

Abstract
Kv channels detect changes in the membrane potential via their voltage-sensing domains (VSDs) that control the status of the S6 bundle crossing (BC) gate. The movement of the VSDs results in a transfer of the S4 gating charges across the cell membrane but only the last 10–20% of the total gating charge movement is associated with BC gate opening, which involves cooperative transition(s) in the subunits. Substituting the proline residue P475 in the S6 of the Shaker channel by a glycine or alanine causes a considerable shift in the voltage-dependence of the cooperative transition(s) of BC gate opening, effectively isolating the late gating charge component from the other gating charge that originates from earlier VSD movements. Interestingly, both mutations also abolished Shaker’s sensitivity to 4-aminopyridine, which is a pharmacological tool to isolate the late gating charge component. The alanine substitution (that would promote a α-helical configuration compared to proline) resulted in the largest separation of both gating charge components; therefore, BC gate flexibility appears to be important for enabling the late cooperative step of channel opening.
Introduction
Voltage-gated potassium (Kv) channels exist as tetramers of α-subunits (1), each containing six membrane spanning helices (S1–S6), and regulate their conducting state as a function of membrane potential. The four S5 through S6 segments form the central K+ pore that can be sealed off by the channel gate; this gate is formed by the bottom parts of the S6 segment (S6c) and localizes at the level of their bundle crossing (BC) (Fig. 1 A, for review see (2)). Through an interaction between the S4-S5 linker and the S6c (3,4), the BC gate is under direct control of the voltage-sensing domains (VSDs) that each contain a positively charged S4 segment (5). The movement of the VSDs and their S4 segments results in a transfer of these positive charges (termed gating charges) across the membrane electric field, generating a transient gating current (6,7). Whereas a membrane depolarization pushes the VSDs outward resulting in BC gate opening, a repolarization forces the VSDs inward and causes BC gate closure (8).
Figure 1.
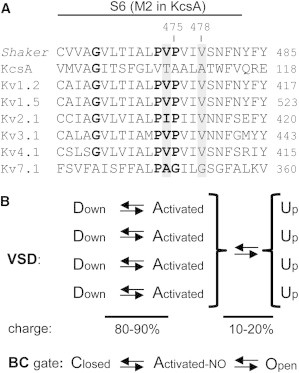
Sequence alignment of Shaker Kv channels with a simplified gating model. (A) The sequence alignment of S6c from different Kv families reveals a highly conserved proline-x-proline motif (PxP), where x is a hydrophobic residue which in Shaker is a valine. It has been proposed that this motif confers flexibility to S6c and forms a hinge region of the BC gate (25). The residues (V474 and V478) that according to a MD simulation (using KcsA as template) are involved in 4-AP binding are highlighted in gray. Interestingly, the Kv7 channels do not possess a full PxP motif and are as KcsA insensitive to 4-AP inhibition. (B) A simplified transition state diagram for the Shaker Kv channel. The different VSD transition states are represented on top with below the corresponding state of the BC gate. Each of the four VSDs can move largely independent from the Down state up to the Activated state. When the VSDs are in their down or activated state the channel is not conducting and the BC gate resides in the closed and activated-not-open (Activated-NO) state, respectively. When all four VSDs have reached their activated state, they transition in a subunit-cooperative way to their Up state, which corresponds with the fully open state of the BC gate. Gating currents provide information on the VSD movements and show that 80–90% of the total gating charges move during their independent reorientation(s) from the down to activated state. The remaining 10–20% of the gating charge is associated with the cooperative transition(s) from Activated to Up, which is accompanied by BC gate opening.
Detailed analysis of both the ionic and gating currents from the Shaker Kv channel has resulted in several kinetic models of channel activation (9–11). Common aspects of these models are that in the activation pathway from closed to fully open, the channel moves through several intermediate states with at least one transition displaying subunit cooperativity (12). It has been proposed that the VSDs move nearly independently from the Down up to their Activated state. When all four VSDs have reached their activated state, the BC gate cooperatively transitions from its activated-not-open to the fully open conducting state (Fig. 1 B). It has been shown that the status of the BC gate in the activated-not-open state differs from both the closed and open state (13). This was further strengthened with voltage-clamp fluorometry, which directly showed that the pathway from a closed to fully open BC gate involves a two-step transition (14). The gating charge component linked to the last cooperative step of channel opening (the transition from activated-not-open to open) could be isolated by several different VSD mutations within the S4 segment (15–18) and S2 segment (19). All these VSD mutations displayed a similar behavior and showed that the last transition in the activation pathway is associated with only 10–20% of the total gating charges being moved. Although this last transition has been linked to BC gate opening, no mutations within the BC gate region have been reported previously that isolate this late gating charge component from earlier VSD movements.
In addition to the introduction of VSD mutations, most Kv channels can also be pharmacologically prevented from passing this last cooperative transition by application of aminopyridine derivatives such as 4-aminopyridine (4-AP) (16). In agreement, gating current analysis showed that 4-AP application resulted in a 10–20% loss in gating charge movement (16,20). Simultaneously, the typical slowing of gating charge return (VSD deactivation), which is due to BC gate opening, is abolished in the presence of 4-AP (20). However, the early VSD movements remain largely unaffected by 4-AP. Mapping the binding site for 4-AP showed the involvement of the S6 segment (21,22) and molecular docking experiments implicated the cytoplasmic entrance of the K+ pore (Fig. 1 A) (23). This 4-AP binding region includes the BC gate and the highly conserved PVP motif that most likely destabilizes S6c to form a hinge point for the BC gate (24,25). Here, we report that substituting the second proline (P475) of the PVP motif by a glycine or an alanine abolishes 4-AP sensitivity in Shaker but at the same time mimics the 4-AP effect by isolating the late gating charge component from earlier VSD movements.
