

Summary
Several enveloped viruses exploit host pathways, such as the cellular endosomal sorting complex required for transport (ESCRT) machinery, for their assembly and release. The influenza A virus (IAV) matrix protein binds to the ESCRT-I complex, although the involvement of early ESCRT proteins such as Tsg101 in IAV trafficking remains to be established. We find that Tsg101 can facilitate IAV trafficking but this is effectively restricted by the interferon (IFN) stimulated protein ISG15. Cytosol from type I IFN-treated cells abolished IAV haemagglutinin (HA) transport to the cell surface in infected semi-intact cells. This inhibition required Tsg101 and could be relieved with deISGylases. Tsg101 is itself ISGylated in IFN-treated cells. Upon infection, intact Tsg101-deficient cells obtained by CRISPR/Cas9 genome editing were defective in surface display of HA and for infectious virion release. These data support the IFN-induced generation of a Tsg101/ISG15-dependent checkpoint in the secretory pathway that compromises influenza virus release.
Introduction
Assembly and release of enveloped viruses is a multi-step process that requires host factors, often hijacked by the virus to execute membrane remodeling and budding. The best characterized are retroviruses, which exploit the cellular endosomal sorting complex required for transport (ESCRT) machinery for budding (Martin-Serrano and Neil, 2011). The matrix protein of many such viruses contain a late-domain sequence (PTAP or L-domain) that binds to components of the ESCRT machinery, typically involved in the formation of vesicles into multi-vesicular bodies. Influenza viruses, on the other hand, are believed to have evolved an ESCRT-independent mode of budding, but the exact mechanism of influenza virus assembly and budding is not clear. The consensus is that the coat proteins initiate the process. HA and NA associate with the virus matrix protein M1 at lipid microdomains, followed by recruitment of M2 – an ion channel capable of altering membrane curvature (Rossman et al., 2010). Mutations introduced into the amphipathic helix of M2 abolish membrane scission and virus release (Rossman and Lamb, 2011; Rossman et al., 2010). Although influenza lacks a conventional L-domain, the matrix protein (M1) binds to the ESCRT-I complex, suggesting a possible role in assembly (Bruce et al., 2009). Budding of filamentous and non-filamentous influenza A requires neither VPS4 nor VPS28 (Bruce et al., 2009). However, involvement of early ESCRT proteins such as Tsg101 in intracellular trafficking of IAV remains to be demonstrated directly.
Host cells impose restrictions on various steps in the virus lifecycle, including entry, replication, assembly and release. Antiviral mechanisms are launched by induction of type I interferon (IFN-I) in the infected cell. ISG15 is one of the most abundantly expressed genes upon IFN-I signaling and has general antiviral effects (Skaug and Chen, 2010; Zhao et al., 2010). Retrovirus release is blocked upon IFN-I treatment or by exogenous expression of ISG15 (Seo and Leis, 2012; Zhao et al., 2013). ISG15 knock-out mice (ISG15−/−) show increased susceptibility to a number of virus infections, including influenza (Hsiang et al., 2009). Expression of ISG15 blocks budding of a number of viruses through conjugation of ESCRT components that are employed during virus infections. For example, budding of Ebola VLP is blocked by ISG15 through inhibition of Nedd4 ligase activity; HIV-1 budding is impaired upon IFN induction due to loss of Tsg101 binding to the HIV Gag protein. Although the non-structural protein 1 (NS1) of influenza B virus directly binds to and antagonizes the activity of ISG15, this is not the case for influenza A virus (Zhao et al., 2013) (Yuan and Krug, 2001) (Hsiang et al., 2009).
Here we use a biochemical assay based on the use of perfringolysin O-perforated semi-intact cells to study flu biogenesis. This preparation faithfully recapitulates intracellular glycoprotein trafficking and virus release. Perfringolysin O (PFO) is a cholesterol-binding pore-forming toxin that selectively perforates the plasma membrane, while leaving intracellular organelles intact. We can thus manipulate composition of the cytoplasm as well as deliver otherwise cell-impermeable reagents such as the non-hydrolysable GTP analog, GTPγS, to determine their effect on cytosol-dependent intracellular trafficking events. Exogenous cytosol from various sources can be delivered to these semi-intact cells through a mild osmotic shock. We previously used this preparation to measure ATP-dependent transport of misfolded glycoproteins from the ER to the cytosol as part of a protein quality control system (Ernst et al., 2011; Sanyal et al., 2012)). We have now extended this approach to understand intracellular protein trafficking routes upon IFN induction during influenza virus infection, and to identify cytosolic components relevant for the process. We show that semi-intact cells support virus assembly and release, with kinetics comparable to those of intact cells. We identified Tsg101 as a component essential for post-Golgi trafficking of HA to the PM, prior to budding of virus, and find that Tsg101 is ISGylated upon exposure of cells to IFN, providing a plausible link between IFN exposure and interference with glycoprotein trafficking. Cas9/CRISPR-mediated inactivation of Tsg101 yielded cells refractory to infection, possibly due to defects in virus entry, underscoring the advantage of the use of semi-intact cells to resolve virus entry, assembly and release processes. Both the use of semi-intact cells and the generation of a knock-out cell line establish the involvement of Tsg101 upon IFN induction and influenza infection. Endogenous glycoprotein transport acquires a pronounced dependency on Tsg101 in IFN-I treated cells, which is negatively regulated by ISG15 and exploited by IAV during infection.
Results
Trafficking of IAV in semi-intact cells requires exogenous cytosol
To demonstrate trafficking and budding of IAV in PFO-permeabilized cells, we infected MDCK cells with influenza A/WSN/33 for 5 hours. Cells were then pulse-labeled with [35S]cysteine/methionine for 10 min., treated with PFO at 0°C, washed briefly to remove excess PFO and then transferred to 37 °C to initiate permeabilization of the plasma membrane. This approach allows us to track the fate of only the wave of newly synthesized viral proteins, whilst ignoring the contribution of unlabeled virus particles poised for release at the time of perforation. The same strategy of cell permeabilization, performed in a small volume to minimize dilution, yielded concentrated cytosol (Figure 1A). We incubated PFO-treated cells with cytosol from MDCK cells at 37°C for the indicated times. At each time point, the supernatant and the pellet fractions were separated by centrifugation. Pellet fractions were lysed in NP40-containing lysis buffer and treated with anti-HA antibodies immobilized on Protein G beads to recover HA. To isolate intact virions released from the PFO-treated cells, supernatants were adsorbed onto chicken erythrocytes, which capture viral particles via the sialic acids on the erythrocytes (Figure 1B). The material was then visualized by SDS-PAGE and autoradiography.
Figure 1. Trafficking of IAV in semi-intact cells requires added cytosol.
