

Abstract
Aim
As the primary relevant tissue (brain) for psychiatric disorders is commonly not available, we aimed to investigate whether blood can be used as a proxy in methylation studies on the basis of two models. In the ‘signature’ model methylation–disease associations occur because a disease-causing factor affected methylation in the blood. In the ‘mirror-site’ model the methylation status in the blood is correlated with the corresponding disease-causing site in the brain.
Materials, methods & results
Methyl-binding domain enrichment and next-generation sequencing of the blood, cortex and hippocampus from four haloperidol-treated and ten untreated C57BL/6 mice revealed high levels of correlation in methylation across tissues. Despite the treatment inducing a large number of methylation changes, this correlation remains high.
Conclusion
Our results show that, consistent with the signature model, factors that affect brain processes (i.e., haloperidol) leave biomarker signatures in the blood and, consistent with the mirror-site model, the methylation status of many sites in the blood mirror those in the brain.
Keywords: antipsychotic, DNA methylation, methyl-CpG binding domain, methylome-wide association study, mirror-site model, next-generation sequencing, signature model
Epigenetic modifications of chromatin provide stability and diversity to the cellular phenotype. These modifications are largely preserved or regenerated during cell division [1–3]. One of the most intensively studied modifications is the methylation of DNA cytosine residues at the carbon 5′ position. This common epigenetic mark is most often, although not exclusively, found in the sequence context of CpG and typically associated with transcriptional repression.
Studies of DNA methylation have considerable potential to complement current studies of variation in genetic sequences [4,5]. Since methylation commonly affects gene expression, knowledge of the methylation status of a gene could aid prediction of disease susceptibility. Indeed, aberrant methylation has already been associated with a variety of human phenotypes, including neurological and psychiatric disorders, such as Alzheimer’s and Parkinson’s diseases [6], Rett’s syndrome [7], schizophrenia and bipolar disorder [8], and post-traumatic stress disorder [9]. Furthermore, methylation may help us to further our understanding of psychiatric disorders. For example, several studies have linked the female sex hormone estrogen to chromatin configuration and DNA methylation profiles at several specific loci in the genome [10,11], suggesting that sex differences observed for many psychiatric conditions may partially be mediated by epigenetic processes [12]. As methylation can be age dependent [13,14] and is dynamic in post-mitotic tissues in the brain [15], it can potentially also account for the different ages of onset or episodic nature of some psychiatric diseases [16]. Finally, methylation sites are potential new drug targets as they are modifiable by pharmaceutical interventions [17] and have good properties from a translational perspective such as being stable and enabling cost-effective assays in biosamples that can be relatively easy to collect [18].
Rapid advances in next-generation sequencing (NGS) now enable assessment of the methylation status of the majority of approximately all 28 million common CpGs in the human genome [1,19,20] allowing methylome-wide association studies (MWAS). This progress resembles the development of genome-wide association studies (GWAS), which resulted in the discovery of many new disease variants [21,22]. A fundamental difference between MWAS and GWAS is that methylation can be tissue specific and analyses are, therefore, ideally performed in the most relevant disease tissue [5]. For psychiatric conditions, where most of the pathogenic processes are likely to involve the brain, the only option would be to use post-mortem tissue. However, the number of available post-mortem samples is typically small, clinical information is limited and confounding factors such as cause of death may distort methylation profiles. The restricted availability of post-mortem brain tissue has made blood the typical tissue used in methylation studies of psychiatric conditions [23–25]. However, as only a smaller number of studies have addressed this topic [26], little is known about the usefulness of blood as a proxy for brain tissue in MWAS.
Figure 1 shows two models explaining how methylation studies in the blood could be informative. It is important to note that in both cases we do not assume that the methylation in the blood is causing the disease. In the ‘signature’ model the association between methylation in the blood and the disease occurs because a disease-related event alters methylation sites (Figure 1A). Traces of this event are preserved and can be detected in the blood [27,28]. Thus, although the sites where the methylation changes occur, differently methylated positions (DMPs), do not affect the psychiatric condition, they implicate an event that contributed to the disease. In the ‘mirror-site’ model the association between the methylation mark in the blood and the phenotype occurs because the methylation status of a site in the blood is correlated with a corresponding site in the brain that may be of relevance for the disease’s etiology (Figure 1B). The mirror sites can occur for several reasons. First, peripheral tissues may reveal methylation marks predating or resulting from the epigenetic reprogramming events affecting germ line cells and embryogenesis [29–32]. Second, blood contains cells that may be modified as they circulate through diseased tissues, and can also include cell-free DNA from those tissues [33]. As such, traces of the aberrant methylation in disease-targeted regions may be present in the blood. Third, genetic polymorphisms can alter methylation marks [34]. Finally, and perhaps most importantly, environmental factors may affect methylation in a similar way across tissues [35–37]. Although these DMPs may only have functional implications in specific tissues, it is very possible that the changes themselves are more systemic and produce similarities in methylation profiles across tissues.
Figure 1. Two path diagrams describing how methylation studies in blood are informative for psychiatric conditions.