Material and Methods
Molecular biology
The N-terminal deletion mutant Δ6–46 Shaker (Shaker-IR), which removes fast inactivation (26), was expressed using a pGW1 expression vector. For gating current recordings the W434F mutation was introduced to generate the nonconducting Shaker-IR-W434F construct (27). The P475A and P475G mutations were introduced in both Shaker-IR and Shaker-IR-W434F by a PCR reaction using mutant primers and the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). After PCR-based mutagenesis, double-strand sequencing of the entire construct confirmed the presence of the desired modification and the absence of unwanted mutations. The plasmid coding for the green fluorescent protein, used to identify transfected cells, was purchased from Clontech (Palo Alto, CA). Plasmid DNA for mammalian expression was obtained by amplification in XL2 Bluescript cells (Stratagene), and then isolated from the bacterial cells with the endotoxin-free Maxiprep kit (Macherey-Nagel, Düren, Germany). The cDNA concentration was determined by ultraviolet absorption.
Electrophysiology and solutions
HEK293 cells were cultured in modified Eagle’s medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% nonessential amino acids (Invitrogen, Carlsbad, CA). Cells were transiently transfected with appropriate concentrations of plasmid DNA using polyethyleneimine (28) that was purchased from Sigma-Aldrich (St Louis, MO); for ionic and gating current measurements 60% confluent culture dishes (6 cm in diameter) were transfected with 0.5 and 6 μg plasmid DNA, respectively. Approximately 20 (for ionic currents) to 48 h (for gating currents) after transfection, cells were harvested by trypsinization and transferred to a recording chamber mounted on the stage of an inverted microscope. Whole-cell current measurements were performed at room temperature (20 to 23°C) with an Axopatch-200B amplifier and the recordings were digitized with a Digidata-1200A acquisition system (Molecular Devices, Sunnyvale, CA). Command voltages and data storage were controlled with pClamp8 software. Patch pipettes were pulled from 1.2 mm quick-fill borosilicate glass capillaries (World Precision Instruments, Sarasota, FL) with a P-2000 puller (Sutter Instrument, Novato, CA) and afterward heat-polished.
For ionic current measurements the cells were constantly superfused with external bath solution that contained (in mM) NaCl 130, KCl 4, CaCl2 1.8, MgCl2 1, HEPES 10, Glucose 10, adjusted to pH 7.35 with NaOH. The patch pipettes were filled with internal solution containing (in mM) KCl 110, K4BAPTA 5, K2ATP 5, MgCl2 1, HEPES 10, adjusted to pH 7.2 with KOH. Gating currents were recorded in the whole-cell configuration by replacing monovalent cations with N-methyl-D-glucamine (NMG+). The bath solution contained (in mM) NMG+ 140, HEPES 10, Glucose 10, MgCl2 1, CaCl2 1.8, titrated to pH 7.35 with HCl. The pipette solution contained (in mM) NMG+ 140, HEPES 10, EGTA 10, MgCl2 1, titrated to pH 7.2 with HCl. For both ionic and gating current measurements the junction potentials were zeroed with the filled pipette in the bath solution and experiments were excluded from analysis if the voltage error estimate exceeded 5 mV after series resistance compensation. For gating current recordings, leak currents and capacitive currents remaining after compensation were subtracted online using a −P/6 method for activation protocols (−95 mV holding potential) and +P/8 for deactivation protocols (using 0 mV holding potential for Shaker-IR-W434F control and +90 mV for both P475A and P475G). Ionic current recordings were not leak-corrected.
4-AP was dissolved in the external recording solution for either ionic or gating current measurements. The pH of the different concentrations was determined and adjusted to 7.35 using HCl, if required. 4-AP solutions were applied to the cells using a pressurized fast perfusion system with quartz micromanifold (ALA Scientific, Farmingdale, NY), allowing rapid exchange of the external solutions.
Data analysis
Details of pulse protocols used to elicit ionic (I) or gating (IQ) currents were adjusted to determine the biophysical properties of each construct adequately and are shown in the figures or described in legends. The conductance versus voltage (GV) curve and the charge versus voltage (QV) curve of Shaker-IR control were fitted with a Boltzmann equation: y = 1/{1 + exp[−(V − V1/2)/k]}, where V represents the applied voltage, V1/2 the midpoint potential at which 50% of the total charge has moved or half of the channels have opened, and k the slope factor. The GV curve of both P475A and P475G were also approximated with a single Boltzmann equation, but their QV curve was fitted with the sum of two Boltzmann distributions to determine V1/2 and k of both components adequately (Table 1). The kinetics of the ionic currents (τ_ Iac and τ_ Ideac) were determined by fitting the rise in current activation (Iac) or decay during deactivation (Ideac) with a single or double exponential function. The time constants of the activating and deactivating gating currents (τ_ IQac and τ_ IQdeac) were determined by fitting the decaying part of the gating current traces from IQac and IQdeac with a single exponential function. Although previous studies showed that IQdeac of Shaker-IR-W434F control was best approximated with a double exponential function (29,30), we used only a single exponential because the slow component could not be resolved; it was hidden within the experimental noise (both for Shaker control and P475A).
Table 1.
Parameters for the GV and QV curves of Shaker control, P475A and P475G
| GV curve (voltage-dependence of BC gate opening) |
||||||
|---|---|---|---|---|---|---|
| V1/2 (mV) | k (mV) | n | ||||
| Shaker-IR | −28.3 ± 2.8 | 4.9 ± 0.4 | 8 | |||
| P475A | 73.3 ± 2.7 | 11.6 ± 0.8 | 8 | |||
| P475G | 17.4 ± 1.2 | 7.8 ± 0.8 | 5 | |||
| QV curve (voltage-dependence of VSD movement) | ||||||
| V1/2 (mV) | k (mV) | n | ||||
| ECS | 4-AP | ECS | 4-AP | ECS | 4-AP | |
| Shaker-IR-W434F | −33.9 ± 2.6 | −25.6 ± 1.2 | 11.5 ± 0.8 | 14.5 ± 1.1 | 8 | 3 |
| P475A: 1st comp | −33.1 ± 0.9 | −34.4 ± 1.5 | 13.3 ± 0.9 | 14.3 ± 3.1 | 5 | 5 |
| 2nd comp | 62.4 ± 3.7 | 60.1 ± 3.4 | 19.0 ± 3.8 | 13.7 ± 3.1 | ||
| P475G: 1st comp | −33.3 ± 1.1 | ND | 13.8 ± 0.9 | ND | 4 | |
| 2nd comp | 18.1 ± 4.6 | 9.5 ± 2.9 | ||||
Values on top represent the half-activation voltage (V1/2) and slope factor (k) for the GV curve of Shaker control, P475A and P475G. n indicates the number of cells analyzed. Bottom values represent the V1/2 and the k values for the QV curves obtained in control conditions (column ECS) or in presence of 1 mM 4-AP (column 4-AP). Both P475A and P457G produced a QV curve with two gating charge components, 1st and 2nd component, respectively. ND: not determined.
