

Abstract
Objective
Pulmonary arterial hypertension, characterized by pulmonary vascular remodelling and vasoconstriction, is associated with excessive proliferative changes in pulmonary vascular walls. However, the role of HDACs in the phenotypic alteration of pulmonary arterial smooth muscle cells (PASMC) is largely unknown.
Material and methods
Pulmonary arterial smooth muscle cells were isolated from newborn sheep. Cell cycle analysis was performed by flow cytometry. mRNA and protein expression were measured by real‐time PCR and Western blot analysis. Wound‐healing scratch assay was used to measure cell migration. Contractility of newborn PASMCs was determined by gel contraction assay. Chromatin immunoprecipitation was used to examine histone modifications along the p21 promoter region. Global DNA methylation was measured by liquid chromatography‐mass spectroscopy.
Results
Inhibition of class I and class II HDACs by apicidin and HDACi VIII suppressed proliferation of newborn PASMC and induced cell cycle arrest in G1 phase. Acetyl H3 levels were higher in newborn PASMC treated with apicidin and HDACi VIII. This was accompanied by increased expression of p21 and reduced expression of CCND1 but not p53. HDAC inhibition altered histone codes around the p21 promoter region in NPASMC. Apicidin inhibited serum‐induced cell migration, and modulated profiling of expression of genes encoding pro‐oxidant and antioxidant enzymes. Contractility and global DNA methylation levels of newborn PASMCs were also markedly modulated by apicidin.
Conclusion
Our results demonstrate that class I HDACs are clearly involved in phenotypic alteration of newborn PASMC.
Introduction
Pulmonary arterial hypertension (PAH), characterized by pulmonary vascular remodelling and vasoconstriction, is associated with proliferative changes in the pulmonary vascular wall 1. Structural alterations within the vessel wall are mainly caused by proliferation and migration of pulmonary arterial smooth muscle cells (PASMC) 2. Although the overall 5‐year survival for PAH patients has significantly increased over the past number of years, none of the approved therapies has shown an ability to cure the disease. Current available treatments are limited, expensive, and often associated with side effects. As such, to further our understanding of mechanisms underlying PAH development, it is important to further our understanding of mechanisms underlying PAH development, so that the improvement of patient management and outcome can occur.
It is being increasingly recognized that environmental factors during prenatal and early postnatal periods influence developmental programming of homoeostatic mechanisms that profoundly impact susceptibility to diseases later in life 3, 4, 5, 6. Neonatal nociception raises basal blood pressure level and attenuates cardiovascular responsiveness to nociceptive stress in adult rats 7. Effects of perinatal insults have most frequently been assessed in animal models of intrauterine growth retardation. Impaired foetal growth is a major cause of perinatal morbidity and has long‐term clinical consequences 8, 9, 10. Perinatal hypoxia increases severity of pulmonary hypertension after reexposure to hypoxia in infant rats 4. It also triggers alterations in K+ channels of adult pulmonary artery smooth muscle cells 5.
Epigenetics can explain that heritable changes in gene expression or cell phenotype are caused by mechanisms other than changes in the underlying DNA sequence. Although knowledge of relationships between PAH and epigenetics is quite limited 11, there are recent studies that show that epigenetic mechanisms are involved in lung development and pulmonary vascular dysfunction 10, 12, 13, 14, 15, 16, 17, 18. Undernutrition during gestation induces pulmonary vascular dysfunction in the offspring through alterations in DNA methylation, whereas administration of HDAC inhibitors to offsprings of restrictive diet during pregnancy normalized pulmonary DNA methylation and vascular function 10. In the rat lung, intrauterine growth retardation induces epigenetic modifications to the PPARr gene, a member of a nuclear receptor family of transcription factors that contributes to epithelial–mesenchymal interactions that are crucial for lung development 15, 16, 17. Moreover, HDAC inhibition has been shown to suppress hypoxia‐induced cardiopulmonary remodelling, improve pulmonary artery acceleration time and reduce systolic notching of the pulmonary artery flow envelope 19, 20. We have recently reported that levels of global DNA methylation and histone acetylation in pulmonary arteries from foetal lung acclimatized to high‐altitude long‐term hypoxia were altered 21.
In this study, we investigated the effects of the inhibition of HDACs using their inhibitors, apicidin, HDACi VIII and Tenovin‐1, on ovine NPASMC proliferation, migration, and expression of cell cycle related genes such as p21, CCND1, CDK4 and p53. Expression of genes encoding pro‐oxidant and antioxidant enzymes was measured. We also determined the effects of inhibition of class I HDACs on NPASMC contractility and global DNA methylation.
Materials and methods
Reagents
Apicidin, HDACi VIII, and Tenovin‐1 were purchased from EMD Millipore, Billerica, MA, USA. Propidium iodide was purchased from Sigma, St. Louis, MO, USA.
Preparation of PASMC from ovine newborn lung
Intrapulmonary arteries, third to fourth generation, were dissected free of parenchyma and kept in ice‐cold modified Krebs‐Ringer bicarbonate buffer, and primary ovine newborn PASMC (NPASMC) were isolated from pulmonary arteries as described previously 22. Cells were maintained in DMEM containing 10% heat‐inactivated FBS and antibiotics. All experiments were performed with cells at passages 4–8.
Cell number and cell viability
Cell number was determined by counting using a haemocytometer. Trypan blue staining was used to distinguish dead from live cells.
