

Abstract
Neuromyelitis optica (NMO) is a disabling autoimmune astrocytopathy characterized by typically severe and recurrent attacks of optic neuritis and longitudinally extensive myelitis. Until recently, NMO was considered an acute aggressive variant of multiple sclerosis (MS), despite the fact that early studies postulated that NMO and MS may be two distinct diseases with a common clinical picture. With the discovery of a highly specific serum autoantibody (NMO‐IgG), Lennon and colleagues provided the first unequivocal evidence distinguishing NMO from MS and other central nervous system (CNS) inflammatory demyelinating disorders. The target antigen of NMO‐IgG was confirmed to be aquaporin‐4 (AQP4), the most abundant water channel protein in the CNS, mainly expressed on astrocytic foot processes at the blood–brain barrier, subpial and subependymal regions. Pathological studies demonstrated that astrocytes were selectively targeted in NMO as evidenced by the extensive loss of immunoreactivities for the astrocytic proteins, AQP4 and glial fibrillary acidic protein (GFAP), as well as perivascular deposition of immunoglobulins and activation of complement even within lesions with a relative preservation of myelin. In support of these pathological findings, GFAP levels in the cerebrospinal fluid (CSF) during acute NMO exacerbations were found to be remarkably elevated in contrast to MS where CSF‐GFAP levels did not substantially differ from controls. Additionally, recent experimental studies showed that AQP4 antibody is pathogenic, resulting in selective astrocyte destruction and dysfunction in vitro, ex vivo and in vivo. These findings strongly suggest that NMO is an autoimmune astrocytopathy where damage to astrocytes exceeds both myelin and neuronal damage. This chapter will review recent neuropathological studies that have provided novel insights into the pathogenic mechanisms, cellular targets, as well as the spectrum of tissue damage in NMO.
Keywords: aquaporin‐4 (AQP4), astrocytopathy, neuromyelitis optica (NMO)
Introduction
Neuromyelitis optica (NMO) is an inflammatory disease of the central nervous system (CNS) clinically characterized by recurrent attacks of severe optic neuritis and transverse myelitis 39, 43, 115. The relationship between NMO and multiple sclerosis (MS) has long been debated 30, 42, 67, 111. Historically, NMO pathological studies emphasized the destructive nature of the lesions, which in contrast to prototypic MS, were characterized by the presence of necrotizing demyelination, widespread axonal swelling and spheroids, cavitation, as well as vascular alterations including thickened vessel walls and hyalinization 2, 30, 50, 52, 96. Lucchinetti et al proposed in 2002 that NMO was a humoral disease targeting a perivascular antigen based on the demonstration of a unique vasculocentric rim and rosette pattern of immune complex deposition and complement activation in active NMO lesions 50. Later studies confirmed that the perivascular antigen targeted by NMO‐IgG was the astrocytic water channel aquaporin‐4 (AQP4), which is concentrated on the perivascular astrocytic foot processes and whose immunoreactivity in the normal CNS had a rim and rosette distribution pattern identical to the vasculocentric pattern of IgG deposition and complement activation observed in NMO lesions 47.
Traditionally, astroglia had been largely considered “glue”‐like supportive components of the nervous tissue, and the detection of reactive gliosis was simply regarded as nonspecific uniform pathologic process 97. However, it has become increasingly clear that astrocytes are more than just inert components of the CNS whose only function is to provide support and protection for neurons. Astrocyte foot processes contact blood vessels and are interconnected to other glial cells via gap junctions. Therefore, they are critically important in the formation and maintenance of the blood–brain barrier (BBB), in maintaining glutamate homeostasis, preserving energy balance and buffering the metabolic load within the CNS 82. Astrocytes envelop synapses and nodes of Ranvier 68, and play essential roles in synaptic transmission within the CNS 97. Astrocytes are also key players in the orchestration of immune responses within the brain and spinal cord, expressing a variety of innate immunity‐related receptors such as toll‐like receptors (TLRs), nucleotide binding oligomerization domains, dsRNA‐dependent protein kinases, scavenger receptors, and mannose receptors 19. When activated, astrocytes synthesize all components of the complement system, and produce both immunomodulatory and immunopathogenic cytokines such as IL‐1, IL‐33, IL‐6, TNF‐α and IL‐10, and chemokines such as RANTES, MCP‐1, IL‐8 and IP‐10 11, 12, 66. Indeed, the astrocyte is located at the interface of brain‐immune interactions and is a critical determinant of the innate‐to‐adaptive transition within the CNS. Astrocytes also release neurotrophic factors and cytokines, which promote glial regeneration 84. In addition to their central role in NMO, astrocyte dysfunction has been associated with a variety of inherited, acquired and metabolic CNS disorders 16.
Anatomical Distribution of NMO Lesions in the CNS
The predilection for NMO to involve the optic nerves and spinal cord is well recognized. Uni‐ or bilateral optic nerve and/or chiasmal involvement are typical of NMO, and lesions may be longitudinally extensive. Spinal cord lesions have a tendency to involve the central cord. In contrast to MS lesions, which are typically located peripherally in the white matter of the spinal cord, NMO lesions often involve the gray matter, with 59% of all spinal cord NMO lesions occupying more than half of the spinal cord cross‐section 58, 60. Acute spinal cord lesions demonstrate diffuse swelling and softening, often extending over multiple cord segments, and occasionally involve the entire spinal cord in a patchy or continuous distribution 72. The presence of a longitudinally extensive spinal cord lesion (≥3 vertebral segments) in an adult is a characteristic magnetic resonance imaging (MRI) feature that may help distinguish myelitis secondary to NMO vs. MS 114.
Although historically, a negative brain MRI was considered an absolute diagnostic criterion for NMO 113, increasing evidence reveals that either asymptomatic or symptomatic, MS‐typical and ‐atypical brain lesions are often seen in NMO 74, and they may even be the presenting feature of the disease 35, 36, 92. Pittock and colleagues reported that 60% of NMO patients show brain abnormalities on MRI, with about 10% developing NMO‐unique hypothalamic, corpus callosal, periventricular or brain stem lesions 74 in regions enriched for the target antigen, AQP4 75, 81, 108. However, in contrast to MS, NMO brain lesions may be reversible.
Although MS may also involve periventricular regions, the lesions are usually small and triangular, in contrast to the more extensive periventricular lesions described in NMO patients 74, 80, 81, 108. In addition, NMO periventricular lesions are generally located directly beneath the ependyma 36, and may extend into the cerebral hemisphere, forming a confluent white matter lesion 35. Extensive hemispheric and large, edematous callosal white matter lesions have also been described in patients with high NMO‐IgG titers 61, 100.
An expanding spectrum of clinical syndromes referred to as NMO spectrum disorders (NMOSD) has been associated with brain lesions and NMO‐IgG seropositivity, in the absence of fulfillment of NMO diagnostic criteria 32, 112. Intractable vomiting and hiccups, oculomotor dysfunction, dysphagia, hearing loss, narcolepsy, central endocrinopathies, central hypotension, posterior reversible encephalopathy and encephalopathic symptoms, especially in children 4, 31, 51 underscore that CNS pathology is widespread in NMO and not restricted to optic nerves and spinal cord.
An interesting case report described symptomatic, radiological and pathological involvement of the hypothalamus in NMO 108. The patient presented with involuntary weight loss, disordered thermoregulation, memory disturbance, excessive daytime somnolence, vomiting and behavioral change. Hypothalmic and brain stem involvement was confirmed on MRI, and brain biopsy of the hypothalamic brain lesion revealed a destructive inflammatory process. The patient ultimately died and autopsy confirmed the presence of opticospinal NMO lesions, as well as involvement of the hypothalamus, midbrain, pons and cerebellum. NMO‐IgG seropositivity was subsequently confirmed.
The brain stem, especially the AQP4‐rich regions adjacent to the fourth ventricle, is often involved in NMO. About 40% of NMO patients have lesions involving the area postrema (AP) and the medullary floor of the fourth ventricle, resulting in a clinical syndrome of intractable hiccups, nausea and/or vomiting. These symptoms may present either as a relapse of NMO or herald the disease onset 4, 56, 79. Recently, a case of isolated dysphagia was described in an NMO patient. The dysphagia was thought caused by the involvement of the nucleus ambiguous, which is more ventrally located than the AP. This case highlights that the spectrum of NMO brain stem symptoms are not necessarily confined to the dorsal medulla, but may extend ventrally 104.
Histopathological Features of NMO Lesions
The basic histopathological features of NMO have previously been described 50, 52; however, recent literature suggests that there are at least two types of NMO lesions 58, 86. The classic acute NMO lesion (Figure 1) is characterized by confluent and/or focal perivascular demyelination, prominent infiltration of myelin‐laden macrophages, severe axonal loss, necrosis of both the gray and white matter of the spinal cord, and pronounced loss of astrocytes. Perivascular inflammation is variable and may include T cells, B cells, plasma cells, neutrophils and eosinophils. Oligodendrocyte loss is evident 50, 53. The second NMO lesion type is characterized by vacuolated myelin in the relative absence of frank demyelination 27, 58, 86, reactive astrocytes, microglial activation, limited axonal injury and variable, typically granulocytic inflammation. Apoptotic oligodendrocytes may also be present. These nondemyelinated lesions with vacuolated myelin do not necessarily progress to destructive demyelinating NMO lesions, given the potential for some NMO lesions to be reversible. In light of the spectrum of both demyelinating and nondemyelinating pathology in NMO, it is difficult to stage NMO lesions according to classification schemes used to stage the demyelinating activity of MS plaques.
Figure 1.
