

Abstract
Src family kinases (SFKs) are signaling enzymes that have long been recognized to regulate critical cellular processes such as proliferation, survival, migration, and metastasis. Recently, considerable work has elucidated mechanisms by which SFKs regulate normal and pathologic processes in vascular biology, including endothelial cell proliferation and permeability. Further, when inappropriately activated, SFKs promote pathologic inflammatory processes and tumor metastasis, in part through their effects on the regulation of endothelial monolayer permeability. In this review, we discuss the roles of aberrantly activated SFKs in mediating endothelial permeability in the context of inflammatory states and tumor cell metastasis. We further summarize recent efforts to translate Src-specific inhibitors into therapy for systemic inflammatory conditions and numerous solid organ cancers.
Keywords: Src family kinases, Inhibitors, Tumor progression, Metastasis, Inflammation
Introduction
In 1911, Francis Peyton Rous discovered that a transmissible virus was the causative agent of a chicken sarcoma (Rous 1911). Many decades passed before the true significance of this discovery was appreciated. Rous sarcoma virus (RSV) harbors a viral oncogene, v-Src, the expression of which is necessary and sufficient for viral-induced malignancy, and was the first of approximately 30 transforming viruses subsequently isolated. Although RSV and other transforming retroviruses were important tools in the study of properties associated with malignant transformation, the real significance of this work was shown in studies of Bishop, Varmus, and colleagues, who demonstrated that the v-Src gene originated from a cellular “protooncogene”, c-Src (Stehelin et al. 1976). Subsequent studies of viral and constitutively active forms of Src have led to major advances in our understanding of the processes involved in malignant transformation. Complementing these endeavors, studies of signaling pathways leading to the activation of Src and its closely related homologs in the Src family of kinases (SFKs) have been central toward understanding normal growth-regulatory processes, including proliferation, apoptosis, cell cycle control, angiogenesis, and cell-cell adhesion and communication (Frame et al. 2002; Summy and Gallick 2003; Yeatman 2004).
The relevance of these studies has become particularly apparent in the last decade, as numerous examples of the aberrant activation of “normal” Src proteins have been associated with pathologic processes. In this review, we focus on specific SFK-mediated processes important in endothelial permeability as they pertain to the inflammatory response and to tumor progression and metastasis. Such roles of SFKs have generated considerable interest in parallel with the development of SFK-selective small molecule inhibitors, now in early stage clinical trials for advanced tumors (Trevino et al. 2006a; Summy and Gallick 2006; Johnson and Gallick 2007; Kopetz et al. 2007). Additionally, clinical trials with SFK inhibitors (SFKIs) have been proposed for pathologic conditions such as osteoporosis and inflammatory conditions. Thus, current and future uses of these inhibitors in the clinic also are discussed in this review.
Structure and regulation of SFKs
SFKs are comprised of nine structurally related molecules, viz., Src, Blk, Fyn, Yes, Lyn, Lck, Hck, Fgr, and Yrk, with conserved peptide domains, termed Src homology (SH) domains. Four conserved SH domains have been defined in all SFKs (Frame 2002). Src homology domain-1 (SH-1) is the enzymatic domain of the molecule and possesses intrinsic tyrosine kinase activity (Hanks et al. 1988). SH-2 and SH-3 domains facilitate intermolecular interactions between Src and proteins with which it forms complexes (Moarefi et al. 1997; Xu et al. 1999). Specifically, the SH-2 domain allows interaction with phosphotyrosine residues on proteins (Moran et al. 1990; Koch et al. 1992b), whereas the SH-3 domain recognizes a pro-x-x-pro motif, present on a wide variety of signaling and structural molecules (Mayer et al. 1988; Stahl et al. 1988; Pawson 1995; Cohen et al. 1995; Moarefi et al. 1997; Birukov et al. 2001). These domains allow SFK participation in a number of signaling complexes and also regulate SFK kinase activity through intramolecular interactions (Sicheri et al. 1997; Xu et al. 1999).
SFK activity is regulated by intramolecular and intermolecular interactions. The SH-2 domain interacts with its own C-terminal tyrosine phosphorylated amino acid (Tyr527) and an enzymatically inactive, “clamped” conformation occurs, reinforced by additional interactions with the SH-3 domain. The phosphorylation of C-terminal tyrosine is catalyzed primarily by C-terminal Src kinase (Csk) and is the principal mechanism through which SFKs are negatively regulated (Brown and Cooper 1996; Irby and Yeatman 2000). The structural basis for Src interaction with Csk has recently been determined (Levinson et al. 2008). Multiple phosphatases, including phosphatase-1B and phosphatase alpha, are capable of removing the 527 phosphate group from the C-terminal tyrosine and “opening” the catalytic SH-1 site, often after the C-terminal phosphorylated tyrosine is “exposed” following intermolecular interaction of Src with binding proteins in signaling complexes (Xu et al. 1997, 1999). Mutations in the C-terminus that delete or alter tyr527, as occurs in v-Src, lead to a transforming protein with constitutive enzymatic activity (Cartwright et al. 1987; Kmiecik and Shalloway 1987; Piwnica-Worms et al. 1987). Autophosphorylation at Tyr416 increases the specific activity of Src (Thomas and Brugge 1997).
Lastly, the NH2-terminal domain, or the SH-4 domain, is myristoylated and responsible for membrane association of SFKs (Resh 1993; Alland et al. 1994). As a whole, regulation through distinct protein domains and post-translational modifications accounts, in part, for the multiple roles Src plays in signaling complexes, where it serves both as a scaffolding protein and as a protein tyrosine kinase.
Function of SFKs in endothelial cells
In endothelial cells, SFKs mediate the tyrosine phosphorylation of numerous molecules involved in endothelial monolayer permeability (Hu et al. 2008). Endothelial cells maintain homeostasis by regulating the passage of cells, fluid, and protein from the vascular space to the interstitial space (Mehta and Malik 2006). Physiologic responses to trauma, infection, and tumor growth, among others, all involve the production of cytokines and growth factors that bind their cognate receptors on endothelial cells. Such receptor binding results in the tyrosine phosphorylation of numerous molecules that effect changes in vascular permeability, including SFKs (Lum and Malik 1994; Nathan 2002). Through their intrinsic tyrosine kinase activity, in turn, SFKs play important roles in the promotion of endothelial permeability through two major mechanisms: paracellular and transcellular transport (Hu et al. 2008).
Paracellular transport results from structural alterations in endothelial cells that change their relative spatial relationship and adhesion to adjacent cells. In response to physiologic stimuli, SFKs regulate cellular architecture through the phosphorylation and activation of proteins that promote cytoskeletal contraction. SFKs also directly affect endothelial monolayer permeability through the phosphorylation and consequent disruption of protein complexes that bind endothelial cells together. Such intercellular connections (junctional complexes) consist of both tight junctions and adherens junctions and are critical in the maintenance of the vascular barrier (Lum and Malik 1994). Lastly, endothelial cell attachments to the extracellular matrix (ECM), so-called focal adhesions, also undergo SFK-dependent tyrosine phosphorylation that alters cellular morphology (Kaplan et al. 1994; Westhoff et al. 2004). As a whole, the net effect of these processes dictates cellular architecture and drives paracellular transport through the formation of intercellular gaps (gap formations) that permit the passive leakage of fluid and solutes into the interstitial space (Mucha et al. 2003).