Cell cycle analysis
Cell cycle distribution was determined by flow cytometric analysis as previously described 23. Briefly, NPASMC (5 × 105 cells) were cultured in serum‐free DMEM medium for 48 h. After starvation, medium containing 10% serum was replaced, and apicidin, HDACi VIII, and Tenovin‐1 were added. Cells were treated for 24 h, then washed in PBS, fixed in 70% ethanol and hypotonically lysed in 500 μl of DNA staining solution [0.05 mg/ml PI (Sigma), 0.1 mg/ml RNase A, and 0.05% Triton X‐100]. NPASMC were incubated, while protected from light at 37 °C for 40 min. Stained cells were washed in PBS and suspended in 300 μl of PBS before analysis. Cell cycle data were analysed using an Epics XL‐MCL flow cytometer with System II (version 3.0) software (Beckman Coulter, Miami, FL, USA). Additional analysis of cell cycle distribution was determined using Modfit LT (Verity Software House, Topsham, ME, USA).
Migration assay
Newborn PASMC were grown to confluence on 35 mm petri dishes. Cells were growth‐arrested for 24 h in DMEM containing 0.1% FBS. A scratch was made in each cell monolayer, medium was replaced by 0.1% FBS medium, baseline (0 h time point) images were captured, and 2.5 μg/ml of apicidin was added for 30 min prior to addition of 10% serum. Photomicrographs were taken at 0 and 24 h, and cell migration distance was determined by subtracting values obtained at 0 h from 24 h. Migration distances were expressed as percentages over control values.
cDNA synthesis and SYBR green real‐time PCR
RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Reverse transcription was performed using Superscript III (Invitrogen) and 50 μm oligo(dT)20 at 50 °C for 50 min. SYBR green real‐time PCR reactions were set up containing 1x Power SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) and 250 nm forward and reverse primers in a 20‐μl reaction. All assays were carried out in 96‐well format. Real‐time fluorescence detection of PCR products was performed using StepOne Plus Real‐Time PCR System (Applied Biosystems) using the following thermocycling conditions: 1 cycle of 95 °C for 10 min; 40 cycles of 95 °C for 30 s, and 60 °C for 1 min. Sequences of primers were designed using Primer Express software (Applied Biosystems). Ovine primer sequences are shown in Table 1. β‐actin was used as an endogenous control for gene expression. For data analysis, the comparative method (∆∆Ct) was used to calculate relative quantities of a nucleic acid sequence.
Table 1.
Ovine primer sequences
| Gene symbol | Assay | Sense | Antisense | Accession |
|---|---|---|---|---|
| SOD1 | Q‐PCR | TCATGGGTTCCACGTCCAT | GAGGGCCTGCACTGGTACAG | NM_001145185 |
| SOD2 | Q‐PCR | CAGGATCCCCTGCAAGGA | CATGCTCCCACACGTCAATC | GQ221055 |
| SOD3 | Q‐PCR | TCACCTTGATTTTTCTCCTTCCTT | TGGCAGAAGTGGTACTCCAGAGT | XM_004009740 |
| PRDX5 | Q‐PCR | AAAAGGAGCCCGGGAACA | GCACTCCCTTCTTGCCTTTG | JX889614 |
| DUOX1 | Q‐PCR | ACCCCGTTTCCACATCTTCTT | ACCAGCTTGTCCCCCACATA | XM_004011029 |
| DUOX2 | Q‐PCR | AGAACTACCGGCGGCACAT | CAAACAGTCCGGCACAGATG | NM_001190392 |
| NOX4 | Q‐PCR | GCTGGAGGCATTGGAGTCA | TTTCCAGTCATCCAGCAGAGTGT | EF369489 |
| p21 | Q‐PCR | CCAGACCAGCATGACAGATTTC | GCTTCCTCTTGGAGCAGATCAG | EE754405 |
| CCND1 | Q‐PCR | TCGAGCACTTCCTCTCCAAAA | GTTTGCGGATGATCTGCTTGT | EU525165 |
| p53 | Q‐PCR | TCTGGGACTTAGTGCCTTTTATGG | CAGTCAGAAACTGTCAAATCATCCA | X81705 |
| cdk4 | Q‐PCR | GCTTGCCAGTGGAGACCATAAA | ATGAAGGAAATCCAGGCCTCTT | NM_001127269 |
| β‐actin | Q‐PCR | GCAGATGTGGATCAGCAAGCA | AGCATTTGCGGTGGACGAT | NM_001009784 |
| p21 | ChIP/Q‐PCR | GAGAGGGAGCGTCCGACTT | TGGCTGGAACACAAGGAAGAC |
Western blot analysis
Total proteins of NPASMC were extracted after lysing cells in cell lysis buffer containing protease and phosphatase inhibitor cocktails (Sigma, St. Louis, MO, USA); protein concentration was determined using a Bradford protein assay kit (Bio‐Rad Laboratories, Hercules, CA, USA). Equal amounts of total protein (10–25 μg) from cells were subjected to SDS‐PAGE. Proteins were transferred to nitrocellulose membrane for 90 min at 100 V. Membranes were blocked for 1 h at room temperature in Tris‐buffered saline containing 5% non‐fat powdered milk, and probed with primary antibody in Tris‐buffered saline with 2.5% non‐fat powdered milk at concentrations from 1:500 to 1:20 000, pre‐incubated overnight according to the manufacturer's instructions, for each antibody. In all cases, secondary antibody labelled with horseradish peroxidase (GE Healthcare Bio‐Sciences, Piscataway, NJ, USA) was used at concentrations from 1:2000 to 1:20 000 for 1 h at room temperature. Immuno‐reactive bands were detected using SuperSignal West Pico Chemi‐luminescent Substrate (Pierce, Rockford, IL, USA) and recorded on photosensitive film. Relative intensities of immuno‐reactive bands detected by Western blot analysis were quantified by densitometry using NIH Image J software, and normalized to β‐actin levels. Primary antibodies used for this study include anti‐Ac‐histone H3 (Millipore), anti‐p21 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti‐β‐actin (Santa Cruz Biotechnology).