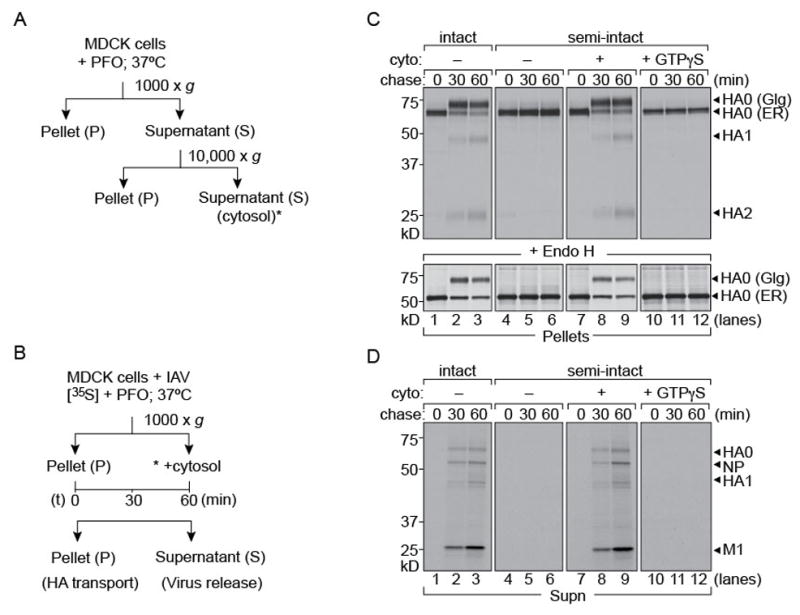
1 × 106 MDCK cells were infected with Influenza A/WSN/33 at an MOI of ~0.5 for 5 hours and radiolabeled with [35S]-cys/met. Cells were left intact or permeabilized with 100 nM PFO, supplemented with concentrated MDCK cytosol and chased for 30 and 60 mins. At each time point pellet and supernatant fractions were separated by centrifugation. (A), (B) Experimental set-up. (C) Upper panel: Pellet fractions were lysed and immunoprecipitated with anti-HA IgG2b on Protein-G beads. Pulse-chase on intact cells (lanes 1–3). Semi-intact cells supplemented with MDCK cytosol (lanes 7–9). Absence of exogenous cytosol or addition of GTPγS arrests HA in the ER (lanes 10–12). Lower panel: EndoH digested HA in the pellet fractions (D) Supernatant fractions from intact or permeabilized cells were treated with chicken erythrocytes to isolate intact virions. Release of virus particles occurred in intact cells (lanes 1–3) and semi-intact cells supplemented with cytosol (lanes 7–9).
We first measured intracellular trafficking of HA from the ER to the Golgi and from there to the plasma membrane in semi-intact cells by monitoring the maturation of N-linked glycans on HA (Figure 1C). The ER-resident high mannose form of HA can be distinguished from its Golgi counterpart by its distinct mobility on SDS-PAGE and by sensitivity of the former to treatment with Endoglycosidase H (EndoH) (Figure 1C; lower panel). Inclusion of trypsin in the incubation medium allows an assessment of the appearance of HA at the cell surface through formation of HA1 and HA2 – the trypsin-cleavage products of HA0. Access of trypsin to the cytosol cannot result in the conversion of intracellular HA0 into HA1 and HA2, because the cleavage site in HA0 is lumenally disposed and therefore not accessible to added protease even when it enters perforated cells. In the absence of added cytosol, intracellular transport of HA ceased (Figure 1C). Kinetics of intracellular transport and surface display of HA in semi-intact cells supplied with cytosol were comparable to those in intact cells, as measured by conversion of HA’s high mannose glycans to the complex type and appearance of HA1 and HA2. Inclusion of GTPγS abolished transport from the ER to Golgi, anticipated because of its inhibition of COP-II mediated vesicular transport (Lee and Miller, 2007). The inclusion of GTPγS is an important control that establishes quantitative permeabilization: residual intact cells would have supported HA maturation. In parallel with transport and surface arrival of HA, semi-intact cells supplemented with concentrated MDCK cytosol released intact virus or virus-like particles recovered by adsorption to chicken red blood cells (Figure 1D). Omission of cytosol or inclusion of GTPγS blocked release of virus particles. Semi-intact cells thus support virus assembly and release when supplemented with cytosol from a compatible exogenous source, and do so at rates comparable to those seen in intact cells. The composition of the released particles – as assessed by SDS-PAGE and autoradiography – is indistinguishable for intact and cytosol-supplemented semi-intact cells.
Cytosol from IAV infected and IFN-I treated cells have distinct effects on virus release
We hypothesized that IAV infection induces host factors in the cytosol that modulate virus assembly and budding. Mammalian cells respond to virus infection by production of IFN-I, which in turn induces expression of host factors with anti-viral properties, a process that takes several hours. Viruses have evolved countermeasures to induce host genes that facilitate virus trafficking and release, either directly or by modification of the existing sets of proteins, while downregulating expression of genes that compromise infection. To recapitulate such a physiological cytosolic environment we used two different sources of cytosol: we extracted cytosol from MDCK cells that had either been infected with IAV or that had been treated with IFN-I. Cytosol obtained from IAV-infected cells soon after infection would contain host proteins that assist in virus assembly and release. On the other hand, cytosol extracted from cells treated with IFN-I would be enriched in host factors that contribute to an antiviral milieu. We supplied these extracts to IAV-infected radiolabeled cells, permeabilized using PFO as described above. Cytosol from uninfected MDCK cells was used as a control (Figure 2A). Neither the abundance of HA nor the kinetics of trafficking from the ER to the Golgi were affected by provision of different cytosol preparations, relative to control cells. However, arrival of HA at the cell surface, as measured by cleavage of HA0 with the concomitant appearance of HA1 and HA2, was impaired by cytosol from IFN-treated cells (Figure 2A). We observed no impairment in particle release into the supernatants from semi-intact cells supplied with cytosol from IAV-infected cells (Figure 2B). Provision of cytosol from IFN-treated cells likewise abolished particle release. Cytosol from IFN-treated cells must therefore contain proteins that prevent arrival of HA at the cell surface and its subsequent incorporation into virions or must lack proteins necessary for this to occur. This block occurs after the acquisition of complex-type glycans. The fraction of viral proteins that appear in released virions is too low to result in a measurable accumulation of intracellular HA upon inhibition of virus release. Release of virions seen upon supplementation with cytosol from IAV-infected cells appears to be a consequence of NS1-mediated inhibition of a restriction factor (supplementary figure S1).
Figure 2. Cytosol from IAV infected and IFN-I treated cells have distinct effects on virus release from semi-intact cells.