Results
P475A mutant displays a QV curve with two gating charge components
Substituting the second proline of the PVP motif by an alanine (mutant P475A, Fig. 1 A) in the fast inactivation removed Shaker channel (Shaker-IR), resulted in voltage-dependent K+ selective currents when expressed in HEK293 cells (Fig. 2, A–C). In our experimental conditions the P475A mutation shifted the voltage-dependence of BC gate opening/closure, shown by the conductance (G) versus voltage GV curve, by ∼100 mV toward more positive potentials (Fig. 2 D, Table 1). Closer inspection of the time course of the ionic current activation (Iac) showed that channel opening was best approximated with a double exponential function (Fig. 2 E). The time constants of both Iac components (τ_Iac_fast and τ_Iac_slow) and also those of ionic current deactivation (τ_Ideac) were markedly slower compared to Shaker-IR control (Fig. 2 F), and this slowing in the kinetics was not only caused by the shift of the GV curve. In addition to the slowing, both Iac and Ideac kinetics displayed a reduced voltage-dependency (mainly τ_Iac), in agreement with the shallower slope of the GV curve. These observations are in good agreement with previous reports on the effect of the corresponding P to A mutation in the human Shaker homologs Kv1.5 (25) and Kv1.4 (38), and the homologous mutation in the Drosophila Shaw2 channel (31).
Figure 2.
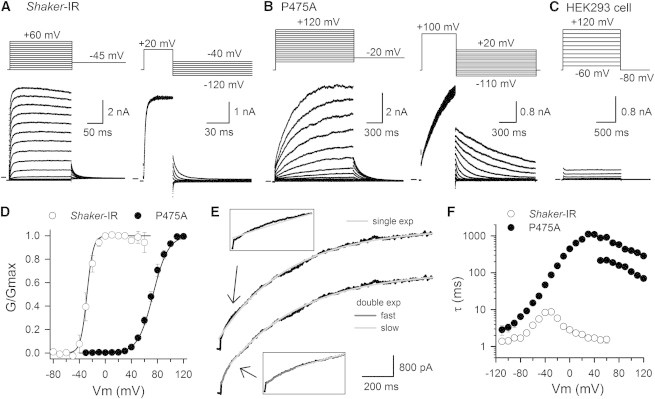
Ionic current measurements of Shaker-IR and P475A. (A) On top are the pulse protocols and below are the corresponding current recordings obtained for Shaker-IR control. The left protocol was used to determine current activation (Iac) and the right protocol for characterizing current deactivation (Ideac). The horizontal bar at the start of the recordings indicates, here and in all other represented ionic current recordings, the zero current level. (B) Representative Iac and Ideac recordings for the mutant P475A are displayed on the left and right, respectively. Both Iac and Ideac were strongly slowed down in P475A compared to control and stronger depolarization were required to reach saturation of the tail currents obtained at −20 mV (note the difference in scale bars and pulse protocols with those of panel A). (C) Panel displays representative current recordings from mock-transfected HEK293 cells (cells were only transfected with the GFP marker). (D) Voltage-dependence of BC gate opening: GV curves for both Shaker-IR control (white circles) and P475A (black circles) were determined from the normalized tail current amplitudes obtained with an activation protocol as shown in panels A and B. Solid lines represent the average fit with a Boltzmann distribution. (E) Displayed on top is an Iac recording of P475A elicited at +100 mV and approximated with a single exponential function. Below the same Iac recording approximated with a double exponential function yielding a clearly better fit with a faster (dark gray) and slower (light gray) component. Both insets show a scaled up view of the first 200 ms of Iac fitted with a single or double exponential function. (F) Time constants of BC gate opening and closure obtained by fitting Iac and Ideac with a single or double (for P475A Iac) exponential function (note the semilogarithmic axis).
To investigate which transition in the activation pathway was affected by the P475A mutation, we introduced the P475A mutant in the nonconducting Shaker-IR pore mutant W434F background that allowed us to perform gating current measurements (27). To determine the voltage-dependence of VSD activation, gating currents were elicited by incrementally pulsing from a constant hyperpolarized initial voltage (−90 mV) to depolarizing voltages (activation protocol, Fig. 3, A and B). Integrating the activating gating currents (IQac) of Shaker-IR-W434F control yielded a charge (Q) versus voltage QV curve with a V1/2 of −33.9 ± 2.6 mV and a slope factor of 11.5 ± 0.8 mV (n = 8) (Fig. 3 C, Table 1). In agreement with the gating scheme in which all VSDs need to be activated before the BC gate opens (Fig. 1 B), the QV curve is located at slightly more negative potentials compared to the GV curve (i.e., VSD movement precedes BC gate opening, Fig. 3 C). Compared to control, the P475A mutation markedly shifted the voltage-dependence of BC gate opening (GV curve) toward more positive potentials (Fig. 2 C), but when the IQac of P475A was integrated, there appeared to be substantial gating charge movement in a voltage range similar to control (Fig. 3 C). Although a previous study on P475A reported this mutation to be nonconducting using Xenopus oocytes as the expression system, they observed similar behavior at the gating current level (32). With more detailed analysis of the gating currents of P475A, our data revealed that this mutation produced a QV curve with two gating charge components, which could be approximated with the sum of two Boltzmann distributions (Fig. 3 C). The first component carried the largest part of the gating charge (82 ±1%, n = 5) and matched the voltage-dependence of gating charge movement in Shaker-IR-W434F control (Table 1). The second component carried the remaining 18 ±1% of gating charge and was shifted by ∼ +95 mV toward more positive potentials such that it matched the voltage-dependence of BC gate opening (the GV curve) quite well. A similar split QV curve with two components was obtained when the deactivating gating currents (IQdeac) of the activation protocol were analyzed instead of IQac (Fig. S1 in the Supporting Material). These data indicated that the P475A mutation did not significantly affect the early VSD movements but isolated a late component of gating charge movement, similar to the reported effects of VSD mutations that dissect the last cooperative step of channel opening (15–19).
