

Abstract
Antibodies are valuable tools for functional studies in vitro, but their use in living cells remains challenging because they do not naturally cross the cell membrane. Here, we present a simple and highly efficient method for the intracytoplasmic delivery of any antibody into cultured cells. By following the fate of monoclonal antibodies that bind to nuclear antigens, it was possible to image endogenous targets and to show that inhibitory antibodies are able to induce cell growth suppression or cell death. Our electrotransfer system allowed the cancer cells we studied to be transduced without loss of viability and may have applications for a variety of intracellular immuno-interventions.
Keywords: monoclonal antibodies, intracellular delivery, electroporation, protein interference, intracellular imaging, living cells
When antibodies specific for endogenous cellular proteins are introduced into intact mammalian cells, it becomes possible to study the functional activity of these proteins in their natural environment. Since antibodies are too large to diffuse passively into cells, several methods have been developed to introduce them into various cell types. Microneedle injection (microinjection) and osmotic lysis of pinocytic vesicles were the first methods used to deliver antibodies in the cytoplasm of cultured cells.1 Although both techniques showed that mature antibodies were stable and functionally active in the cytoplasm, the microinjection approach was too time-consuming when large numbers of cells had to be injected. Osmotic lysis of vesicles results in massive cell damage, requiring several days of incubation for cellular repair, which severely limits the readout assays when individual cells are observed microscopically. Proteins have also been introduced into eukaryotic cells using liposome-mediated delivery and fusion of red cell ghosts loaded with protein,2 but this method was difficult to control. More recently, it was shown that a small subset of anti-DNA autoantibodies can penetrate into cells due to an unusual high frequency of basic residues in the CDRs, which favors their interaction with macromolecules at the surface of the cells.3,4 It is likely that these observations stimulated the development of synthetic vectors for delivering a larger fraction of antibodies into cells by protein transfection, i.e., by so-called “profection,” an approach similar to that used for DNA and siRNA transfection. Such a direct delivery of proteins following in vitro association of protein and transfection reagent is more challenging than DNA transfection since proteins vary in structure and charge much more than nucleic acids. Intracellular protein delivery by means of cationic polymers has also been reported,5 although most commercially-available protein transfection systems use lipid-based delivery agents.6,7 We previously tried to deliver neutralizing antibodies that recognize HPV16 E6 protein expressed in keratinocytes, using various cationic lipid formulations and found that only a few antibodies were able to interact with cationic lipids and could be successfully delivered to the cytoplasm.8 Depending on the formulation, only up to 30% of treated cells were positive, indicating that this approach was not sufficiently robust to allow antibody-mediated intracellular targeting.
An alternative delivery technique is electroporation, which utilizes a brief electric pulse of high field strength to create transient pores in the cell membrane. This technique has been used successfully for many years to introduce DNA and RNA in bacterial and yeast cells, but is rarely used for transfecting animal cells because of the very large number of cells that must be treated to recover enough viable cells. Although a few groups succeeded in delivering antibodies into mammalian cells by electroporation,9-11 the method has not been widely used for the intracellular transfer of proteins, possibly because there is no convenient way to distinguish viable from damaged cells after incorporation of the protein of interest.
There recently has been renewed interest in the electroporation of nucleic acids for delivering genes to mammalian cells that are difficult to transfect with conventional synthetic vectors. Several companies currently propose improved devices for the electroporation of nucleic acids that achieve a high cell viability in a wide range of cell lines. To deliver antibodies into cells by electroporation, we used a device for gene transfection that does not involve classical cuvettes (Neon®, Life Technologies) and adapted it to the electrotransfer of monoclonal antibodies (mAbs) to cultured cells. We show here that this method of intracytoplasmic delivery of antibodies retains a high degree of cell viability and is extremely effective for targeting endogenous nuclear proteins.