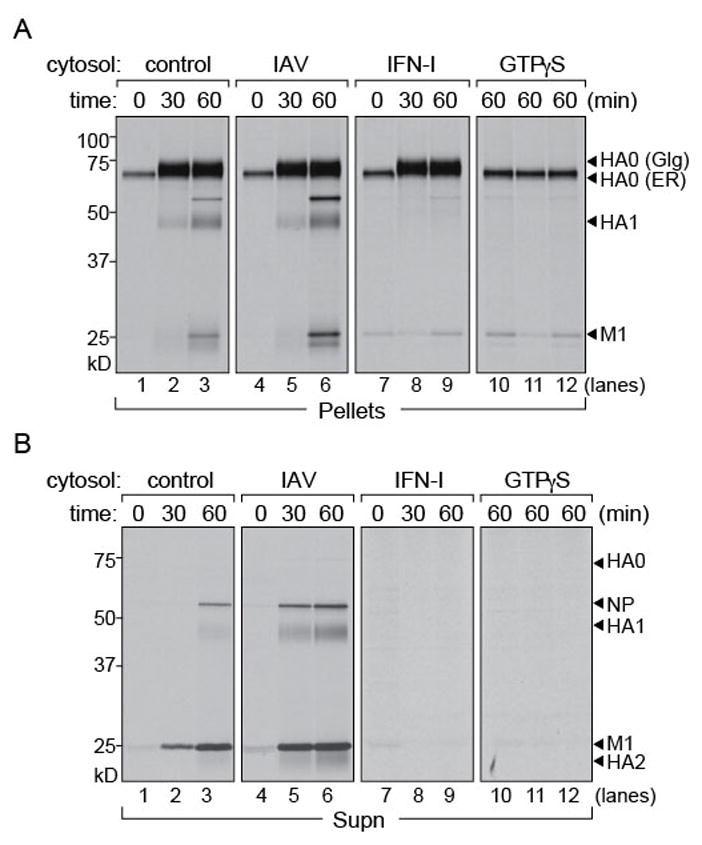
1 × 106 MDCK cells were infected with Influenza A/WSN/33, MOI of ~0.5 for 5 hours and radiolabeled with [35S]-cys/met. Permeabilized cells were supplemented with (i) concentrated MDCK cytosol, (ii) MDCK cytosol from IAV infected cells, (iii) MDCK cytosol treated with IFN-I and chased for 30 and 60 mins. At each time point pellet and supernatant fractions were separated by centrifugation. (A) Pellet fractions were lysed and immunoprecipitated with anti-HA IgG2b on Protein-G beads. Control cytosol (lanes 1–3) and cytosol from IAV infected MDCK cells (lanes 4–6). Cytosol from IFN treated MDCK cells (lanes 7–9) does not support HA transport from the Golgi to the PM. Cytosol supplemented with GTPγS (lanes 10–12). (B) Supernatant fractions from permeabilized cells were treated with chicken erythrocytes to isolate intact virions. Release of virus particles with control cytosol (lanes 1–3), cytosol from IAV infected cells (lanes 4–6), with IFN-treated cytosol (lanes 7–9) and GTPγS supplemented cytosol (lanes 10–12). Figure 2, related to figure S1.
ISG15 inhibits a late step in IAV glycoprotein transport and blocks virus release
ISG15 is an abundantly expressed gene, induced immediately upon exposure to type I IFN. We hypothesized that the observed blockade in HA transport to the cell surface in IFN-I exposed cytosolic extracts was due to ISGylation of host components that in their unmodified form facilitate virus trafficking. We therefore expressed Ubp43 (a specific de-ISGylase) (Malakhov et al., 2002) or CCHFV-L Otu (L protein of Crimean-Congo Hemorrhagic fever virus containing an ovarian-tumor domain that antagonizes ISG15 activity) (Frias-Staheli et al., 2007) in HEK 293T cells and prepared cytosol fractions by PFO treatment. Cytosol from IFN-I treated cells was mixed with that of either Ubp43 or CCHFV-L Otu-containing cytosol in a 1:1 ratio and supplied to semi-intact cells to measure HA transport and virus production. An inactive mutant of CCHFV-L Otu served as control. To control for dilution of factors essential for transport, we mixed in a 1:1 ratio cytosol from IFN-treated cells with that of HEK293T cells expressing an empty vector. Provision of this mixture inhibited arrival of HA at the cell surface, indicating no loss of activity of the IFN-induced factors upon two-fold dilution (Figure 3A). For cytosol from IFN-I treated cells supplemented with either Ubp43 or CCHFV-L Otu, transport of HA to the cell surface was restored, as judged from the appearance of HA1 and HA2. The catalytically inactive mutant of CCHFV-L Otu failed to relieve the inhibition observed upon supply of cytosol from IFN-I treated cells. These observations were corroborated by measuring the release of virus particles (Figure 3B). Block in virus release imposed upon IFN-I treatment was restored in samples incubated with either Ubp43 or CCHFV-L Otu, but not with inactive CCHFV-L Otu. ISG15 must therefore modify either a viral or a host protein that inhibits a late step in the assembly of progeny virions and their subsequent release from cells.
Figure 3. ISG15 blocks a late step in glycoprotein transport.

1 × 106 MDCK cells were infected with Influenza A/WSN/33 at an MOI of ~0.5 for 5 hours and radiolabeled with [35S]-cys/met. Permeabilized cells were supplemented with (i) concentrated MDCK cytosol, (ii) cytosol treated with IFN-I mixed with HEK 293T cytosol expressing an empty vector control, (iii) IFN-I treated cytosol mixed with Ubp43 expressed in HEK 293T cells (iv) IFN-I treated cytosol mixed with CCHFVL-Otu (OtuL) expressed in HEK 293T cells (v) IFN-I treated cytosol mixed with catalytically dead variant of CCHFVL-Otu (mOtuL) expressed in HEK 293T cells and chased for 30 and 60 mins. (A) Pellet fractions were lysed and immunoprecipitated with anti-HA IgG2b on Protein-G beads. Control cytosol (lanes 1–3); IFN treated cytosol with empty control vector (lanes 4–6); cytosol from IFN-treated cells mixed with Ubp43 (lanes 7–9), with CCHFVL-Otu (lanes 10–12) or mutant CCHFVL-Otu (lanes 13–15). (B) Supernatant fractions from permeabilized cells were treated with chicken erythrocytes to isolate intact virions. Virus particles released with control cytosol (lanes 1–3), with that from IFN-I treatment mixed with either Ubp43 (lanes 7–9) or CCHFVL-Otu (lanes 10–12). A block in release occurred with IFN-treated cytosol mixed with either control empty vector (lanes 4–6) or mutant CCHFVL-Otu (lanes 12–15). (C) Intact A549 cells either mock treated or with IFN-I for 8 hours (upper left panel) was infected with HA-srt virus (A/WSN/33 with a sortaggable HA) for 5 hours through a brief acid shock at pH 5, pulse labeled with [35S]-cys/met and chased for indicated time intervals. HA at the cell surface was biotinylated with Sortase A and GGGK-biotin and isolated on NeutrAvidin beads whereas total HA in the cell was immunoprecipitated on anti-HA antibodies. A549 cells transfected with either empty control vector or Ubp43 were IFN-I treated and infected with HA-Srt as above (upper right panel). Surface HA was biotinylated, total HA was immunoprecipitated on anti-HA antibodies. (Lower panels): Amount of HA reaching the cell surface was quantitated through densitometric analysis of the ratio of biotinylated HA to the total (intracellular + surface) HA [(HAbiotin/HATotal) *100]. Error bars are from three independent experiments. (D) Radiolabeled particles released into the media from intact A549 cells were adsorbed onto chicken erythrocytes and resolved by SDS-PAGE.
To corroborate our findings in intact cells, we used the human lung epithelial cell line A549. We introduced either a control vector or an Ubp43 construct into these cells and treated them with IFN-I for 8 hours. Since IFN-I treatment renders cells refractory to infection, we forced fusion of virus (HA-Srt; MOI of 1) at the plasma membrane by a brief exposure to pH 5.5 to deliver vRNPs to the cytosol (White et al., 1981). The HA-Srt virus generated from A/WSN/33 contains an LPETG motif on its HA that can be selectively biotinylated using Sortase A (Popp et al., 2012). Upon incubation (4 hours) to allow synthesis of viral proteins, we performed a pulse-chase experiment. At each time point, cell surface-displayed HA was biotinylated using Sortase A and isolated on NeutrAvidin beads, whereas total HA was immunoprecipitated using anti-HA antibodies. We observed a block in arrival of HA at the cell surface in IFN-I treated cells, restored in transfectants that express Ubp43, (Figure 3C). Supernatants released from these cells were adsorbed onto chicken erythrocytes and resolved by SDS-PAGE (Figure 3D). As observed with the surface appearance of HA, release of intact virions was blocked upon IFN-I treatment and restored by expression of Ubp43. The presence of radiolabeled M1 at the later time points in IFN-I treated cells is most likely attributable to non-specific adsorption due to release of viral components by dying cells.