Figure 3.
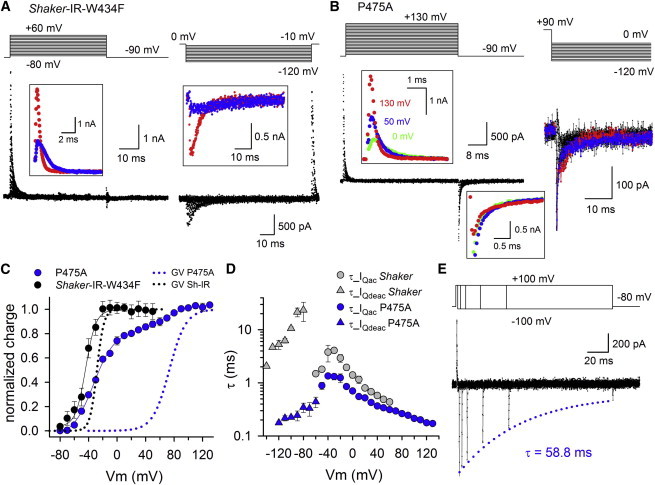
Gating current measurements of Shaker-IR-W434F and P475A. (A) Gating current recordings of Shaker-IR-W434F control obtained with activation (left) or a deactivation (right) protocol. On the left, a scaled up view of the activating gating currents (IQac) at +20 mV (blue) and +60 mV (red) are shown as inset. With increasing depolarization strengths the IQac kinetics accelerate causing the IQac tracings to cross each other. Inset on the right shows a scaled up view of the deactivating gating currents (IQdeac) at −90 mV (blue) and −120 mV (red). Similar to IQac, IQdeac decay accelerated with stronger repolarizations. (B) Representative IQac (left) and IQdeac (right) recordings of P475A. Insets show a scaled up view of both IQac and IQdeac obtained with the activation protocol. Similar to control (panel A), IQac accelerated with stronger depolarizations, which is highlighted by coloring the recordings at 0 mV (green), +50 mV (blue), and +130 mV (red). Scale up view of the IQdeac recordings at −90 mV shows that IQdeac did not slow down up to prepulse depolarizations of +50 mV in strength (note the overlap of the green and blue recording, which were obtained upon a 0 and +50 mV prepulse, respectively). However, upon a +130 mV prepulse (red trace) there was a reduction in IQdeac amplitude suggesting that the IQdeac kinetics were slowed down (larger scale of this scaled up view is shown in the Supporting Material). Right panel displays IQdeac recordings that were elicited with a deactivation pulse protocol by stepping after a 150-ms +90 mV prepulse to potentials between 0 and −120 mV. The blue and the red trace were obtained at −80 mV and −120 mV, respectively. (C) QV curves (symbols and line, which represent average fit with a single or sum of two Boltzmann distributions) of Shaker-IR-W434F (black) and P475A (blue) obtained by integrating and normalizing the IQac recordings. For comparison, the GV curves are represented in dotted lines. Note, the QV curve of P475A displayed two gating charge components whereby the first component matched the QV curve of Shaker-IR-W434F, whereas the second component was shifted by ∼ +95 mV and corresponded to the shift in the GV curve. (D) Voltage-dependency of the IQac (circles) and IQdeac (triangles) time constants ± SE for Shaker-IR-W434F (gray symbols, n = 8) and P475A (blue symbols, n = 6). (E) Envelope pulse protocol to determine in P475A the effect of prolonging the prepulse depolarization on the speed of IQdeac decay (τ_IQdeac), which was elicited by repolarizing to −100 mV. With longer depolarization times at +100 mV there was a gradual decrease in IQdeac amplitude, most likely because of a slowing down in τ_IQdeac. Approximating the decay in IQdeac amplitude with a single exponential function (blue dotted line) yielded a time constant of 58.8 ms in the represented recording.
BC gate opening slows VSD deactivation in both P475A and Shaker control
Fitting the decay of both activating IQac and deactivating IQdeac gating currents, which were elicited with a deactivation protocol (Fig. 3, A and B), yielded the voltage-dependent time constants of VSD activation (τ_IQac) and deactivation (τ_IQdeac, Fig. 3 D). For Shaker-IR control it has been well established that τ_IQac matches τ_Iac (reflecting BC gate opening), and that τ_IQdeac slows down when the amplitude and duration of the depolarizing prepulse are sufficient to cause BC gate opening (Fig. 3 A) (9). This slowing in IQdeac kinetics reflects the stabilization of the VSD in the Up state, which is imposed by BC gate opening (33). As reported previously, the τ_IQdeac kinetics was on average slower than the kinetics of BC gate closure (29).
Analyzing the IQac currents of P475A showed that the τ_IQac kinetics were faster than the ionic current kinetics τ_Iac (Fig. 2, F and Fig. 3 D). Most likely these τ_IQac kinetics report largely on the early VSD movements. To test whether BC gate opening still stabilized the VSD in the Up state in P475A, we analyzed IQdeac decay upon stronger and prolonged depolarizations. From the activation protocol with 50-ms step depolarizations (Fig. 3 B), we observed slowing of τ_IQdeac when the depolarization exceeded +50 mV (inset of Fig. 3 B). To estimate the time constant of this slowing process in τ_IQdeac, the prepulse duration at +90 mV was varied (Fig. 3 E). Longer prepulse depolarizations resulted in a gradual decrease of IQdeac amplitude, which indicates an immobilization of VSD deactivation that corresponds with a slowing in τ_IQdeac (34). This slowing in τ_IQdeac developed with a time constant of 56.6 ± 5.6 ms (n = 7) that indeed matched τ_Iac at +90 mV (Fig. 3 E). These observations indicated that BC gate opening stabilized the VSD in the Up state in P475A, as was the case in Shaker control. Accordingly, to determine the τ_IQdeac kinetics of P475A the prepulse depolarization at +90 mV was prolonged to 150 ms to assure that most channels have reached the fully open conformation. Using this protocol the τ_IQdeac kinetics were still markedly faster than the control values and nicely followed the τ_IQac kinetics creating a symmetrical bell-shaped voltage-dependence (Fig. 3 D). Based on the observed slowing in τ_IQdeac (detected as a reduction in amplitude, Fig. 3 E), this suggests there is a slow IQdeac component that is hidden within the electrical noise and consequently cannot be resolved.
