ultrastructure of osteogenesis and resorption in osteocytic (cellular) and anosteocytic (acellular) bones. Calcif Tissue Int. 1979;28(1):43–56. doi: 10.1007/BF02441217. [DOI] [PubMed] [Google Scholar]
- 40.Glowacki J, Cox KA, O’sullivan J, Wilkie D, Deftos LJ. Osteoclasts can be induced in fish having an acellular bony skeleton. Proc Natl Acad Sci U S A. 1986;83(11):4104–4107. doi: 10.1073/pnas.83.11.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Witten PE, Huysseune A. A comparative view on mechanisms and functions of skeletal remodelling in teleost fish, with special emphasis on osteoclasts and their function. Biol Rev Camb Philos Soc. 2009;84(2):315–346. doi: 10.1111/j.1469-185X.2009.00077.x. [DOI] [PubMed] [Google Scholar]
- 42.Moss ML. Studies of the acellular bone of teleost fish. V. Histology and mineral homeostasis of fresh-water species. Acta Anat (Basel). 1965;60:262–276. doi: 10.1159/000142652. [DOI] [PubMed] [Google Scholar]
- 43.Kim SW, et al. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J Bone Miner Res. 2012;27(10):2075–2084. doi: 10.1002/jbmr.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terauchi M, et al. T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab. 2009;10(3):229–240. doi: 10.1016/j.cmet.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prisby R, et al. Intermittent PTH(1-84) is osteoanabolic but not osteoangiogenic and relocates bone marrow blood vessels closer to bone-forming sites. J Bone Miner Res. 2011;26(11):2583–2596. doi: 10.1002/jbmr.459. [DOI] [PubMed] [Google Scholar]
- 46.Jiang Y, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418(6893):41–49. doi: 10.1038/nature00870. [DOI] [PubMed] [Google Scholar]
- 47.He X, Semenov M, Tamai K, Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/β-catenin signaling: arrows point the way. Development. 2004;131(8):1663–1677. doi: 10.1242/dev.01117. [DOI] [PubMed] [Google Scholar]
- 48.Itasaki N, Hoppler S. Crosstalk between Wnt and bone morphogenic protein signaling: a turbulent relationship. Dev Dyn. 2010;239(1):16–33. doi: 10.1002/dvdy.22009. [DOI] [PubMed] [Google Scholar]
- 49.Polo S, Di Fiore PP. Endocytosis conducts the cell signaling orchestra. Cell. 2006;124(5):897–900. doi: 10.1016/j.cell.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 50.Li C, et al. Disruption of LRP6 in osteoblasts blunts the bone anabolic activity of PTH. J Bone Miner Res. 2013;28(10):2094–2108. doi: 10.1002/jbmr.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oshimori N, Fuchs E. The harmonies played by TGF-β in stem cell biology. Cell Stem Cell. 2012;11(6):751–764. doi: 10.1016/j.stem.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Worthington JJ, Klementowicz JE, Travis MA. TGFβ: a sleeping giant awoken by integrins. Trends Biochem Sci. 2011;36(1):47–54. doi: 10.1016/j.tibs.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Dallas SL, Park-Snyder S, Miyazono K, Twardzik D, Mundy GR, Bonewald LF. Characterization and autoregulation of latent transforming growth factor beta (TGF β) complexes in osteoblast-like cell lines. Production of a latent complex lacking the latent TGF β-binding protein. J Biol Chem. 1994;269(9):6815–6821. [PubMed] [Google Scholar]
- 54.Yang T, et al. E-selectin ligand 1 regulates bone remodeling by limiting bioactive TGF-β in the bone microenvironment. Proc Natl Acad Sci U S A. 2013;110(18):7336–7341. doi: 10.1073/pnas.1219748110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seyedin SM, Thomas TC, Thompson AY, Rosen DM, Piez KA. Purification and characterization of two cartilage-inducing factors from bovine demineralized bone. Proc Natl Acad Sci U S A. 1985;82(8):2267–2271. doi: 10.1073/pnas.82.8.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hering S, et al. TGFbeta1 and TGFβ2 mRNA and protein expression in human bone samples. Exp Clin Endocrinol Diabetes. 2001;109(4):217–226. doi: 10.1055/s-2001-15109. [DOI] [PubMed] [Google Scholar]