In contrast, transcellular transport is an active process and entails the transportation of macromolecules, including albumin, from luminal to basal endothelial cell surfaces (Rippe et al. 2002). Such transcellular transport of macromolecules maintains colloid osmotic pressure while delivering important vascular solutes to the interstitial space. The transport of macromolecules across the endothelial monolayer is dependent upon on the formation and release of caveolae, i.e., small plasma membrane invaginations that form vesicles and mediate endocytosis and transcytosis of macromolecules from the vascular compartment (Tuma and Hubbard 2003). Through the tyrosine phosphorylation of caveolin-1 (the primary protein structural component of caveolae), Src regulates caveolae formation and their detachment from the plasma membrane as endocytic vesicles (Parton et al. 1994; Li et al. 1996; Tiruppathi et al. 1997). The caveolae are then transported through the intracellular space and fuse with the basal endothelial surface, releasing their contents into the interstitial space (Tuma and Hubbard 2003). We detail the roles of SFKs in each of these transport mechanisms below.
SFKs contribute to paracellular transport
Cytoskeletal effects of SFKs
Src is important in the initiation of endothelial cytoskeletal contraction in endothelial cells. Putative SH-2- and/or SH-3-binding regions near the N-terminus of myosin light chain kinase (MLCK) allow Src/MLCK interactions, leading to Src-mediated phosphorylation and activation of MLCK at Tyr464 and Tyr471 (Garcia et al. 1999; Birukov et al. 2001). Activated MLCK is a regulatory motor protein that phosphorylates the myosin light chain at Ser19, thereby initiating crossbridge formation between myosin and actin (Garcia et al. 1995, 1999; Shi et al. 2000; Birukov et al. 2001). As major contractile mechanisms within the cell, activated myosin filaments pull actin filaments together in an ATP-dependent process, altering the actin cytoskeleton and resulting in morphologic changes that promote intercellular gap formation and increased permeability (Garcia et al. 1995; Yuan et al. 1997). Although the mechanisms governing initial Src activation in this setting are still under investigation (and presumably involve the release of cytokines from surrounding cells), its subsequent phosphorylation and activation of MLCK is critical in regulating the formation of intercellular gaps through cytoskeletal contraction (Lambeng et al. 2005).
Junctional complex effects of SFKs
Whereas Src is an important regulator of the vascular barrier through cytoskeletal contraction, SFKs also affect endothelial monolayer permeability through the regulation of intercellular connections. Junctional complexes, consisting both of tight junctions and adherens junctions, bind endothelial cells and function as semi-permeable barriers to the passive efflux of fluid and solutes. In endothelial cells, vascular endothelial cadherin (VE-cadherin) is the primary component of adherens junctions (Dejana et al. 1999; Bazzoni and Dejana 2001). Through its extracellular and cytoplasmic domains, VE-cadherin interacts with neighboring endothelial cells and the actin cytoskeleton, respectively.
Src associates with adherens junctions by interacting with VE-cadherin (Lambeng et al. 2005). Upon association in protein complexes, Src can phosphorylate VE-cadherin; however, recent experiments indicate that Src-dependent phosphorylation and activation of VE-cadherin is dependent upon complex formation between Src, Flk (vascular endothelial growth factor receptor 2; VEGFR2), and VE-cadherin (Fig. 1; Weis et al. 2004b). VEGF-A is produced under physiologic conditions by numerous cells, including tumor cells, and signals through receptor tyrosine kinases via autocrine and paracrine mechanisms (Bellamy et al. 1999; Dias et al. 2000, 2001; Masood et al. 2001). Upon VEGF-A binding to its cognate receptors (VEGFR1 and VEGFR2 in vascular endothelial cells), the intrinsic tyrosine kinase activity of these receptors is activated, leading to trans-phosphorylation and direct interaction with many SH-2-containing signaling molecules, including Src (Ito et al. 1998). Once bound to VEGFR1 or VEGFR2, Src undergoes a conformational change (see below) leading to its activation and the subsequent phosphorylation of VE-cadherin (Millauer et al. 1993; Chou et al. 2002). Such Src-dependent activation of VE-cadherin results in its dissociation from proteins in VE-cadherin-mediated signaling complexes, including VEGFR and β-catenin, and disruption of the endothelial cell barrier (Weis et al. 2004b). β-Catenin links the cytoplasmic portion of VE-cadherin to the actin cytoskeleton while serving as an important signaling molecule, performing multiple functions outside the scope of this review (Gumbiner 1995), and is phosphorylated by Src on Y654 and by Fyn, a related SFK, on Y142 (Lilien and Balsamo 2005). Importantly, both VE-cadherin and β-catenin are subject to phosphorylation by SFKs, resulting in the disruption of intracellular cadherin-actin complexes and alterations in endothelial permeability (Aberle et al. 1996; Roura et al. 1999; Wong et al. 1999).
Fig. 1.
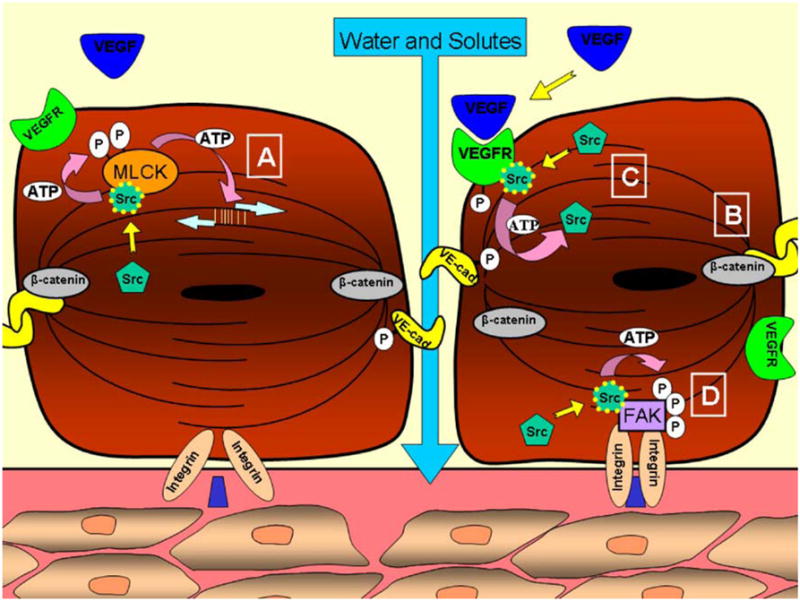
Mechanism of paracellular transport in endothelial cells. Boxed A Activated Src binds to SH-2 and/or SH-3 regions of myosin light chain kinase (MLCK), leading to its phosphorylation and activation. Myosin-actin crossbridge formation ensues followed by cytoskeletal contraction. Boxed B VE-cadherin and β-catenin in resting complex formation. Upon binding of vascular endothelial growth factor (VEGF) ligand (boxed C), VEGFR undergoes trans-phosphorylation and binds Src at a SH-2 site, resulting in Src phosphorylation and activation. Src then phosphorylates VE-cadherin, resulting in complex dissociation and increased vascular permeability. Boxed D Engagement of integrins with extracellular matrix components results in auto-phosphorylation of focal adhesion kinase (FAK) and an SH-2-binding site to which Src binds. FAK then undergoes Src-dependent phosphorylation and activation at multiple sites, initiating downstream events related to actin assembly and focal adhesion turnover
Recent evidence from Ha and colleagues (2008) provides an alternate mechanism of VE-cadherin-mediated endothelial permeability. Using human umbilical vein endothelial cells and bovine aortic endothelial cells, this group has proposed a model in which Csk and c-Src remain associated with VE-cadherin in resting states. Within this complex, Csk is able to exert its negative regulatory effects on Src by maintaining Tyr-527 phosphorylation. Upon activation by VEGF, VEGFR2 phosphorylates VE-cadherin, initiating the recruitment of the phosphatase SHP-2 and the release of bound Csk. In the absence of Csk, in turn, Tyr 527 is dephosphorylated, Src is activated, and the VE-cadherin/SHP-2/Src signaling module activates downstream Akt/eNOS, resulting in disruptions in endothelial cell-cell junctions. Thus, although the precise mechanisms whereby Src affects endothelial permeability remain uncertain, the importance of Src in this process is very clear. It should be further noted that numerous additional activators of Src exist that result in increased permeability, including hydrogen peroxide, tumor necrosis factor-alpha (TNF-α), and thrombin, among others (reviewed in Hu et al. 2008).