Initial experiments were done with two mAbs, 4C6 and 7C2, that bind to the HPV16 E6 oncoprotein8 and the C-terminal domain of the RNA polymerase II largest subunit12 (RNA Pol II), respectively. By varying several parameters of the electroporation protocol, we obtained optimal conditions for delivering the two mAbs to HeLa cells (Table S1). Their presence in the cells was revealed using a fluorescent anti-mouse antibody, 24 h post- treatment. As shown in Figure 1A, the 4C6 antibody was detected in the cell cytoplasm, indicating that the nuclear membrane had not been permeabilized by the electric shock, whereas the 7C2 antibody was found almost exclusively in the nucleus. This result suggests that the 7C2 antibody binds to de novo synthesized RNA Pol II in the cytoplasm and is translocated into the nucleus in a piggyback fashion with the nuclear enzyme.13 The same result was obtained with the 7G5 mAb that recognizes another epitope on the same RNA Pol II subunit (Fig. S1). These findings are in agreement with previous observations obtained after microinjection of the 3E9 antibody, which binds to native β-importin.14 Since HeLa cells do not contain HPV E6 protein, the 4C6 mAb remained in the cytoplasmic compartment, as expected. It should be noted that all treated cells were positive (compare DAPI and anti-mouse IgG staining in Figure 1A) and that almost no floating dead cells were detectable 24 h post-treatment.
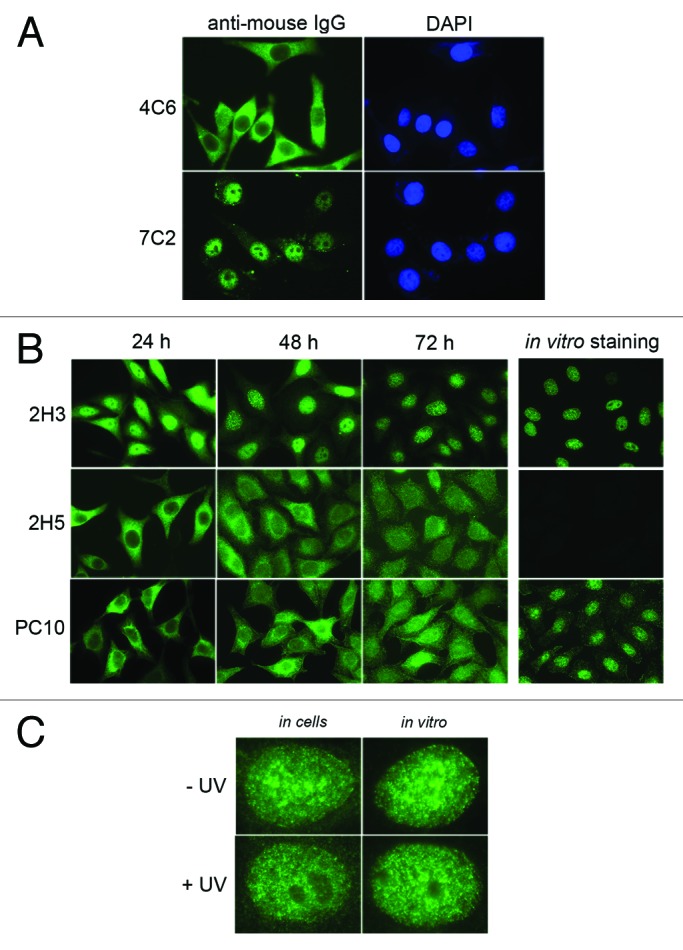
Figure 1. Intracellular localization of delivered antibodies as probed by immunofluorescence microscopy. (A) Micrographs show typical HeLa cells transduced with either 4C6 or 7C2 antibody. The delivered antibodies were revealed with Alexa 448-labeled goat anti-mouse immunoglobulins 24 h post-transduction. Both antibodies are IgG1κ. Magnification: 630 × . (B) The pictures correspond to typical fields of HeLa cells transduced with the indicated antibodies at 24, 48 and 72 h post-treatment. The delivered antibodies were revealed as in A. The panels on the right correspond to the staining by indirect immunofluorescence of HeLa cells incubated with the relevant antibody (in vitro staining). Magnification: 630 × . (C) The pictures show typical nuclei of HeLa cells either transduced and fixed at 48 h post-treatment (in cells) or stained by indirect immunofluorescence (in vitro) with 2H3 antibody without (- UV) or with (+ UV) UVC irradiation 8 h before analysis. Magnification: 100 × .