HA transport to the cell surface is Vps4-independent but Tsg101-dependent
An obvious target of ISGylation is the endosomal sorting complex required for transport (ESCRT) machinery. Components of the ESCRT machinery can be ISGylated and have been implicated in budding of retroviruses such as HIV-I (Martin-Serrano et al., 2001; Okumura et al., 2006). For the biogenesis of IAV, involvement of the ESCRT complex was considered unlikely, since a dominant negative version of Vps4, an effector protein on which the ESCRT complexes converge, did not affect virus budding (Watanabe and Lamb, 2010). Instead, IAV budding is facilitated by M2, an ion channel equipped with an amphipathic helix that can alter membrane curvature (Chen et al., 2008; Rossman et al., 2010). We prepared cytosol from MDCK cells by permeabilization using PFO and depleted it of either Vps4 or Tsg101 (Figure 4A). Only trace amounts of Vps4 or Tsg101 remained, as verified by immunoblotting. Levels of GAPDH remained unchanged, verifying that depletion was specific. Cytosol that either contained Vps4 or that had been depleted of Vps4 was applied to semi-intact cells infected with IAV to measure trafficking of HA. Both Vps4-replete and Vps4 depleted cytosol supported transport of HA to the cell surface (Figure 4B).
Figure 4. HA transport to the cell surface is Vps4-independent but Tsg101-dependent.

(A) Immunodepletions from MDCK cytosol performed with anti-Vps4 or anti-Tsg101 antibodies on Protein A beads. Beads without antibodies were used as control. Equivalent fractions of input and flow-through were probed for Vps4 and Tsg101 respectively. GAPDH was used as a loading control to verify that depletion was specific. (B) 1 × 106 MDCK cells were infected with Influenza A/WSN/33 at an MOI of ~0.5 for 5 hours and radiolabeled with [35S]-cys/met. Cells were permeabilized with 100 nM PFO and supplemented with (i) concentrated MDCK cytosol, (ii) MDCK cytosol treated with control beads, (iii) cytosol depleted of Vps4 (iv) cytosol supplemented with GTPγS and chased for 30 and 60 mins. Control cytosol (lanes 1–3), cytosol containing Vps4 (lanes 4–6) and cytosol lacking Vps4 (lanes 7–9). Cytosol supplemented with GTPγS (lanes 10–12). (C) 1 × 106 MDCK cells were infected with Influenza A/WSN/33 at an MOI of ~0.5 for 5 hours and radiolabelled with [35S]-cys/met. Permeabilized cells were supplemented with (i) concentrated MDCK cytosol either mock depleted or Tsg101-deficient (ii) MDCK cytosol treated with IFN-I (iii) MDCK cytosol treated with IFN-I mixed with Ubp43 and either mock depleted or Tsg101-deficient (iv) cytosol supplemented with GTPγS and chased for 30 and 60 mins. Untreated cytosol containing Tsg101 (lanes 1–3); cytosol lacking Tsg101 (lanes 4–6). Cytosol from IFN treated cells treated with Ubp43, containing Tsg101 (lanes 10–12) or Tsg101 deficient cytosol (lanes 13–15). Cytosol supplemented with GTPγS (lanes 16–18). Autoradiograms are representative of atleast 3 independent experiments. (D) HEK293T cells were co-transfected with HA-ISG15 and either empty vector or Ubp43 de-ISGylase. Transfected cells were either untreated or treated with type-I IFN for 6 hours (100 U/ml) and lysed in NP-40. Lysates were immunoblotted with anti-HA antibodies. ISGylated products were immunoprecipitated with anti-HA and probed with anti-Tsg101.
Tsg101 is a component of the ESCRT-I complex involved in intracellular transport and assembly of a number of viruses such as HIV and Ebola (Martin-Serrano et al., 2001). To test the involvement of Tsg101 in transport and externalization of HA, we immunodepleted cytosol with anti-Tsg101 as described for Vps4 (Figure 4C). We delivered control and Tsg101-deficient cytosol to [35S]cysteine/methionine labeled semi-intact cells infected with IAV. Samples that received mock-depleted cytosol displayed unaltered kinetics of HA transport to the cell surface. Tsg101-depleted cytosol supported ER to Golgi transport but not post-Golgi traffic of HA to the PM. Our results indicate that Tsg101 is required for HA to reach the cell surface from the Golgi network.
We next determined whether the Tsg101 dependence observed in infected cells also applies to cytosol from cells treated with IFN-I. Samples were supplemented with cytosol transduced with Ubp43 to relieve transport of HA to the cell surface and tested in the presence or absence of Tsg101. Samples that received mock-depleted cytosol from IFN-I treated cells supplemented with Ubp43 supported post-Golgi HA transport, whereas Tsg101-deficient extracts failed to do so, regardless of the presence of Ubp43. GTPγS supplemented cytosol was used as control for permeabilization. We conclude that intracellular traffic of HA relies on a Tsg101-dependent step and is sensitive to ISGylation. We examined the modification status of Tsg101 in transfectants that express HA-tagged ISG15. Covalent modification of Tsg101 with ISG15 is observed only in cells exposed to IFN-I, required for induction of the ISG15 conjugation apparatus, and is reversed by cotransfection with the deISGylase Ubp43 (Figure 4D).
IFN-I imposes a block in transport of HA to the cell surface in Cas9/CRISPR-mediated Tsg101 knock out cells
We generated Tsg101-deficient A549 cells using the Cas9/CRISPR method (Cong et al., 2013; Hwang et al., 2013; Mali et al., 2013) as well as siRNA-mediated knock-downs (data not shown). Although ablation of Tsg101 is embryonic lethal (Wagner et al., 2003), we obtained several clones of Tsg101−/− A549 cells. These displayed reduced growth rates compared to wild type cells, but showed no gross morphological defects as seen by light microscopy. We isolated five clones for which deletion of Tsg101 was verified by immunoblotting of cell lysates (Fig 5A). Tsg101−/− cells showed no residual Tsg101 and were refractory to infection by IAV, possibly due to aberrations in the endocytic machinery imposed by the Tsg101 deficiency. This observation underscores the advantage of using semi-intact cells to separate entry and assembly defects that are otherwise difficult to identify in genome-wide screens applied to intact cells. In perforated cells, the only distinction between the two experimental settings is the presence or absence of Tsg101 from the delivered cytosol during these short incubations, all intracellular organelles being the same. Indeed, Tsg101 did not surface as a significant host factor in any of the published RNAi screens (Brass et al., 2009; Karlas et al., 2010; Shapira et al., 2009). To circumvent the resistance of Tsg101−/− cells to IAV infection through the standard endosomal entry pathway we resorted to low pH fusion of IAV to deliver vRNPs directly into the cytosol and performed a pulse-chase experiment. At each time-point, surface-disposed HA was biotinylated using sortase A (Figure 5B; upper panel). Total intracellular HA was immunoprecipitated using anti-HA antibodies (Figure 5B; lower panel). We observed a complete block in arrival of HA at the cell surface in Tsg101−/− cells, confirming our findings with permeabilized cells (Fig 5B). Release of intact virions into the supernatant was likewise blocked in Tsg101−/− cells as observed by the absence of radiolabeled virus particles (Fig 5D). Inhibition of release of infectious virus from Tsg101−/− cells was essentially complete, as confirmed by plaque assays (Fig 5C).