Focal adhesion effects of SFKs
In addition to the above-described mechanisms, SFKs affect vascular permeability through the regulation of cell-extracellular matrix connections (Guo et al. 2005). The endothelial cytoskeleton is bound to the extracellular matrix through focal adhesion complexes consisting of integrins, focal adhesion kinase (FAK), and multiple adaptor proteins (Aplin et al. 1998; Geiger et al. 2001). Integrins are transmembrane proteins and principal components of focal adhesions, serving as both adhesive and signaling receptors (Luscinskas and Lawler 1994). As studied primarily in fibroblasts, FAK upon integrin engagement undergoes autophosphorylation at Tyr397, and resultant conformational changes lead to SFK association through the Src SH-2 domain, leading to the phosphorylation of FAK at several tyrosine sites, including 861 (Schlaepfer et al. 1994; Calalb et al. 1995, 1996; Eide et al. 1995; Schlaepfer and Hunter 1996). These additional phosphorylations of FAK enhance the assembly of a calpain2/FAK/p42 ERK complex that then affects actin fiber assembly and focal adhesion formation/turnover (Westhoff et al. 2004). Thus, SFKs in focal adhesion complexes affect not only cellular migration, but also endothelial cell “shape” and vascular permeability (Riveline et al. 2001).
Src contributes to transcellular transport
A principal role of Src in transcellular transport is to coordinate protein complexes that form and internalize caveolae. The formation of caveolae, in turn, requires the tyrosine phosphorylation of caveolin-1, a membrane protein that acts as the primary structural component of caveolae (Li et al. 1996; Tiruppathi et al. 1997; Drab et al. 2001; Razani et al. 2001; Shajahan et al. 2004b). Upon binding of albumin to its receptor, gp60, at the endothelial surface, caveolin-1 interacts with clustered gp60, and Src is autophosphorylated at tyrosine 416 (Fig. 2; Parton et al. 1994; Li et al. 1996; Minshall et al. 2000). Activated Src then phosphorylates caveolin-1 on tyrosine 14, initiating caveolae fission from the plasma membrane (Shajahan et al. 2004a, 2004b). Importantly, caveolin-1 “knockout” mice fail to form caveolae and demonstrate impaired albumin uptake and transport (Drab et al. 2001; Razani et al. 2001; Schubert et al. 2001).
Fig. 2.
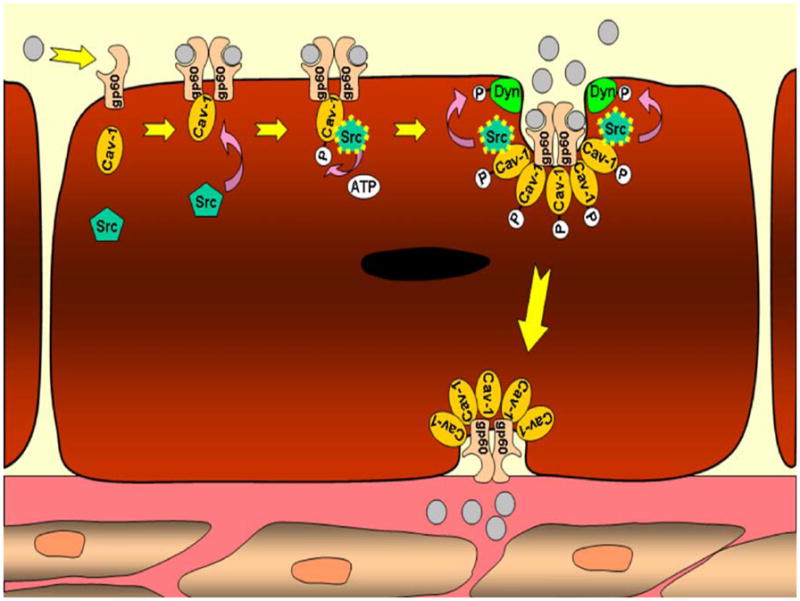
Mechanism of transcellular transport in endothelial cells. Albumin, the prototypical macromolecule involved in transcellular transport, binds its receptor, gp60. The gp60 receptors bound to albumin form clusters and interact with calveolin-1. Src then binds the calveolin-1 scaffolding domain and phosphorylates calveolin-1 and gp60. Additional caveolin is activated, as is dynamin-2, initiating vesicle fission and transcellular transport of albumin. Vesicle contents are released on the basolateral surface of the endothelial cell where they affect colloid osmotic pressure
Activated Src also phosphorylates caveolin-2, which can form hetero-oligomers with caveolin-1. These hetero-oligomers are thought to be important in the regulation of caveolae size (Li et al. 1998). Phosphorylation of both caveolin-1 and caveolin-2 may disrupt hetero-oligomer interactions and favor the formation of caveolae. In addition, Src phosphorylates dynamin-2, a GTPase protein that binds caveolin-1 and localizes to the neck of caveolae, mediating their release from the plasma membrane as intracellular vesicles. Thus, through the activation of Src kinase and its phosphorylation of caveolin-1, caveolin-2, and dynamin-2, Src plays a major role in obligatory steps of caveolae fission and caveolae-mediated endocytosis (Sverdlov et al. 2007; Hu et al. 2008; Thomas and Smart 2008).
SFKs promote endothelial permeability in inflammatory processes
As Src plays such a key role in normal endothelial cell barrier functions, it is not surprising that pathologic processes in which endothelial cell function are perturbed also involve SFK activation. One such state is inflammation. Fundamentally, the inflammatory response protects against stimuli related to infection, trauma, ischemia, burns, toxic agents, foreign antigens, and autoimmune processes (Nathan 2002). The clinical spectrum of inflammatory responses ranges from benign to overwhelming; excessive inflammatory responses attributable to vast cytokine and growth factor release by leukocytes may result in vascular endothelial damage and microvessel injury (Lehr et al. 2000). Such harm to the vascular endothelium may result in the dysfunction of paracellular and transcellular transport mechanisms, leading to a diminished ability to regulate the trans-endothelial passage of fluid, protein, and solute. Resultant “leaky vessels” and hemodynamic instability from such endothelial damage characterize systemic inflammatory syndromes and confer significant patient morbidity and mortality (Sibbald et al. 1995; Brun-Buisson 2000).