Gel contraction assay
Collagen contraction assay was performed as described previously 24. Briefly, collagen gels were prepared according to the manufacturer's instructions to a final collagen concentration of 1.5 mg/ml (Bacton Dickinson, Franklin Lanes, NJ, USA). NPASMC were seeded into gel mixtures at 2 × 105/ml in the presence or absence of apicidin, and gels were allowed to polymerize for 20 min at 37 °C in 24‐well plates. Thereafter, gels were mechanically loosened from the sides of wells. Three gels were analysed for each condition in each individual experiment.
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation assay was performed as previously described 25. Briefly, NPASMC (5 × 106) were treated with 1% formaldehyde for 10 min to crosslink histones to DNA. After washing in cold PBS, cell pellets were resuspended in cell lysis buffer (10 mm Tris, pH 8.0, 10 mm NaCl, 0.2% NP40). Nuclei were resuspended in nuclei lysis buffer (50 mm Tris pH 8.0, 10 mm EDTA, 1% SDS) and sonicated. Soluble chromatin fractions were collected, and 5 μl antibody of acetyl‐H3 (Millipore), acetyl‐H4 (Abcam, Cambridge, MA, USA), or normal rabbit IgG (Santa Cruz Biotechnology) was added. After incubation, chromatin‐antibody complexes were collected using salmon sperm DNA/protein A/G agarose beads (Millipore). After washing, immunoprecipitated DNA was treated with RNase and crosslinks were reversed by heating samples at 65 °C for 6 h. DNA was extracted using QIAquick PCR Purification kit (Qiagen, Valencia, CA, USA) and analysed by SYBR green real‐time PCR. The primer pair used for ChIP assay is shown in Table 1.
Liquid chromatography‐mass spectroscopy (LC/MS)
Total cytosine methylation was performed by LC/MS as described previously 26. Briefly, DNA was hydrolysed to nucleosides by adding 5 U nuclease P1 (Sigma) at 37 °C for 2 h, 0.002 units venom phosphodiesterase I (Sigma) at 37 °C for 2 h, 0.5 units alkaline phosphatase at 37 °C for 1 h. Stock solutions of 2′‐deoxycytidine (Sigma) and 5‐methyl‐2′‐deoxycytidine (ChemGenes, Wilmington, MA, USA) were prepared in water. Eight‐point standard stock mixture was carefully prepared to provide exact known concentration ratio of 2′‐deoxycytidine and 5‐methyl‐2′‐deoxycytidine. The concentration of 2′‐deoxycytidine and 5‐methyl‐2′‐deoxycytidine in each sample was calculated from the standard curve. Each DNA sample was analysed in triplicate. A quantity of 25 μl (80 ng) per sample was injected into LC and run through an Atlantis DC18 sillica column (Waters Corporation, Milford, MA, USA). Identification of 2′‐deoxycytidine and 5‐methyl‐2′‐deoxycytidine was obtained by mass spectra of chromatographic peaks.
Statistical analysis
Statistical analysis of the data was performed using standard two‐sample Student's t‐test, assuming unequal variances of the two data sets. Statistical significance was determined by two‐tailed distribution assumption and was set at 5% level (P < 0.05).
Results
Effect of HDAC inhibition on cell proliferation and cell cycle arrest in NPASMC
To determine which class or classes of HDACs are involved in NPASMC proliferation, HDAC inhibitors mainly targeting HDAC class I (apicidin), class II (HDACi VIII) and class III (Tenovin‐1), were used individually to treat NPASMC. As shown in Fig. 1a, cell accounting assay showed that apicidin, an HDAC inhibitor mainly targeting class I, strongly inhibited NPASMC proliferation at concentrations ranging from 0.5 to 10 μg/ml. In addition, HDACi VIII also inhibited cell proliferation in a dose‐dependent manner (Fig. 1b), indicating that class I and class II HDACs play important roles in contributing to pulmonary vascular SMC proliferation. However, Tenovin‐1 had no marked effect on proliferation of NPASMC (Fig. 1c).
Figure 1.
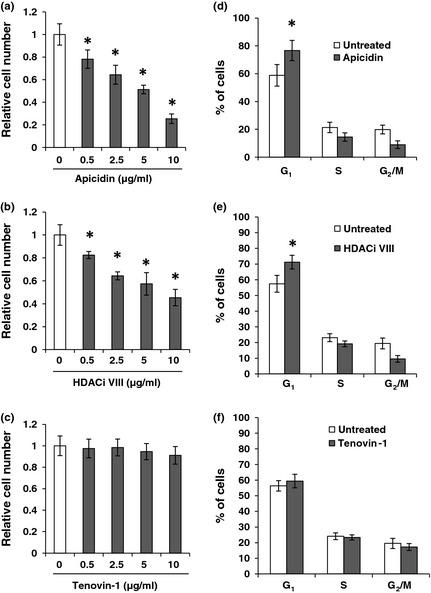
Effect of HDAC inhibition on proliferation and cell cycle of NPASMC . Cell proliferation was assessed by cell counting. NPASMC (2 × 105) were treated with various concentrations of apicidin (a), HDACi VIII (b), and Tenovin‐1 (c) for 24 h. All experiments were performed in triplicate and mean ± SEM of three independent experiments is shown. Results are expressed as a ratio of untreated cells. *P < 0.05 compared to untreated cells. For cell cycle analysis, PASMC (5 × 105 cells) of newborn sheep were serum‐starved for 48 h and treated with or without 5 μg/ml apicidin (d), HDACi VIII (e) or Tenovine‐1 (f) for 24 h in the presence of 10% serum, and subjected to flow cytometric analysis to determine cell cycle distribution. *P < 0.05 compared to untreated PASMC of newborn sheep.