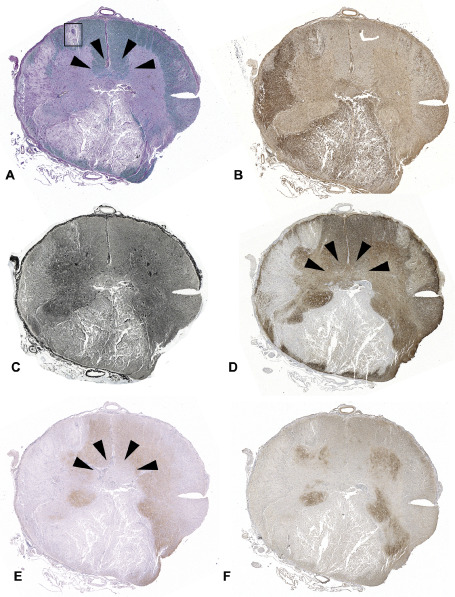
NMO spinal cord lesion. (A) Spinal cord cross‐section demonstrates extensive demyelination involving both the gray and white matter (LFB/PAS). (B) Extensive macrophage/microglia infiltration is present in the lesion (Kim1p). (C) Severe axonal loss is present in some areas of the lesion (Bielschowsky's staining). The lesion shows obvious loss of GFAP (D), AQP4 (E) and the glutamate transporter, EAAT2 (F). AQP4 loss is also evident in an area of the central spinal cord (E, arrowheads) where demyelination (A) and GFAP loss (D) are absent.
Remyelination, evidenced by thinly myelinated axons, is occasionally present at the edge of demyelinated spinal cord NMO lesions. Interestingly, in NMO spinal cord lesions, remyelination can be driven by Schwann cells that extend from the peripheral spinal nerve into the spinal cord parenchyma 29. Astrocytes normally prevent the entrance of Schwann cells into the CNS parenchyma 1. Therefore, the ability of the Schwann cells to enter the spinal cord and remyelinate may reflect the underlying dysfunction of astrocytes in NMO 29.
Chronic NMO lesions are characterized by gliosis, cystic degeneration, cavitation, and atrophy of the spinal cord and optic nerves. An apparent increase in the number and prominence of blood vessels with thickened and hyalinized walls has been observed in necrotic and peri‐necrotic spinal cord areas. Fibrotic thickening of the vessel walls in the absence of fibrinoid necrosis or vasculitis is common 50.
Immunopathological Features of NMO Lesions
Systematic immunopathological analysis of active NMO lesions described a unique vasculocentric pattern of complement activation, and eosinophil/neutrophil infiltration (Figure 2), distinct from MS 50. The finding of deposits of IgG and IgM colocalizing with products of complement activation in a vasculocentric pattern around thickened hyalinized blood vessels, supported a pathogenic role for humoral immunity targeting an antigen in the perivascular space 50. These early pathological findings also emphasized an important role for complement activation along the classical pathway and warrant the use of complement inhibitors as a potential treatment for NMO 76. The presence of C9neo in a subset of NMO lesions clearly indicated that the terminal lytic complement complex (ie, membrane attack complex) was activated preferentially at perivascular sites. The characteristic rim and rosette pattern of perivascular Ig deposition and complement C9neo activation on the adluminal vasculature surface was observed in active NMO lesions, and precisely corresponds to the localization of astrocytic endfeet that envelop the blood vessels 50. Subsequent studies confirmed that the target antigen was AQP4, a perivascular antigen concentrated at the astrocyte foot process in these regions.
Figure 2.
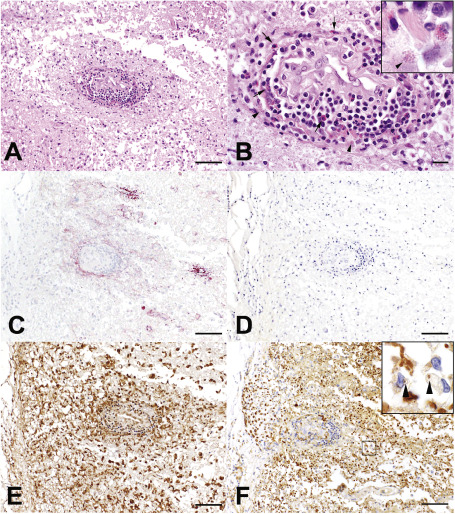
Perivascular NMO spinal cord lesion (boxed region in Figure 1). (A) A focal NMO lesion shows perivascular inflammation and tissue destruction in (H/E). (B) Lymphocytes and numerous perivascular eosinophils (arrows) are present, with evidence of eosinophil degranulation (arrowhead; H/E). (C) Complement C9neo deposition (C9neo IHC) is found in perivascular region of AQP4 loss (D, AQP4 IHC). (E) Prominent macrophage infiltration and microglial activation is present, (E, Kim1p IHC), with evidence of active demyelination defined by the presence of myelin‐laden macrophages (inset; F, MAG IHC; bar in A, C, D and E = 100 μm, bar in F = 50 μm, bar in B = 20 μm).
Granulocytes
The presence of both neutrophils and eosinophils have been reported in early NMO lesions 50, 57, but their pathogenic role has not been clear. Saadoun et al found that neutropenia reduced neuroinflammation, whereas neutrophilia, induced by granulocyte colony‐stimulating factor (GCSF), increased the severity of experimental NMO lesions induced by intracerebral injection of AQP4 antibody and human complement into rats 91. Administration of the neutrophil protease inhibitor Sivelestat ameliorated pathological findings of early experimental NMO‐like lesions. The potentially pathogenic implication of neutrophils in NMO was further highlighted in a recent report of a patient whose first NMO episode was exacerbated by inadvertent administration of GCSF, which stimulated the function and proliferation of granulocytes 33.
Degranulated CCR3+ eosinophils have been identified both in NMO meninges and early lesions (Figure 2) 50. Eosinophils, like astrocytes, are critical innate cellular immune responders. Although traditionally viewed as endstage effector cells involved in parasitic immunity and in allergic and atopic diseases, a newly emerging paradigm portrays eosinophils as pleiotropic, multifunctional leukocytes involved in the initiation and propagation of diverse inflammatory responses, as well as modulators of innate and adaptive immunity 88. The varied functions of eosinophils include tissue injury via release of cytokines, chemokines, lipid mediators, eicosanoids, oxygen burst components and cytotoxic granule cationic proteins [ie, major basic protein (MBP); eosinophil cationic protein (ECP), eosinophil peroxidase (EPO) and eosinophil‐derived neurotoxin (EDN)], as well as immunologic roles including antigen processing and presentation, induction of antigen‐specific T cell proliferation, polarization of T cells to a Th2 phenotype, expression of Th2 cytokines, priming of naïve T and B cells, and regulation of other innate cells (dendritic cells, mast cells, basophils and neutrophils) 7, 28, 95.
Recognition of the multifunctional roles of eosinophils makes it doubtful that the early eosinophilic infiltration of CNS tissues in NMO is merely a downstream chemotactic response to complement activation and/or tissue necrosis following NMO‐IgG binding to the astrocyte. Interestingly, cerebrospinal fluid (CSF) levels of eotaxin‐2, eotaxin‐3 and ECP are reportedly elevated in NMO patients in comparison to healthy controls (HC) and MS patients 13. These findings are consistent with recruitment and degranulation of activated eosinophils in the CNS. IL‐6 is also markedly elevated in the CSF of NMO patients, and is known to be released by degranulated eosinophils 13. It is noteworthy that drugs with potential to promote eosinophil infiltration, such as interferon beta, paradoxically worsen NMO 69. A recent report described the clinical, molecular and neuropathological findings in an AQP4 seropositive patient who developed extensive brain lesions prior to death which were associated with early and prominent eosiophil perivascular inflammation 3. Histopathological examination revealed numerous relatively small perivascular early lesions associated with numerous eosinophils and few lymphocytes, which were not visible either radiographically or grossly. AQP4 was lost and demyelination not obvious. More advanced lesions contained fewer eosinophils, incomplete demyelination and numerous round glial fibrillary acidic protein (GFAP)‐positive profiles suggesting swollen processes of reactive and degenerating astrocytes. Scattered macrophages were GFAP positive. Focally vesiculated myelin sheaths were evident. In the most advanced lesion, there was complete demyelination, no perivascular eosinophils and innumerable GFAP‐positive globules and swollen bizarre GFAP‐positive cellular profiles in the lesion, consistent with astrocytes in the final stage of disintegration. The massive disintegration of astrocytes that was evident in both acute and chronic lesions, supports early targeting of the astrocyte in NMO.
Lymphocyte infiltration
Both T and B lymphocytes are present in NMO lesions to varying degrees. T cells from NMO patients demonstrate a greater proliferation in the presence of AQP4 than those from normal controls 106. Some T cells from NMO patients specifically recognize the AQP4 peptide (p. 61–80) 106. These AQP4‐specific T cells exhibit a Th17 bias that can promote neutrophil infiltration through IL‐17. Furthermore, elevated levels of IL‐17 have been associated with NMO, supporting the involvement of Th17 cells 105, 110 in disease pathogenesis. Bradl et al demonstrated that AQP4‐specific T cells can induce experimental brain inflammation in the rat, with selective targeting of the astrocytic glia limitans thereby facilitating the entry of pathogenic AQP4 antibodies to induce NMO‐like CNS lesions 54, 77.
Expansion of the CD19+ B‐cell population has also been reported in the CSF of NMO patients 15. In addition, a clonally expanded plasma cell population has been identified in the CSF of NMO patients, and the monoclonal recombinant antibodies generated from paired heavy and light chain sequences of these cells demonstrated AQP4 specificity 6. Rituximab, a chimeric monoclonal antibody recognizing CD20 that induces apoptosis and prolonged absence of circulating B cells 71, has been shown to be very effective in relapse prevention in NMO 14, 23, 24. This finding suggests B lymphocytes play a key role in the NMO relapse. However, given that rituximab also has a direct impact on B cell–T cell interactions and suppresses T cell proliferation in response to antigenic stimulation, it is plausible that its therapeutic efficacy in NMO may in part also be attributable to suppression of AQP4‐specific T cells in disease pathogenesis 14.