SFK function in endothelial permeability during pathologic states has been studied in mouse strains in which Src is functionally deleted (Src “knock-out” mice). Such mice have shown marked reductions in inflammatory responses to a variety of physiologic insults including endotoxemia and reperfusion injury. Paul and coworkers (2001) have demonstrated reduced VEGF-induced infarction volumes and associated edema in Src knockout mice following stroke, indicating the relevance of SFKs in mediating vascular permeability in the presence of VEGF. Similarly, Weis and colleagues (2004b) have shown that Flk (VEGF-R2)/VE-cadherin/beta-catenin complexes require Src for VEGF-induced permeability; upon genetic or pharmacologic blockade of Src, brain infarct volume and vascular permeability are significantly diminished. Using the same strategy, other Src family members have been implicated in the inflammatory response. Lowell and Berton (1998) have demonstrated resistance to lipopolysaccharide (LPS)-induced shock with an 80% reduction in mortality in mice functionally inactivated in the SFKs, Hck and Fgr.
The above studies have led to an examination of potential roles of Src inhibitors in important disease scenarios involving the inflammatory response. Utilizing PP2 (an SFKI) in rats, Lennmyr and coworkers (2004) have revealed a 50% reduction in brain infarct size on magnetic resonance imaging and staining with tri-phenyl tetrazolium chloride (a histologic marker for infarcted tissue) after transient middle cerebral artery occlusion. Reductions in acute lung injury following shock states have also been reported with SU6656, a selective SFKI. Severgnini et al. (2005) have demonstrated that after pre-treatment with SU6656, mortality in mice is reduced by 70% after LPS challenge. Khadaroo et al. (2004) have also reported modest reductions in acute lung injury with PP2 treatment after induced shock/resuscitation and subsequent LPS challenge. In the same study, this group has further demonstrated decreased alveolar macrophage priming to LPS activation after induced hypovolemic shock. The SFKI, PP1, has been shown to reduce vascular permeability, infarct size, and cerebral edema following stroke (Paul et al. 2001). Weis et al. (2004a) have translated the protective effects of PP1 into the arena of cardiovascular disease, demonstrating reduced edema and myocardial injury following myocardial infarction (MI). These authors have also reported the improvement in long-term survival and cardiac function in mice treated with PP1 after MI. Mechanistically, the authors propose that Src inhibition blocks the VEGF-induced disruption of a protein signaling complex consisting of Flk, VE-cadherin, beta-catenin, and Src, i.e. the same complex discussed above by which normal permeability is affected. Taken together, these data confirm the role of aberrant SFK activation, through Flk, on enhanced endothelial permeability and the negative impact of their activation on myocardial function following MI.
The above studies suggest that Src inhibitors have some efficacy in the treatment of certain ischemic/inflammatory conditions. Which conditions might benefit from these inhibitors requires further study. Several questions remain as to how SFKIs might be used in these conditions. First, are the responses to SFKIs seen in murine models attributable more to effects on vascular permeability or to the inhibition of leukocytes and their release of inflammatory mediators? Will the inhibition of endothelial permeability mechanisms confer greater benefit than the inhibition of leukocyte recruitment/activation, or vice versa? If so, should our pharmacologic targets be focused more on paracellular and/or transcellular transport mechanisms or on leukocyte recruitment and activation? Although not discussed in this review, others have detailed the important roles of SFKs in the recruitment and activation of neutrophils and monocytes (Baruzzi et al. 2008). These questions may not be as important in the administration of non-specific SFKIs so long as they prove therapeutic. However, as our understanding of the contributions of individual SFK members in different locations and organ systems increases, the true targets of therapy and the development of selective SFK member inhibitors may reinforce the relevance of these issues.
Next, relative to the progression of ischemic/inflammatory disease processes, would the efficacy of SFKIs vary with the timing of their administration? Specifically, when in the timeline of an ischemic/inflammatory disease process will SFKIs work best, if at all? Since preclinical evidence indicts SFKs as important mediators of endothelial permeability and leukocyte adhesion/activation, SFKIs may not prove efficacious once these processes have been robustly activated. Clinical trials involving anti-TNF- α therapy in the setting of systemic inflammatory conditions have failed to demonstrate significant efficacy, presumably because of the effects already imparted by inflammatory mediators before treatment (Reinhart and Karzai 2001). However, as the side effects of SFKIs are limited, their selective administration to “high-risk” patients early in evolving or anticipated ischemic/inflammatory complications may prove helpful. Examples of such clinical scenarios involve reperfusion procedures to ischemic tissues/limbs and may include acute MI, stroke, intracranial hemorrhage, and coronary artery bypass surgery.
Lastly, in what clinical contexts and groups/subgroups of patients are treatment with SFKIs indicated? Will SFKIs prove more efficacious in certain pathologic states than others? SFKs are important in leukocyte recruitment and activation; their inhibition in the setting of infectious processes may attenuate immune responses and worsen patient outcomes. However, SFKIs may prove extremely efficacious in states of tissue ischemia where the depression of immune responses may correlate with improved patient outcomes. Most importantly, will the administration of SFKIs truly result in reductions in morbidity and mortality? For example, if SFKIs reduce edema and infarct size during cerebral infarction, it is questionable whether a sufficient reduction can be obtained to make a meaningful impact on function or clinical outcome.
Although no clinical trials have yet been established to evaluate SFKIs in ischemic/inflammatory settings, their toleration by patients thus far in the treatment of neoplastic conditions has been acceptable. Further pre-clinical testing is required to evaluate the efficacy and timing of SKF inhibitors in animal models prior to their translation into treatment for ischemic/inflammatory conditions. Nonetheless, SFKIs remain compelling targets of therapy in such states and should be actively pursued.
Src mediates metastasis through VEGF-dependent alterations in the endothelial monolayer
Endothelial cell permeability affects two important steps in cancer metastasis: intravasation and extravasation of tumor cells from the vascular space (for a review, see Langley and Fidler 2007). Activation of SFKs in both tumor and endothelial cells is critical in these processes. In tumor cells, Src promotes the expression of pro-angiogenic factors, such as VEGF-A and VEGF-C. VEGF also activates SFKs in endothelial cells and affects endothelial cell permeability as described above. The actions of Src in tumor cells that then affect roles of Src in endothelial cell permeability are discussed in this section.
Src regulates VEGF expression in tumor cells and VEGF-induced endothelial permeability
Many studies have demonstrated important roles for Src activation mediating VEGF expression in both normal and tumor cells (Summy and Gallick 2003). Src was first shown to be important for VEGF expression in experiments of Mukhopadhyay and colleagues (1995) who demonstrated the attenuation of hypoxia-induced VEGF expression in Src-null fibroblasts. Recent work has provided additional evidence that Src directly regulates VEGF expression. In tumor cells, constitutive Src activation is important in VEGF deregulation. Evaluating the effects of Src knockdown in human colon cancer cells, investigators have demonstrated a 50-fold reduction in VEGF expression in response to hypoxic conditions (Ellis et al. 1998). In ovarian tumor cells, the expression of antisense constructs to Src decreases VEGF expression (Wiener et al. 1999); these investigators have also shown that VEGF expression requires Src activation in both prostate and pancreatic cancer cell lines under hypoxic conditions (Gray et al. 2005). Lastly, several studies have demonstrated that Src contributes to constitutive and EGF-induced VEGF expression in pancreatic cancer cell lines (Summy et al. 2005).