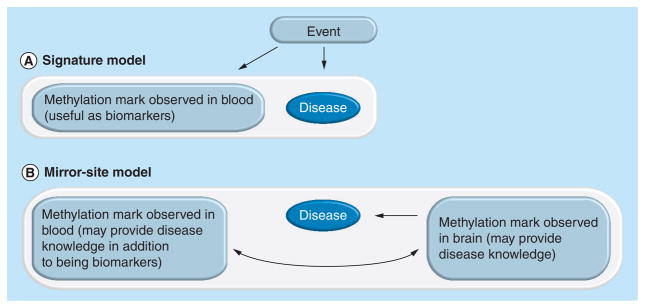
In neither model do we assume that the methylation mark observed in blood is causing the development of the disease. The white boxes indicate an association between features. The curved arrow indicates correlation. (A) The signature model: an event causes a methylation change in blood that is associated with the disease. (B) The mirror-site model: a methylation change in blood is associated with the disease. The site is also correlated with the methylation mark for the corresponding site in the brain, which may help reveal functional information related to the disease development.
To study the plausibility of the two models in Figure 1, in other words, whether factors that affect brain processes can leave signatures in the blood (signature model) and whether the methylation status of DMPs in the blood can mirror those in the brain (mirror-site model), we performed methylome-wide profiling using three tissues (blood, cortex and hippocampus) from 14 male C57BL/6 mice. Four of these mice were administered the highly potent anti-psychotic haloperidol, while the remaining ten were untreated. Haloperidol affects processes in the brain and has previously been associated with global changes in methylation [23]. Thus, the purpose of this treatment is not to specifically study the effects of haloperidol but to introduce a methylation change and investigate whether there is an overlap in effect in different tissues. The mice were raised in a controlled environment with minimal variation, the three tissues from each mouse were collected at a single time point, all mice were of the same age and, as we used an inbred mouse strain, the methylation differences were minimal. This maximizes our statistical power to detect induced changes and study the correspondence of subsequent changes in the blood and brain. To avoid drawing conclusions based on a limited number of preselected sites, we addressed our questions on a methylome-wide scale using methyl-CpG binding domain (MBD) protein-based enrichment in combination with NGS (MBD-seq).
Materials & methods
Study sample
The present study includes ten untreated mice and four haloperidol-treated mice. Adult C57BL/6 male mice (Jackson Laboratory, ME, USA) were housed five per cage on a 12-h/12-h light/dark cycle in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited animal facility with continuous access to food and water. At 11–12 weeks of age the mice were anesthetized with 4% isoflurane followed by cardiac puncture. A parallel set of mice were housed and treated as described with the exception that subcutaneous pellets (Innovative Research of America, FL, USA) releasing haloperidol at a continuous rate of 5 mg/kg/day for 28 days were implanted at approximately 8 weeks, which yields a plasma level in C57BL/6 that corresponds to the optimal therapeutic dosage (10 ng/ml) in humans [38]. A similar administration approach has previously been used for haloperidol [39,40].
Blood samples from heart punctures were collected in Microtainer® tubes with ethylene-diaminetetraacetic acid (Becton Dickinson, NJ, USA) and stored for <24 h at 4°C prior to DNA extraction. The cortex and hippocampus were extracted from all mice by a skilled technician, frozen in liquid nitrogen and stored at −80°C until DNA extraction. All procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals guidelines [41] and were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University (VA, USA).
Methylome-wide profiling
We used MethylMiner™ (Invitrogen, CA, USA), which employs MBD protein to enrich for the methylated genomic DNA fraction, followed by NGS (MBD-seq) on the Applied Biosystems SOLiD™ 4 platform (Life Technologies, CA, USA). Methods were standard and based upon manufacturers’ recommendations. Briefly, genomic DNA was fragmented with ultra sonication to a median fragment size of 150 bp. We extracted the methylated fraction of the genome using an elution buffer of 0.5 M NaCl. This concentration has previously been proven to yield a balanced representation of methylated fragments from regions with high and low CpG density [42]. The eluted material was used as input material for barcoded fragment libraries. Each library was sequenced using single-end chemistry and 50-bp read length. The MBD-seq approach has already been demonstrated to be highly specific, sensitive and applicable in identifying differently methylated regions [43–48].
We have recently developed an analysis pipeline for methylome-wide investigations that is specifically designed for MBD-seq [42]. In short, the sequenced reads were aligned to the C57BL/6 genome (build 9/NCBI37) using BioScope™ 1.2 (Life Technologies) that aligns in color space and takes full advantage of the enhanced ability of SOLiD’s two-base encoding to distinguish errors from sequence variation [49]. In the case of MBD-seq, only fragments with methylated CpGs can be extracted. Given that we know exactly where the CpGs are located in the reference genome, there is no need for algorithms [50,51] that search for read peaks to find methylated sites. Instead, we calculated the coverage for approximately 20.3 million autosomal CpG sites in the C57BL/6 genome [52]. A standard procedure is to count the sequence reads covering each CpG. Owing to the methylation of any CpG in the entire fragment potentially leading to its capture, the read length is sometimes extended to the expected fragment length. However, because not all of the fragments are exactly the same size, there may be variation between samples and the fragment pool obtained after shearing may not be identical to the pool that is successfully sequenced (e.g., smaller fragments may be more likely to be extracted by the enrichment protocol); therefore, this procedure can be imprecise. Thus, rather than assuming an identical predetermined fragment size for all fragments and samples, we estimated the fragment size distribution for each sample from the empirical sequencing data [52]. The sample-specific estimated fragment size distributions were used to calculate the probability for each read that the fragment it is tagging covers the CpG under consideration. Coverage for each CpG can then be calculated by taking the sum of the probabilities that all fragments in its neighborhood cover the CpG.