AQP4 Expression in NMO
The localization of AQP4 in the astrocytic foot processes surrounding endothelial cells is consistent with the role of astrocytes in the development, function and integrity of the interface between brain parenchyma and perivascular space, and between brain and CSF, where they serve to mediate water flux 62. The distribution of AQP4 at glial‐fluid interfaces coincides with sites to which NMO‐IgG binds, and is similar to the deposition pattern of Ig and complement activation products observed in actively demyelinating NMO lesions 47, 50. These observations make a compelling but circumstantial case for AQP4‐IgG being a primary effector of NMO lesions.
A comparison of the expression patterns of AQP4 immunoreactivity in NMO optic nerve and spinal cord lesions with patterns observed in normal CNS; in acute, subacute and chronic CNS infarcts; and in acute and chronic MS lesions revealed that AQP4 IHC is lost in active demyelinating NMO lesions. Furthermore, AQP4 loss was observed at sites of vasculocentric Ig deposition and complement activation even in NMO lesions that showed a relative preservation of myelin (Figure 1) 57, 86, 100. Areas of AQP4 loss tended to extend beyond regions of GFAP loss (Figure 1) 58. In contrast, active and remyelinated MS lesions demonstrated increased AQP4 immunoreactivity, particularly around blood vessels and enhanced cytoplasmic staining of astrocytes. Inactive, completely demyelinated as well as destructive and cystic MS and NMO lesions, both showed loss of AQP4 86.
The loss of AQP4 immunoreactivity in active demyelinating NMO lesions supports a targeted attack against AQP4. This study also provided a plausible explanation for the rim and rosette pattern of Ig and complement activation product deposits that were previously reported as distinctive characteristics of NMO lesions 50. The finding of an identical staining pattern of AQP4 in normal brain, optic nerve and spinal cord localizing to astrocytic endfeet in the perivascular glia limitans, suggests that the rim and rosette pattern of Ig and terminal complement deposition reflects the regional density of targeted AQP4 molecules 64, 107. The widespread expression of AQP4 in the brain is paradoxical in face of the typically optic‐spinal predominant locations of NMO lesions and predilection for brain stem. Regional differences in AQP4 concentration could contribute to this paradox, as well as regional differences in the spatial distribution or molecular orientation of AQP4 epitopes on astrocytic endfeet that might preclude efficient complement activation or intermolecular cross‐linking at some sites 45, 46, 48.
The Pathology of NMO Brain Lesions
Brain lesions
MRI detects brain lesions in 60% of NMO patients, which include NMO‐specific brain lesions in regions of high AQP4 expression (eg, AP, hypothalamus), as well as supratentorial (ST) lesions, which are either nonspecific or have an MS‐like appearance. The presence of these supratentorial brain lesions has suggested a possible pathologic overlap of NMO and MS, or NMO and ADEM. However, comparison of ST NMO lesions with opticospinal (OS) NMO lesions, as well as ST MS and ADEM lesions, suggests that the pathology of NMO ST lesions resembles NMO OS lesions with respect to type of inflammation, perivascular immune complex deposits and AQP4 loss. These findings suggest that NMO ST and OS lesions have a shared pathogenesis. In addition, a subset of NMO supraspinal lesions demonstrate a preferential loss of myelin‐associated glycoprotein (MAG) in regions corresponding to AQP4 loss, as well as evidence of vasculocentric complement deposition 9.
Area postrema involvement in NMO
Intractable, but reversible nausea associated with hiccups and/or vomiting have also been reported to be characteristic symptoms of NMO 41, 55, 56, 79, 96, 101, and can even be the initial and isolated presenting symptom 4, 56. The AP consists of two symmetrical narrow structures at the floor of the rhomboid fossa and is known as a “chemoreceptor trigger zone” and the center for the emetic reflex. Like the other circumventricular organs, the loose tissue of the AP consists of glia and neurons, has a thin ependymal cover and contains numerous convoluted capillaries that lack tight junctions between endothelial cells forming a permeable BBB. The AP has important connections with hypothalamic and brain stem nuclei regulating the fluid balance, osmoregulation, immunomodulation, cardiorespiratory functions, feeding and metabolism and thermoregulation. Like other periventricular areas, the medullary floor of the fourth ventricle and AP are sites rich in AQP4 expression and thus, sites of predilection for NMO lesions and, indeed, inflammatory, nondemyelinated, non‐necrotic NMO lesions have been shown to involve this region 86. Immunohistopathological characterization of the neuropathological features of NMO at the medullary floor of the fourth ventricle and AP in 15 NMO cases revealed six NMO cases (40%) demonstrated unilateral or bilateral lesions involving the AP and the medullary floor of the fourth ventricle. These lesions were characterized by tissue rarefaction, blood vessel thickening, no obvious neuronal or axonal pathology and, in the subependymal medullary tegmentum, preservation of myelin. All six cases showed loss and/or marked reduction of AQP4, moderate to marked perivascular and parenchymal lymphocytic inflammatory infiltrates, prominent microglial activation and in three cases, eosinophils. Complement deposition in astrocytes, macrophages and/or perivascularly, and a prominent astroglial reaction were also variably present. The presence of AP lesions increased the odds of nausea/vomiting being present in NMO patients 16‐fold (95% CI 1.43–437, P = 0.02). These findings suggest the AP may be a preferential and selective target of the disease process, and are compatible with clinical reports of nausea and vomiting preceding episodes of optic neuritis and transverse myelitis or representing the heralding symptom of the disease.
Cerebral cortex
AQP4 is also heavily expressed in normal cortex; however, unlike MS, no cortical demyelinated lesions are present in NMO 78. Nevertheless, cortical gray matter abnormalities have been described on MRI 20, 85, 117, 118, including mild cortical thinning 10. Pathologically, NMO cortical lesions are characterized by prominent cortical gliosis, mostly involving interlaminar astrocytes, as well as pyknotic neurons 78. Reactive astrocytes with swollen cell bodies, clear cytoplasm, prominent nuclei, and multiple, elongated and abundant thin processes were present in all cortical layers, but cortical activation of complement was not seen in NMO. A recent study suggested that AQP4 immunoreactivity was lost on most astrocytes in cortical layer I, but preserved on reactive astrocytes in layers II to VI. Quantitative analysis showed that the number of GFAP‐positive astrocytes was decreased in layer I, but increased in layers II to VI. A prominent microglial reaction was present in cortical layer II and a significant loss of cortical neurons was also seen in layers II, III and IV 93. The loss of AQP4 and/or GFAP expression limited to cortical layer I was thought to reflect a unique sublytic astrocyte injury in NMO 93.
NMO Pathology Outside CNS
AQP4 expression is not restricted to the CNS, and can also be found in other locations including distal collecting tubules of the kidney, parietal cells of the gut, as well as on the cytoplasmic surface of fast‐twitch skeletal muscle. There are three case reports of AQP4 antibody seropositive NMO patients who developed hyperCKemia during the 2 weeks before the onset of CNS symptoms (ie, hiccup, optic neuritis or myelitis), and two of the three cases had interstitial pneumonia 17, 18, 34, 99, 116. The pathogenic role of AQP4 antibody outside the CNS however remains unclear.
Spectrum of Astrocytic Pathology in NMO
A spectrum of astrocytic pathology has been observed in NMO lesions (Figure 3). Astrocyte loss and dystrophic astrocytic profiles have been described 58, 70, as has the presence of GFAP‐laden macrophages suggesting phagocytosis of lytic astrocytes by macrophages 5. The presence of astrocytic clasmatodendrosis in NMO lesions has also been reported 59. These astrocytes are characterized by massively enlarged perinuclear cytoplasm, the presence of intracytoplasmic vacuoles and retraction, as well as beading and clumping of the cell processes (Figure 3). These degenerating astrocytes show condensed nuclei with fragmentation of DNA suggesting apoptosis 9. Astrocytic proliferation has also been described in NMO 9, 70. The presence of GFAP‐positive, but AQP4‐negative unipolar or bipolar astrocyte progenitor cells or small elongated naïve astrocytes suggests they may take part in reparative processes in NMO 70.
Figure 3.
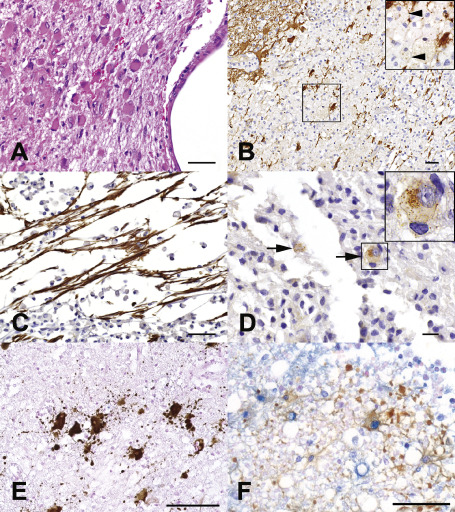
Astrocytic pathology in NMO. (A) Prominent gliosis in a nondestructive periependymal NMO lesion (HE). (B) Astrocyte dystrophy is present in destructive regions of the lesion and GFAP debris within macrophages (arrowheads) suggests astrocyte lysis (GFAP IHC). (C) Bipolar and unipolar GFAP‐positive progenitor cells present in a destructive region (GFAP IHC). (D) AQP4 internalization with loss of astrocyte surface immunoreactivity is compatible with in situ evidence of antigenic modulation (arrows; AQP4 IHC). (E) Disintegrated astrocyte foot processes in AQP4‐loss cerebral white matter NMO lesion (GFAP IHC). (F) Double‐staining of astrocyte (brown; GFAP IHC) and TUNEL staining (blue) in cervical cord NMO lesion suggests apoptosis (bar in A, B and C = 50 μm, bar in D = 20 μm; bar in E = 100 μm; bar in F = 50 μm).