We recently isolated new mAbs against the proliferating cell nuclear antigen (PCNA), a nuclear protein involved in DNA replication and genome integrity surveillance. To demonstrate that these mAbs are useful for detecting endogenous nuclear proteins in living cells, we analyzed the behavior of two mAbs, 2H3 and 2H5, that bind in vitro exclusively to native and denatured PCNA, respectively (Fig. S2). Both antibodies, as well as the anti-PCNA reference mAb PC10,15 were transduced in HeLa cells and their presence was detected at different times post-treatment. After three days incubation, the 2H3 mAb exhibited a nuclear staining pattern indistinguishable from that observed when it was used as primary antibody to immunostain fixed cells in vitro (Fig. 1B). Since all 2H3 antibodies were located in the nucleus, this suggests that the corresponding PCNA epitope is accessible in the cytoplasm and that the antibodies, which are diluted after each cell division, remain fully active after several days of incubation. By contrast, the transduced 2H5 mAb was present essentially in the cytoplasmic compartment after 24 h and subsequently spread within the entire cell with no specific nuclear enrichment. Although mAb PC10 easily revealed the PCNA protein both by western blot (Fig. S2) and by conventional in vitro immunostaining (Fig. 1B), this antibody did not co-localize with PCNA in living cells after transduction, even after three days of incubation. This suggests that the PCNA epitope recognized by mAb PC10, which has been mapped precisely,16 is not accessible in the cellular context, in contrast to the one recognized by mAb 2H3. These results demonstrate that our procedure allowed antibodies to bind to the accessible epitopes of nuclear proteins in living cells. We also established that mAb 2H3 was successfully electrotransferred and specifically stained PCNA in other cell lines such as CaSki, H1299, MEL501 and U2OS (Fig. S3). We also investigated how UV irradiation of treated cells affected the nuclear pattern of PCNA revealed by antibodies. Since UV irradiation blocks cellular proliferation,17 the typical S phase PCNA pattern obtained with transduced 2H3 disappeared while the nucleoli became clearly visible in nearly all UV-treated HeLa cells (Fig. S4). This UV-induced change was observed both by staining in the cells (after transduction) and after conventional immunostaining in vitro (Fig. 1C). The PCNA contained within replication/repair foci could therefore be precisely localized under physiological conditions using this mAb delivery system.
PCNA detection was facilitated by the fact that it is an abundant nuclear protein18 (~5x105 molecules per cell). To determine whether this “in cellulo” staining protocol could also be applied for targeting nuclear proteins that are less abundant, we decreased the amount of delivered antibodies and measured the amount of antibody remaining in the cells as a function of the amount added in the transduction mixture. Interestingly, the residual amount was proportional to the amount of antibody added (Fig. S5). We estimate that about 105 antibody molecules were present per cell when using 1 μg of mAb in the reaction mixture. The same result was obtained when we tested three different antibody preparations, indicating the general applicability of our protocol. When we used 2 μg of the 2H3 antibody (Fig. 1B), we found that nearly all molecules were translocated into the nucleus, in agreement with the conclusion that the PCNA in this case was in excess. It seems likely, therefore, that our protocol could be applied to the specific imaging of other nuclear proteins simply by adjusting the antibody concentration in the delivery mixture. Since 105 cells are able to integrate about 1010 antibody molecules, this represents a very low percentage of the antibody added in the reaction mixture (~4.1012 molecules/μg). When we tried to improve the delivery efficiency by increasing the number of electric pulses, we found that cell viability was severely impaired.