Continuous methylation measurements
MBD-seq is a semi-quantitative approach in the sense that it yields estimates that are directly comparable between samples for a specific site, where each site is (on average) covered by multiple reads and, therefore, gives an estimate of the methylation level [42]. However, when using methylation enrichment-based approaches, such as MBD-seq, the number of fragments covering a particular CpG depends not only on the methylation status of that site but also on the number of methylated CpGs in the region [43]. Prior comparisons involving multiple CpGs were performed for normalization of the coverage estimates using the local CpG density as a proxy for the number of methylated CpGs in the region [53,54]. For this purpose we calculated a so-called calibration curve [54] and normalized coverage estimates for each site by subtracting the appropriate expected mean. This normalization makes coverage estimates more comparable across sites and improves the correlation with actual methylation levels.
Dichotomized methylation measurements
Following a similar approach as was previously proposed by Harris et al. [43], we, in addition to the continuous measure, also used a dichotomize methylation measure. We first selected sites that were at least 400 bp away from the nearest CpG. As MBD-seq can only enrich for methylation occurring at CpGs, the coverage observed at these non-CpG sites cannot be due to methylation but reflects a ‘noise’ coverage level. Whereas unmethylated CpGs are expected to have a mean coverage level similar to the noise coverage level, methylated CpGs will on average have higher coverage. We called a CpG ‘likely methylated’ if its mean coverage level was higher than the 99th percentile of the coverage estimates at non-CpG sites. Thus, this method ensures that only 1% of the unmethylated CpGs are mistakenly called likely methylated.
Statistical analyses
To assess whether a treatment that affects methylation in the brain could cause similar changes in the blood we used a number of indices. In these analyses we treat the methylation measures both as a continuous variable (which we use to calculate correlations) and as a dichotomous variable (which we use to calculate concordances).
For each mouse we calculated the correlation between the continuous coverage estimates across tissues. This so-called sample correlation reflects the extent to which the coverage estimates for all CpG sites in an individual are ‘ranked’ in the same way across tissues. We also calculated the sample concordance, which measures the concordance in methylation status across tissues, using the dichotomized measure. With this measurement we calculated the concordance in methylation status between the different tissues.
If haloperidol causes DMPs in different sites across tissues, the sample correlation will be lower than in cases where haloperidol was not administered (Supplementary Equation 1; see www.future-medicine.com/doi/suppl/10.2217/epi.13.36). Therefore, to investigate whether the different tissues were similarly affected by the treatment we compared the sample correlations among untreated and treated mice.
To further examine whether processes that affect the brain cause corresponding changes in the blood, we calculated the site correlation using all four haloperidol-treated mice and four randomly selected untreated mice. Using an equal number of treated and untreated mice ensures a balanced design. Thus, for each CpG site we studied whether the coverage estimates for the mice ranked in the same way across two tissues. As we used an inbred mouse strain that has been raised in a controlled environment, the variation in methylation across mice was minimal. This maximizes our power to detect the effect of the treatment and study whether coverage changes in the blood mirror those of the corresponding sites in the brain. In contrast to the sample correlations, where all CpGs are considered for a single individual, the site correlations include one site at a time but data from eight mice. Therefore, the local CpG density normalization does not affect the site correlation calculations (these correlations are not affected by linear transformations), and the results are identical with or without this normalization.
The CpGs sites are probably of two types: those that have uncorrelated coverage between tissues and those that have correlated coverage levels across tissues. To disentangle these components, we fitted a two-component mixture model to the site correlations. We assumed a mixture of normal distributions (i.e., assuming conditional normality of the site correlations) and used the normalmixEM function in the statistical computational tool of the R project, which employs an expectation–maximization algorithm to estimate the model parameters: the mixing proportions, means and variances.
Finally, we also calculated site concordance, which is a dichotomous measure to explore whether corresponding changes could be observed in the brain and blood. If a change in methylation (or a lack of change) for a specific site was detected in two tissues we considered this site concordant. However, for a change to be considered, the direction of the effect was required to be the same (i.e., both tissues needed to show increased methylation or decreased methylation). A difference in methylation was defined as an absolute difference in mean coverage between untreated and treated mice greater than three-times the standard deviation of the untreated mice. If a specific site was methylated in only one of the three tissues, this site was considered to be a tissue-specific differently methylated region(T-DMR).
Bioinformatics
To study properties of different sets of CpGs that were concordantly or disconcordantly methylated in various tissues, we downloaded data from the UCSC genome browser [101]. The features that were considered included: CpG islands (CGIs; CGIs track), CGI shores (defined as 1000 bp on either side of a CGI), transcription factor binding sites (Stan/Yale transcription factor binding sites track), transcript boundary, information about the location of exons, introns and untranslated regions (RefSeq Gene track), potential gene promoter (represented by a region 2000-bp upstream of the transcription start site), repeats (Repeat-Masker track), and whether the CpG was located within an evolutionary conserved region (Euarch El track).
Results
The estimated confidence of each base call in the sequences is high. The quality value (QV) for a particular call is related to its probability of error:
The average QV across the samples were 23.4 (standard deviation [SD]: 1.06). This value can be compared with a QV of 20, which is considered to be a good quality standard. We generated on average 51.2 million (SD: 12.3 million) sequenced reads per sample that were used for alignment. When aligning the reads to the C57BL/6 genome (build 9/NCBI37) 74.2% of the sequenced reads were aligned.