A recent report proposed a classification scheme to define the spectrum of lesion pathology that could be found in NMO (Table 1; Figure 4). Comparative immunohistochemistry of AQP4, AQP1 and GFAP was performed. AQP1 has a similar role in regulating water transport and is also expressed in human brain astrocytes as well as rodent CNS choroid plexus 59, 63, 94. Type 1 lesions were characterized by the presence of complement deposition at the surface of astrocytes, granulocyte infiltration and astrocyte necrosis 57, 58. Lesions were typically cystic or cavitary. Type 2 lesions were further characterized by the presence of GFAP‐positive debris, beading, ragged fibers or disintegrated astrocytes, and immature bipolar astrocytes could occasionally be observed 70. Type III lesions showed signs of Wallerian degeneration in the lesion‐related tracts, in which extensive loss of myelin, oligodendrocytes and axons were present together with profound fibrillary gliosis characterized by densely packed GFAP and AQP1‐reactive astrocytic processes. AQP4 loss was occasionally observed in these lesions 59. Type IV lesions lacked AQP4; however, both AQP1 and GFAP were relatively preserved suggesting a sublethal injury to the astrocyte. Type V NMO lesions were defined by the presence of clasmatodendrosis of astrocytes, and were associated with internalization of AQP4 and AQP1 and astrocyte apoptosis in the absence of complement deposition. Extensive astrocyte loss was typically observed in these lesions in the absence of obvious demyelination and/or axonal loss 59. Type VI lesions were characterized by a variable degree of astrocyte dystrophic changes including clasmatodendrosis, associated with plaque‐like primary demyelination and variable oligodendrocyte apoptosis. The authors suggest that these diverse lesion outcomes may occur sequentially. The extent of pathological changes in astrocytes as observed in NMO are strikingly different than those described in MS, and further reinforce the concept that NMO is a primary astrocytic disorder. Furthermore, not all the pathology seen in NMO is associated with complement deposition or astrocytic lysis; therefore, future therapies must also consider upstream events in lesion pathogenesis before irreversible astrocytic injury ensues.
Table 1.
Key pathological features of different lesion types in NMO. Abbreviations: lesion type, T = T cells; Gr = granulocytes; C9n = complement C9neo antigen; AG = astroglia pathology; Demy = demyelination; OG loss = loss of oligodendrocytes; Ax loss = axonal loss.
| Les. type | T | Gr | C9n | AG | Demy | OG loss | Ax loss |
|---|---|---|---|---|---|---|---|
| 1 | ++ | +++ | +++ | Necrosis | + | Apo | + |
| AQP4 variable | |||||||
| AQP1 variable | |||||||
| GDAP variable | |||||||
| 2 | +/++ | +/− | +/− | AQP4 loss | Complete | +++ | +++ |
| AQP1 loss | |||||||
| GFAP loss | |||||||
| 3 | – | – | – | React. gliosis | +++ | +++ | +++ |
| 4 | +/− | – | – | AQP4 loss | – | – | – |
| Clasmatodendr. + | |||||||
| 5 | ++ | – | – | Clasmatodenr. +++ | – | – | – |
| AQP4 loss | |||||||
| AQP1 loss | |||||||
| GFAP loss | |||||||
| 6 | ++ | – | – | Clasmatodenr. +/++ | Complete | +++ | ++ |
| AQP4 loss variable | |||||||
| AQP1 loss variable | |||||||
| GFAP loss +/− |
±: minor or absent; +: minor; ++, moderate; +++, severe.
Sources: Acta Neuropathologica 125:815–827. Misu T, Hoftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S, Nakashima I, Konno H, Bradl M, Garzuly F, Itoyama Y, Aoki M, Lassmann H Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Copyright (2013), with permission from Springer.
Figure 4.
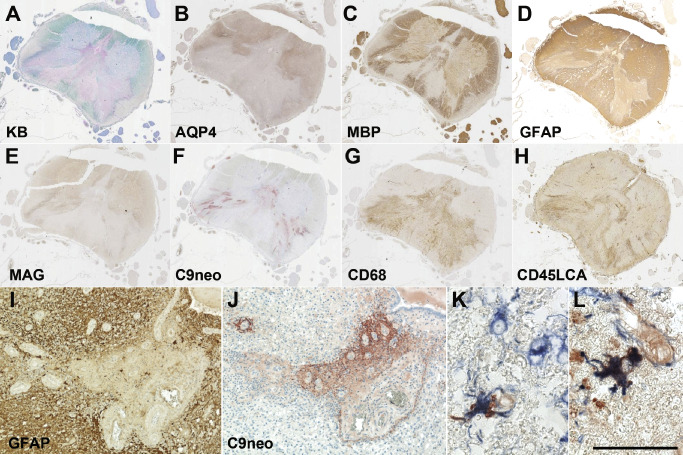
Diverse patterns of tissue injury in NMO. (A) Centrally located NMO spinal cord demyelinated lesion [Kluver‐Bucy (KB) myelin stain]. (B–D) A large area of loss of AQP4 immunoreactivity is present (B), corresponding to both demyelinated areas, as well as extending into regions with relatively preserved MBP immunoreactivity (C). (D) GFAP IR is variably reduced. (E) Loss of myelin‐associated glycoprotein (MAG) is found in areas associated with AQP4 loss. (F) Vasculocentric C9neo deposition is prominent in areas associated with GFAP loss. (G‐H) Monocytes (G; CD68) and lymphocytes (H; CD45LCA) are present in the spinal cord. (I) Perivascular GFAP immunoreactivity is lost in the region of C9neo deposition. C9neo deposition is found both perivascularly in a rosette pattern (J), as well as on the surface of scattered astrocyte membranes [K; C9neo IHC (red); GFAP IHC (blue)] and cytoplasm [L; C9neo IHC (red); GFAP IHC (blue)]. Bar = 50 μm.
Serum and CSF Biomarkers Related to Astrocyte Damage
To observe the damage of astrocytes, myelin and neurons in the CNS during the acute phase of NMO, Misu et al measured the concentrations of GFAP, S100B, MBP and neurofilament heavy chain (NF‐H) in the CSF by ELISA in patients with NMO and MS, ADEM, spinal infarction and other neurologic diseases (OND) 61, 102, 103 (Table 2). Regardless of NMO lesions' localization (myelitis, brain lesions and optic neuritis), the CSF‐GFAP levels during relapse in NMO were significantly higher than those in MS, ADEM and OND (Figure 5). These were referred to as “volcanic ash” in the accompanying editorial. CSF‐S100B in NMO was also elevated but the difference was less remarkable. The ratio of CSF‐GFAP to CSF‐MBP was also higher in NMO than MS and OND 21. After high‐dose corticosteroid therapy, CSF‐GFAP levels rapidly decreased to normal levels, while CSF‐MBP remained high. CSF‐GFAP, CSF‐S100B or CSF‐MBP levels strongly correlated with the expanded disability status scale (EDSS) or spinal lesion length in the acute phase of NMO, but only CSF‐GFAP correlated with EDSS at 6‐month follow‐up 103. This may be explained by the fact that GFAP is highly specific to astrocytes, while S100B is also expressed by microglia and oligodendrocytes.
Table 2.
CSF levels of GFAP, S100b, MBP and NF‐H. Abbreviations: GFAP = glial fibrillary acidic protein; MBP = myelin basic protein; NF‐H = neurofilament heavy chain; NMOSD = neuromyelitis optica spectrum disorder; MS = multiple sclerosis; ADEM = acute disseminated encephalomyelitis; OND = other neurologic disease
| NMOSD | MS | ADEM | Neuro‐Behçet's disease | Meningitis | Spinal infarction | OND | |
|---|---|---|---|---|---|---|---|
| GFAP (ng/mL) | 2476.6 ± 8815.0 a,b,c,d | 0.8 ± 0.4 | 14.1 ± 27.4 | 0.4 ± 0.3 | 45.7 ± 134.1 | 354.7 ± 459.0 | 0.7 ± 0.5 |
| S100B (pg/mL) | 3444.0 ± 10938.1 a | 160.3 ± 70.7 | 905.1 ± 1897.5 | 102.9 ± 40.7 | 1667.8 ± 4313.1 | 940.4 ± 944.6 | 134.7 ± 61.2 |
| MBP (pg/mL) | 705.8 ± 1132.2 d | 106.2 ± 171.9 | 614.0 ± 961.6 | 16.6 ± 30.1 | 168.2 ± 366.7 e | 324.5 ± 443.5 | 10.3 ± 8.2 |
| NF‐H (ng/mL) | 0.2 ± 0.4 | 0.4 ± 1.1 | 3.7 ± 6.2 f | 0.8 ± 0.8 | 2.2 ± 3.4 e | 0.2 ± 0.3 | 0 ± 0 |
The values are mean ± SD. P‐values in multiple comparison: *P < 0.01.
a P: NMOSD vs. MS; b P: NMOSD vs. Neuro‐Behçet's disease; c P: NMO vs. meningitis; d P: NMO vs. OND; e P: meningitis vs. OND; f P: ADEM vs. OND (modified from Neurology 75: 208–216. Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y. Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Copyright (2010), with permission from Wolters Kluwer Health).
Figure 5.
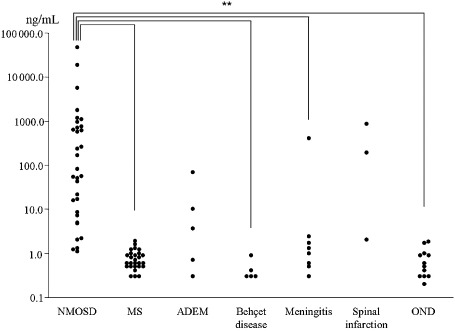
Change of CSF‐GFAP levels in NMO, MS and other CNS diseases. The CSF‐GFAP levels in NMO were significantly higher than those in OND, MS and Behcet's disease. The scales of Y‐axis are logarithmic (modified from Neurology 75: 208–216. Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y. Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Copyright (2010), with permission from Wolters Kluwer Health).