Figure 5. Cas9/CRISPR mediated knock-out of Tsg101 in A549 cells.

(A) Clonal isolates of A549 cells were immunoblotted for Tsg101 (+: control Tsg101 proficient cells, 1–9: different clonal isolates after limiting dilution). (B) Wild-type and Tsg101−/− cells were infected with HA-Srt for 5 hours (MOI = 1) through a brief acid shock at pH 5.0 followed by pulse-labeling with [35S]-cys/met for 10 mins and chase for the indicated time intervals. At each time point cell surface HA was biotinylated through Sortase A and GGGK-biotin and isolated on NeutrAvidin beads. Remaining intracellular HA was immunoprecipitated on anti-HA antibodies. (C) Supernatants from infected (MOI 1 at pH 5.0; 4 hours) wild-type or Tsg101 knock out cells (3 independent clones) were used to infect MDCK cells in 10 fold serial dilutions. MDCK cells were plated and overlaid with agar. Following two days of infection cells were stained with biotin conjugated anti-NP VHH for visualization. Viral titers were quantitated from serial dilution of triplicates. Error bars represent mean ± SD. (D) Radiolabeled particles released into the media from wild-type and Tsg101−/− were adsorbed onto chicken erythrocytes and resolved by SDS-PAGE. (E) Wild-type and Tsg101−/− A549 cells were infected with IAV (pH 5.0; MOI = 1.0) and incubated for 5 hours. Fixed and permeabilized cells were stained with anti-HA to identify virus particles and anti-TGN46 to define the trans Golgi network. Images were acquired with a Perkin Elmer spinning disk confocal microscope.
To identify the site at which the block in HA transport occurs, we prepared control and Tsg101−/− samples infected with IAV at low pH for 5 hours. Fixed and permeabilized cells were stained with TGN46 and anti-HA antibodies and examined by confocal microscopy (Fig 5E). In wild-type A549 cells, HA was distributed throughout the cell in a typical punctate pattern whereas in Tsg101−/− cells, HA was found in large accumulations that co-localized with the TGN network. Our results in Tsg101−/− cells, combined with those of permeabilized cells, show that Tsg101 plays a critical role in transport of HA from the TGN to the plasma membrane.
Specific host proteins traffic via Tsg101-dependent pathway upon IFN-I induction
Tsg101-dependent transport of HA to the cell surface could either be specific for IAV proteins or could be a more general consequence of IFN-I exposure, which might affect other glycoproteins as well. We asked whether trafficking of at least a subset of endogenous glycoproteins would display the same properties as that of HA upon IFN-I induction. We took advantage of two glycoprotein reporters, CD40L and transferrin receptor (TfR), both engineered to contain an LPETG tag, which can be biotinylated on the surface of intact cells in a sortase A-catalyzed reaction (Popp et al., 2007; 2009). We generated MDCK cells that stably express either CD40L or TfR with an LPETG and an HA epitope tag placed at their C-terminus for quantitation of total protein levels. We first tested whether trafficking characteristics of these reporters were altered upon IFN-I induction. Intact MDCK cells expressing either CD40L or TfR were exposed to IFN-I for 8 hours, radiolabeled and chased for various time intervals. Arrival at the cell surface was monitored through sortase A-catalyzed biotinylation. We observed a clear difference between transport characteristics of CD40L and TfR to the cell surface upon IFN-I treatment. Arrival of CD40L at the cell surface was abolished (Figure 6A) whereas that of TfR was unaffected (Figure 6A) by IFN-I treatment. To confirm these results we measured transport of TfR in permeabilized cells, supplemented with cytosol from control or IFN-I treated cells, either proficient or deficient in Tsg101. Under all conditions, TfR transport to the cell surface was unaffected (Figure 6B).
Figure 6. Tsg101-dependent transport of endogenous host proteins.
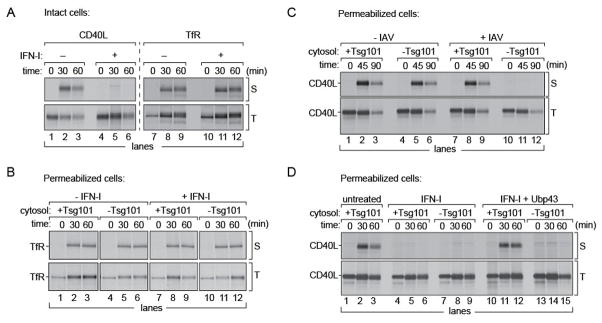
(A) MDCK cells stably expressing either CD40L or TfR were treated with IFN-I for 8 hours, radiolabeled with [35S]-cys/met for 15 mins and chased for indicated time intervals. At each time point surface displayed CD40L or TfR were biotinylated. Total CD40L or TfR were immunoprecipitated on anti-HA beads. (B) TfR expressing cells were radiolabeled, permeabilized and supplemented with (i) untreated MDCK cytosol either Tsg101-replete or Tsg101-deficient (ii) IFN-I treated, Tsg101-replete or Tsg101-deficient cytosol. At each time point cell pellets were biotinylated through Sortase A and GGGK-biotin and isolated on NeutrAvidin beads. Total TfR was immunoprecipitated on anti-HA beads. (C) MDCK cells expressing CD40L were either mock infected or infected with A/WSN/33 virus, MOI of 0.5 for 5 hours and radiolabeled. Permeabilized cells were supplemented with Tsg101-replete or Tsg101-deficient cytosol. At each time point, pellets were treated with Sortase A and biotin for 1 hour on ice, and immunoprecipitated on Neutravidin beads (S: surface displayed). Intracellular CD40L was immunoprecipitated on immobilized HA beads (T: total). Uninfected cells (lanes 1–3, 4–6) For flu-infected cells, transport of CD40L to the PM was abolished in the absence of Tsg101. (D) CD40L expressing cells were radiolabeled, permeabilized and supplemented with (i) untreated MDCK cytosol (ii) IFN-I treated, Tsg101-replete or Tsg101-deficient cytosol (iii) IFN-I treated Ubp43-expressing cytosol, either Tsg101-replete or Tsg101-deficient. At each time point, pellets from these semi-intact cells were (a) treated with Sortase A and biotin (S: surface displayed). (b) Lysed and immunoprecipitated on immobilized HA beads (T: total). Uninfected cells, (lanes 1–3, 4–6), IFN-I treated cells in the presence or absence of Tsg101 (lanes 4–6, 7–9), IFN-I treated cytosol supplemented with Ubp43 in the presence of Tsg101 (lanes 10–12) and the absence of Tsg101 (lanes 13–15). Autoradiograms are representative of atleast 3 independent experiments.