Next, we investigated whether HDAC inhibitors reduced cell proliferation via cell cycle arrest. We resorted to flow cytometry to determine if HDAC inhibition caused a fall in cell proliferation of PASMC from newborn sheep lung. After 48 h serum starvation, NPASMC were cultured for 24 h in 10% FBS with or without 2.5 μg/ml apicidin, HDACi VIII or Tenovine‐1. Cells were stained with propidium iodide to study the cell cycle progression. As shown in Fig. 1d and 1e, treatment of NPASMC with apicidin and HDACi VIII induced cell cycle arrest in G1 phase. Apicidin showed more potent cell cycle arrest at G1. However, Tenovine‐1 had no effect on cell cycle arrest (Fig. 1f).
Effect of HDAC inhibition on expression of cell cycle‐related genes and histone H3 acetylation levels
To determine which genes were involved in apicidin‐ and HDACi VIII‐induced cell cycle arrest, expression of p21, a potent cyclin‐dependent kinase inhibitor, was initially examined (Fig. 2). As shown in Fig. 2a, both apicidin and HDACi VIII markedly increased p21 expression. Higher levels of p21 were induced in apicidin‐treated NPASMC compared to HDACi VIII‐treated ones. Also, treatment of NPASMC with apicidin and HDACi VIII increased accumulation of hyperacetylated histone H3 (Fig. 2a), suggesting that histone hyperacetylation modulated by apicidin and HDACi VIII was responsible for inhibitory effects of pulmonary vascular SMC proliferation via selective induction of cell cycle‐related genes such as p21. Induced p21 expression was not seen in Tenovine‐1‐treated NPASMC, as is shown in Fig. 2b.
Figure 2.
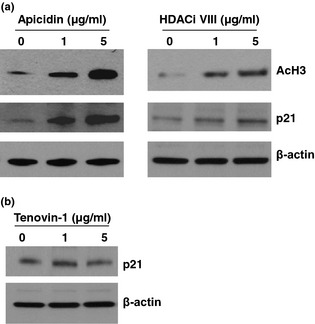
Effect of HDAC inhibition on protein expression level of cell cycle‐related genes and acetyl‐H3. (a) Cell lysates from untreated and apicidin‐ and HDACi VIII‐treated NPASMC were prepared. Levels of p21 and AcH3 were examined by Western blot analysis using antibodies against p21 and AcH3, respectively. β‐actin was used as an endogenous control. (b) Cell lysates from untreated and Tenovine‐1‐treated NPASMC were prepared. Levels of p21 were examined by Western blot analysis using antibody against p21. β‐actin was used as an endogenous control.
To determine whether increased p21 protein expression is regulated at transcriptional level in NPASMC after treatment with apicidin and HDACi VIII, RNA expression of p21 was also determined. As shown in Fig. 3a, RNA expression of p21 was markedly increased in a dose‐dependent manner in NPASMC treated with both apicidin and HDACi VIII. Higher levels of p21 RNA were observed in apicidin‐treated NPASMC compared to HDACi VIII‐treated NPASMC. There was no marked induction of p21 expression in NPASMC treated with Tenovin‐1. In addition, RNA expression of cyclinD1, which forms a complex with and functions as a regulatory subunit of CDK4 or CDK6, was reduced in NPASMC treated with apicidin and HDACi VIII; however, there was no remarkable change in cyclinD1 expression in NPASMC treated with Tenovin‐1 (Fig. 3b). Moreover, RNA expression of p53 and CDK4 were not markedly altered in NPASMC treated with apicidin, HDACi VIII, or Tenovin‐1 (Fig. 3c,d).
Figure 3.

Effect of HDAC inhibitors on RNA expression of cell cycle‐related genes in NPASMC s. NPASMCs were treated with 2.5 and 10 μg/ml of apicidin, HDACi VIII, and Tenovin‐1 for 24 h. RNA was isolated using Trizol reagent. cDNA was synthesized and subjected to quantitative RNA expression of p21 (a), CCND1 (b), p53 (c) and CDK4 (d) by SYBR green q‐PCR. β‐actin was used as an endogenous control.
HDAC inhibition altered histone modifications around the p21 promoter
To determine the relationship between p21 expression and histone modifications, we tested effects of apicidin, HDACi VIII and Tenovin‐1 on histone marks (AcH3 and AcH4) around the p21 promoter region. As shown in Fig. 4a and 4b, apicidin and HDACi VIII modulated histone codes. Following treatment of the NPASMC with apicidin, levels of AcH3 and AcH4 along the p21 promoter region were markedly enriched after 2 d treatment with apicidin. In addition, enrichment of AcH3 and AcH4 was also observed in cells treated with HDACi VIII (Fig. 4a,b). Moreover, more enrichment of AcH3‐bound DNA was observed in apicidin‐treated ovine NPASMC compared to HDACi VIII‐treated ones. However, levels of AcH3 and AcH4 along the p21 promoter were not markedly changed in NPASMC treated with Tenovin‐1. In addition, enrichment of IgG‐bound DNA was not markedly changed between treated and untreated groups around the p21 promoter region (Fig. 4a). Our data suggest that elevated levels of p21 expression is due to recruitment of AcH3 and AcH4 to the p21 promoter region in these ovine NPASMC.
Figure 4.