Correspondence in methylation changes after haloperidol administration
Haloperidol changed the methylation status of 42.2% of the sites in at least one of the investigated tissues. The highest number of DMPs was observed in the hippocampus (23.2%), followed by the blood (21.1%) and cortex (19.4%). The fact that approximately the same number of DMPs were observed in each of the three tissues indicates that the factors that affect brain processes, here caused by haloperidol administration, could also alter methylation marks in the blood.
Figure 2 shows the sample correlations for the three tissue combinations for each of the untreated and the treated mice. The average sample correlation, with/without normalization on local CpG density, across all 14 mice is high for the three tissue combinations. The highest average correlation with/without normalization is observed for the cortex versus hippocampus (mean: 0.68/0.72; SD: 0.5/0.05) followed by the blood versus the cortex (mean: 0.60/0.68; SD: 0.07/0.07) and the blood versus the hippocampus (mean: 0.55/0.60; SD: 0.07/0.08). This shows that the coverage estimates for all CpG sites are ranked in a similar way across the three tissues. This effect was only slightly smaller between the blood and either of the brain tissues versus between the two brain tissues.
Figure 2. Sample correlations for each mouse.

Sample correlations for the three tissue combinations for each of the untreated (1–10) and treated (11–14) mice are shown on the left and right side of the vertical line, respectively. Sample correlations are given with (solid lines) and without (dashed lines) CpG density normalization.
As mentioned in the ‘Materials & methods’ section, if the treatment causes methylation changes in different sites across tissues, we would expect the sample correlation to be lower than in the case where the drug was not administered. When comparing the sample correlation between the groups of untreated and treated mice we did not observe any significant decrease in correlation. The p-values ranged from 0.09 to 0.95/0.06 to 0.96 for the three tissue combinations with/without normalization. Thus, the effect of the treatment did not significantly lower the sample correlations, which indicates that the tissues were partly similarly affected by the treatment.
Using the dichotomous definition (see the ‘Material & methods’ section) of whether a site is likely methylated, we found that the proportion of likely methylated sites to be the highest in the hippocampus (72.4 and 84.8%) followed by the cortex (64.4 and 75.4%) and blood (51.5 and 63.6%) in the untreated and treated mice, respectively. Furthermore, for untreated/treated mice 13.8/13.8% of all CpGs are methylated in the two brain regions, but not in the blood. Only 1.3/1.1% of CpGs are methylated in the blood, but not in any of the brain tissues. Further details are provided in Supplementary Tables 1 & 2. The pairwise sample concordances were very similar in untreated/treated mice. The highest pairwise concordance was observed for the cortex versus the hippocampus (88.1 and 87.6%), followed by the blood versus the cortex (82.5 and 82.5%) and the blood versus the hippocampus (75.4 and 75.0%) for untreated and treated mice, respectively. In addition, the total sample concordance rate for all three tissues (sites that have concordant methylation profiles in all three tissues) were very similar (73.0 and 72.7%) in untreated and treated mice, respectively. Although drug administration changed the methylation status of 42.2% of the sites in at least one of the investigated tissues and the percentage of likely methylated sites increased in treated mice, the sample concordance between tissues remained, which indicates that the tissues were similarly affected by the treatment.
Figure 3 shows the site correlations for all CpGs. The distributions of the site correlations are similar for all three tissue combinations. They are all clearly left skewed (the Pearson χ2 and Kolmogorov–Smirnov tests for normality were both highly significant; p < 1 × 10−15 for all three combinations), indicating an over-representation of strong positive correlations as compared with what is expected under the null hypothesis. The site correlation distributions in Figure 3 will comprise a mixture of uncorrelated and correlated sites. The mixture model estimated that the mean site correlations for components one and two were −0.01 and 0.62 for the cortex versus the hippocampus, −0.14 and 0.51 for the blood versus the cortex, and −0.08 and 0.54 for the blood versus the hippocampus, respectively. The site correlations for the second component indicated the presence of sites where knowledge of the methylation levels of a site in one tissue can be used to predict the methylation levels of that site in another tissue. For all three comparisons, the estimated percentage of sites in the second component (mean correlations ranging from 0.51 to 0.62) was 57–65% of the sites where we observed a change in methylation in at least one tissue. These percentages suggest that, given that a change is observed, the majority of sites show correlated methylation profiles across tissues. Assuming that all changes are uncorrelated across tissues we would expect, by chance, to see <5% (as compared with >7.5% observed) overlap between any two tissues and <1% (as compared with 4.5% observed) overlap across all three tissues. Thus, the observed overlap of DMPs is significantly different from what is expected by chance (p < 2.2 × 10−16). The pairwise site concordances (DMPs that changed in the same direction between different tissues or sites that did not change in any tissue) were similar for all three tissue combinations (Table 1). Highest pairwise site concordance was observed in the cortex versus the hippocampus (67.2%), followed by the blood versus the hippocampus (66.9%) and the blood versus the cortex (65.3%). In addition, the total site concordance for all three tissues was 62.3%. These percentages suggest that the majority of sites show concordant methylation profiles across tissues.
Figure 3. Site correlation for the three tissue combinations for all methylated CpGs in a balanced set of untreated and treated mice.

The x-axis shows the site correlation and the y-axis shows the density of the distribution.
Table 1.