We used this new transfer method to assess the activity of several inhibitory mAbs delivered to living cells such as CaSki cells, which express the HPV16 E6 oncogene that mediates the degradation of the p53 protein. We have previously shown that the 4C6 mAb, when delivered in CaSki cells with cationic lipids, is able to inhibit the E6-mediated p53 degradation that contributes to cell growth arrest.8 Following electrotransfer of 4C6 in CaSki cells, we found that p53 levels, which were undetectable in untreated cells, were restored in all cells to the level observed after anti-E6 siRNA treatment (Fig. 2A). Since E6 is preferentially localized in the nucleus of cervical carcinoma cells,19 we chemically modified the 4C6 antibody to direct it to the nucleus after it entered the cell. This was done by conjugating a peptide corresponding to the SV40 nuclear localization signal (NLS) to the 4C6 mAb (4C6- NLS; Fig. S6). The 4C6-NLS conjugate accumulated exclusively in the nuclei and 48 h post-transduction, the p53 levels were slightly raised compared with what was observed with unmodified 4C6 (Fig. 2A), indicating that the antibodies that moved to the nucleus were still active. The biological effect of this procedure was confirmed by clonogenic assays (Fig. S7), indicating that it is possible to target a nuclear antigen.
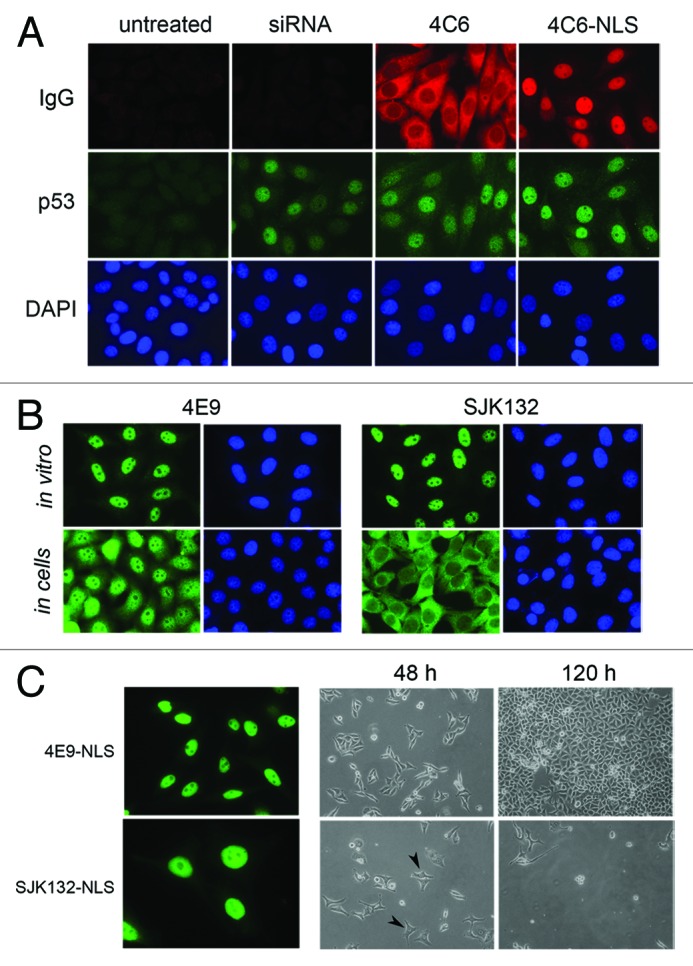
Figure 2. Biological effect of delivered anti-E6 and anti-DNA polymerase α antibodies. (A) Micrographs of typical CaSki cells following transfection with anti-E6 siRNA or transduction with 4C6 antibody. At 48 h post-treatment, the 4C6 antibodies were revealed with Alexa 568-labeled goat anti-mouse immunoglobulins, in parallel with p53 as indicated in the method section. Magnification: 630 × . (B) Micrographs of HeLa cells either stained by indirect immunofluorescence (in vitro) or after transduction at 48 h (in cells) with the indicated antibody. The monoclonal antibodies were detected as indicated in the legend of Figure 1. Magnification: 630 × . (C) The panels on the left represent typical HeLa cells after transduction of 4E9-NLS and SJK123-NLS conjugates and stained with Alexa 488-labeled goat anti-mouse immunoglobulins as in B (in cells). Magnification: 630 × . The micrographs on the right represent typical fields of cells after 2 or 5 d post-treatment with each conjugate observed under the optical microscope. Typical enlarged nuclei are ticked with black arrows. Magnification: 200 × .