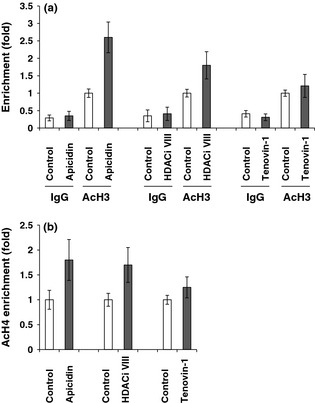
Alteration of histone marks around the p21 promoter region. ChIP/quantitative PCR was performed around the p21 promoter region in NPASMC in the presence or absence of apicidin, HDACi VIII, or Tenovin‐1. (a) ChIP/quantitative PCR was performed to determine AcH3 levels around the p21 promoter region in apicidin and HDACi VIII‐treated and non‐treated cells. Normal rabbit IgG was used as negative control. (b) ChIP/quantitative PCR was performed to determine the AcH4 levels around the p21 promoter region in apicidin and HDACi VIII‐treated and non‐treated NPASMCs.
Class I HDAC inhibition attenuated serum‐induced migration of NPASMC
As migration of vascular smooth muscle cells is involved in vascular remodelling, in the next experiment, we examined if class I HDAC inhibition reduced NPASMC migration induced by serum, as class I HDAC inhibition showed more potent cell cycle arrest. The effect of class I HDAC inhibition on serum‐induced NPASMC migration using the wound‐healing model/scratch assay was performed. As shown in Fig. 5a and 5b, there was minimal migration of NPASMC observed in medium containing 0.1% serum at 24 h compared to the 0 h time point; 10% serum caused over 2.8‐fold increase in cell migration compared to untreated controls. However, inhibition of HDAC activity with 2.5 μg/ml apicidin resulted in ∼25% reduction in serum‐induced cell migration (P < 0.05).
Figure 5.
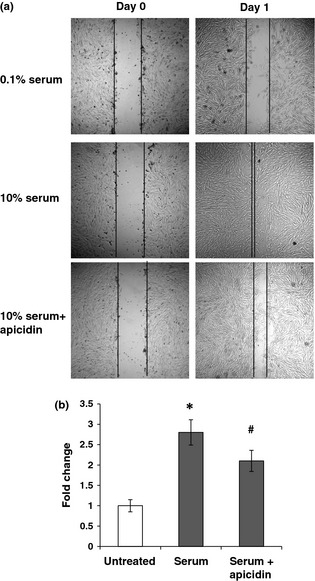
Effect of class I HDAC inhibition on serum‐induced migration of NPASMC s. (a) Representative images of serum‐induced NPASMC migration in the presence and absence of apicidin. (b) Cell migration into the wound areas was examined using microscopy, and photomicrographs were taken at 0 and 24 h. Cell migration distance was determined by subtracting values obtained at 0 h from 24 h. Migration distances were expressed as percentages over control values. *P < 0.05 compared to untreated control. #P < 0.05 compared to serum group.
Effect of class I HDAC inhibition on NPASMC‐mediated collagen gel contraction
As apicidin showed more potent anti‐proliferation effects, we next determined the consequences of apicidin on contractility of NPASMC using a collagen gel contraction assay. Surface area of 24‐well dishes containing no cells was defined as 100%. In the presence of 10% FBS, untreated NPASMCs had significant collagen gel contractility after 24 h culture (Fig. 6a,b, *P < 0.01 compared to the group without cells). Contractility of NPASMCs was significantly attenuated by apicidin at concentrations of 0.5 and 5 μg/ml (#P < 0.05 compared to untreated group).
Figure 6.
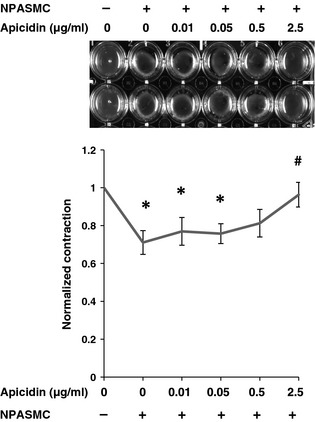
Effect of class I HDAC inhibition on contractility of NPASMC as assessed by collagen gel contraction assay. NPASMC were cultured in 3‐D collagen gels for 24 h in the presence of various concentrations of apicidin. NPASMC‐mediated collagen gel contraction was assessed by measuring gel surface area using Imagine J software. (a) Collagen gels were photographed. (b) Contractility of collagen gel without NPASMC was defined as 1. Relative contractility of apicidin‐treated and untreated NPASMCs was calculated. *P < 0.05 compared to no cells group; #P < 0.05 compared to untreated cells group.
Effect of class I HDAC inhibition on expression of genes encoding pro‐oxidant and antioxidant enzymes
Previous studies have documented that HDACs are involved in oxidative stress‐induced pulmonary hypertension. To determine whether HDAC inhibition alters expression of pro‐oxidant and antioxidant enzymes which regulate the redox state, real‐time PCR analysis was performed; our data reveal that apicidin modulated mRNA levels of pro‐oxidant and antioxidant enzymes. As shown in Fig. 7a, apicidin treatment exhibited marked reduction in expression of genes encoding pro‐oxidant enzymes, including DUOX1 and DUOX2. On the other hand, apicidin treatment exhibited increased expression of genes encoding antioxidant enzymes including SOD2 and SOD3 as shown in Fig. 7b. Increased expression of SOD1 and PRDX5 proteins was not seen. Our studies suggest that HDAC inhibition may confer substantial protection against oxidative stress.
Figure 7.