Two other groups independently confirmed the significant elevation of CSF‐GFAP levels during relapse of NMO, supporting that astrocytes are destroyed in the acute stages of NMO and suggesting that GFAP may be a clinically useful CSF biomarker for NMO 73. Serum GFAP and S100B levels are also increased during relapse in some patients with NMO, but differences are less striking than those in CSF, and may be of less diagnostic value 22, 98, 103.
In Vitro Studies Support Pathogenicity of AQP4‐IgG
Complement‐dependent pathogenesis
In vitro studies demonstrate that NMO‐IgG binds selectively to the surface of living target cell membranes expressing AQP4, a prerequisite for IgG to effect organ‐specific pathogenicity. This binding initiates two potentially competing outcomes: (i) rapid downregulation of AQP4 via endocytosis/degradation and (ii) activation of the lytic complement cascade 25. The relative predominance of antigenic modulation and complement activation, which represent competing sequelae of IgG binding to the surface AQP4, may determine an individuals clinical presentation, response to therapy and disease course. The rapid endocytosis and degradation of surface AQP4 initiated by IgG binding coupled with the rapid replenishment of newly synthesized AQP4 also supports a potentially reversible insult, at least during early disease stages, and may explain two clinicopathologic observations in NMO: (i) loss of AQP4 in inflammatory NMO lesions that lack demyelination 86 and (ii) reversible NMO‐typical radiologic lesions observed in the AP and other circumventricular organs rich in AQP4 74.
Confocal microscopy recently confirmed that paranodal astrocytic endfeet in both spinal cord and optic nerve highly express AQP4 25. Already in the early 1970s, freeze fracture studies defined assemblies of particles as an ultrastructural feature of astrocytic endfeet surrounding axons at the nodes of Ranvier 44. These assemblies correspond to the recently identified orthogonal arrays formed by AQP4 homotetramers in transfected cells 83. The extensive array of Fc domains presented by IgG bound to complementary homotetramers of AQP4 in astrocytic endfeet would fulfill the precise geometric alignment required to accommodate the requisite first component of the classic complement cascade, C1q 25, 109. This array of epitopes is analogous to the close packing of nicotinic receptors in the postsynaptic membrane of skeletal muscle that enables explosive activation of complement in acute passive transfer experimental models of myasthenia gravis 46. The activation of complement cascade in NMO would be expected to increase membrane permeability and promote influx of serum immunoglobulins, which would further amplify inflammation at astrocytic endfeet targeted by AQP4‐specific IgG and thus perpetuate complement activation 25.
Complement‐independent pathogenesis
A recent study further demonstrated that in the absence of complement, astrocyte membranes remained intact, but AQP4 was endocytosed with concomitant loss of Na‐dependent glutamate transport and loss of the excitatory transporter, EAAT2 26. These findings suggest that EAAT2 and AQP4 exist in astrocytic membranes as a macromolecular complex. Furthermore, NMO lesions demonstrated marked reduction in EAAT2 in regions of AQP4 loss. In summary, binding of NMO‐IgG to astrocytic AQP4 initiates not only complement activation, but AQP4 and EAAT2 downregulation, which would be expected to disrupt glutamate homeostasis. This could lead to injury of oligodendrocytes that express calcium‐permeable glutamate receptors. Another in vitro study that examined how AQP4 antibody altered the morphology and function of primary rat astrocytes and oligodendrocytes showed that in the absence of complement, AQP4 antibody reduced membrane expression of AQP4 and glutamate transporter type 1 on cultured astrocytes 53. This treatment also reduced oligodendrocytic cell processes and induced their cell death both in vivo and ex vivo. These findings further support the hypothesis of a glutamate‐mediated excitotoxic death of oligodendrocytes as a consequence of AQP4‐IgG binding to astrocytes 26. Oligodendrocytes in the spinal cord and optic nerve are highly sensitive to changes in glutamate concentration, and increases in extracellular glutamate would be expected to render oligodendrocytes additionally susceptible to Ig‐dependent complement attack.
Another recent study identified two additional novel properties of NMO‐IgG 27. First, the binding of NMO‐IgG to AQP4 had isoform‐specific outcomes. The M1 isoform was completely internalized, but M23 resisted internalization and was aggregated into larger order orthogonal arrays of particles that activate complement more effectively than M1 when bound by NMO‐IgG. Second, NMO‐IgG binding to either isoform impaired water flux directly, independently of antigen downregulation or complement activation. Although this conclusion has not been universally accepted 27, 49, 87, the presence of reactive astrocytes with persistent foci of surface AQP4 and the presence of vacuolation in adjacent myelin consistent with edema, are both observed in NMO tissues and support these in vitro findings. The vacuolated myelin was usually seen in the periplaque white matter and thought to be the pathological outcome of water homeostasis disturbance caused by NMO‐IgG binding to AQP4 27. Differences in the nature and anatomical distribution of NMO lesions, and in the clinical and imaging manifestations of disease documented in pediatric and adult patients, may be influenced by regional and maturational differences in the ratio of M1 to M23 proteins in astrocytic membranes.
In summary, there are several possible mechanisms that might initiate demyelination in NMO 25: (i) oligodendrocytes are more susceptible than astrocytes to lethal injury by noxious stimuli, and would be expected to be injured at the paranode where they directly contact AQP4 containing astrocytic foot processes; (ii) demyelination could be secondary as a result of alterations in the ionic microenvironment at the internode leading to myelinolysis; and (iii) glutamate toxicity may contribute to demyelination.
Comparison of Human NMO and Experimental Animal Models
Attempts to generate experimental NMO models have relied on three basic approaches:
-
(i)
Experimental autoimmune encephalomyelitis (EAE)/NMO‐IgG models: These models involve immunizing animals with CNS antigen or passive transfer of CNS antigen‐specific T cells, in order to first induce CNS inflammation and BBB breakdown. In a next step, NMO‐IgG is passively transferred into the animals in order to induce an NMO‐like pathology. Experimental lesions are characterized by AQP4 loss, variable neutrophil recruitment, and perivascular immunoglobulin and complement deposition.
-
(ii)
NMO‐IgG/human complement models: This system involves the direct intracerebral injection of NMO‐IgG or recombinant AQP4‐IgG and human complement into animals and produces an NMO‐like pathology consisting of AQP4 loss, granulocyte infiltration, perivascular complement deposition and acute axonal injury.
-
(iii)
Cytokine/NMO‐IgG models: A third approach has involved the injection of different cytokines and chemokines together with NMO‐IgG. Although tumor necrosis factor alpha, interleukin‐6, gamma interferon and CXCL2 failed to induce NMO‐like pathology, interleukin‐1 beta triggered an NMO‐IgG‐dependent pathology characterized by AQP4 loss and neutrophil infiltration.
Table 3 summarizes and compares current animal models with human NMO pathology. Although each model recapitulates some of the features of the human NMO lesion, a number of factors may limit direct comparisons. These include the following: (i) astrocytes in human CNS are more complex and diverse than rodents 65. Human protoplasmic astrocytes manifest a threefold larger diameter and have 10‐fold more primary process than their rodent counterparts; (ii) the NMO‐IgG used to induce NMO animal models is of human origin. Whereas it can activate rat complement, it does not activate mouse complement. Furthermore the co‐injection of human complement into murine models does not activate mouse complement inhibitors, which may underestimate the relevance of complement inhibitor function in NMO inflammatory pathology; and (iii) EAE models in rats are Th1‐cell‐mediated, whereas AQP4‐specific T cells in NMO reportedly show a preferential Th17 bias 106.
Table 3.
Comparison of NMO human pathology to current experimental animal models
| Model (ref) | Human NMO 58, 86 | IL‐1 beta/NMO‐IgG model 40 | NMO‐IgG/human complement injection model 89, 90, 91 | EAE/NMO‐IgG model 8 | EAE/recombinant human AQP4 Ab model 6 | AQP4‐specific T cell/NMO‐IgG 77 | EAE/NMO‐IgG model 37 | NMO‐IgG/CFA pretreated models 38 |
|---|---|---|---|---|---|---|---|---|
| Species | Human | Lewis rat | CD1 mice, AQP4 KO mice, nude mice | Lewis rat | Lewis rat | Lewis rat | Lewis rat | Lewis rat |
| Method | Human disease | Intracerebral injection of cytokine and NMO‐IgG | Repeated intracerebral injection of NMO‐IgG and human complement into brain parenchyma and ventricles | Passive transfer of MBP T cell line (passive EAE) + NMO‐IgG | Recombinant human AQP4 Ab into the retrobulbar venous plexus of MBP EAE | Intraperitoneal injecting AQP4‐specific T cells/intraperitoneal NMO‐IgG | Immunization with MBP inducing EAE/intraperitoneal NMO‐IgG | CFA treatment; intraperitoneal injection of NMO‐IgG |
| CNS trauma | No | Yes | Yes | No | No | No | No | No |
| Demyelination | Yes | No | Yes | No | No (only perivascular myelin vacuolization) | No | Unknown | Unknown |
| T lymphocyte inflammation | Yes | Yes | Yes/(No in nude mice) | Yes | Yes | Yes | Yes | Limited |
| Macrophage/microglial reaction | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Granulocyte infiltration | Neutrophil and eosinophil | Neutrophil | Neutrophil | Neutrophil | Unknown | Neutrophil | Neutrophil and eosinophil | Granulocytes |
| AQP4 loss | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| GFAP loss | Yes (variable) | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Other astrocyte pathology | Both astrocytic lytic and sublytic changes | Loss of astrocytes | Loss of astrocytes | Swollen astrocyte, astrocytic destruction | Loss of astrocytic bodies and processes | Unknown | Balloon‐like dying astrocyte | Swelling of astrocytes |
| Immune complex deposition | Perivascular complement and IgG deposition | Perivascular and astrocytic complement deposition | Vasculocentric complement deposition | Perivascular IgG and complement deposition | Perivascular IgG and complement deposition | Perivascular IgG and complement deposition | Perivascular deposition of Ig and complement | Perivascular deposition of Ig and complement |
| Axonal pathology | Yes (severe) | Unknown | Yes | No | Unknown | Unknown | Unknown | Unknown |
Conclusions
NMO is a primary astrocytopathy with secondary demyelination, which is different from MS, and clinical, paraclinical and pathological criteria that can differentiate between NMO and MS are now available. The astrocytic water channel, APQ4, has been proven to be the target antigen in NMO, and recent pathological observations support the pathogenic role of AQP4 autoantibody: (i) NMO shows an early and primary astrocytic pathology; (ii) NMO active lesions exhibit a characteristic loss of AQP4, while AQP4 is increased in active MS lesions; (iii) NMO lesion exhibit deposition of Ig and activation of complement in a vasculocentric pattern that coincides with normal AQP4 distribution; and (iv) NMO lesions preferentially involve the regions with high AQP4 expression.