Percentages of autosomal differently methylated positions altered by treatment.
| Blood | Cortex | Hippocampus | Frequency (%) | CGI (%) | CGI shore (%) | Upstream (%) | Gene (%) | UTR (%) | Exon (%) | Intron (%) | TFBS (%) | Isolated CpG (%) | Repeat (%) | Cons. (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| – | – | – | 57.8 | 5.9 | 3.2 | 4.3 | 46.0 | 2.9 | 9.3 | 37.2 | 5.4 | 0.9 | 37.1 | 1.9 |
| Δ | Δ | Δ | 4.5 | 0.9 | 1.4 | 2.2 | 40.2 | 1.5 | 3.1 | 37.3 | 2.2 | 0.2 | 43.3 | 0.5 |
| Δ | Δ | – | 3.0 | 3.2 | 3.0 | 3.8 | 44.2 | 2.2 | 4.7 | 39.7 | 4.5 | 0.8 | 36.5 | 1.3 |
| Δ | – | Δ | 4.6 | 2.1 | 2.5 | 3.2 | 41.4 | 1.7 | 3.4 | 38.2 | 3.7 | 0.6 | 38.9 | 1.0 |
| – | Δ | Δ | 4.9 | 2.1 | 2.3 | 3.1 | 39.9 | 1.7 | 3.5 | 36.6 | 3.6 | 0.6 | 42.2 | 1.0 |
| Δ | – | – | 9.0 | 5.0 | 3.4 | 4.5 | 43.4 | 2.5 | 5.3 | 38.5 | 5.6 | 1.3 | 35.9 | 1.8 |
| – | Δ | – | 7.0 | 7.0 | 4.1 | 5.6 | 44.9 | 3.2 | 6.6 | 38.7 | 7.1 | 1.3 | 35.2 | 2.5 |
| – | – | Δ | 9.2 | 5.0 | 3.6 | 4.7 | 42.0 | 2.4 | 4.8 | 37.6 | 5.9 | 1.1 | 37.0 | 1.9 |
| Δ in at least one tissue | 42.2 | 4.1 | 3.1 | 4.1 | 42.4 | 2.3 | 4.7 | 38.0 | 5.0 | 1.0 | 37.9 | 1.6 | ||
| In the methylome | 100 | 5.2 | 3.1 | 4.2 | 44.5 | 2.7 | 7.4 | 37.6 | 5.2 | 0.9 | 37.4 | 1.8 | ||
Frequency indicates the percentage of the methylome that is included using the given selection criteria. Note that a CpG site can overlap with more than one biological feature. CGI shore is the 1000-bp region flanking the CGI. Upstream is a region 2000-bp upstream of transcription start. Isolated CpG is an isolated CpG site ≥400 bp from the closest CpG site.
A methylation change was detected after haloperidol treatment; −: No change was detected; CGI: CpG island; Cons.: Evolutionary conserved region; Repeat: Site overlapping with RepeatMasker; TFBS: Transcription factor binding site.
Biological features overlapping with changing sites
Approximately half of the 20.3 million autosomal CpGs in the mouse reference genome are located in genes (44.5%) or within 2000 bp upstream of the genes (4.2%). The commonly studied CGIs and CGI shores make up 5.2 and 3.1% of the CpGs in the reference genome, respectively. Furthermore, 5.2% of the CpGs overlap with transcription factor binding sites, 1.8% of the sites have been conserved during evolution and 37.4% of the regions were marked by RepeatMasker (Table 1). The proportions of methylated sites that overlapped with the biological features are shown in Supplementary Tables 1 & 2. The distributions of the biological features that overlap with the DMP are given in Table 1.
Discussion
To study the plausibility of the two models in Figure 1, indicating how methylation studies in the blood can be informative for psychiatric conditions, we performed methylome-wide profiling with MBD-seq using three tissues (blood, cortex and hippocampus) from 14 male C57BL/6 mice. Four of these mice were administered the highly potent antipsychotic haloperidol and ten mice were untreated. We found that the treatment affected methylation in a large number (42.2%) of CpG sites. The changes were not limited to the brain tissues (21.1% of sites changed in the blood). This suggests that factors that are strongly associated with brain function may leave methylation signatures in the blood. Furthermore, as much as approximately 9% of the sites were DMP in the blood but not in the brain tissues. Thus, in agreement with the signature model (Figure 1A), the methylation status of specific sites detected in blood may potentially be useful as biomarkers for psychiatric conditions.
In support of methylation studies in the blood potentially contributing valuable pathogenic information for psychiatric disorders, a recent study reported high correlations between blood and brain tissues (0.66 and 0.76 for the cerebellum and cortex, respectively) for sites that demonstrate significant ‘between-individual’ variation in DNA methylation [26]. In the current study, we show further support for the use of blood as a proxy for brain tissue. We show that the sample correlations for all tissue combinations were similar (~0.6) in both untreated and treated mice, suggesting that the methylation status of a substantial number of sites in the blood may mirror those in the brain. To further investigate if DMP in blood can predict the methylation status in the brain, we studied the site correlations for each CpG. We found that for all three tissue combinations, more than 57% of DMPs had mean site correlations greater than 0.51. This means that more than half of the sites where a methylation change occurred or approximately a quarter of all investigated CpGs may show non-tissue-specific marks that are likely to be mirrored between different brain tissues and between blood and brain tissues. Thus, findings in the blood may have value as a proxy for brain tissue, as suggested by the mirror-site model (Figure 1B), which may give direct indications about functional relevance.