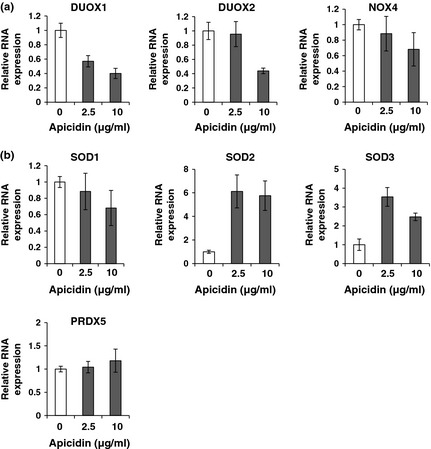
Effect of class I HDAC inhibition on expression of genes encoding pro‐oxidant and antioxidant enzymes. (a) mRNA levels from genes encoding pro‐oxidant enzymes including DUOX1, DUOX2 and NOX4 were measured by real‐time PCR analysis. (b) mRNA levels from genes encoding antioxidant enzymes including SOD1, SOD2, SOD3 and PRDX5 were measured by real‐time PCR analysis. β‐actin was used as an endogenous control.
Class I HDAC inhibition modulated global DNA methylation in NPASMC
It has been reported that interplay between histone modification and DNA methylation occurs in a variety of cells 25, 27, 28, 29, thus, we determined whether class I HDAC inhibition altered overall DNA methylation in NPASMC. The cells were treated with 0.05, 0.5 and 2.5 μg/ml apicidin for 48 h, and global DNA methylation was measured by LC/MS. As shown in Fig. 8, treatment of NPASMC by apicidin at concentrations of 0.5 and 2.5 μg/ml resulted in over 2‐fold increase in overall 5‐methylcytosine methylation in a dose‐dependent manner (P < 0.05). However, apicidin at a lower concentration (0.05 μg/ml) reduced global DNA methylation levels (P < 0.05), indicating that the pattern of global DNA methylation was modulated by apicidin in a dose‐dependent manner.
Figure 8.
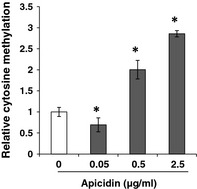
Effect of class I HDAC inhibition on global DNA methylation level in NPASMC . DNA methylation was determined by LC/MS. Genomic DNA was isolated from untreated and apicidin‐treated NPASMC (0.05, 0.5 and 2.5 μg/ml for 48 h). DNA was then subjected to digestion with nuclease P1, venom phosphodiesterase I, and alkaline phosphatase, respectively. Concentration of 2′‐deoxycytidine and 5‐methyl‐2′‐deoxycytidine in each sample was calculated from the standard curve. Each genomic DNA sample was analysed in triplicate. *P < 0.05 compared to untreated control.
Discussion
Excessive cell proliferation of PASMC contributes to vascular remodelling in patients with PAH 30. In this study, we demonstrated that inhibition of HDACs reduced serum‐induced PASMC proliferation. Apicidin is able to block serum‐stimulated cell proliferation in a dose‐dependent manner. It is possible that inhibitory effects of apicidin were exerted by changing the balance of CDK‐cyclin and CDK inhibitors. CDK‐cyclin complex and CDK inhibitor play a central role in regulation of cell proliferation during vascular remodelling 21, 31. In this study, we examined expression level of p21, CDK4, p53 and cyclinD1 in NPASMC treated with HDACi. Expression of CDK inhibitor p21 was markedly increased in cells treated with apicidin and HDACi VIII compared to vehicle control, which correlated with increased expression of acetylated histone H3. Inhibition of HDAC activity by apicidin and HDACi VIII significantly induced p21 expression, but without induction of p53, indicating that apicidin‐ and HDACi VIII‐induced p21 induction is p53‐independent. This is consistent with previous reports showing that apicidin arrests cancer cell population growth through induction of p21 32, 33. On the other hand, apicidin reduced expression of cyclinD1 in the NPASMC. Although the molecular mechanism underlying apicidin‐induced reduction of cyclinD1 expression here needs to be investigated, our ChIP data suggest that p21 upregulation in apicidin and HDACi VIII‐treated cells is due to recruitment of AcH3 and AcH4 to the p21 promoter region in ovine PASMC.
It has been reported that HDACs modulate vascular smooth muscle cell migration induced by cyclic mechanical strain 34. Cyclic strain upregulates levels of acetylated histone H3 and HDAC7 while downregulating levels of HDAC3/4 in VSMCs. Furthermore, mechanically induced VSMC migration was diminished by treatment with tributyrin, another HDAC inhibitor 34. However, whether HDACs take part in modulating migration of newborn PVSMC is poorly understood. We have demonstrated that in addition to its inhibitory effect on PASMC proliferation, inhibition of class I HDACs attenuated serum‐induced cell migration. Mechanisms of apicidin‐induced inhibitory effects on migration of PASMC require further investigation; however, our previous studies have shown that Trichostatin A significantly reduced phosphorylation of ERK 21. These results provide convincing evidence that HDACs are involved in proliferation and migration of PASMC induced by serum through chromatin remodelling. Thus, inhibition of HDAC may be beneficial in preventing migration of PASMC in treating proliferative vascular diseases.
Oxidative stress is involved in contributing to the development and progression of PAH 35, 36, 37. In this study, we determined whether inhibition of class I HDAC altered expression of genes encoding pro‐oxidant and antioxidant enzymes in NPASMC. Real‐time PCR analysis revealed increased expression of genes encoding antioxidant enzymes including SOD2 and SOD3 proteins in cells treated with apicidin. In addition, apicidin reduced expression of genes encoding pro‐oxidant enzymes including DUOX1. Our studies are in accordance with recently published results showing that β‐hydroxybutyrate, an endogenous histone deacetylase inhibitor, activated genes encoding oxidative stress resistance factors FOXO3A and MT2. Treatment of cells with β‐hydroxybutyrate increased histone acetylation at the Foxo3a and Mt2 promoters, and both genes were activated by selective depletion of HDAC1 and HDAC2. Consistent with increased FOXO3A and MT2 activity, treatment of mice with βOHB conferred substantial protection against oxidative stress 38.