To explore whether the defect in CD40L transport upon IFN-I treatment was also a Tsg101-dependent process, we first tested whether trafficking of CD40L was altered by virus infection. Transport of CD40L to the cell surface was monitored in IAV-infected samples and in mock-infected controls (Figure 6C). For both samples, cells were first radiolabeled, permeabilized and incubated with exogenous MDCK cytosol containing Tsg101, or immunodepleted of Tsg101. At 45 and 90 min., pellet fractions were collected and the surface-disposed pool of CD40L was modified by sortase A-catalyzed biotinylation followed by immunoprecipitation on NeutrAvidin beads. Since the LPETG tag is luminally oriented, it remains protected from Sortase A unless exposed at the cell surface. Transport of CD40L to the surface and subsequent biotinylation was not affected in mock-infected control cells, irrespective of Tsg101 status. However, surface exposure of CD40L in cells infected with IAV required Tsg101 (Figure 6C; upper panel). Expression levels of CD40L remained constant for all samples (Figure 6C; lower panel). We conclude that post-Golgi protein transport to the cell surface of CD40L becomes Tsg101-dependent during IAV infection.
Next we tested whether alteration of CD40L trafficking patterns was the result of an antiviral response that ensues upon virus infection. We used MDCK cells that stably express CD40L and radiolabeled them in a 15 min pulse. We permeabilized these cells and incubated them with cytosol from IFN-I treated cells, either Tsg101-replete or immunodepleted with anti-Tsg101 antibodies. At 30 and 60 min, pellet fractions were collected and the surface-disposed fraction of CD40L was modified by sortase A-catalyzed biotinylation as described. Samples incubated with untreated MDCK cytosol supported CD40L transport and biotinylation at the cell surface (Figure 6D; upper panel). In the absence of any viral infection, but when supplying IFN-I treated cytosol, transport of CD40L to the cell surface was compromised, irrespective of the presence of Tsg101. Results with CD40L thus recapitulate the characteristics of HA transport under IFN-I treatment, and CD40L presumably suffers a similar ISG15-imposed arrest. To relieve ISG15-mediated inhibition of transport, we prepared cytosol from IFN-I treated cells and mixed it with Ubp43-containing cytosol (as described above), followed by immunodepletion of Tsg101. In cells that were incubated with cytosol from IFN-I exposed cells supplemented with Ubp43, transport of CD40L to the cell surface was restored in the presence of Tsg101. Cytosol depleted of Tsg101 failed to support appearance of CD40L at the cell surface. Total levels of CD40L remained constant as detected by immunoprecipitation via the HA-epitope (Figure 6D; lower panel). We conclude that upon treatment with IFN-I, CD40L trafficking patterns are altered such that they become dependent on Tsg101 and sensitive to ISGylation, whereas that of CD71 remains unaffected due to distinct routes of transport.
Discussion
Cells exploit two major trafficking pathways for internalization and export of cargo – the endocytic and the secretory pathways respectively. Proteins destined for the extracellular environment enter the secretory pathway, mostly by co-translational translocation into the ER and subsequent vesicular transport to the Golgi and finally to the cell surface. However, during viral or bacterial infections, resulting in IFN induction, intracellular host protein trafficking routes must necessarily be altered to regulate cytokine secretion and transport of other host proteins. Viral glycoproteins likely possess intrinsic determinants for selecting sites of assembly, transport routes and budding (Rossman and Lamb, 2011; Schmitt and Lamb, 2005). Amongst other host proteins typically involved in viral assembly, the family of proteins of the endosomal sorting complex required for transport (ESCRT) interact with the Gag and matrix proteins of a number of viruses, such as HIV and Ebola (Martin-Serrano et al., 2001) (Jouvenet et al., 2011) (Freed, 2004). These proteins include Tsg101 and those of the Nedd4 ligase family (Okumura et al., 2008). Tsg101 functions in the ESCRT-I complex as part of the vacuolar sorting machinery, where it helps select cargo for incorporation into vesicles that bud into multivesicular bodies (MVBs) (Razi and Futter, 2006). This process requires the formation of vesicles that bud away from the cytosol into the topological equivalent of extracellular space, a process essentially similar to virus budding at the PM (Hanson and Cashikar, 2012). Tsg101 is homologous to a ubiquitin E2 conjugating enzyme, but lacks the ability to engage in ubiquitin transfer, notwithstanding its ability to interact tightly with ubiquitin.
Influenza viruses exit from discrete sites at the plasma membrane (Popp et al., 2012; Rossman and Lamb, 2011), enriched in cholesterol and sphingolipids. HA is the most abundant of the three envelope proteins, with critical determinants for lipid association in its transmembrane domain (Ruigrok et al., 2000; Tafesse et al., 2013; Tsurudome et al., 1992). Because a mutant version of the AAA ATPase Vps4 devoid of enzymatic activity does not inhibit IAV release, involvement of the ESCRT complex in IAV budding was considered unlikely (Bruce et al., 2009; Watanabe and Lamb, 2010). In addition, M2 alters membrane curvature in vitro, suggesting that the final step of membrane scission can be executed without involvement of Vps4 (Rossman et al., 2010). Both intracellular transport and surface display of HA, as reconstituted here in semi-intact cells, support the lack of involvement of Vps4 in intracellular trafficking of HA or release of virus. In contrast, depletion of Tsg101 abolishes HA transport to the cell surface and subsequent release of virus. In the semi-intact cell system, Tsg101-deficient cytosol is added to infected, but otherwise normal cells. There is therefore no reason to assume that the secretory pathway in these cells is massively defective, an argument supported by the ability to transport HA and allow the release of virus particles. Indeed, the provision of control cytosol is required to have transport proceed, while Tsg101-depleted cytosol is ineffective. Although the final stages of virus budding may indeed depend on M2, Tsg101 appears to play a critical role in recruitment of HA to the cell surface, prior to release of IAV. Our demonstration that Tsg101 is itself ISGylated in IFN-treated cells provides a conceptual link between Tsg101’s ability to orchestrate membrane trafficking, and the consequences of IFN-I treatment on the intracellular movement of glycoproteins. Transport of an engineered CD40L, shows a remarkable similarity to that of IAV HA, whereas an unrelated type II membrane protein, TfR, appears to follow a distinct trafficking route. The Tsg101-mediated mode of transport is either not active or redundant under normal physiological conditions, as confirmed by depletion from regular cytosol with no inhibitory effect on CD40L transport. The features of structure that confer this regulatory property on membrane proteins remain to be determined, but do not easily segregate according to their type I or type II transmembrane topology. We speculate that this property might extend to secreted proteins as well. Accordingly we propose that factors induced by IFN-I treatment differentially affect aspects of the secretory pathway, the details of which remain to be determined.
The M1 protein encoded by Influenza A virus contains a putative late domain motif, YKRL, and interacts with the UEV domain of Tsg101 (Diaz et al., 2009). M1 could facilitate influenza budding similar to the well-established interaction of HIV Gag or Ebola VP-40 with Tsg101. Virus infection of host cells elicits the production of IFN and presumably also attenuates or alters secretory activity. We speculate that transport vesicles are generated in a Tsg101-dependent fashion from the Golgi network, while induction of IFN-I affects such trafficking (Figure 7). ISGylation of Tsg101 and possibly other components of the ESCRT machinery would interfere with normal operation of at least some of these transport pathways. The trafficking characteristics of CD40L and TfR upon IFN-I induction might depend on features of their transmembrane segments, requiring specific lipid-protein interactions, as observed for Influenza HA and sphingomyelin (Tafesse et al, 2013). These transport vesicles may be akin to sorting endosomes or the ESCRT-dependent MVBs typically generated from late endosomes – a multi-subunit complex with several components modified by ISG15. Hijacking of these transport vesicles by IAV with its glycoproteins anchored to the limiting membrane would allow arrival at the PM, followed by M2-mediated budding. The operation of this trafficking route is partially supported by the finding that Rab11 – a protein enriched in the TGN and recycling endosomes/MVBs – is required for IAV budding (Bruce et al., 2010; Cox et al., 2000; Eisfeld et al., 2011). Our results predict that IAV must be able to either exploit host de-ISGylases or employ NS1 to prevent ISG15 levels from reaching a threshold to be able to overcome the inhibition imposed by IFN induction. Future work will determine whether or not that occurs.