The pathogenesis of NMO is complex as binding of NMO‐IgG to AQP4 in the human CNS can cause direct water blockade, AQP4 internalization with secondary water blockade, EAAT2 downregulation and glutamate exictotoxicity, and/or activation of the lytic complement cascade with destruction of astrocytes. Therefore, a complex therapeutic approach is likely needed in order to target the diverse mechanisms of tissue injury in NMO. Whereas present rodent animal models have provided useful information about the pathogenesis of an NMO‐like disease in rodents, they all fail to reproduce all the pathological features of the human disease. Therefore, the development of an animal model that truly mirrors the human disease clinically, paraclinically and pathologically would be a tremendous advancement that might allow researchers to not only decipher the role that each mechanism of tissue injury plays in the formation and progression of NMO lesions, but also test various therapeutic approaches in the hope that one day, this devastating disease will be cured.
Acknowledgments
Financial disclosure
Dr. Lucchinetti may accrue revenue for a patent re: aquaporin‐4 associated antibodies for diagnosis of neuromyelitis optica; receives royalties from the publication of Blue Books of Neurology: Multiple Sclerosis 3 (Saunders Elsevier, 2010).
Grant support
Dr. Lucchinetti receives research support from the NIH (NS49577‐R01; principal investigator), the Guthy Jackson Charitable Foundation (principal investigator) and the National MS Society (RG 3185 B‐3; principal investigator). Dr. Popescu receives research support from the Saskatchewan Health Research Foundation (principal investigator) and the Canada Research Chairs program (principal investigator). Drs. Misu, Fujihara and Itoyama receive support from the Grants‐in‐Aid for Scientific Research (#22229008) from the Ministry of Education, Science and Technology of Japan and by the Grants‐in‐Aid for Scientific Research (for Neuroimmunological Diseases) from the Ministry of Health, Welfare and Labor of Japan.
References
- 1. Afshari FT, Kwok JC, White L, Fawcett JW (2010) Schwann cell migration is integrin‐dependent and inhibited by astrocyte‐produced aggrecan. Glia 58:857–869. [DOI] [PubMed] [Google Scholar]
- 2. Allen IV, Millar JH, Kirk J, Shillington RK (1979) Systemic lupus erythematosus clinically resembling multiple sclerosis and with unusual pathological and ultrastructural features. J Neurol Neurosurg Psychiatry 42:392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Almekhlafi MA, Clark AW, Lucchinetti CF, Zhang Y, Power C, Bell RB (2011) Neuromyelitis optica with extensive active brain involvement: an autopsy study. Arch Neurol 68:508–512. [DOI] [PubMed] [Google Scholar]
- 4. Apiwattanakul M, Popescu BF, Matiello M, Weinshenker BG, Lucchinetti CF, Lennon VA et al (2010) Intractable vomiting as the initial presentation of neuromyelitis optica. Ann Neurol 68:757–761. [DOI] [PubMed] [Google Scholar]
- 5. Barnett MH, Prineas JW, Buckland ME, Parratt JD, Pollard JD (2012) Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult Scler 18:108–112. [DOI] [PubMed] [Google Scholar]
- 6. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C et al (2009) Intrathecal pathogenic anti‐aquaporin‐4 antibodies in early neuromyelitis optica. Ann Neurol 66:617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blanchard C, Rothenberg ME (2009) Biology of the eosinophil. Adv Immunol 101:81–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M et al (2009) Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo . Ann Neurol 66:630–643. [DOI] [PubMed] [Google Scholar]
- 9. Bruck W, Popescu B, Lucchinetti CF, Markovic‐Plese S, Gold R, Thal DR, Metz I (2012) Neuromyelitis optica lesions may inform multiple sclerosis heterogeneity debate. Ann Neurol 72:385–394. [DOI] [PubMed] [Google Scholar]
- 10. Calabrese M, Oh MS, Favaretto A, Rinaldi F, Poretto V, Alessio S et al (2012) No MRI evidence of cortical lesions in neuromyelitis optica. Neurology 79:1671–1676. [DOI] [PubMed] [Google Scholar]
- 11. Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD (2005) Differential activation of astrocytes by innate and adaptive immune stimuli. Glia 49:360–374. [DOI] [PubMed] [Google Scholar]
- 12. Chakraborty S, Kaushik DK, Gupta M, Basu A (2010) Inflammasome signaling at the heart of central nervous system pathology. J Neurosci Res 88:1615–1631. [DOI] [PubMed] [Google Scholar]
- 13. Correale J, Fiol M (2004) Activation of humoral immunity and eosinophils in neuromyelitis optica. Neurology 63:2363–2370. [DOI] [PubMed] [Google Scholar]
- 14. Cree BA, Lamb S, Morgan K, Chen A, Waubant E, Genain C (2005) An open label study of the effects of rituximab in neuromyelitis optica. Neurology 64:1270–1272. [DOI] [PubMed] [Google Scholar]
- 15. Dale RC, Tantsis E, Merheb V, Brilot F (2011) Cerebrospinal fluid B‐cell expansion in longitudinally extensive transverse myelitis associated with neuromyelitis optica immunoglobulin G. Dev Med Child Neurol 53:856–860. [DOI] [PubMed] [Google Scholar]
- 16. De Keyser J, Mostert JP, Koch MW (2008) Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J Neurol Sci 267:3–16. [DOI] [PubMed] [Google Scholar]
- 17. Deguchi S, Deguchi K, Sato K, Yunoki T, Omote Y, Morimoto N et al (2012) HyperCKemia related to the initial and recurrent attacks of neuromyelitis optica. Intern Med 51:2617–2620. [DOI] [PubMed] [Google Scholar]
- 18. Di Filippo M, Franciotta D, Massa R, Di Gregorio M, Zardini E, Gastaldi M et al (2012) Recurrent hyperCKemia with normal muscle biopsy in a pediatric patient with neuromyelitis optica. Neurology 79:1182–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol 28:138–145. [DOI] [PubMed] [Google Scholar]
- 20. Filippi M, Rocca MA, Moiola L, Martinelli V, Ghezzi A, Capra R et al (1999) MRI and magnetization transfer imaging changes in the brain and cervical cord of patients with Devic's neuromyelitis optica. Neurology 53:1705–1710. [DOI] [PubMed] [Google Scholar]
- 21. Fujihara K, Misu T, Nakashima I, Takahashi T, Bradl M, Lassmann H et al (2012) Neuromyelitis optica should be classified as an astrocytopathic disease rather than a demyelinating disease. Clin Exp Neuroimmunol 3:58–73. [Google Scholar]
- 22. Fujii C, Tokuda T, Ishigami N, Mizuno T, Nakagawa M (2011) Usefulness of serum S100B as a marker for the acute phase of aquaporin‐4 autoimmune syndrome. Neurosci Lett 494:86–88. [DOI] [PubMed] [Google Scholar]
- 23. Gredler V, Mader S, Schanda K, Hegen H, Di Pauli F, Kuenz B et al (2013) Clinical and immunological follow‐up of B‐cell depleting therapy in CNS demyelinating diseases. J Neurol Sci 328:77–82. [DOI] [PubMed] [Google Scholar]
- 24. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ et al (2008) B‐cell depletion with rituximab in relapsing‐remitting multiple sclerosis. N Engl J Med 358:676–688. [DOI] [PubMed] [Google Scholar]
- 25. Hinson SR, Pittock SJ, Lucchinett CF, Roemer SF, Fryer JP, Kryzer TJ, Lennon VA (2007) Pathologic potential of IgG binding to water channel exrtacellular domain in neuromyelitis optica. Neurology 69:1–11. [DOI] [PubMed] [Google Scholar]
- 26. Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Kryzer TJ, Chamberlain JL et al (2008) Aquaporin‐4‐binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down‐regulating EAAT2. J Exp Med 205:2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H et al (2012) Molecular outcomes of neuromyelitis optica (NMO)‐IgG binding to aquaporin‐4 in astrocytes. Proc Natl Acad Sci U S A 109:1245–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hogan SP, Rosenberg HF, Moqbel R, Phipps S, Foster PS, Lacy P et al (2008) Eosinophils: biological properties and role in health and disease. Clin Exp Allergy 38:709–750. [DOI] [PubMed] [Google Scholar]
- 29. Ikota H, Iwasaki A, Kawarai M, Nakazato Y (2010) Neuromyelitis optica with intraspinal expansion of Schwann cell remyelination. Neuropathology 30:427–433. [DOI] [PubMed] [Google Scholar]
- 30. Ikuta F, Zimmerman HM (1976) Distribution of plaques in seventy autopsy cases of multiple sclerosis in the United States. Neurology 26:26–28. [DOI] [PubMed] [Google Scholar]
- 31. Iorio R, Lucchinetti CF, Lennon VA, Costanzi C, Hinson S, Weinshenker BG, Pittock SJ (2011) Syndrome of inappropriate antidiuresis may herald or accompany neuromyelitis optica. Neurology 77:1644–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jacob A, McKeon A, Nakashima I, Sato DK, Elsone L, Fujihara K, de Seze J (2013) Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry 84:922–930. [DOI] [PubMed] [Google Scholar]