Tissue specific methylation was fairly modest with, for example, only 1.3% of methylated sites in the blood being unmethylated in the cortex and hippocampus. Although the portion of T-DMRs observed in our study is slightly lower (1.3–8.9% of sites were uniquely methylated in one tissue on a methylome-wide scale) than what has been reported elsewhere, it is in line with previous studies suggesting that T-DMRs constitute only a limited set of all methylated sites [55,56]. The reason for our slightly lower estimate is that our analysis reports the subset of T-DMRs where one tissue is methylated and the other two are unmethylated.
The three investigated tissues each consisted of several cell types. Therefore, the methylation measures represent averages of all cells in each investigated tissue. Given that the mice are from an inbred strain, are of the same age and have experienced similar environments, it is reasonable to believe that the tissues are similar in cell composition across individuals. Furthermore, given that the treatment used is known to alter methylation but not the cellular composition itself, it is not likely that the treatment would cause shifts in the cellular composition. With these assumptions, it is reasonable to believe that the cellular heterogeneity in the investigated tissues will not affect the outcome of the analysis.
When evaluating of the blood as a surrogate for the brain in methylation investigations, it is important to note that there are not just differences between the blood and brain but also between brain tissues. For example, if we were to study the hippocampus, with the goal of inferring methylation status in the cortex, we would have an 8.9% chance of detecting a signal that is uniquely methylated in the surrogate tissue (hippocampus). This risk would be lower, only 1.3%, if we instead used blood as a surrogate for the cortex. The implication is that unless there is a precise idea about where the relevant methylation changes occur in the brain, it may be equally useful to study methylation in the blood as it is to use the ‘wrong’ brain tissue (i.e., not the most relevant brain tissue for a specific phenotype).
Both proposed models imply potential biomarkers that may be of clinical relevance to improve treatment, diagnosis and prognosis of disease. However, sites that are consistent with the mirror-site model can also provide information about pathogenic processes. In practice, a challenge is to determine which sites are mirror sites. To follow-up promising marks directly in human post-mortem brain tissues is one way to tackle this uncertainty or, if human brain tissue is not available, an animal model may potentially reveal whether methylation mark found in the blood is mirrored in the brain. Another approach involves data integration [57,58]. For example, integrating GWAS data can be helpful as observing an association signal with a SNP at the same chromosomal location makes it more likely that the methylation site has functional relevance and integration with mRNA brain expression data can help determine if the methylation signal directly affects gene expression. The idea is that CpGs/loci that are also enhanced by other data types showing disease associations at the same location are more likely to represent biologically meaningful results and, therefore, are more likely to be mirror sites (Figure 1B).
Taken together, our data suggest that the result of haloperidol treatment on DMPs are correlated between tissues for a large number of sites. This shows that the factors altering methylation in brain may also alter methylation in the blood and vice versa. It is reasonable to assume that the specific treatment (the drug and the stress related to the administration procedure) used in this study is not the only environmental effect that may cause this type of correlated change. It is also reasonable to assume that not all environmental effects cause the same correlation patterns between tissues. However, as described in this study using blood as a proxy for brain tissue is a promising approach that is likely to reveal potentially useful biomarkers for the studied phenotype, but further investigations are required to identify specifically which biomarkers are functional mirror sites that can be used to increase our understanding of brain function.
It is important to note that in this investigation we used antipsychotic treatment to alter methylation to study whether correlated changes can be observed across multiple tissues (brain and blood). Thus, the primary goal of this study was not to investigate the direct effect of the specific drug. We believe that the nature of drug-specific changes is also a very interesting research topic. However, the design of the present study does not include placebo-administered individuals exposed to vehicles only and, therefore, the current study is not suitable to detect haloperidol-specific changes.
Conclusion
For psychiatric disorders and other brain-related conditions, the primary relevant tissue (brain) is commonly not available. For those investigations, our results show, consistent with the signature model (Figure 1A), that factors affecting brain processes (here haloperidol) leave biomarker signatures in the blood and, consistent with the mirror-site model (Figure 1B), that the methylation status of many sites in the blood mirror those in the brain. These findings support the use of blood as a surrogate tissue.
Future perspective
Studies of DNA methylation have considerable potential to complement current studies of variation in genetic sequences. Owing to this potential and the rapidly developing technology that allows for methylome-wide approaches, MWAS investigations are likely to become a standard tool to identify biomarkers for complex traits. Given that blood is fairly easy to collect, it is likely to be used as a proxy for the primary disease tissue (e.g., brain for psychiatric conditions) and become the most prominent tissue for these analyses. While methylation markers detected in blood may directly be useful as biomarkers, integration with other data types (e.g., expression data, genotype data and information about biological networks) is probably required to indentify markers that are functional mirror sites of biological relevance in the primary disease tissue.
Supplementary Material
Executive summary.
Background
As the primary relevant tissue for psychiatric disorders is commonly not available, we used a methyl-binding domain enrichment and next-generation sequencing approach to investigate whether blood can be used as a proxy for the brain tissue in methylation studies.
Materials & methods
We performed methylome-wide profiling using three tissues (blood, cortex and hippocampus) from mice. Four mice were administered the highly potent antipsychotic haloperidol, while the remaining ten were untreated.
Results
A similar percentage (19.4–23.2%) of treatment-induced methylation changes were observed in the cortex, hippocampus and blood, with a total of 42.2% of CpGs changing in at least one tissue.