It has been considered that DNA methylation and histone modifications appear to act in a coordinated fashion 39, 40. Although HDAC inhibition by apicidin in the range of 0.05–2.5 μg/ml is capable of modulating global DNA methylation levels in NPASMC, lower concentration of apicidin at 0.05 μg/ml reduced global DNA methylation level. On the other hand, relatively high concentrations of apicidin (0.5 and 2.5 μg/ml) increased global DNA methylation level, which is reverse‐correlated with cell proliferation, suggesting that the dose‐dependent effect of apicidin on alteration of DNA methylation pattern in the genome of NPASMC occurred. Our data provide evidence showing that the interplay between histone modification and DNA methylation may be involved in the process of gene transcription and aberrant silencing related to PASMC behaviour. One may expect that HDAC inhibition may modulate abnormal distribution of DNA methylation in PASMCs leading to increased risk of abnormal cell proliferation.
Taken together, inhibition of class I HDACs results in modulation of the epigenome in NPASMC, with changes in global acetylation and DNA methylation levels. HDAC inhibition is capable of inhibiting PASMC proliferation and cell cycle progression through chromatin remodelling. This change in NPASMC behaviour is associated with alteration in p21 and CCND1 expression. p21 upregulation in apicidin‐ and HDACi VIII‐treated NPASMC is due to recruitment of AcH3 and AcH4 to the p21 promoter region in ovine PASMC. Moreover, HDAC inhibition attenuates migration of NPASMC in response to serum stimulation. HDAC inhibition modulates gel contraction ability and mRNA levels of pro‐oxidant and antioxidant enzymes. Finally, HDAC inhibition modulated global DNA methylation in NPASMC. Our studies suggest that epigenetic mechanisms of histone acetylation may have significant mechanistic and therapeutic implications in human PAH, and histone acetylation modifiers may be considered as a new target for therapy for vascular disease.
Disclosures
No conflicts of interest, financial or otherwise, are declared by the author(s).
Acknowledgements
This work was supported in part by National Institutes of Health grants R01 HL075187 and R01 HL059435 to J. Usha Raj. We thank Ms. Ziyan Lu and Tala Akkawi for technical assistance.
References
- 1. Gao Y, Raj JU (2010) Regulation of the pulmonary circulation in the fetus and newborn. Physiol. Rev. 90, 1291–1335. [DOI] [PubMed] [Google Scholar]
- 2. Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M (2006) Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda) 21, 134–145. [DOI] [PubMed] [Google Scholar]
- 3. Nanduri J, Makarenko V, Reddy VD, Yuan G, Pawar A, Wang N, et al (2012) Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc. Natl. Acad. Sci. USA 109, 2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tang JR, Le Cras TD, Morris KG Jr, Abman SH (2000) Brief perinatal hypoxia increases severity of pulmonary hypertension after reexposure to hypoxia in infant rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 278, L356–L364. [DOI] [PubMed] [Google Scholar]
- 5. Marino M, Beny JL, Peyter AC, Bychkov R, Diaceri G, Tolsa JF (2007) Perinatal hypoxia triggers alterations in K+ channels of adult pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L1171–L1182. [DOI] [PubMed] [Google Scholar]
- 6. Sartori C, Allemann Y, Trueb L, Delabays A, Nicod P, Scherrer U (1999) Augmented vasoreactivity in adult life associated with perinatal vascular insult. Lancet 353, 2205–2207. [DOI] [PubMed] [Google Scholar]
- 7. Chu YC, Yang CC, Lin HT, Chen PT, Chang KY, Yang SC, et al (2012) Neonatal nociception elevated baseline blood pressure and attenuated cardiovascular responsiveness to noxious stress in adult rats. Int. J. Dev. Neurosci. 30, 421–426. [DOI] [PubMed] [Google Scholar]
- 8. Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD (2000) Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am. J. Physiol. Endocrinol. Metab. 279, E83–E87. [DOI] [PubMed] [Google Scholar]
- 9. Goyal R, Goyal D, Leitzke A, Gheorghe CP, Longo LD (2010) Brain renin‐angiotensin system: fetal epigenetic programming by maternal protein restriction during pregnancy. Reprod. Sci. 17, 227–238. [DOI] [PubMed] [Google Scholar]
- 10. Rexhaj E, Bloch J, Jayet PY, Rimoldi SF, Dessen P, Mathieu C, et al (2011) Fetal programming of pulmonary vascular dysfunction in mice: role of epigenetic mechanisms. Am. J. Physiol. Heart Circ. Physiol. 301, H247–H252. [DOI] [PubMed] [Google Scholar]
- 11. Stenmark KR, Frid MG, Yeager M, Li M, Riddle S, McKinsey T, et al (2012) Targeting the adventitial microenvironment in pulmonary hypertension: a potential approach to therapy that considers epigenetic change. Pulm. Circ. 2, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, et al (2010) Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121, 2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu XF, Ma XL, Shen Z, Wu XL, Cheng F, Du LZ (2010) Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J. Hypertens. 28, 2227–2235. [DOI] [PubMed] [Google Scholar]
- 14. Archer SL, Roudebush WE (2013) Enhancement of sperm motility using pentoxifylline and platelet‐activating factor. Methods Mol. Biol. 927, 241–245. [DOI] [PubMed] [Google Scholar]