Recent work has revealed that Src activation is also important in the endothelial cell response to Src-mediated VEGF expression in tumors. As described above in the section on paracellular transport, VEGF binding to its cognate receptors results in VEGFR trans-phosphorylation and interactions with SFKs through the cytoplasmic SH-2 domains of these receptors. Subsequent phosphorylation of Src and its substrates (e.g., VE-cadherin and β-catenin) leads directly to changes in the permeability of the endothelial cell layer. This increased vascular permeability promotes the trans-endothelial migration of cancer cells and subsequent dissemination through the vascular system. Recent studies have shown that RNAi downregulation of c-Src in pancreatic cancer cells results in a significantly decreased incidence of VEGF-A expression in these cells, leading to a significant reduction of metastasis in orthotopic nude mouse models (Trevino et al. 2006b); in the same study, this group has also demonstrated that pharmacologic inhibition of Src in orthotopic pancreatic tumors results in decreased tumor size and metastatic spread, reaffirming the important role of Src activation and metastatic properties of many solid tumors.
The importance of Src function in endothelial cells in response to VEGF has also been demonstrated. Weis et al. (2004a) have shown that hematogenous injection of VEGF-expressing tumor cells in mice harboring genetic inactivation of Src or Yes results in a reduced incidence of lung and liver metastases compared with controls. Criscuoli et al. (2005) have also found a significantly reduced number of metastatic lung lesions in Src knockout mice upon subcutaneous and hematogenous injection of syngeneic lung cancer cells. Taken together, these data firmly support a role for SFKs in mediating VEGF expression in tumor cells and the extravasation of tumor cells from the vascular space. Such findings highlight the important roles of SFKs in metastasis while implicating SFKIs as potential agents that may inhibit these processes.
SFKIs as anti-metastatic agents in cancer
As discussed above, SFKs represent attractive targets for therapy because they have been shown to modify endothelial layer permeability through the regulation of VEGF and to mediate key steps of the metastatic cascade, namely tumor cell migration and extravasation (Weis et al. 2004a; Criscuoli et al. 2005). Whereas the specific mechanisms of SFK-related tumor cell migration through the endothelial layer remain to be fully elucidated, inhibitors of SFK activity and/or interactions through SH-2/SH-3 domains may attenuate important SFK-mediated functions related to metastasis. Specifically, such inhibitors may reduce tumor cell secretion and the endothelial cell response to VEGF, which as described above are important in mediating metastasis. The anti-metastatic effects of Src inhibitors may also be bolstered by additional anti-angiogenic effects that have been well documented in the literature (Park et al. 2007). For example, Src has been shown to mediate the expression of interleukin-8, an important cytokine involved in angiogeneis and tumor progression (Koch et al. 1992a; Strieter et al. 1995; Murdoch et al. 1999; Shi et al. 1999).
Currently, four SFKIs are under study in clinical trials in the USA: KX2–391, AZD0530, bosutinib, and dasatinib (Kopetz et al. 2007). KX2–391 inhibits substrate binding of SFK substrates, whereas the other agents competitively inhibit ATP binding. These agents are being tested as both single agents and in combination with other cytotoxic, anti-hormonal, and growth factor receptor inhibitory agents (Kopetz et al. 2007).
At the time of tumor detection, patients present in various stages of their disease. In cases where a patient's disease is clearly confined and curable through surgical excision (such as in situ lesions), clinical suspicion for regional or distant spread is low, and anti-metastatic agents may not be indicated. Likewise, patients who present with a large tumor burden and clear evidence of metastatic disease are likely not to benefit from therapeutic agents that function solely to inhibit metastasis. Between these two scenarios of benign and malignant disease, anti-metastatic agents might have an application that is currently undefined. However, the determination of SFKI efficacy in such roles may represent a significant challenge because of the frequent pre-existence of micro-metastatic disease at the time of diagnosis.
The ability of modern radiographic imaging to discern metastatic lesions remains limited for microscopic lesions. Often, patients are presumed to harbor sub-clinical metastasis based on the size or type of their primary tumor, providing the rationale for neoadjuvant therapy. Likewise, lymph nodes harboring tumor cells are often non-palpable on clinical examination. Even pathologic evaluation of lymph nodes has limitations and occasionally does not assuage patient and physician suspicions of local and/or distant disease. Such clinical situations lead to important questions regarding the introduction of anti-metastatic agents. Specifically, should anti-metastatic agents be administered to patients who (1) probably possess sub-clinical metastases, or (2) might develop metastases? Most importantly, will the administration of anti-metastatic agents, alone or in combination with current therapy, result in improved survival relative to current therapy alone? At present, the answers to these questions remain unknown. As clinical trials involving SFKIs progress, and their clinical impact in various patient groups/subgroups emerge, such potential anti-metastatic effects will eventually be evaluated. Finally, another use of Src inhibitors in the treatment of solid tumors is emerging. In several pre-clinical tumor models, Src activation is associated with chemo-resistance, which is overcome by SFKIs (Kopetz et al. 2007). Thus, Src inhibitors may have value in other settings than the prevention of metastases.
Concluding remarks
The roles of SFKs in mediating endothelial permeability are many. Through the tyrosine phosphorylation of both structural and signaling molecules, SFKs promote cytoskeletal contraction and the remodeling of cell-cell/cell-ECM connections. These changes in cell morphology and intercellular/cell-matrix interactions produce gap formations between neighboring endothelial cells and promote the paracellular transport of fluid and solutes. Such results with SFK inhibition have led to enormous interest in the therapeutic potential of Src inhibitors in the treatment of inflammation-related illness and neoplasms. To our knowledge, no clinical trials have yet tested the efficacy of SFKIs in ischemic/inflammatory conditions. However, given the correlation of Src activity with tumor progression, numerous Src inhibitors have been enrolled in clinical trials to evaluate for efficacy against cancers. The results of such trials are highly anticipated and will undoubtedly reveal much about the efficacy of SFKIs and their appropriate clinical application. Concurrent with these clinical trials, further studies must be undertaken into the specific mechanisms of SFK function; these will undoubtedly result in additional clinical targets and therapies.
Acknowledgments
The authors' own research was supported in part by NIH U54 CA 090810 and P20 CA101936 (G.E.G) and NIH T32 CA 09599 (M.P.K.)