- 57.Wan M, et al. Injury-activated transforming growth factor beta controls mobilization of mesenchymal stem cells for tissue remodeling. Stem Cells. 2012;30(11):2498–2511. doi: 10.1002/stem.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pfeilschifter J, Wolf O, Naumann A, Minne HW, Mundy GR, Ziegler R. Chemotactic response of osteoblastlike cells to transforming growth factor beta. J Bone Miner Res. 1990;5(8):825–830. doi: 10.1002/jbmr.5650050805. [DOI] [PubMed] [Google Scholar]
- 59.Zwijsen A, Verschueren K, Huylebroeck D. New intracellular components of bone morphogenetic protein/Smad signaling cascades. FEBS Lett. 2003;546(1):133–139. doi: 10.1016/S0014-5793(03)00566-0. [DOI] [PubMed] [Google Scholar]
- 60.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 61.Attisano L, Wrana JL. Signal transduction by the TGF-β superfamily. Science. 2002;296(5573):1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 62.Kretzschmar M, Massague J. SMADs: mediators and regulators of TGF-β signaling. Curr Opin Genet Dev. 1998;8(1):103–111. doi: 10.1016/S0959-437X(98)80069-5. [DOI] [PubMed] [Google Scholar]
- 63.Heldin CH, Miyazono K, ten DP. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390(6659):465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 64.Miyazono K. Positive and negative regulation of TGF-β signaling. J Cell Sci. 2000;113(pt 7):1101–1109. doi: 10.1242/jcs.113.7.1101. [DOI] [PubMed] [Google Scholar]
- 65.Derynck R. TGF-β-receptor-mediated signaling. Trends Biochem Sci. 1994;19(12):548–553. doi: 10.1016/0968-0004(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 66.Cohen PT. Protein phosphatase 1 — targeted in many directions. J Cell Sci. 2002;115(pt 2):241–256. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- 67.Mullen AC, et al. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 2011;147(3):565–576. doi: 10.1016/j.cell.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alliston T, Choy L, Ducy P, Karsenty G, Derynck R. TGF-β-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J. 2001;20(9):2254–2272. doi: 10.1093/emboj/20.9.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim JS, et al. Transforming growth factor-β1 regulates macrophage migration via RhoA. Blood. 2006;108(6):1821–1829. doi: 10.1182/blood-2005-10-009191. [DOI] [PubMed] [Google Scholar]
- 70.Zhang L, et al. A transforming growth factor β-induced Smad3/Smad4 complex directly activates protein kinase A. Mol Cell Biol. 2004;24(5):2169–2180. doi: 10.1128/MCB.24.5.2169-2180.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Janssens K, et al. Mutations in the gene encoding the latency-associated peptide of TGF-β 1 cause Camurati-Engelmann disease. Nat Genet. 2000;26(3):273–275. doi: 10.1038/81563. [DOI] [PubMed] [Google Scholar]
- 72.Kinoshita A, et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet. 2000;26(1):19–20. doi: 10.1038/79128. [DOI] [PubMed] [Google Scholar]
- 73.Janssens K, ten DP, Ralston SH, Bergmann C, Van HW. Transforming growth factor-β 1 mutations in Camurati-Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J Biol Chem. 2003;278(9):7718–7724. doi: 10.1074/jbc.M208857200. [DOI] [PubMed] [Google Scholar]
- 74.Saito T, et al. Domain-specific mutations of a transforming growth factor (TGF)-β 1 latency-associated peptide cause Camurati-Engelmann disease because of the formation of a constitutively active form of TGF-β1. J Biol Chem. 2001;276(15):11469–11472. doi: 10.1074/jbc.C000859200. [DOI] [PubMed] [Google Scholar]
- 75.Janssens K, et al. Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet. 2006;43(1):1–11. doi: 10.1136/jmg.2005.033522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whyte MP, Totty WG, Novack DV, Zhang X, Wenkert D, Mumm S. Camurati-Engelmann disease: unique variant featuring a novel mutation in TGFβ1 encoding transforming growth factor β 1 and a missense change in TNFSF11 encoding RANK ligand. J Bone Miner Res. 2011;26(5):920–933. doi: 10.1002/jbmr.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bondestam J, Mayranpaa MK, Ikegawa S, Marttinen E, Kroger H, Makitie O. Bone biopsy and densitometry findings in a child with Camurati-Engelmann disease. Clin Rheumatol. 2007;26(10):1773–1777. doi: 10.1007/s10067-006-0511-z. [DOI] [PubMed] [Google Scholar]