Approximately the same number of changes was observed in each of the three tissues. This indicates that factors that affect brain processes, here caused by haloperidol treatment, could also alter the methylation marks in blood.
We show that the sample correlations for all tissue combinations were similar (~0.6) in both untreated and treated mice, which indicates that the tissues were partly similarly affected by the treatment.
Although treatment changed the methylation status of 42.2% of the sites in at least one of the investigated tissues and the percentage of likely methylated sites increased in treated mice, the sample concordance between tissues remained, which indicates that the tissues were similarly affected by the treatment.
We found that for all three tissue combinations more than 57% of CpGs that changed had mean site correlations greater than 0.51, suggesting that these sites may show non-tissue-specific marks that are likely to be mirrored between different tissues.
The total site concordance for all three tissues was 62.3%, which suggests that the majority of sites show concordant methylation profiles across tissues.
Conclusion
In conclusion, consistent with the ‘signature model’, factors that affect brain processes (i.e., haloperidol) leave biomarker signatures in the blood and, consistent with the ‘mirror-site’ model, the methylation status of many sites in the blood mirror those in the brain.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Financial & competing interests disclosure
This work was supported by a grant from the National Institute of Mental Health (RC2MH089996). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
- 1.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11(3):191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 2.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330(6004):612–616. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293(5532):1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 4.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465(7299):721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- 5.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 6.Kwok JB. Role of epigenetics in Alzheimer’s and Parkinson’s disease. Epigenomics. 2010;2(5):671–682. doi: 10.2217/epi.10.43. [DOI] [PubMed] [Google Scholar]
- 7.Samaco RC, Neul JL. Complexities of Rett syndrome and MeCP2. J Neurosci. 2011;31(22):7951–7959. doi: 10.1523/JNEUROSCI.0169-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mill J, Tang T, Kaminsky Z, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82(3):696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith AK, Conneely KN, Kilaru V, et al. Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(6):700–708. doi: 10.1002/ajmg.b.31212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jost JP, Saluz HP, Pawlak A. Estradiol down regulates the binding activity of an avian vitellogenin gene repressor (MDBP-2) and triggers a gradual demethylation of the mCpG pair of its DNA binding site. Nucleic Acids Res. 1991;19(20):5771–5775. doi: 10.1093/nar/19.20.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokomori N, Moore R, Negishi M. Sexually dimorphic DNA demethylation in the promoter of the Slp (sex-limited protein) gene in mouse liver. Proc Natl Acad Sci USA. 1995;92(5):1302–1306. doi: 10.1073/pnas.92.5.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaminsky Z, Wang SC, Petronis A. Complex disease, gender and epigenetics. Ann Med. 2006;38(8):530–544. doi: 10.1080/07853890600989211. [DOI] [PubMed] [Google Scholar]
- 13.Cooney CA. Are somatic cells inherently deficient in methylation metabolism? A proposed mechanism for DNA methylation loss, senescence and aging. Growth Dev Aging. 1993;57(4):261–273. [PubMed] [Google Scholar]
- 14.Hernandez DG, Nalls MA, Gibbs JR, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20(6):1164–1172. doi: 10.1093/hmg/ddq561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53(6):857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 16.Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004;20(8):350–358. doi: 10.1016/j.tig.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 17.Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet. 2000;24(1):88–91. doi: 10.1038/71750. [DOI] [PubMed] [Google Scholar]
- 18.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3(4):253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 19.Beck S, Rakyan VK. The methylome: approaches for global DNA methylation profiling. Trends Genet. 2008;24(5):231–237. doi: 10.1016/j.tig.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12(8):529–541. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sklar P, Ripke S, Scott LJ, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43(10):977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ripke S, Sanders AR, Kendler KS, et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23▪.Melas PA, Rogdaki M, Osby U, Schalling M, Lavebratt C, Ekstrom TJ. Epigenetic aberrations in leukocytes of patients with schizophrenia: association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB J. 2012;26(6):2712–2718. doi: 10.1096/fj.11-202069. Demonstrates that haloperidol can induce DNA methylation changes. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Zhang J, Zhang L, Shen Y, Xu Q. Effects of MAOA promoter methylation on susceptibility to paranoid schizophrenia. Hum Genet. 2012;131(7):1081–1087. doi: 10.1007/s00439-011-1131-5. [DOI] [PubMed] [Google Scholar]
- 25.Dempster EL, Pidsley R, Schalkwyk LC, et al. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Hum Mol Genet. 2011;20(24):4786–4796. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26▪.Davies MN, Volta M, Pidsley R, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13(6):R43. doi: 10.1186/gb-2012-13-6-r43. Compares DNA methylation in the blood and brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murgatroyd C, Patchev AV, Wu Y, et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009;12(12):1559–1566. doi: 10.1038/nn.2436. [DOI] [PubMed] [Google Scholar]