Contributor Information
M. P. Kim, Department of Cancer Biology, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Box no. 173, Houston, TX 77030–4009, USA, Department of Surgical Oncology, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX, USA
S. I. Park, Department of Cancer Biology, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Box no. 173, Houston, TX 77030–4009, USA
S. Kopetz, Department of Cancer Biology, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Box no. 173, Houston, TX 77030–4009, USA, Department of Gastrointestinal Medical Oncology, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX, USA
G. E. Gallick, Email: ggallick@mdanderson.org, Department of Cancer Biology, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Box no. 173, Houston, TX 77030–4009, USA
References
- Aberle H, Schwartz H, Kemler R. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem. 1996;61:514–523. doi: 10.1002/(SICI)1097-4644(19960616)61:4%3C514::AID-JCB4%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Alland L, Peseckis SM, Atherton RE, Berthiaume L, Resh MD. Dual myristylation and palmitylation of Src family member p59fyn affects subcellular localization. J Biol Chem. 1994;269:16701–16705. [PubMed] [Google Scholar]
- Aplin AE, Howe A, Alahari SK, Juliano RL. Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol Rev. 1998;50:197–263. [PubMed] [Google Scholar]
- Baruzzi A, Caveggion E, Berton G. Regulation of phagocyte migration and recruitment by Src-family kinases. Cell Mol Life Sci. 2008;65:2175–2190. doi: 10.1007/s00018-008-8005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni G, Dejana E. Pores in the sieve and channels in the wall: control of paracellular permeability by junctional proteins in endothelial cells. Microcirculation. 2001;8:143–152. doi: 10.1038/sj/mn/7800084. [DOI] [PubMed] [Google Scholar]
- Bellamy WT, Richter L, Frutiger Y, Grogan TM. Expression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer Res. 1999;59:728–733. [PubMed] [Google Scholar]
- Birukov KG, Csortos C, Marzilli L, Dudek S, Ma SF, Bresnick AR, Verin AD, Cotter RJ, Garcia JG. Differential regulation of alternatively spliced endothelial cell myosin light chain kinase isoforms by p60(Src) J Biol Chem. 2001;276:8567–8573. doi: 10.1074/jbc.M005270200. [DOI] [PubMed] [Google Scholar]
- Brown MT, Cooper JA. Regulation, substrates and functions of src. Biochim Biophys Acta. 1996;1287:121–149. doi: 10.1016/0304-419x(96)00003-0. [DOI] [PubMed] [Google Scholar]
- Brun-Buisson C. The epidemiology of the systemic inflammatory response. Intensive Care Med. 2000;26(Suppl 1):S64–S74. doi: 10.1007/s001340051121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calalb MB, Zhang X, Polte TR, Hanks SK. Focal adhesion kinase tyrosine-861 is a major site of phosphorylation by Src. Biochem Biophys Res Commun. 1996;228:662–668. doi: 10.1006/bbrc.1996.1714. [DOI] [PubMed] [Google Scholar]
- Cartwright CA, Eckhart W, Simon S, Kaplan PL. Cell transformation by pp60c-src mutated in the carboxy-terminal regulatory domain. Cell. 1987;49:83–91. doi: 10.1016/0092-8674(87)90758-6. [DOI] [PubMed] [Google Scholar]
- Chou MT, Wang J, Fujita DJ. Src kinase becomes preferentially associated with the VEGFR, KDR/Flk-1, following VEGF stimulation of vascular endothelial cells. BMC Biochem. 2002;3:32. doi: 10.1186/1471-2091-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen GB, Ren R, Baltimore D. Modular binding domains in signal transduction proteins. Cell. 1995;80:237–248. doi: 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- Criscuoli ML, Nguyen M, Eliceiri BP. Tumor metastasis but not tumor growth is dependent on Src-mediated vascular permeability. Blood. 2005;105:1508–1514. doi: 10.1182/blood-2004-06-2246. [DOI] [PubMed] [Google Scholar]
- Dejana E, Bazzoni G, Lampugnani MG. Vascular endothelial (VE)-cadherin: only an intercellular glue? Exp Cell Res. 1999;252:13–19. doi: 10.1006/excr.1999.4601. [DOI] [PubMed] [Google Scholar]
- Dias S, Hattori K, Zhu Z, Heissig B, Choy M, Lane W, Wu Y, Chadburn A, Hyjek E, Gill M, et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest. 2000;106:511–521. doi: 10.1172/JCI8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias S, Hattori K, Heissig B, Zhu Z, Wu Y, Witte L, Hicklin DJ, Tateno M, Bohlen P, Moore MA, et al. Inhibition of both paracrine and autocrine VEGF/VEGFR-2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias. Proc Natl Acad Sci USA. 2001;98:10857–10862. doi: 10.1073/pnas.191117498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Eide BL, Turck CW, Escobedo JA. Identification of Tyr-397 as the primary site of tyrosine phosphorylation and pp60src association in the focal adhesion kinase, pp125FAK. Mol Cell Biol. 1995;15:2819–2827. doi: 10.1128/mcb.15.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis LM, Staley CA, Liu W, Fleming RY, Parikh NU, Bucana CD, Gallick GE. Down-regulation of vascular endothelial growth factor in a human colon carcinoma cell line transfected with an antisense expression vector specific for c-src. J Biol Chem. 1998;273:1052–1057. doi: 10.1074/jbc.273.2.1052. [DOI] [PubMed] [Google Scholar]
- Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602:114–130. doi: 10.1016/s0304-419x(02)00040-9. [DOI] [PubMed] [Google Scholar]
- Frame MC, Fincham VJ, Carragher NO, Wyke JA. v-Src's hold over actin and cell adhesions. Nat Rev Mol Cell Biol. 2002;3:233–245. doi: 10.1038/nrm779. [DOI] [PubMed] [Google Scholar]
- Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995;163:510–522. doi: 10.1002/jcp.1041630311. [DOI] [PubMed] [Google Scholar]
- Garcia JG, Verin AD, Schaphorst K, Siddiqui R, Patterson CE, Csortos C, Natarajan V. Regulation of endothelial cell myosin light chain kinase by Rho, cortactin, and p60(src) Am J Physiol. 1999;276:L989–L998. doi: 10.1152/ajplung.1999.276.6.L989. [DOI] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix–cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS, Gallick GE. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24:3110–3120. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Signal transduction of beta-catenin. Curr Opin Cell Biol. 1995;7:634–640. doi: 10.1016/0955-0674(95)80104-9. [DOI] [PubMed] [Google Scholar]
- Guo M, Wu MH, Granger HJ, Yuan SY. Focal adhesion kinase in neutrophil-induced microvascular hyperpermeability. Microcirculation. 2005;12:223–232. doi: 10.1080/10739680590905251. [DOI] [PubMed] [Google Scholar]
- Ha CH, Bennett AM, Jin ZG. A novel role of vascular endothelial cadherin in modulating c-Src activation and downstream signaling of vascular endothelial growth factor. J Biol Chem. 2008;283:7261–7270. doi: 10.1074/jbc.M702881200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Hu G, Place AT, Minshall RD. Regulation of endothelial permeability by Src kinase signaling: vascular leakage versus transcellular transport of drugs and macromolecules. Chem Biol Interact. 2008;171:177–189. doi: 10.1016/j.cbi.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–5642. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- Ito N, Wernstedt C, Engstrom U, Claesson-Welsh L. Identification of vascular endothelial growth factor receptor-1 tyrosine phosphorylation sites and binding of SH2 domain-containing molecules. J Biol Chem. 1998;273:23410–23418. doi: 10.1074/jbc.273.36.23410. [DOI] [PubMed] [Google Scholar]