- 78.Neptune ER, et al. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33(3):407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 79.Loeys BL, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 80.Carmignac V, et al. In-frame mutations in exon 1 of SKI cause dominant Shprintzen-Goldberg syndrome. Am J Hum Genet. 2012;91(5):950–957. doi: 10.1016/j.ajhg.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Doyle AJ, et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet. 2012;44(11):1249–1254. doi: 10.1038/ng.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Prunier C, Pessah M, Ferrand N, Seo SR, Howe P, Atfi A. The oncoprotein Ski acts as an antagonist of transforming growth factor-beta signaling by suppressing Smad2 phosphorylation. J Biol Chem. 2003;278(28):26249–26257. doi: 10.1074/jbc.M304459200. [DOI] [PubMed] [Google Scholar]
- 83.Reed JA, Bales E, Xu W, Okan NA, Bandyopadhyay D, Medrano EE. Cytoplasmic localization of the oncogenic protein Ski in human cutaneous melanomas in vivo: functional implications for transforming growth factor beta signaling. Cancer Res. 2001;61(22):8074–8078. [PubMed] [Google Scholar]
- 84.Nomura T, et al. Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and thyroid hormone receptor. Genes Dev. 1999;13(4):412–423. doi: 10.1101/gad.13.4.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rhodes SD, et al. Hyperactive transforming growth factor-β1 signaling potentiates skeletal defects in a neurofibromatosis type 1 mouse model. J Bone Miner Res. 2013;28(12):2476–2489. doi: 10.1002/jbmr.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhen G, et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;19(6):704–712. doi: 10.1038/nm.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ameye L, Young MF. Mice deficient in small leucine-rich proteoglycans: novel in vivo models for osteoporosis, osteoarthritis, Ehlers-Danlos syndrome, muscular dystrophy, and corneal diseases. Glycobiology. 2002;12(9):107R–116R. doi: 10.1093/glycob/cwf065. [DOI] [PubMed] [Google Scholar]
- 88.Patel J, Whiting J, Jones D. Secondary knee osteoarthritis due to neurofibromatosis type 1 treated with above the knee amputation: a case report. Case Rep Orthop. 2013;2013:782106. doi: 10.1155/2013/782106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wischmeijer A, et al. Thoracic aortic aneurysm in infancy in aneurysms-osteoarthritis syndrome due to a novel SMAD3 mutation: further delineation of the phenotype. Am J Med Genet A. 2013;161A(5):1028–1035. doi: 10.1002/ajmg.a.35852. [DOI] [PubMed] [Google Scholar]
- 90.Hunter DJ, et al. Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res Ther. 2009;11(1):R11. doi: 10.1186/ar2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Suri S, Walsh DA. Osteochondral alterations in osteoarthritis. Bone. 2012;51(2):204–211. doi: 10.1016/j.bone.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 92.Park D, et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell. 2012;10(3):259–272. doi: 10.1016/j.stem.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31(3):266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Almeida M, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282(37):27285–27297. doi: 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou S, et al. Age-related intrinsic changes in human bone-marrow-derived mesenchymal stem cells and their differentiation to osteoblasts. Aging Cell. 2008;7(3):335–343. doi: 10.1111/j.1474-9726.2008.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stenderup K, Justesen J, Clausen C, Kassem M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone. 2003;33(6):919–926. doi: 10.1016/j.bone.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 97.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9(2):81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 98.Tilton FE, Degioanni JJ, Schneider VS. Long-term follow-up of Skylab bone demineralization. Aviat Space Environ Med. 1980;51(11):1209–1213. [PubMed] [Google Scholar]