- 28.Nestler EJ. Epigenetics: stress makes its molecular mark. Nature. 2012;490(7419):171–172. doi: 10.1038/490171a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monk M, Boubelik M, Lehnert S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development. 1987;99(3):371–382. doi: 10.1242/dev.99.3.371. [DOI] [PubMed] [Google Scholar]
- 30.Efstratiadis A. Parental imprinting of autosomal mammalian genes. Curr Opin Genet Dev. 1994;4(2):265–280. doi: 10.1016/s0959-437x(05)80054-1. [DOI] [PubMed] [Google Scholar]
- 31.Yeivin A, Razin A. Gene methylation patterns and expression. EXS. 1993;64:523–568. doi: 10.1007/978-3-0348-9118-9_24. [DOI] [PubMed] [Google Scholar]
- 32.Rakyan VK, Preis J, Morgan HD, Whitelaw E. The marks, mechanisms and memory of epigenetic states in mammals. Biochem J. 2001;356(Pt 1):1–10. doi: 10.1042/0264-6021:3560001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lavon I, Refael M, Zelikovitch B, Shalom E, Siegal T. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neuro Oncol. 2010;12(2):173–180. doi: 10.1093/neuonc/nop041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kerkel K, Spadola A, Yuan E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40(7):904–908. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- 35.McGowan PO, Meaney MJ, Szyf M. Diet and the epigenetic (re)programming of phenotypic differences in behavior. Brain Res. 2008;1237:12–24. doi: 10.1016/j.brainres.2008.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pilsner JR, Liu X, Ahsan H, et al. Genomic methylation of peripheral blood leukocyte DNA: influences of arsenic and folate in Bangladeshi adults. Am J Clin Nutr. 2007;86(4):1179–1186. doi: 10.1093/ajcn/86.4.1179. [DOI] [PubMed] [Google Scholar]
- 37.Sutherland JE, Costa M. Epigenetics and the environment. Ann NY Acad Sci. 2003;983:151–160. doi: 10.1111/j.1749-6632.2003.tb05970.x. [DOI] [PubMed] [Google Scholar]
- 38.Ulrich S, Neuhof S, Braun V, Meyer FP. Therapeutic window of serum haloperidol concentration in acute schizophrenia and schizoaffective disorder. Pharmacopsychiatry. 1998;31(5):163–169. doi: 10.1055/s-2007-979322. [DOI] [PubMed] [Google Scholar]
- 39.Crowley JJ, Kim Y, Szatkiewicz JP, et al. Genome-wide association mapping of loci for antipsychotic-induced extrapyramidal symptoms in mice. Mamm Genome. 2012;23(5–6):322–335. doi: 10.1007/s00335-011-9385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crowley JJ, Adkins DE, Pratt AL, et al. Antipsychotic-induced vacuous chewing movements and extrapyramidal side effects are highly heritable in mice. Pharmacogenomics J. 2012;12(2):147–155. doi: 10.1038/tpj.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guide for the Care and Use of Laboratory Animals guidelines. Institute of Laboratory Animal Resources, National Academy Press; Washington DC, USA: 1996. [Google Scholar]
- 42▪.Aberg KA, McClay JL, Nerella S, et al. MBD-seq as a cost-effective approach for methylome-wide association studies: demonstration in 1500 case-control samples. Epigenomics. 2012;4(6):605–621. doi: 10.2217/epi.12.59. Describes the methyl-binding domain enrichment and next-generation sequencing analysis pipeline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43▪.Harris RA, Wang T, Coarfa C, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2010;28(10):1097–1105. doi: 10.1038/nbt.1682. Compares methylome-wide approaches and provides a description of the original dichotomization strategy of data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hogart A, Lichtenberg J, Ajay SS, Anderson SM, Margulies EH, Bodine DM. Genome-wide DNA methylation profiles in hematopoietic stem and progenitor cells reveal over-representation of ETS transcription factor binding sites. Genome Res. 2012;22(8):1407–1418. doi: 10.1101/gr.132878.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lan X, Adams C, Landers M, et al. High resolution detection and analysis of CpG dinucleotides methylation using MBD-Seq technology. PLoS One. 2011;6(7):e22226. doi: 10.1371/journal.pone.0022226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li N, Ye M, Li Y, et al. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods. 2010;52(3):203–212. doi: 10.1016/j.ymeth.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 47.Nair SS, Coolen MW, Stirzaker C, et al. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics. 2011;6(1):34–44. doi: 10.4161/epi.6.1.13313. [DOI] [PubMed] [Google Scholar]
- 48▪.Serre D, Lee BH, Ting AH. MBD-isolated genome sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38(2):391–399. doi: 10.1093/nar/gkp992. Evaluates methyl-binding domain enrichment and next-generation sequencing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McKernan KJ, Peckham HE, Costa GL, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19(9):1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat Methods. 2009;6(Suppl 11):S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van den Oord E, Bukszar J, Rudolf G, et al. Estimation of CpG coverage in whole methylome next-generation sequencing studies. BMC Bioinformatics. 2013;14(1):50. doi: 10.1186/1471-2105-14-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Down TA, Rakyan VK, Turner DJ, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008;26(7):779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chavez L, Jozefczuk J, Grimm C, et al. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res. 2010;20(10):1441–1450. doi: 10.1101/gr.110114.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Christensen BC, Houseman EA, Marsit CJ, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5(8):e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eckhardt F, Lewin J, Cortese R, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38(12):1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aberg KA, Liu Y, Bukszár J, et al. A comprehensive family-based replication study of schizophrenia genes. JAMA Psychiatry. 2013;70(2):1–9. doi: 10.1001/jamapsychiatry.2013.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ayalew M, Le-Niculescu H, Levey DF, et al. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol Psychiatry. 2012;17(9):887–905. doi: 10.1038/mp.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Website
- 101.University of California. Santa Cruz genome browser. http://genome.ucsc.edu.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.