To examine if the same effect occurred with other well-characterized inhibitory antibodies, we tested two validated mAbs, 4E9 and SJK132–20 (SJK132), that specifically bind to DNA polymerase α, a nuclear enzyme essential for the replication fork progression in mammalian cells. Both antibodies allow the enzyme to be detected in the nucleus of HeLa cells (Fig. 2B; in vitro). Although both antibodies are able to immunoprecipitate DNA polymerase α from whole-cell extracts (E. Weiss, unpublished observation), only SJK132 was found to block DNA synthesis in an in vitro replication reaction assay.20 After transducing both antibodies into HeLa cells, we found that after two days, mAb 4E9 was mainly localized in the nucleus whereas mAb SJK132 remained in the cytoplasm (Fig. 2B, in cells). In view of what we observed earlier with mAb PC10, we hypothesized that the epitope recognized by SJK132 in de novo synthesized DNA polymerase α might be masked in the cytoplasm and becomes accessible only after the molecule reaches the nucleus. This possibility was tested by preparing NLS peptide-antibody conjugates (4E9-NLS and SJK132-NLS) to bring them closer to the polymerase antigen. At 24 h post-transduction, both 4E9-NLS and SJK-NLS were exclusively present in the nuclei of treated cells (Fig. 2C). However, whereas the cells transduced with 4E9-NLS were growing at the same constant rate as those containing 4E9 or SJK132, the cells treated with SJK132-NLS stopped growing shortly after incubation started. This was easily observed using an optical microscope. After 48 h, the average size of the nuclei of SJK132-NLS- treated cells had increased considerably compared with the nuclei of 4E9-NLS-treated cells (from 16 +/− 2 μm to 25 +/− 3 μm; Figure 2C). This is similar to what is observed when hydroxyurea, a potent and selective inhibitor of replication, is added to cell culture medium.21 After prolonged incubation, nearly all SJK132-NLS-treated cells rounded up (Fig. S8) and became detached from the plastic. After four days incubation, there were almost no cells left on the dishes whereas cells transduced in parallel with 4E9-NLS were nearly confluent (Fig. 2C). Similar results were obtained with U2OS cells, which are p53-positive tumor cells (not shown). These findings show that our protocol makes it possible to target nuclear antigens with antibodies and that this can lead to cell death by apoptosis, if the antibody interferes with an essential biological activity. Our results also suggest that the active region of DNA polymerase α is not accessible to the SJK132 antibody following its de novo synthesis in the cytoplasm.
The present study shows that mAbs can be efficiently transduced into living cancer cells using an electroporation device initially designed for nucleic acid transfection. Since our protocol was successful with all the cells we used and allows all treated cells to be transduced, irrespective of the amount (μg range) of antibody that was added in the electroporation mixture, we were able in all cases to ascertain the intracellular fate of the delivered antibodies. This is a major advantage compared with methods that use synthetic vectors such as cationic lipids, which make it difficult to control endosomal escape after endocytosis, that are likely to limit efficient delivery. It also suggests that the delivered antibodies remain stable in the dividing cells and are not subjected to proteolysis or degradation that would be easily detectable. To date, we have tested 17 different antibodies and five different cell lines and have not observed any difference in the transduction efficiency, suggesting that the method may be applicable to many commercially-available antibodies. Furthermore, since chemically modified antibody molecules can also be electrotransferred, the method may be useful for validating targets either with neutralizing antibodies or with antibody-drug conjugates that could allow the specific intracellular delivery of small molecules. Another possible application is the imaging of sites of endogenous nuclear proteins that are accessible in living cells, using fluorophore-labeled antibodies. Since we have shown that antibodies that inhibit the activity of an antigen in vitro often possess the same functional activity in living cells, it is also likely that the same approach could be used as a functional screening test for isolating neutralizing antibodies able to bind to intracellular targets of therapeutic interest.