P475A reduces Shaker’s sensitivity to 4-AP but not quinidine
Shaker (and other Kv channels) can be stabilized pharmacologically in the activated-not-open state by 4-AP (16). Molecular docking experiments proposed that 4-AP binds at the cytoplasmic entrance of the K+ pore, in the vicinity of the BC gate and the PVP motif (23). Therefore, we tested whether the P475A mutant was still sensitive to 4-AP. As reported previously (20), Shaker-IR is inhibited by 4-AP in a concentration-dependent manner (Fig. 4 A). Fig. 4 B shows the concentration-dependence of ionic current inhibition, which was approximated with a Hill function, yielding an IC50 value of 47.7 ± 2.4 μM (concentration that induces 50% of the maximal effect) and Hill coefficient of 1.11 ± 0.29 (n = 9). Remarkably, applying up to 10 mM 4-AP had no effect on the ionic currents from P475A (Fig. 4, C and D).
Figure 4.
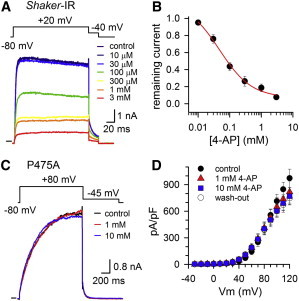
Effect of 4-AP on the ionic currents of Shaker-IR and P475A. (A) Concentration-dependent inhibition of the ionic currents of Shaker-IR; shown for a +20 mV depolarization in presence of different 4-AP concentrations. (B) Concentration-effect curve of 4-AP for Shaker-IR obtained by plotting the normalized Iac currents from pulse protocols shown in panel A as a function of 4-AP concentration; the solid line represents the fit with the Hill equation. (C) Ionic currents of P475A elicited using the pulse protocol shown on top. Represented recordings were the steady-state currents after repetitive pulsing to +90 mV in control condition, in presence of 1 mM and 10 mM 4-AP. In contrast to Shaker-IR control (panel A), there was no inhibition of the current amplitude even with a 4-AP concentration of 10 mM. (D) Current density of P475A (obtained by dividing the current amplitude by the cell capacitance) in control condition (black circles), in presence of 1 mM (triangles), and 10 mM (squares) 4-AP, and upon subsequent wash-out of 4-AP (white circles) (n = 4). To see this figure in color, go online.
At the gating current level 4-AP accelerates τ_IQdeac (and to a smaller extent also τ_IQac) (20) and immobilizes gating charge (16). Consequently, wash in of 1 mM 4-AP (a concentration to reach ∼95% inhibition in Shaker-IR) induced two noticeable changes in the gating currents of Shaker-IR-W434F: a), IQdeac decay accelerated (Fig. 5 A), and b), there was a loss in gating charge of 10–20% (Fig. 5 C). Such effects were not observed for P475A where application of up to 10 mM 4-AP had no effect on the gating currents and τ_IQdeac still slowed down upon prolonged depolarizations (Fig. 5 B). This slowing in τ_IQdeac developed with a time constant of 59.9 ± 10.6 ms (n = 3), which was similar to the rate of this process in control conditions. Consequently, in P475A no changes in the QV curve or time constants of both IQac and IQdeac decay were observed after 4-AP application (Fig. 5, C and D). This indicated that converting the PVP motif to PVA drastically decreased the channel’s sensitivity to 4-AP modulation (at least 300-fold).
Figure 5.
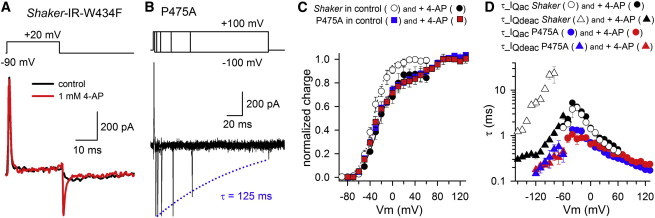
Effect of 4-AP on the gating currents of Shaker-IR-W434F and P475A. (A) IQac and IQdeac current of Shaker-IR-W434F in control condition (black) and presence of 1 mM 4-AP (red) elicited with a depolarization to +20 mV and subsequent repolarization to −90 mV. Note that IQdeac decayed markedly faster in the presence of 4-AP. (B) To determine whether 4-AP prevented the slowing of τ_IQdeac in P475A, we used a similar envelope pulse protocol (represented on top) as in Fig. 3E. The gating current recordings obtained in the presence of 10 mM 4-AP clearly showed a reduction in IQdeac amplitude (indicative of a slowing in τ_IQdeac) upon prolonged duration at +100 mV. This slowing in τ_IQdeac developed in the represented recording with a time constant of 125 ms, which was similar to control condition (absence of 4-AP, Fig. 3E). (C) QV curve (obtained from integrating IQac) of Shaker-IR-W434F in control conditions (white circles) and in presence of 1 mM 4-AP (black circles). Normalizing the total charge movement in presence of 4-AP to the charge movement in control conditions showed a 15 ± 5% (n = 3) loss in gating charge. In contrast, P475A displayed a similar QV curve with two gating charge components in control conditions (blue circles) and in presence of up to 10 mM 4-AP (red circles). Fitting the QV curves with a sum of two Boltzmann distributions indicated that V1/2 and slope factor values were not changed significantly after 4-AP application (Table 1). Note, that the amplitude of the second gating charge component of P475A matched the loss in gating charge observed in Shaker-IR-W434F upon 4-AP application. (D) τ_IQac (circles) and τ_IQdeac (triangles) ± SE for Shaker-IR-W434F (n = 3) in control conditions (white) and in presence of 1 mM 4-AP (black). τ_IQac (circles) and τ_IQdeac (triangles) kinetics of P475A in control conditions and in presence of 10 mM 4-AP are represented in blue and red, respectively (n = 5). In contrast to Shaker-IR-W434F, both the τ_IQac and τ_IQdeac kinetics were not affected by 4-AP in P475A. Note that the τ_IQdeac kinetics were markedly accelerated in Shaker-IR-W434F after 4-AP application and that these faster τ_IQdeac values (black) matched the τ_IQdeac kinetics of P475A.