- Johnson FM, Gallick GE. SRC family nonreceptor tyrosine kinases as molecular targets for cancer therapy. Anti-cancer Agents Med Chem. 2007;7:651–659. doi: 10.2174/187152007784111278. [DOI] [PubMed] [Google Scholar]
- Kaplan KB, Bibbins KB, Swedlow JR, Arnaud M, Morgan DO, Varmus HE. Association of the amino-terminal half of c-Src with focal adhesions alters their properties and is regulated by phosphorylation of tyrosine 527. EMBO J. 1994;13:4745–4756. doi: 10.1002/j.1460-2075.1994.tb06800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khadaroo RG, He R, Parodo J, Powers KA, Marshall JC, Kapus A, Rotstein OD. The role of the Src family of tyrosine kinases after oxidant-induced lung injury in vivo. Surgery. 2004;136:483–488. doi: 10.1016/j.surg.2004.05.029. [DOI] [PubMed] [Google Scholar]
- Kmiecik TE, Shalloway D. Activation and suppression of pp60c-src transforming ability by mutation of its primary sites of tyrosine phosphorylation. Cell. 1987;49:65–73. doi: 10.1016/0092-8674(87)90756-2. [DOI] [PubMed] [Google Scholar]
- Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992a;258:1798–1801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- Koch CA, Moran MF, Anderson D, Liu XQ, Mbamalu G, Pawson T. Multiple SH2-mediated interactions in v-src-transformed cells. Mol Cell Biol. 1992b;12:1366–1374. doi: 10.1128/mcb.12.3.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopetz S, Shah AN, Gallick GE. Src continues aging: current and future clinical directions. Clin Cancer Res. 2007;13:7232–7236. doi: 10.1158/1078-0432.CCR-07-1902. [DOI] [PubMed] [Google Scholar]
- Lambeng N, Wallez Y, Rampon C, Cand F, Christe G, Gulino-Debrac D, Vilgrain I, Huber P. Vascular endothelial-cadherin tyrosine phosphorylation in angiogenic and quiescent adult tissues. Circ Res. 2005;96:384–391. doi: 10.1161/01.RES.0000156652.99586.9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley RR, Fidler IJ. Tumor cell-organ microenvironment interactions in the pathogenesis of cancer metastasis. Endocr Rev. 2007;28:297–321. doi: 10.1210/er.2006-0027. [DOI] [PubMed] [Google Scholar]
- Lehr HA, Bittinger F, Kirkpatrick CJ. Microcirculatory dysfunction in sepsis: a pathogenetic basis for therapy? J Pathol. 2000;190:373–386. doi: 10.1002/(SICI)1096-9896(200002)190:3<373::AID-PATH593>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Lennmyr F, Ericsson A, Gerwins P, Akterin S, Ahlstrom H, Terent A. Src family kinase-inhibitor PP2 reduces focal ischemic brain injury. Acta Neurol Scand. 2004;110:175–179. doi: 10.1111/j.1600-0404.2004.00306.x. [DOI] [PubMed] [Google Scholar]
- Levinson NM, Seeliger MA, Cole PA, Kuriyan J. Structural basis for the recognition of c-Src by its inactivator Csk. Cell. 2008;134:124–134. doi: 10.1016/j.cell.2008.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Seitz R, Lisanti MP. Phosphorylation of caveolin by src tyrosine kinases. The alpha-isoform of caveolin is selectively phosphorylated by v-Src in vivo. J Biol Chem. 1996;271:3863–3868. [PubMed] [Google Scholar]
- Li S, Galbiati F, Volonte D, Sargiacomo M, Engelman JA, Das K, Scherer PE, Lisanti MP. Mutational analysis of caveolin-induced vesicle formation. Expression of caveolin-1 recruits caveolin-2 to caveolae membranes. FEBS Lett. 1998;434:127–134. doi: 10.1016/s0014-5793(98)00945-4. [DOI] [PubMed] [Google Scholar]
- Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol. 2005;17:459–465. doi: 10.1016/j.ceb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Lowell CA, Berton G. Resistance to endotoxic shock and reduced neutrophil migration in mice deficient for the Src-family kinases Hck and Fgr. Proc Natl Acad Sci USA. 1998;95:7580–7584. doi: 10.1073/pnas.95.13.7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum H, Malik AB. Regulation of vascular endothelial barrier function. Am J Physiol. 1994;267:L223–L241. doi: 10.1152/ajplung.1994.267.3.L223. [DOI] [PubMed] [Google Scholar]
- Luscinskas FW, Lawler J. Integrins as dynamic regulators of vascular function. FASEB J. 1994;8:929–938. doi: 10.1096/fasebj.8.12.7522194. [DOI] [PubMed] [Google Scholar]
- Masood R, Cai J, Zheng T, Smith DL, Hinton DR, Gill PS. Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor-positive human tumors. Blood. 2001;98:1904–1913. doi: 10.1182/blood.v98.6.1904. [DOI] [PubMed] [Google Scholar]
- Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332:272–275. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NP, Risau W, Ullrich A. High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell. 1993;72:835–846. doi: 10.1016/0092-8674(93)90573-9. [DOI] [PubMed] [Google Scholar]
- Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE, Malik AB. Endothelial cell-surface gp60 activates vesicle formation and trafficking via G(i)-coupled Src kinase signaling pathway. J Cell Biol. 2000;150:1057–1070. doi: 10.1083/jcb.150.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee CH, Kuriyan J, Miller WT. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385:650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- Moran MF, Koch CA, Anderson D, Ellis C, England L, Martin GS, Pawson T. Src homology region 2 domains direct protein-protein interactions in signal transduction. Proc Natl Acad Sci USA. 1990;87:8622–8626. doi: 10.1073/pnas.87.21.8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha DR, Myers CL, Schaeffer RC., Jr Endothelial contraction and monolayer hyperpermeability are regulated by Src kinase. Am J Physiol. 2003;284:H994–H1002. doi: 10.1152/ajpheart.00862.2002. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Tsiokas L, Zhou XM, Foster D, Brugge JS, Sukhatme VP. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature. 1995;375:577–581. doi: 10.1038/375577a0. [DOI] [PubMed] [Google Scholar]
- Murdoch C, Monk PN, Finn A. Cxc chemokine receptor expression on human endothelial cells. Cytokine. 1999;11:704–712. doi: 10.1006/cyto.1998.0465. [DOI] [PubMed] [Google Scholar]
- Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- Park SI, Shah AN, Zhang J, Gallick GE. Regulation of angiogenesis and vascular permeability by Src family kinases: opportunities for therapeutic treatment of solid tumors. Expert Opin Ther Targets. 2007;11:1207–1217. doi: 10.1517/14728222.11.9.1207. [DOI] [PubMed] [Google Scholar]
- Parton RG, Joggerst B, Simons K. Regulated internalization of caveolae. J Cell Biol. 1994;127:1199–1215. doi: 10.1083/jcb.127.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul R, Zhang ZG, Eliceiri BP, Jiang Q, Boccia AD, Zhang RL, Chopp M, Cheresh DA. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med. 2001;7:222–227. doi: 10.1038/84675. [DOI] [PubMed] [Google Scholar]
- Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- Piwnica-Worms H, Saunders KB, Roberts TM, Smith AE, Cheng SH. Tyrosine phosphorylation regulates the biochemical and biological properties of pp60c-src. Cell. 1987;49:75–82. doi: 10.1016/0092-8674(87)90757-4. [DOI] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, et al. Caveolin-1 null mice are viable but show evidence of hyper-proliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- Reinhart K, Karzai W. Anti-tumor necrosis factor therapy in sepsis: update on clinical trials and lessons learned. Crit Care Med. 2001;29:S121–125. doi: 10.1097/00003246-200107001-00037. [DOI] [PubMed] [Google Scholar]
- Resh MD. Interaction of tyrosine kinase oncoproteins with cellular membranes. Biochim Biophys Acta. 1993;1155:307–322. doi: 10.1016/0304-419x(93)90012-2. [DOI] [PubMed] [Google Scholar]
- Rippe B, Rosengren BI, Carlsson O, Venturoli D. Transendothelial transport: the vesicle controversy. J Vasc Res. 2002;39:375–390. doi: 10.1159/000064521. [DOI] [PubMed] [Google Scholar]
- Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, Narumiya S, Kam Z, Geiger B, Bershadsky AD. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153:1175–1186. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274:36734–36740. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- Rous FP. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med. 1911;13:397–411. doi: 10.1084/jem.13.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, Geer van der P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schubert W, Frank PG, Razani B, Park DS, Chow CW, Lisanti MP. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J Biol Chem. 2001;276:48619–48622. doi: 10.1074/jbc.C100613200. [DOI] [PubMed] [Google Scholar]
- Severgnini M, Takahashi S, Tu P, Perides G, Homer RJ, Jhung JW, Bhavsar D, Cochran BH, Simon AR. Inhibition of the Src and Jak kinases protects against lipopolysaccharide-induced acute lung injury. Am J Respir Crit Care Med. 2005;171:858–867. doi: 10.1164/rccm.200407-981OC. [DOI] [PubMed] [Google Scholar]
- Shajahan AN, Timblin BK, Sandoval R, Tiruppathi C, Malik AB, Minshall RD. Role of Src-induced dynamin-2 phosphorylation in caveolae-mediated endocytosis in endothelial cells. J Biol Chem. 2004a;279:20392–20400. doi: 10.1074/jbc.M308710200. [DOI] [PubMed] [Google Scholar]
- Shajahan AN, Tiruppathi C, Smrcka AV, Malik AB, Minshall RD. Gbetagamma activation of Src induces caveolae-mediated endocytosis in endothelial cells. J Biol Chem. 2004b;279:48055–48062. doi: 10.1074/jbc.M405837200. [DOI] [PubMed] [Google Scholar]
- Shi Q, Abbruzzese JL, Huang S, Fidler IJ, Xiong Q, Xie K. Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin Cancer Res. 1999;5:3711–3721. [PubMed] [Google Scholar]
- Shi S, Garcia JG, Roy S, Parinandi NL, Natarajan V. Involvement of c-Src in diperoxovanadate-induced endothelial cell barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2000;279:L441–L451. doi: 10.1152/ajplung.2000.279.3.L441. [DOI] [PubMed] [Google Scholar]
- Sibbald WJ, Doig G, Inman KJ. Sepsis, SIRS and infection. Int Care Med. 1995;21:299–301. doi: 10.1007/BF01705407. [DOI] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- Stahl ML, Ferenz CR, Kelleher KL, Kriz RW, Knopf JL. Sequence similarity of phospholipase C with the non-catalytic region of src. Nature. 1988;332:269–272. doi: 10.1038/332269a0. [DOI] [PubMed] [Google Scholar]
- Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170–173. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- Strieter RM, Polverini PJ, Arenberg DA, Walz A, Opdenakker G, Van Damme J, Kunkel SL. Role of C-X-C chemokines as regulators of angiogenesis in lung cancer. J Leukoc Biol. 1995;57:752–762. doi: 10.1002/jlb.57.5.752. [DOI] [PubMed] [Google Scholar]
- Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- Summy JM, Gallick GE. Treatment for advanced tumors: SRC reclaims center stage. Clin Cancer Res. 2006;12:1398–1401. doi: 10.1158/1078-0432.CCR-05-2692. [DOI] [PubMed] [Google Scholar]
- Summy JM, Trevino JG, Baker CH, Gallick GE. c-Src regulates constitutive and EGF-mediated VEGF expression in pancreatic tumor cells through activation of phosphatidyl inositol-3 kinase and p38 MAPK. Pancreas. 2005;31:263–274. doi: 10.1097/01.mpa.0000178280.50534.0c. [DOI] [PubMed] [Google Scholar]
- Sverdlov M, Shajahan AN, Minshall RD. Tyrosine phosphorylation-dependence of caveolae-mediated endocytosis. J Cell Mol Med. 2007;11:1239–1250. doi: 10.1111/j.1582-4934.2007.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Thomas CM, Smart EJ. Caveolae structure and function. J Cell Mol Med. 2008;12:796–809. doi: 10.1111/j.1582-4934.2008.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiruppathi C, Song W, Bergenfeldt M, Sass P, Malik AB. Gp60 activation mediates albumin transcytosis in endothelial cells by tyrosine kinase-dependent pathway. J Biol Chem. 1997;272:25968–25975. doi: 10.1074/jbc.272.41.25968. [DOI] [PubMed] [Google Scholar]
- Trevino JG, Summy JM, Gallick GE. SRC inhibitors as potential therapeutic agents for human cancers. Mini Rev Med Chem. 2006a;6:681–687. doi: 10.2174/138955706777435724. [DOI] [PubMed] [Google Scholar]
- Trevino JG, Summy JM, Lesslie DP, Parikh NU, Hong DS, Lee FY, Donato NJ, Abbruzzese JL, Baker CH, Gallick GE. Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am J Pathol. 2006b;168:962–972. doi: 10.2353/ajpath.2006.050570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma PL, Hubbard AL. Transcytosis: crossing cellular barriers. Physiol Rev. 2003;83:871–932. doi: 10.1152/physrev.00001.2003. [DOI] [PubMed] [Google Scholar]
- Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol. 2004a;167:223–229. doi: 10.1083/jcb.200408130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, McSharry H, Iwakura A, Yoon YS, Himes N, et al. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004b;113:885–894. doi: 10.1172/JCI20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhoff MA, Serrels B, Fincham VJ, Frame MC, Carragher NO. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol Cell Biol. 2004;24:8113–8133. doi: 10.1128/MCB.24.18.8113-8133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener JR, Nakano K, Kruzelock RP, Bucana CD, Bast RC, Jr, Gallick GE. Decreased Src tyrosine kinase activity inhibits malignant human ovarian cancer tumor growth in a nude mouse model. Clin Cancer Res. 1999;5:2164–2170. [PubMed] [Google Scholar]
- Wong RK, Baldwin AL, Heimark RL. Cadherin-5 redistribution at sites of TNF-alpha and IFN-gamma-induced permeability in mesenteric venules. Am J Physiol. 1999;276:H736–H748. doi: 10.1152/ajpheart.1999.276.2.H736. [DOI] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- Yeatman TJ. A renaissance for SRC. Nat Rev. 2004;4:470–480. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Huang Q, Wu HM. Myosin light chain phosphorylation: modulation of basal and agonist-stimulated venular permeability. Am J Physiol. 1997;272:H1437–H1443. doi: 10.1152/ajpheart.1997.272.3.H1437. [DOI] [PubMed] [Google Scholar]