Materials and Methods
Generation and purification of anti-PCNA monoclonal antibodies
BALB/c mice were immunized with purified human PCNA protein overexpressed in BL21 cells. After 3 injections of 0.1 mg protein/mouse mixed with poly I-poly C polymer, the spleen cells of the responding animals were fused with SP2/0-Ag14. Growing clones were screened by ELISA using microtiter plates coated with pure PCNA protein (0.5 μg/well) and by cell staining using fixed HeLa cells. The positive cultures were cloned twice in soft agar. All antibodies were purified from either ascitic fluid or hybridoma supernatant on Protein G Sepharose (GE Healthcare). They were subsequently dialysed against PBS and concentrated with centrifugal filtration units (Merck Millipore) to obtain antibody samples of 2–5 mg/mL.
Preparation of antibody-NLS peptide conjugates
Purified antibody (1 mg) in PBS was incubated with a 20-fold molar excess of sulfosuccinimidyl-4-(N-maleimidomethyl)- cyclohexane-1-carboxylate (sulfo-SMCC; Thermo Scientific) for 1 h at room temperature. Excess crosslinker was removed using a Zeba Desalt spin column (2 ml; Thermo Scientific). The antibodies recovered in the flow-trough were mixed with a 5- to 15-fold molar excess of the NLS-encoding synthetic peptide (CAGPKKKRKVED) containing a cysteine residue at the N-terminal end. After incubation for 1 h at room temperature, the antibody-NLS peptide conjugates were purified on desalting columns as described above and stored at 4°C at a concentration of 2 mg/mL.
Cell culture and electroporation
The HeLa, CaSki, Mel501 and U2OS cell lines were grown according to ATCC guidelines and maintained in Dulbecco’s modified Eagle’s tissue culture medium (DMEM; Life Technologies) supplemented with L-glutamine (2 mM), gentamicin (50 μg/ml) and 10% heat-inactivated fetal calf serum at 37°C in a humidified 5% CO2 atmosphere. Fresh cells were thawed from frozen stocks after 12 passages. Transduction experiments with purified mAbs were performed according to the NeonR transfection system protocol (Life Technologies). Typically, 105 cells in R buffer (provided in the kit) were mixed with 2−5 µg of antibody in a total volume of 12 μl. The mixture was then transferred in a NeonR pipette tip chamber (10 µl) and subjected to electroporation in the system pipette station by applying the following optimized parameters: 1550 V, 10 ms and 3 pulses. The treated samples were deposited into 24-well plates containing sterile glass coverslips in 500 µl pre-warmed culture medium without the antibiotic. After incubation for 24 h at 37°C, the culture medium was replaced with complete medium. Where indicated, the cells were irradiated with 20 J/m2 UVC 8 h before harvesting. For the electroporation of siRNA (final concentration 50 nmol/L), settings were 1005 V, 35 ms and 2 pulses.
Immunofluorescence microscopy
The electroporated cells were fixed at the indicated time with 4% paraformaldehyde (w/v) for 30 min and washed twice with PBS. After permeabilization with 0.2% Triton X100 for 5 min, they were incubated for 1 h with AlexaFluor 488 or 568 labeled-anti-mouse immunoglobulins (Molecular Probes, Eugene, USA). Following several washes with PBS, the coverslips were mounted with 4’,6’-diamino-2 phenylindole (DAPI) Fluoromount-G (SouthernBiotech). Staining of the p53 protein was done with rabbit antiserum as previously described.8 The cells were imaged with a Leica DM5500 microscope equipped with FITC and Cy3 filters. Images were collected with a 63 × or a 100 × objective and analyzed with Photoshop software.