Previous studies in Shaker-type Kv channels reported that S6 residues just above and below the PVP motif control the affinity for the open channel blocker quinidine (35), and it was suggested that both binding sites overlap partially (36,37). However, quinidine blocked the ionic currents of P475A in a concentration-dependent manner as in Shaker-IR control (Fig. 6, A and B). Approximating the concentration effect curve with a Hill function yielded for Shaker-IR control (n = 7) and P475A (n = 10) a IC50 value of 64.4 ± 5.6 μM and 1.4 ± 0.3 μM with a Hill coefficient of 1.1 ± 0.1 and 1.0 ± 0.2, respectively (Fig. 6 C). Thus, in contrast to 4-AP, the P475A mutant channel was still blocked by the open channel blocker quinidine and displayed a sensitivity that was even 45-fold higher compared to Shaker-IR control.
Figure 6.
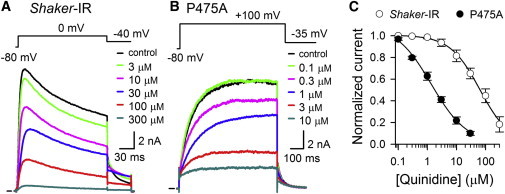
Quinidine block of Shaker-IR and P475A. (A) Concentration-dependent inhibition of ionic currents of Shaker-IR by quinidine. Represented traces are the steady-state currents at 0 mV depolarization in presence of different quinidine concentrations. (B) Representative ionic current inhibition for P475A by different concentrations of quinidine. (C) Concentration-effect curve of Shaker-IR (open circles) and P475A (black circles) for quinidine obtained by plotting the normalized Iac currents from pulse protocols shown in panels A and B as a function of quinidine concentration. The solid line represents the fit with the Hill equation. To see this figure in color, go online.
P475G behaves as P475A but results in a weaker isolation of the late gating charge component
Previously, we proposed that the proline residues of the PVP motif confer flexibility to S6c in the human Shaker-homolog Kv1.5, a feature that is important for BC gate operation (24,25). In that study, a glycine substitution was better tolerated than alanine and we proposed that this difference was because the alanine would promote a rigid α-helix, whereas the glycine tends to destabilize an α-helix and could adopt a proline-like configuration. To test whether this BC gate flexibility is important in dissecting the late gating charge component from the early VSD movements, the P475G mutation was introduced in Shaker-IR and Shaker-IR-W434F for ionic and gating current measurements, respectively (Fig. 7, A and B). At the ionic current level the P475G mutation shifted the GV curve by ∼ +45mV toward more positive potentials (Fig. 7 C, Table 1) and the kinetics of BC gate opening and closure were slowed down (Fig. 7D). These observations, at the ionic current level, were in good agreement with previous reports on the P475G mutation in both Shaker and the Shaker-type Kv1.4 and Kv1.5 channels (25,32,38). Detailed analysis of the gating currents showed that P475G behaved similar to P475A and resulted in a QV curve with two gating charge components (Fig. 7 C). Approximating the QV curve with the sum of two Boltzmann distributions showed that the voltage-dependence of the first component was similar to that of Shaker-IR-W434F control and that the second component matched the voltage-dependence of the GV curve (Fig. 7 C, Table 1). Furthermore, the first component carried 78 ± 3% (n = 4) of the gating charge and the second component the remaining 22 ± 3% that is associated with BC gate opening (Fig. 7 C). The resolved τ_IQac kinetics were substantially faster than the τ_Iac kinetics (Fig. 7 D), as was the case in P475A. Monitoring the IQdeac decay upon prolonged depolarizations showed that BC gate opening slowed down τ_IQdeac (Fig. 7 E), similar to P475A and Shaker-IR-W434F control. This slowing in τ_IQdeac developed at +40 mV depolarization with a time constant of 13.6 ± 6.8 ms (n = 3), which matched the speed of BC gate opening (τ_Iac at +40 mV). Thus, the P475G mutation behaved similar to P475A but the separation of both gating charge components was less pronounced. The voltage difference between the V1/2 of the first and second component was in P475G only 50 mV, whereas in P475A it was 95 mV (Table 1). These data are in good agreement with our expectations if isolating the late gating charge component from the early one(s) is related to BC gate flexibility. Testing the effect of 4-AP showed that P475G displayed a substantially decreased sensitivity, similar to P475A, and up to 10 mM 4-AP failed to inhibit the ionic currents (Fig. 7, F and G).
Figure 7.