- 15. Joss‐Moore LA, Albertine KH, Lane RH (2011) Epigenetics and the developmental origins of lung disease. Mol. Genet. Metab. 104, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simon DM, Arikan MC, Srisuma S, Bhattacharya S, Tsai LW, Ingenito EP, et al (2006) Epithelial cell PPAR[gamma] contributes to normal lung maturation. FASEB J. 20, 1507–1509. [DOI] [PubMed] [Google Scholar]
- 17. Gluckman PD, Cutfield W, Hofman P, Hanson MA (2005) The fetal, neonatal, and infant environments‐the long‐term consequences for disease risk. Early Hum. Dev. 81, 51–59. [DOI] [PubMed] [Google Scholar]
- 18. Joss‐Moore LA, Metcalfe DB, Albertine KH, McKnight RA, Lane RH (2010) Epigenetics and fetal adaptation to perinatal events: diversity through fidelity. J. Anim. Sci. 88, E216–E222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cavasin MA, Demos‐Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, et al (2012) Selective class I histone deacetylase inhibition suppresses hypoxia‐induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ. Res. 110, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, et al (2012) Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 126, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang Q, Lu Z, Ramchandran R, Longo LD, Raj JU (2012) Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high altitude long‐term hypoxia: role of histone acetylation. Am. J. Physiol. Lung Cell. Mol. Physiol. 303, L1001–L1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Golovina VA, Blaustein MP (2006) Preparation of primary cultured mesenteric artery smooth muscle cells for fluorescent imaging and physiological studies. Nat. Protoc. 1, 2681–2687. [DOI] [PubMed] [Google Scholar]
- 23. Yang QW, Liu S, Tian Y, Salwen HR, Chlenski A, Weinstein J, et al (2003) Methylation‐associated silencing of the thrombospondin‐1 gene in human neuroblastoma. Cancer Res. 63, 6299–6310. [PubMed] [Google Scholar]
- 24. Yang Q, Lu Z, Singh D, Raj JU (2012) BIX‐01294 treatment blocks cell proliferation, migration and contractility in ovine foetal pulmonary arterial smooth muscle cells. Cell Prolif. 45, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang Q, Tian Y, Ostler KR, Chlenski A, Guerrero LJ, Salwen HR, et al (2010) Epigenetic alterations differ in phenotypically distinct human neuroblastoma cell lines. BMC Cancer 10, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shah MY, Vasanthakumar A, Barnes NY, Figueroa ME, Kamp A, Hendrick C, et al (2010) DNMT3B7, a truncated DNMT3B isoform expressed in human tumors, disrupts embryonic development and accelerates lymphomagenesis. Cancer Res. 70, 5840–5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu JL, Zhou BO, Zhang RR, Zhang KL, Zhou JQ, Xu GL (2009) The N‐terminus of histone H3 is required for de novo DNA methylation in chromatin. Proc. Natl. Acad. Sci. USA 106, 22187–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ikegami K, Ohgane J, Tanaka S, Yagi S, Shiota K (2009) Interplay between DNA methylation, histone modification and chromatin remodeling in stem cells and during development. Int. J. Dev. Biol. 53, 203–214. [DOI] [PubMed] [Google Scholar]
- 29. Vaissiere T, Sawan C, Herceg Z (2008) Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 659, 40–48. [DOI] [PubMed] [Google Scholar]
- 30. Stenmark KR, Fagan KA, Frid MG (2006) Hypoxia‐induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ. Res. 99, 675–691. [DOI] [PubMed] [Google Scholar]
- 31. Mizuno S, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez‐Arroyo J, Voelkel NF, et al (2011) p53 Gene deficiency promotes hypoxia‐induced pulmonary hypertension and vascular remodeling in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 300, L753–L761. [DOI] [PubMed] [Google Scholar]
- 32. Kwon SH, Ahn SH, Kim YK, Bae GU, Yoon JW, Hong S, et al (2002) Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J. Biol. Chem. 277, 2073–2080. [DOI] [PubMed] [Google Scholar]
- 33. Han JW, Ahn SH, Park SH, Wang SY, Bae GU, Seo DW, et al (2000) Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Res. 60, 6068–6074. [PubMed] [Google Scholar]
- 34. Yan ZQ, Yao QP, Zhang ML, Qi YX, Guo ZY, Shen BR, et al (2009) Histone deacetylases modulate vascular smooth muscle cell migration induced by cyclic mechanical strain. J. Biomech. 42, 945–948. [DOI] [PubMed] [Google Scholar]
- 35. Kim GH, Ryan JJ, Archer SL (2013) The role of redox signaling in epigenetics and cardiovascular disease. Antioxid. Redox Signal. 18, 1920–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gupte SA, Wolin MS (2008) Oxidant and redox signaling in vascular oxygen sensing: implications for systemic and pulmonary hypertension. Antioxid. Redox Signal. 10, 1137–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wolin MS, Gupte SA, Neo BH, Gao Q, Ahmad M (2010) Oxidant‐redox regulation of pulmonary vascular responses to hypoxia and nitric oxide‐cGMP signaling. Cardiol. Rev. 18, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al (2013) Suppression of oxidative stress by beta‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hagarman JA, Motley MP, Kristjansdottir K, Soloway PD (2013) Coordinate regulation of DNA methylation and H3K27me3 in mouse embryonic stem cells. PLoS One 8, e53880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim GH, Ryan JJ, Marsboom G, Archer SL (2011) Epigenetic mechanisms of pulmonary hypertension. Pulm. Circ. 1, 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]