- 99.Vico L, et al. Effects of long-term microgravity exposure on cancellous and cortical weight-bearing bones of cosmonauts. Lancet. 2000;355(9215):1607–1611. doi: 10.1016/S0140-6736(00)02217-0. [DOI] [PubMed] [Google Scholar]
- 100.Carmeliet G, Nys G, Stockmans I, Bouillon R. Gene expression related to the differentiation of osteoblastic cells is altered by microgravity. Bone. 1998;22(5 suppl):139S–143S. doi: 10.1016/S8756-3282(98)00007-6. [DOI] [PubMed] [Google Scholar]
- 101.Smith SM, et al. Bone markers, calcium metabolism, and calcium kinetics during extended-duration space flight on the mir space station. J Bone Miner Res. 2005;20(2):208–218. doi: 10.1359/JBMR.041105. [DOI] [PubMed] [Google Scholar]
- 102.Meyers VE, Zayzafoon M, Douglas JT, McDonald JM. RhoA and cytoskeletal disruption mediate reduced osteoblastogenesis and enhanced adipogenesis of human mesenchymal stem cells in modeled microgravity. J Bone Miner Res. 2005;20(10):1858–1866. doi: 10.1359/JBMR.050611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- 104.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guise TA, et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin Cancer Res. 2006;12(20 pt 2):6213s–6216s. doi: 10.1158/1078-0432.CCR-06-1007. [DOI] [PubMed] [Google Scholar]
- 106.Juarez P, Guise TA. TGF-β in cancer and bone: implications for treatment of bone metastases. Bone. 2011;48(1):23–29. doi: 10.1016/j.bone.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 107.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350(16):1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 108.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 109.Ikushima H, Miyazono K. TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10(6):415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 110.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11(6):411–425. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Balooch G, et al. TGF-β regulates the mechanical properties and composition of bone matrix. Proc Natl Acad Sci U S A. 2005;102(52):18813–18818. doi: 10.1073/pnas.0507417102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Edwards JR, et al. Inhibition of TGF-β signaling by 1D11 antibody treatment increases bone mass and quality in vivo. J Bone Miner Res. 2010;25(11):2419–2426. doi: 10.1002/jbmr.139. [DOI] [PubMed] [Google Scholar]
- 113.Smith AL, Robin TP, Ford HL. Molecular pathways: targeting the TGF-β pathway for cancer therapy. Clin Cancer Res. 2012;18(17):4514–4521. doi: 10.1158/1078-0432.CCR-11-3224. [DOI] [PubMed] [Google Scholar]
- 114.Biswas S, et al. Anti-transforming growth factor ss antibody treatment rescues bone loss and prevents breast cancer metastasis to bone. PLoS One. 2011;6(11):e27090. doi: 10.1371/journal.pone.0027090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ganapathy V, et al. Targeting the Transforming Growth Factor-β pathway inhibits human basal-like breast cancer metastasis. Mol Cancer. 2010;9:122. doi: 10.1186/1476-4598-9-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bellido M, et al. Improving subchondral bone integrity reduces progression of cartilage damage in experimental osteoarthritis preceded by osteoporosis. Osteoarthritis Cartilage. 2011;19(10):1228–1236. doi: 10.1016/j.joca.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 117.Orth P, Cucchiarini M, Zurakowski D, Menger MD, Kohn DM, Madry H. Parathyroid hormone [1-34] improves articular cartilage surface architecture and integration and subchondral bone reconstitution in osteochondral defects in vivo. Osteoarthritis Cartilage. 2013;21(4):614–624. doi: 10.1016/j.joca.2013.01.008. [DOI] [PubMed] [Google Scholar]