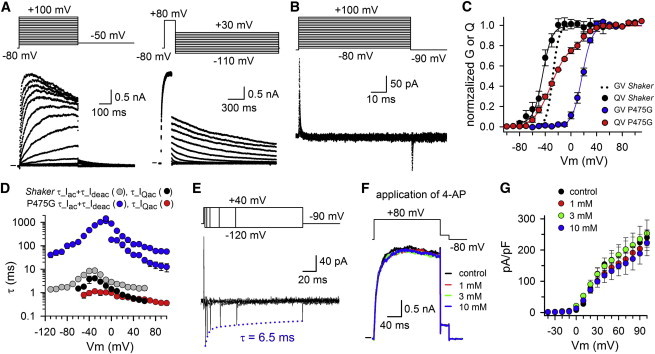
Biophysical properties of the P475G mutation. (A) Representative Iac and Ideac recordings for the mutant P475G are displayed on the left and right, respectively. (B) Representative IQac recording of P475G using the activation pulse protocol shown on top. (C) Voltage-dependence of BC gate opening (GV curve) and VSD movement (QV curve) for P475G. The GV curve of P475G (blue circles, n = 5) was shifted by ∼ +45 mV toward more positive potentials compared to Shaker-IR control (black dotted line). The blue line represents the average fit with a single Boltzmann equation. The QV curve of P475G (red circles, n = 4) was obtained by integrating and normalizing the IQac recordings obtained with activation protocol shown in panel B. Note, P475G displayed, similar to P475A, a QV curve with two gating charge components whereby the first component carried ∼78% of the gating charge and superposed on the QV curve of Shaker-IR-W434F control (black). The second component carried the remaining 22% of the gating charge and matched the GV curve (blue line). (D) Voltage-dependent time constants of BC gate opening and closure in P475G (blue) obtained by fitting Iac with a double and Ideac with a single exponential function. The τ_Iac (even the fast one) and τ_Ideac kinetics were clearly slower than those of Shaker-IR control (gray). Voltage-dependency of τ_IQac ± SE for P475G (red, n = 8) and Shaker-IR-W434F (black). (E) Increasing gradually the conditioning prepulse duration at +40 mV (pulse protocol is shown on top) resulted in P475G in a gradual decrease in IQdeac amplitude, which reflects a slowing down of τ_IQdeac. Approaching the gradual decrease in IQdeac with a single exponential (blue dotted line) yielded a time constant of 6.5 ms. (F) Ionic currents of P475G after pulsing to +80 mV (pulse protocol shown on top) in control condition (black), and in presence of 1 mM (red), 3 mM (green), and 10 mM (blue) 4-AP. (G) Current density of P475G in control condition (black), in presence of 1 mM (red), 3 mM (green), and 10 mM (blue) 4-AP (n = 4). Over the voltage range tested, no difference in the current density was observed upon 4-AP application.
Discussion
P475A and P475G isolate the late gating charge component from VSD movements earlier in the activation pathway from closed to open
The status of the BC gate is under tight control of the VSDs: the VSDs translate changes in membrane potential into BC gate opening/closure through an electromechanical coupling (for review see (3,4)). Although the Down state of the VSD is still debated (39–45), it is generally accepted that the VSDs move largely independently from the Down state up to the Activated state (Fig. 1 B). When all four VSDs have reached this activated state, the BC gate converts in a subunit-cooperative manner to the open conducting state (14), which at the same time is accompanied by the movement of the VSDs from the Activated to their Up state. Most of the gating charge displacement (80–90%) is associated with the independent VSD movements, whereas the late subunit-cooperative transition in the activation pathway carries the remaining 10–20% of the total gating charge. Many VSD mutations have been described to isolate this late gating charge component (15–19), but direct evidence for the contribution of the BC gate was still lacking. Therefore, our report that substituting the second proline of the PVP motif in the BC gate region by an alanine or glycine isolates the late gating charge component is, to our knowledge, the first direct evidence for a contribution of the BC gate to this state transition.
The steepness (slope factor) of a channel’s conducting GV curve depends on the number of gating charges, but also on the degree of cooperativity between the subunits (12). For the ILT VSD mutant it has been shown that the late cooperative transition in the activation pathway becomes rate limiting and manifests itself by a shallow slope factor (46). Similarly, both the P475A and P475G mutant displayed a shallower GV curve compared to Shaker-IR control (Fig. 2 D and Fig. 7 D), supporting that both mutations affect a rate-limiting cooperative step. At the gating current level, both the P475A and P475G mutants resulted in a split of the QV curve where the first component reflected the early VSD movements that carry ∼80% of the total gating charge. The second component reports then on the late VSD transition(s) linked to BC gate opening and carry the remaining 20% of gating charge. Analysis of the activating gating currents (IQac) showed that P475A displays IQac kinetics that resembled the kinetics of Shaker-IR-W434F control after 4-AP inhibition (Fig. 5 D). Because 4-AP isolates the late gating charge component from earlier state transitions (16), this strengthens the proposal that the P475A mutation impairs only this late VSD transition. In agreement with the suggestion that the rate constants of the late cooperative step are less voltage-dependent than those of the early VSD transitions (9–11), the τ_Iac kinetics that report on BC gate opening displayed a weaker voltage-dependence in the P475A mutant compared to Shaker-IR control (Fig. 2 F).
The activation kinetics of the ionic currents (τ_Iac) for both P475A and P475G were best approximated by a double exponential function (Fig. 2 E). This yielded a fast and a slow component that both displayed a reduced voltage-dependency and were both slower compared to control values (Fig. 2 F, Fig. 6 D). It has been shown that the kinetics of IQac decay (τ_IQac) match τ_Iac in Shaker control (29). In P475A and P475G the τ_IQac kinetics were substantially faster than those of τ_Iac, possibly because the IQac component that reflects BC gate opening is too slow to be resolved and is hidden within the experimental noise. However, if this was the case we would hardly detect the second gating charge component in the QV curve obtained from either the IQac or IQdeac gating currents of the activation protocol (Fig. 3 C, Fig. S1). It has been shown that the Shaker channel has multiple open states and we propose that in both P475A and P475G only a later one is fully conductive. The transition to these later (subsequent) open gate configurations, such as the relaxed state, is not associated with gating charge movement but can be detected by a slowing of τ_IQdeac due to the stabilization of the VSD in the Up state (29,33). Because both P475A and P475G displayed ionic currents their BC gate must have opened and τ_IQdeac indeed slowed down upon prolonged depolarizations (Fig. 3 C, Fig. 7 C). This slowing in τ_IQdeac developed with a time constant that reasonably well matched the fast Iac component (τ_Iac_fast) of BC gate opening (Fig. 2, F, Fig. 7 D). Consequently, for both P475A and P475G, the IQac kinetics report mainly on the VSD movements that are linked to the early state transitions. Because these VSD movements display similar kinetics and a voltage-dependency like Shaker control upon 4-AP inhibition (Fig. 5 D), we conclude that the P475A mutation and most likely also the P475G mutation leave the early VSD transitions largely unaffected.