Figure 7. Proposed model for intracellular influenza trafficking and budding.

Proteins destined for the cell surface are synthesized at ER bound ribosomes and traffic through the secretory pathway to arrive at the extracellular space. Influenza virus infection results in induction of IFNs α/β, which in turn create an antiviral state in the host cell by regulating gene expression and protein translation. Tsg101 functions as part of the ESCRT complex to form multivesicular bodies; induction of IFNs activates an ISG15 regulated Tsg101 dependent pathway for re-routing proteins to the plasma membrane. This pathway is exploited by Influenza glycoproteins along with other membrane proteins such as CD40L to traffic to the plasma membrane upon IFN induction.
Experimental procedures
Antibodies, cell lines, constructs
MDCK, Vero, A549 and HEK293T cells were purchased from American Type Culture Collection (ATCC) and cultured. Serum containing IgG against A/WSN/33 Influenza HA was from a transnuclear mouse generated in the laboratory. Antibody against the HA-epitope was purchased from Roche (3F-10). Plasmid encoding PFO was provided by Art Johnson (Texas A&M University, TX). Antibodies against Tsg101, Vps4, IAV M1 were purchased from Abcam. GTPγS was purchased from Sigma. Construct for recombinant Perfringolysin O (PFO) and its purification have been described before (Sanyal 2012, Flanagan et al., 2009). HA-agarose beads were purchased from Roche Applied Science and Protein A-agarose beads were from RepliGen Bioprocessing. Generation of HA-srt virus has been described before (Popp et al., 2012).
HA-transport assays and virus budding in PFO-permeabilized cells
MDCK cells were grown to 80–90% confluency, infected with WSN/33/A virus for 5 hours and labeled with [35S]cysteine/methionine at 37°C in suspension. Labeled cells were treated with 0.1 μM PFO (in HBSS) on ice as described above. Excess PFO was removed by diluting in HBSS and centrifugation. For measuring IAV trafficking and budding, ~2.5 × 106 PFO treated semi-intact cells were incubated with ~100 μg of concentrated MDCK cytosol in 50 μl and incubated at 37°C for 30 and 60 min. For all samples, cytosol preps used were normalized to total protein concentrations. At each time point cell suspensions were centrifuged at 1000 × g for 3 min to separate the pellet and supernatant fractions. The pellets were washed once with cold PBS and lysed (0.5% NP-40, 150 mM NaCl, 5mM MgCl2, 25 mM Tris, pH 7.5). The supernatants were diluted to 500 μl in PBS centrifuged further at 2000 rpm for 5 min to remove any contaminants and treated with chicken erythrocytes to isolate released virus particles. HA was recovered with serum from transnuclear mouse immobilized on Protein G-sepharose. Immunoprecipitated material was resolved by SDS-PAGE and visualized by autoradiography. Chicken erythrocytes with adsorbed virions were lysed in resolving sample buffer and loaded directly for SDS-PAGE.
Designing CRISPR target sequence and Prediction of Potential off-targets
Potential target sequence for CRISPR interference were found using the rules outline in (Mali et al., 2013). The seed sequence preceding PAM motif was found in the exon of Tsg101 as follows:
Tsg101 target sequence: CTGTTCTGTTTTCAGGCCGAGG
Potential off-target effects of the seed sequence was confirmed using NCBI human nucleotide blast.
Procedure for Generating CRISPR RNA Expressing Vector
The sense and anti-sense were designed to incorporate into the restriction enzymatic site BbsI of pX330 bicistronic expression vector expressing Cas9 and sgRNA (Cong et al., 2013) as follows.
Sense oligo: caccgCTGTTCTGTTTTCAGGCCG
Anti-sense oligo: aaacCGGCCTGAAAACAGAACAGc
The oligo DNAs were annealed, phosphorylated and incorporated into pX330 vector linearized with BbsI restriction enzyme.
Limiting dilution to generate Tsg101 knockout cell lines
A549 cells were transfected with the Tsg101-targeting pX330 using FuGENE6 reagent (Promega) according to the manufacturer’s instructions. Twelve hours after transfection, the cells were re-plated to a 96 well plate at a density of 0.5 cells per well. The individual colonies were picked, and the expressions of Tsg101 were checked by immunoblotting.
Plaque assay
Serial dilutions of supernatants from infected cells were overlaid on MDCK cells and incubated for 30 min. Cells were washed and plaque media was added to the cells (MEM, 0.8% agar, 1μg/mL trypsin) and placed at 37°C. 48 h after infections, cells were fixed in 4% paraformaldehyde and the agar plug was removed. Cells were blocked in 5% BSA, 1% goat serum and probed first with anti-NP VHH54-biotin followed by streptavidin-HRP (GE Cat. No. RPN1231V). Plaques were subsequently visualized using True Blue Peroxidase Substrate (KPL Cat. No. 50-78-02).
Immunodepletions of Tsg101 and Vps4
100 μl of concentrated cytosol from MDCK cells (500 μg protein) isolated by PFO treatments were subjected to either control beads, anti-Vps4 or anti-Tsg101 immobilized on Protein A-sepharose beads. Following incubation for 3 hours at 4°C, flow-through fractions were collected from mock-depleted, Vps4-deficient or Tsg101-deficient samples and subjected to immunoblotting to confirm depletion. GAPDH was used as a loading control to verify that depletion was specific. Immunodepleted cytosol was incubated with semi-intact cells to measure HA transport and virus budding as described.
Supplementary Material
Highlights.