Western blot analysis
For the quantification of the delivered antibodies, cells were electroporated in the presence of varying amounts (0.5 to 5 μg) of antibody and incubated for 24 h in 24-well plates as above. Following recovery by trypsinization and counting, the cells were lysed in RIPA buffer and soluble extracts corresponding to 5.105 cells were analyzed by western blotting, in parallel with calibrated antibody preparations. The heavy chain content of these extracts was finally quantitated after incubation with horseradish peroxidase-conjugated anti-mouse globulins and ECL reagent (GE Healthcare) and analysis with the ImageQuantLAS 4000 imager (GE Healthcare). The analysis of the binding activity of the anti-PCNA antibodies was essentially performed as above, except that extracts from untreated cells were used. The actin polypeptide was revealed with rabbit polyclonal antibody A206 (Sigma-Aldrich).
Clonogenic survival assay
Clonogenic survival assays were done essentially as described.8 Briefly, CaSki cells were plated in triplicate in 12-well plates (105 cells/well) and, after 24 h, treated with siRNA (25 pmol) or antibody (3 μg) as described above. After 48 h of incubation at 37°C, the cells were harvested by trypsinization and 1/30 of the pooled cell suspension was replated in triplicate in 60 mm-diameter tissue culture dishes. After 6−7 d of incubation in complete medium, the growing cells were counted manually.
Supplementary Material
Acknowledgments
We thank J.L. Galzi and B. Chatton for constant encouragement, L. Tora for stimulating discussions and advice, A. Stoessel for excellent technical assistance, H. de Rocquigny for providing the synthetic NLS peptide. This work was supported by the Ligue contre le Cancer (Comités de la Marne et du Bas-Rhin). GF was supported by a fellowship of the Direction Générale de l’Armement (DGA) and the Conseil Régional d’Alsace.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/25084
References
- 1.Chakrabarti R, Wylie DE, Schuster SM. Transfer of monoclonal antibodies into mammalian cells by electroporation. J Biol Chem. 1989;264:15494–500. [PubMed] [Google Scholar]
- 2.Ozawa K, Hosoi T, Tsao CJ, Urabe A, Uchida T, Takaku F. Microinjection of macromolecules into leukemic cells by cell fusion technique: search for intracellular growth-suppressive factors. Biochem Biophys Res Commun. 1985;130:257–63. doi: 10.1016/0006-291X(85)90410-3. [DOI] [PubMed] [Google Scholar]
- 3.Avrameas A, Ternynck T, Nato F, Buttin G, Avrameas S. Polyreactive anti-DNA monoclonal antibodies and a derived peptide as vectors for the intracytoplasmic and intranuclear translocation of macromolecules. Proc Natl Acad Sci U S A. 1998;95:5601–6. doi: 10.1073/pnas.95.10.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song YC, Sun GH, Lee TP, Huang JC, Yu CL, Chen CH, et al. Arginines in the CDR of anti-dsDNA autoantibodies facilitate cell internalization via electrostatic interactions. Eur J Immunol. 2008;38:3178–90. doi: 10.1002/eji.200838678. [DOI] [PubMed] [Google Scholar]
- 5.Futami M, Watanabe Y, Asama T, Murata H, Tada H, Kosaka M, et al. Uniformly cationized protein efficiently reaches the cytosol of mammalian cells. Bioconjug Chem. 2012;23:2025–31. doi: 10.1021/bc300030d. [DOI] [PubMed] [Google Scholar]