- 33. Jacob A, Saadoun S, Kitley J, Leite M, Palace J, Schon F, Papadopoulos MC (2012) Detrimental role of granulocyte‐colony stimulating factor in neuromyelitis optica: clinical case and histological evidence. Mult Scler 18:1801–1803. [DOI] [PubMed] [Google Scholar]
- 34. Jeret JS, Suzuki N, Takahashi T, Fujihara K (2010) Neuromyelitis optica preceded by hyperCKemia episode. Neurology 75:2253–2254. [DOI] [PubMed] [Google Scholar]
- 35. Kim W, Kim SH, Lee SH, Li XF, Kim HJ (2011) Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 17:1107–1112. [DOI] [PubMed] [Google Scholar]
- 36. Kim W, Park MS, Lee SH, Kim SH, Jung IJ, Takahashi T et al (2010) Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin‐4 autoimmunity. Mult Scler 16:1229–1236. [DOI] [PubMed] [Google Scholar]
- 37. Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T et al (2009) Neuromyelitis optica: passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun 386:623–627. [DOI] [PubMed] [Google Scholar]
- 38. Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T et al (2010) Anti‐aquaporin‐4 antibody induces astrocytic cytotoxicity in the absence of CNS antigen‐specific T cells. Biochem Biophys Res Commun 394:205–210. [DOI] [PubMed] [Google Scholar]
- 39. Kira J (2003) Multiple sclerosis in the Japanese population. Lancet Neurol 2:117–127. [DOI] [PubMed] [Google Scholar]
- 40. Kitic M, Hochmeister S, Wimmer I, Bauer J, Misu T, Mader S et al (2013) Intrastriatal injection of interleukin‐1 beta triggers the formation of neuromyelitis optica‐like lesions in NMO‐IgG seropositive rats. Acta Neuropathol Commun 1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kobayashi Z, Tsuchiya K, Uchihara T, Nakamura A, Haga C, Yokota O et al (2009) Intractable hiccup caused by medulla oblongata lesions: a study of an autopsy patient with possible neuromyelitis optica. J Neurol Sci 285:241–245. [DOI] [PubMed] [Google Scholar]
- 42. Kuroiwa Y, Igata A, Itahara K, Koshijima S, Tsubaki T (1975) Nationwide survey of multiple sclerosis in Japan. Clinical analysis of 1084 cases. Neurology 25:845–851. [DOI] [PubMed] [Google Scholar]
- 43. Kuroiwa Y, Shibasaki H (1973) Clinical studies of multiple sclerosis in Japan. I. A current appraisal of 83 cases. Neurology 23:609–617. [DOI] [PubMed] [Google Scholar]
- 44. Landis DMD, Reese TS (1974) Arrays of freeze‐fractured astrocytic membranes. J Cell Biol 60:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lennon VA (1978) Myasthenia gravis: a prototype immunopharmacological disease. In: The Menarini Series on Immunopathology. Miescher PA (ed.), pp. 178–198. Schwabe & Co.: Basel. [Google Scholar]
- 46. Lennon VA, Lambert EH, Griesmann GE (1984) Membrane array of acetylcholine receptors determines complement‐dependent mononuclear phagocytosis in experimental myasthenia gravis. Fed Proc 43:1764. [Google Scholar]
- 47. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K et al (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 48. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR (2005) IgG marker of optic‐spinal MS binds to the aquaporin‐4 water channel. J Exp Med 202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lennon VA, Hinson SR, Romero MF, Fallier‐Becker P, Lucchinetti CF (2012) Reply to Rossi et al: Immunohistopathological findings in neuromyelitis optica concur with immunobiological observations in vitro . PNAS 109:E1512. [Google Scholar]
- 50. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM et al (2002) A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 125:1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Magana SM, Matiello M, Pittock SJ, McKeon A, Lennon VA, Rabinstein AA et al (2009) Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology 72:712–717. [DOI] [PubMed] [Google Scholar]
- 52. Mandler RN, Davis LE, Jeffery DR, Kornfeld M (1993) Devic's neuromyelitis optica: a clinicopathological study of 8 patients. Ann Neurol 34:162–168. [DOI] [PubMed] [Google Scholar]
- 53. Marignier R, Nicolle A, Watrin C, Touret M, Cavagna S, Varrin‐Doyer M et al (2010) Oligodendrocytes are damaged by neuromyelitis optica immunoglobulin G via astrocyte injury. Brain 133:2578–2591. [DOI] [PubMed] [Google Scholar]
- 54. Matsuya N, Komori M, Nomura K, Nakane S, Fukudome T, Goto H et al (2011) Increased T‐cell immunity against aquaporin‐4 and proteolipid protein in neuromyelitis optica. Int Immunol 23:565–573. [DOI] [PubMed] [Google Scholar]
- 55. Misu T, Fujihara K, Nakashima I, Miyazawa I, Okita N, Takase S, Itoyama Y (2002) Pure optic‐spinal form of multiple sclerosis in Japan. Brain 125:2460–2468. [DOI] [PubMed] [Google Scholar]
- 56. Misu T, Fujihara K, Nakashima I, Sato S, Itoyama Y (2005) Intractable hiccup and nausea with periaqueductal lesions in neuromyelitis optica. Neurology 65:1479–1482. [DOI] [PubMed] [Google Scholar]
- 57. Misu T, Fujihara K, Nakamura M, Murakami K, Endo M, Konno H, Itoyama Y (2006) Loss of aquaporin‐4 in active perivascular lesions in neuromyelitis optica: a case report. Tohoku J Exp Med 209:269–275. [DOI] [PubMed] [Google Scholar]
- 58. Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S et al (2007) Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 130:1224–1234. [DOI] [PubMed] [Google Scholar]
- 59. Misu T, Hoftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S et al (2013) Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol 125:815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nakamura M, Miyazawa I, Fujihara K, Nakashima I, Misu T, Watanabe S et al (2008) Preferential spinal central gray matter involvement in neuromyelitis optica. An MRI study. J Neurol 255:163–170. [DOI] [PubMed] [Google Scholar]
- 61. Nakamura M, Misu T, Fujihara K, Miyazawa I, Nakashima I, Takahashi T et al (2009) Occurrence of acute large and edematous callosal lesions in neuromyelitis optica. Mult Scler 15:695–700. [DOI] [PubMed] [Google Scholar]
- 62. Nicchia GP, Nico B, Camassa LMA, Mola MG, Loh N, Dermietzel R et al (2004) The role of aquaporin‐4 in blood‐brain barrier development and integrity: studies in animal and cell culture models. Neuroscience 129:935–945. [DOI] [PubMed] [Google Scholar]
- 63. Nielsen S, Smith BL, Christensen EI, Agre P (1993) Distribution of the aquaporin CHIP in secretory and resorptive epithelia and capillary endothelia. Proc Natl Acad Sci U S A 90:7275–7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nielsen S, Nagelhus EA, Amiry‐Moghaddam M, Bourque C, Agre P, Ottersen OP (1997) Specialized membrane domains for water transport in glial cells: high resolution immunogold cytochemistry of aquaporin‐4 in rat brain. J Neurosci 17:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Oberheim NA, Wang X, Goldman S, Nedergaard M (2006) Astrocytic complexity distinguishes the human brain. Trends Neurosci 29:547–553. [DOI] [PubMed] [Google Scholar]
- 66. Oh JW, Schwiebert LM, Benveniste EN (1999) Cytokine regulation of CC and CXC chemokine expression by human astrocytes. J Neurovirol 5:82–94. [DOI] [PubMed] [Google Scholar]
- 67. Okinaka S, Tsubaki T, Kuroiwa Y, Toyokura Y, Imamura Y (1958) Multiple sclerosis and allied diseases in Japan; clinical characteristics. Neurology 8:756–763. [DOI] [PubMed] [Google Scholar]
- 68. Orthmann‐Murphy JL, Abrams CK, Scherer SS (2008) Gap junctions couple astrocytes and oligodendrocytes. J Mol Neurosci: MN 35:101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Palace J, Leite MI, Nairne A, Vincent A (2010) Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol 67:1016–1017. [DOI] [PubMed] [Google Scholar]
- 70. Parratt JD, Prineas JW (2010) Neuromyelitis optica: a demyelinating disease characterized by acute destruction and regeneration of perivascular astrocytes. Mult Scler 16:1156–1172. [DOI] [PubMed] [Google Scholar]
- 71. Pescovitz MD (2006) Rituximab, an anti‐cd20 monoclonal antibody: history and mechanism of action. Am J Transplant 6:859–866. [DOI] [PubMed] [Google Scholar]
- 72. Petelin Gadze Z, Hajnsek S, Basic S, Sporis D, Pavlisa G, Nankovic S (2009) Patient with neuromyelitis optica and inflammatory demyelinating lesions comprising whole spinal cord from C2 level till conus: case report. BMC Neurol 9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Petzold A, Marignier R, Verbeek MM, Confavreux C (2011) Glial but not axonal protein biomarkers as a new supportive diagnostic criteria for Devic neuromyelitis optica? Preliminary results on 188 patients with different neurological diseases. J Neurol Neurosurg Psychiatry 82:467–469. [DOI] [PubMed] [Google Scholar]
- 74. Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG (2006) Brain abnormalities in neuromyelitis optica. Arch Neurol 63:390–396. [DOI] [PubMed] [Google Scholar]
- 75. Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA (2006) Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 63:964–968. [DOI] [PubMed] [Google Scholar]