IFN-I-treated cell cytosol abolishes IAV HA cell surface transport in semi-intact cells
IFN-dependent HA transport inhibition requires Tsg101 and is relieved by deISGylases
Intact Tsg101-deficient cells are defective for cell surface HA and virion release
Tsg101 is itself ISGylated in IFN-treated cells
Acknowledgments
This work was funded by NIH (RO1 grants AI033456 and AI087879 to HLP), Sanofi Pasteur (HLP) and partially by Center for Research on Influenza Pathogenesis, NIAID CEIRS contract HHSN266200700010C and NIAID grant U19 AI083025 (AG-S). For technical assistance and discussions, the authors acknowledge Richard Cadagan, Fikadu Tafesse, Carla Guimaraes and Lee Kim Swee.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce EA, Digard P, Stuart AD. The Rab11 pathway is required for influenza A virus budding and filament formation. Journal of Virology. 2010;84:5848–5859. doi: 10.1128/JVI.00307-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce EA, Medcalf L, Crump CM, Noton SL, Stuart AD, Wise HM, Elton D, Bowers K, Digard P. Budding of filamentous and non-filamentous influenza A virus occurs via a VPS4 and VPS28-independent pathway. Virology. 2009;390:268–278. doi: 10.1016/j.virol.2009.05.016. [DOI] [PubMed] [Google Scholar]
- Chen BJ, Leser GP, Jackson D, Lamb RA. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. Journal of Virology. 2008;82:10059–10070. doi: 10.1128/JVI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D, Lee DJ, Dale BM, Calafat J, Greenberg S. A Rab11-containing rapidly recycling compartment in macrophages that promotes phagocytosis. Proc Natl Acad Sci USA. 2000;97:680–685. doi: 10.1073/pnas.97.2.680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Cassella, Bonavia Duan, Santos Fesseha, Goldblatt, Kinch Recruitment of the TSG101/ESCRT-I Machinery in Host Cells by Influenza Virus: Implications for Broad-Spectrum Therapy. Antiviral Research. 2009;82:0–0. [Google Scholar]
- Eisfeld AJ, Kawakami E, Watanabe T, Neumann G, Kawaoka Y. RAB11A is essential for transport of the influenza virus genome to the plasma membrane. Journal of Virology. 2011;85:6117–6126. doi: 10.1128/JVI.00378-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R, Claessen JHL, Mueller B, Sanyal S, Spooner E, van der Veen AG, Kirak O, Schlieker CD, Weihofen WA, Ploegh HL. Enzymatic blockade of the ubiquitin-proteasome pathway. PLoS Biol. 2011;8:e1000605. doi: 10.1371/journal.pbio.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO. HIV-1 and the host cell: an intimate association. Trends Microbiol. 2004;12:170–177. doi: 10.1016/j.tim.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Frias-Staheli N, Giannakopoulos NV, Kikkert M, Taylor SL, Bridgen A, Paragas J, Richt JA, Rowland RR, Schmaljohn CS, Lenschow DJ, et al. Ovarian Tumor Domain-Containing Viral Proteases Evade Ubiquitin- and ISG15-Dependent Innate Immune Responses. Cell Host & Microbe. 2007;2:404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Cashikar A. Multivesicular body morphogenesis. Annu Rev Cell Dev Biol. 2012;28:337–362. doi: 10.1146/annurev-cellbio-092910-154152. [DOI] [PubMed] [Google Scholar]
- Hsiang TY, Zhao C, Krug RM. IFN-induced ISG15 conjugation inhibits influenza A virus gene expression and replication in human cells. Journal of Virology. 2009;83:5971–5977. doi: 10.1128/JVI.01667-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JRJ, Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature Biotechnology. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Simon SM, Bieniasz PD. Visualizing HIV-1 assembly. J Mol Biol. 2011;410:501–511. doi: 10.1016/j.jmb.2011.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–822. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- Lee MCS, Miller EA. Molecular mechanisms of COPII vesicle formation. Semin Cell Dev Biol. 2007;18:424–434. doi: 10.1016/j.semcdb.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277:9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7:1313–1319. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- Martin-Serrano J, Neil SJD. Host factors involved in retroviral budding and release. Nat Rev Microbiol. 2011;9:519–531. doi: 10.1038/nrmicro2596. [DOI] [PubMed] [Google Scholar]
- Okumura A, Lu G, Pitha-Rowe I, Pitha PM. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc Natl Acad Sci USA. 2006;103:1440–1445. doi: 10.1073/pnas.0510518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura A, Pitha PM, Harty RN. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc Natl Acad Sci USA. 2008;105:3974–3979. doi: 10.1073/pnas.0710629105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL. Sortagging: a versatile method for protein labeling. Nature Chemical Biology. 2007;3:707–708. doi: 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- Popp MW-L, Antos JM, Ploegh HL. Site-specific protein labeling via sortase-mediated transpeptidation. Curr Protoc Protein Sci. 2009;Chapter 15(Unit15.3) doi: 10.1002/0471140864.ps1503s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MWL, Karssemeijer RA, Ploegh HL. Chemoenzymatic Site-Specific Labeling of Influenza Glycoproteins as a Tool to Observe Virus Budding in Real Time. PLoS Pathog. 2012;8:e1002604. doi: 10.1371/journal.ppat.1002604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razi M, Futter CE. Distinct roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol Biol Cell. 2006;17:3469–3483. doi: 10.1091/mbc.E05-11-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Lamb RA. Influenza virus assembly and budding. Virology. 2011;411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Jing X, Leser GP, Lamb RA. Influenza Virus M2 Protein Mediates ESCRT-Independent Membrane Scission. Cell. 2010;142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok RWH, Barge A, Durrer P, Brunner J, Ma K, Whittaker GR. Membrane Interaction of Influenza Virus M1 Protein. Virology. 2000;267:289–298. doi: 10.1006/viro.1999.0134. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Claessen JHL, Ploegh HL. A viral deubiquitylating enzyme restores dislocation of substrates from the endoplasmic reticulum (ER) in semi-intact cells. Journal of Biological Chemistry. 2012;287:23594–23603. doi: 10.1074/jbc.M112.365312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AP, Lamb RA. Influenza virus assembly and budding at the viral budozone. Adv Virus Res. 2005;64:383–416. doi: 10.1016/S0065-3527(05)64012-2. [DOI] [PubMed] [Google Scholar]
- Seo EJ, Leis J. Budding of Enveloped Viruses: Interferon-Induced ISG15-Antivirus Mechanisms Targeting the Release Process. Advances in Virology. 2012;2012:532723. doi: 10.1155/2012/532723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira SD, Gat-Viks I, Shum BOV, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, et al. A Physical and Regulatory Map of Host-Influenza Interactions Reveals Pathways in H1N1 Infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaug B, Chen ZJ. Emerging role of ISG15 in antiviral immunity. Cell. 2010;143:187–190. doi: 10.1016/j.cell.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafesse FG, Sanyal S, Ashour J, Guimaraes CP, Hermansson M, Somerharju P, Ploegh HL. Intact sphingomyelin biosynthetic pathway is essential for intracellular transport of influenza virus glycoproteins. Proceedings of the National Academy of Sciences. 2013;110:6406–6411. doi: 10.1073/pnas.1219909110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurudome M, Glück R, Graf R, Falchetto R, Schaller U, Brunner J. Lipid interactions of the hemagglutinin HA2 NH2-terminal segment during influenza virus-induced membrane fusion. J Biol Chem. 1992;267:20225–20232. [PubMed] [Google Scholar]
- Wagner KU, Krempler A, Qi Y, Park K, Henry MD, Triplett AA, Riedlinger G, Rucker EB, III, Hennighausen L. Tsg101 Is Essential for Cell Growth, Proliferation, and Cell Survival of Embryonic and Adult Tissues. Mol Cell Biol. 2003;23:150–162. doi: 10.1128/MCB.23.1.150-162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Lamb RA. Influenza virus budding does not require a functional AAA+ ATPase, VPS4. Virus Res. 2010;153:58–63. doi: 10.1016/j.virusres.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, Matlin KK, Helenius AA. Cell fusion by Semliki Forest, influenza, and vesicular stomatitis viruses. Journal of Cell Biology. 1981;89:674–679. doi: 10.1083/jcb.89.3.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. Embo J. 2001;20:362–371. doi: 10.1093/emboj/20.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Collins M, Hsiang T-Y, Krug RM. Interferon-induced ISG15 pathway: an ongoing virus–host battle. Trends Microbiol. 2013:1–6. doi: 10.1016/j.tim.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Hsiang TY, Kuo RL, Krug RM. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proceedings of the National Academy of Sciences. 2010;107:2253–2258. doi: 10.1073/pnas.0909144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.