- 6.Dalkara D, Zuber G, Behr JP. Intracytoplasmic delivery of anionic proteins. Mol Ther. 2004;9:964–9. doi: 10.1016/j.ymthe.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Marschall AL, Frenzel A, Schirrmann T, Schüngel M, Dübel S. Targeting antibodies to the cytoplasm. MAbs. 2011;3:3–16. doi: 10.4161/mabs.3.1.14110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Courtête J, Sibler AP, Zeder-Lutz G, Dalkara D, Oulad-Abdelghani M, Zuber G, et al. Suppression of cervical carcinoma cell growth by intracytoplasmic codelivery of anti-oncoprotein E6 antibody and small interfering RNA. Mol Cancer Ther. 2007;6:1728–35. doi: 10.1158/1535-7163.MCT-06-0808. [DOI] [PubMed] [Google Scholar]
- 9.Berglund DL, Starkey JR. Introduction of antibody into viable cells using electroporation. Cytometry. 1991;12:64–7. doi: 10.1002/cyto.990120109. [DOI] [PubMed] [Google Scholar]
- 10.Lukas J, Bartek J, Strauss M. Efficient transfer of antibodies into mammalian cells by electroporation. J Immunol Methods. 1994;170:255–9. doi: 10.1016/0022-1759(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 11.Baron S, Poast J, Rizzo D, McFarland E, Kieff E. Electroporation of antibodies, DNA, and other macromolecules into cells: a highly efficient method. J Immunol Methods. 2000;242:115–26. doi: 10.1016/S0022-1759(00)00242-8. [DOI] [PubMed] [Google Scholar]
- 12.Puvion-Dutilleul F, Besse S, Diaz JJ, Kindbeiter K, Vigneron M, Warren SL, et al. Identification of transcription factories in nuclei of HeLa cells transiently expressing the Us11 gene of herpes simplex virus type 1. Gene Expr. 1997;6:315–32. [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson ME. BRCA1 16 years later: nuclear import and export processes. FEBS J. 2010;277:3072–8. doi: 10.1111/j.1742-4658.2010.07733.x. [DOI] [PubMed] [Google Scholar]
- 14.Marg A, Meyer T, Vigneron M, Vinkemeier U. Microinjected antibodies interfere with protein nucleocytoplasmic shuttling by distinct molecular mechanisms. Cytometry A. 2008;73A:1128–40. doi: 10.1002/cyto.a.20635. [DOI] [PubMed] [Google Scholar]
- 15.Waseem NH, Lane DP. Monoclonal antibody analysis of the proliferating cell nuclear antigen (PCNA). Structural conservation and the detection of a nucleolar form. J Cell Sci. 1990;96:121–9. doi: 10.1242/jcs.96.1.121. [DOI] [PubMed] [Google Scholar]
- 16.Roos G, Jiang Y, Landberg G, Nielsen NH, Zhang P, Lee MY. Determination of the epitope of an inhibitory antibody to proliferating cell nuclear antigen. Exp Cell Res. 1996;226:208–13. doi: 10.1006/excr.1996.0220. [DOI] [PubMed] [Google Scholar]
- 17.Jiang K, Pereira E, Maxfield M, Russell B, Goudelock DM, Sanchez Y. Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J Biol Chem. 2003;278:25207–17. doi: 10.1074/jbc.M300070200. [DOI] [PubMed] [Google Scholar]
- 18.Naryzhny SN. Proliferating cell nuclear antigen: a proteomics view. Cell Mol Life Sci. 2008;65:3789–808. doi: 10.1007/s00018-008-8305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masson M, Hindelang C, Sibler AP, Schwalbach G, Travé G, Weiss E. Preferential nuclear localization of the human papillomavirus type 16 E6 oncoprotein in cervical carcinoma cells. J Gen Virol. 2003;84:2099–104. doi: 10.1099/vir.0.18961-0. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka S, Hu SZ, Wang TS, Korn D. Preparation and preliminary characterization of monoclonal antibodies against human DNA polymerase alpha. J Biol Chem. 1982;257:8386–90. [PubMed] [Google Scholar]
- 21.Rybaczek D, Kowalewicz-Kulbat M. Premature chromosome condensation induced by caffeine, 2-aminopurine, staurosporine and sodium metavanadate in S-phase arrested HeLa cells is associated with a decrease in Chk1 phosphorylation, formation of phospho-H2AX and minor cytoskeletal rearrangements. Histochem Cell Biol. 2011;135:263–80. doi: 10.1007/s00418-011-0793-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.