- 76. Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF et al (2013) Eculizumab in AQP4‐IgG‐positive relapsing neuromyelitis optica spectrum disorders: an open‐label pilot study. Lancet Neurol 12:554–562. [DOI] [PubMed] [Google Scholar]
- 77. Pohl M, Fischer MT, Mader S, Schanda K, Kitic M, Sharma R et al (2011) Pathogenic T cell responses against aquaporin 4. Acta Neuropathol (Berl) 122:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Popescu BF, Parisi JE, Cabrera‐Gomez JA, Newell K, Mandler RN, Pittock SJ et al (2010) Absence of cortical demyelination in neuromyelitis optica. Neurology 75:2103–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Popescu BF, Lennon VA, Parisi JE, Howe CL, Weigand SD, Cabrera‐Gomez JA et al (2011) Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology 76:1229–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Poppe AY, Lapierre Y, Melancon D, Lowden D, Wardell L, Fullerton LM, Bar‐Or A (2005) Neuromyelitis optica with hypothalamic involvement. Mult Scler 11:617–621. [DOI] [PubMed] [Google Scholar]
- 81. Qiu W, Raven S, Wu JS, Bundell C, Hollingsworth P, Carroll WM et al (2011) Hypothalamic lesions in multiple sclerosis. J Neurol Neurosurg Psychiatry 82:819–822. [DOI] [PubMed] [Google Scholar]
- 82. Ransom B, Behar T, Nedergaard M (2003) New roles for astrocytes (stars at last). Trends Neurosci 26:520–522. [DOI] [PubMed] [Google Scholar]
- 83. Rash JE, Davidson KG, Yasamura T, Furman CS (2004) Freeze‐fracture and immunogold analysis of aquaportin‐4 square arrays with models of AQP4 lattice assembly. Neuroscience 129:915–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ridet JL, Malhotra SK, Privat A, Gage FH (1997) Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 20:570–577. [DOI] [PubMed] [Google Scholar]
- 85. Rocca MA, Agosta F, Mezzapesa DM, Martinelli V, Salvi F, Ghezzi A et al (2004) Magnetization transfer and diffusion tensor MRI show gray matter damage in neuromyelitis optica. Neurology 62:476–478. [DOI] [PubMed] [Google Scholar]
- 86. Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W et al (2007) Pattern‐specific loss of aquaporin‐4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 130:1194–1205. [DOI] [PubMed] [Google Scholar]
- 87. Rossi A, Ratelade J, Papadopoulos MC, Bennett JL, Verkman AS (2012) Consequences of NMO‐IgG binding to aquaporin‐4 in neuromyelitis optica. Proc Natl Acad Sci U S A 109:E1511–E1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rothenberg ME, Hogan SP (2006) The eosinophil. Annu Rev Immunol 24:147–174. [DOI] [PubMed] [Google Scholar]
- 89. Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2010) Intra‐cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 133:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Saadoun S, Waters P, Macdonald C, Bridges LR, Bell BA, Vincent A et al (2011) T cell deficiency does not reduce lesions in mice produced by intracerebral injection of NMO‐IgG and complement. J Neuroimmunol 235:27–32. [DOI] [PubMed] [Google Scholar]
- 91. Saadoun S, Waters P, MacDonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2012) Neutrophil protease inhibition reduces neuromyelitis optica‐immunoglobulin G‐induced damage in mouse brain. Ann Neurol 71:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Saiki S, Ueno Y, Moritani T, Sato T, Sekine T, Kawajiri S et al (2009) Extensive hemispheric lesions with radiological evidence of blood‐brain barrier integrity in a patient with neuromyelitis optica. J Neurol Sci 284:217–219. [DOI] [PubMed] [Google Scholar]
- 93. Saji E, Arakawa M, Yanagawa K, Toyoshima Y, Yokoseki A, Okamoto K et al (2013) Cognitive impairment and cortical degeneration in neuromyelitis optica. Ann Neurol 73:65–76. [DOI] [PubMed] [Google Scholar]
- 94. Satoh J, Tabunoki H, Yamamura T, Arima K, Konno H (2007) Human astrocytes express aquaporin‐1 and aquaporin‐4 in vitro and in vivo . Neuropathology 27:245–256. [DOI] [PubMed] [Google Scholar]
- 95. Shi H‐Z (2004) Eosinophils function as antigen‐presenting cells. J Leukoc Biol 76:520–527. [DOI] [PubMed] [Google Scholar]
- 96. Shibasaki H, Kuroiwa Y (1969) Statistical analysis of multiple sclerosis and neuromyelitis optica based on autopsied cases in Jan. Folia Psychiatr Neurol Jpn 23:1–10. [DOI] [PubMed] [Google Scholar]
- 97. Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol (Berl) 119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Storoni M, Petzold A, Plant GT (2011) The use of serum glial fibrillary acidic protein measurements in the diagnosis of neuromyelitis optica spectrum optic neuritis. PLoS ONE 6:e23489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Suzuki N, Takahashi T, Aoki M, Misu T, Konohana S, Okumura T et al (2010) Neuromyelitis optica preceded by hyperCKemia episode. Neurology 74:1543–1545. [DOI] [PubMed] [Google Scholar]
- 100. Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M et al (2007) Anti‐aquaporin‐4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 130:1235–1243. [DOI] [PubMed] [Google Scholar]
- 101. Takahashi T, Miyazawa I, Misu T, Takano R, Nakashima I, Fujihara K et al (2008) Intractable hiccup and nausea in neuromyelitis optica with anti‐aquaporin‐4 antibody: a herald of acute exacerbations. J Neurol Neurosurg Psychiatry 79:1075–1078. [DOI] [PubMed] [Google Scholar]
- 102. Takano R, Misu T, Takahashi T, Izumiyama M, Fujihara K, Itoyama Y (2008) A prominent elevation of glial fibrillary acidic protein in the cerebrospinal fluid during relapse in neuromyelitis optica. Tohoku J Exp Med 215:55–59. [DOI] [PubMed] [Google Scholar]
- 103. Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y (2010) Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology 75:208–216. [DOI] [PubMed] [Google Scholar]
- 104. Turkoglu R, Kiyat‐Atamer A, Tuzun E, Akman‐Demir G (2012) Isolated dysphagia caused by aquaporin‐4 autoimmunity. Turk J Gastroenterol 23:804–805. [DOI] [PubMed] [Google Scholar]
- 105. Uzawa A, Mori M, Arai K, Sato Y, Hayakawa S, Masuda S et al (2010) Cytokine and chemokine profiles in neuromyelitis optica: significance of interleukin‐6. Mult Scler 16:1443–1452. [DOI] [PubMed] [Google Scholar]
- 106. Varrin‐Doyer M, Spencer CM, Schulze‐Topphoff U, Nelson PA, Stroud RM, Cree BA, Zamvil SS (2012) Aquaporin 4‐specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize clostridium ABC transporter. Ann Neurol 72:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Venero JL, Vizuete ML, Ilundain AA, Machado A, Echevarria M, Cano J (1999) Detailed localization of aquaporin‐4 messenger RNA in the CNS: preferential expression in periventricular organs. Neuroscience 94:239–250. [DOI] [PubMed] [Google Scholar]
- 108. Viegas S, Weir A, Esiri M, Kuker W, Waters P, Leite MI et al (2009) Symptomatic, radiological and pathological involvement of the hypothalamus in neuromyelitis optica. J Neurol Neurosurg Psychiatry 80:679–682. [DOI] [PubMed] [Google Scholar]
- 109. Walport MJ (2001) Advances in immunology: complement. NEJM 344:1058–1066. [DOI] [PubMed] [Google Scholar]
- 110. Weiner HL (2012) Role of T cells in neuromyelitis optica. Ann Neurol 72:6–8. [DOI] [PubMed] [Google Scholar]
- 111. Weinshenker BG (2003) Neuromyelitis optica: what it is and what it might be. Lancet 361:889–890. [DOI] [PubMed] [Google Scholar]
- 112. Weinshenker BG, Wingerchuk DM (2008) Neuromyelitis optica: clinical syndrome and the NMO‐IgG autoantibody marker. Curr Top Microbiol Immunol 318:343–356. [DOI] [PubMed] [Google Scholar]
- 113. Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG (1999) The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 114. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG (2006) Revised diagnostic criteria for neuromyelitis optica. Neurology 66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 115. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG (2007) The spectrum of neuromyelitis optica. Lancet Neurol 6:805–815. [DOI] [PubMed] [Google Scholar]
- 116. Yokoyama N, Niino M, Takahashi T, Matsushima M, Maruo Y (2012) Seroconversion of neuromyelitis optica spectrum disorder with hyperCKemia: a case report. Eur J Neurol 19:e143. [DOI] [PubMed] [Google Scholar]
- 117. Yu C, Lin F, Li K, Jiang T, Qin W, Sun H, Chan P (2008) Pathogenesis of normal‐appearing white matter damage in neuromyelitis optica: diffusion‐tensor MR imaging. Radiology 246:222–228. [DOI] [PubMed] [Google Scholar]
- 118. Yu CS, Lin FC, Li KC, Jiang TZ, Zhu CZ, Qin W et al (2006) Diffusion tensor imaging in the assessment of normal‐appearing brain tissue damage in relapsing neuromyelitis optica. AJNR Am J Neuroradiol 27:1009–1015. [PMC free article] [PubMed] [Google Scholar]