Passing the subunit-cooperative transition of BC gate opening is governed by S6 flexibility
Not only the VSD, but also the BC gate may display different sequential conformations during the transition from the closed to the open state (13,14). Even after channel opening the BC gate can reside in several open states, as manifested by a slowing in the kinetics of both VSD deactivation and BC gate closure (29). For these BC gate reorientations to occur, a flexible hinge region is required that allows for the swiveling and bending motions of S6c (2). In Shaker-type Kv channels this S6 flexibility, as shown by molecular dynamics (MD) simulations, may be conferred by the PVP motif (Fig. 1 A) that is located near the narrow part of the putative BC gate (47–49). The three-dimensional crystal structure of the Kv1.2 channel indeed showed that the S6 segment bends at the level of the PVP motif (5) and MD simulations on this structure showed that S6 kinks at this motif to seal off the K+ flow (40). The observation that in the human Shaker homolog Kv1.5 substituting the PVP motif by AVPP sequence yielded channels with two gating modes supports that this region indeed reorients during BC gate opening and closure (24). In agreement with our previous results (25), a glycine substitution that tends to destabilize the S6 α-helix like a proline (31,32) had a milder impact on the channel kinetics and isolated the late gating charge component to a lesser extent than the alanine substitution that favors an α-helical configuration (Table 1). A recent MD simulation study predicted that the S6 helix of Shaker-type Kv channels is indeed less kinked when the second proline of the PVP motif (P475) is substituted by an alanine (50). Accordingly, the hinge of the BC gate is most likely stiffer in the P475A mutant than in P475G making it harder for the BC gate to reorient. Although the P475A and P475G mutations may affect the overall orientation of S6c and its interaction with the S4-S5 linker, we propose that the flexibility conferred by the PVP motif is an important mechanistic component during the late (final) cooperative step of BC gate opening.
P475A and P475G both affect and mimic the gating modification by 4-AP
The observation that mutations in the BC gate affect the VSD movements strengthens the concept that the coupling between VSD and BC gate is strong in Shaker-type Kv channels. Whereas decoupling the BC gate from the VSD facilitates gating charge movement (33,51,52), we show that restricting BC gate movement, by lowering the flexibility of S6c, impairs the VSD movement(s), which is associated with the late subunit-cooperative transition(s) in the activation pathway. In addition to isolating the late (final) cooperative step of channel opening, substituting the PVP motif by a PVA or PVG sequence also drastically reduced the channel’s sensitivity to 4-AP, which is a quite universal Kv channel inhibitor that acts from the cytoplasmic side (53) and stabilizes the channels in the activated-not-open state (16). 4-AP supposedly binds in the vicinity of the PVP motif (Fig. 1 A) (21–23), and shares its binding site, at least partially, with that of the open channel blockers such as quinidine (36,37). In contrast to the loss in 4-AP affinity, sensitivity for quinidine was 45-fold higher in the P475A mutation than in Shaker-IR control (Fig. 6 C). Although residue P475 might be directly involved in the binding of 4-AP and quinidine, we propose, based on the contrasting effects in 4-AP and quinidine affinity, that both an alanine and glycine substitution affect the binding affinity of both compounds in an allosteric manner by changing the conformation of S6c. An alternative explanation for losing only the 4-AP modulation, rather than specifically affecting binding affinity, is that both the mutations and 4-AP alter the same late (final) cooperative step of channel opening (i.e., the mutation mimics the 4-AP effect). Because we propose that both mutations exert their effect by making the hinge region of the BC gate less flexible, we speculate that 4-AP inhibits channel opening by restricting the final movements of the BC gate hinge upon binding. In addition to affecting 4-AP modulation, the P475A and P475G mutations might also impair the action of other gating modifiers such as guanidine compounds that seem to operate in a similar way as 4-AP (54).
Interestingly, the closely related silent Kv channels, which are unable to form functional homotetramers but coassemble with subunits of the Kv2 family, do not possess a full PVP motif and might therefore alter the 4-AP sensitivity (55). Only a small subset of the silent Kv channels has been tested for their role in 4-AP sensitivity, but so far no marked changes have been observed. Because the silent Kv subunits only exist as heterotetramers with Kv2, this may indicate that lacking a full PVP motif in only some subunits may be insufficient to abolish 4-AP modulation. On the other hand, the Kv7 (KCNQ) channels, which can exist as homotetramers, possess neither a full PVP motif (Fig. 1 A) nor 4-AP sensitivity (56,57).
In conclusion, we report that substituting the second proline of the conserved PVP motif in Shaker-type Kv channels by an alanine or glycine isolates the late gating charge component linked to BC gate opening. The degree of separating this component from the earlier VSD movements depended on the reduction in the flexibility of S6c because an alanine substitution resulted in a stronger separation of both gating charge components in the QV curve compared to a glycine substitution. These data suggest that BC gate flexibility is an important molecular determinant for enabling the late (final) subunit-cooperative transition that leads to channel opening.
Acknowledgments
We thank Dr. H. C. Hyde for the critical review of the manuscript.
This work was supported by the Mexican National Council for Science and Technology CONACyT #203936 (to E.M.M.), and the Belgian Federal Science Policy Office FWO (Fonds voor Wetenschappelijk Onderzoek Vlaanderen) grants G.0449.11 and G0433.12N (to D.J.S.) and 1.5.087.11N (to A.J.L.).
Supporting Material
References
- 1.MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- 2.Labro A.J., Snyders D.J. Being flexible: the voltage-controllable activation gate of kv channels. Front Pharmacol. 2012;3:168. doi: 10.3389/fphar.2012.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blunck R., Batulan Z. Mechanism of electromechanical coupling in voltage-gated potassium channels. Front Pharmacol. 2012;3:166. doi: 10.3389/fphar.2012.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vardanyan V., Pongs O. Coupling of voltage-sensors to the channel pore: a comparative view. Front Pharmacol. 2012;3:145. doi: 10.3389/fphar.2012.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long S.B., Campbell E.B., Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 6.Aggarwal S.K., MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 7.Seoh S.A., Sigg D., Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 8.Larsson H.P., Baker O.S., Isacoff E.Y. Transmembrane movement of the shaker K+ channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 9.Bezanilla F., Perozo E., Stefani E. Gating of Shaker K+ channels: II. The components of gating currents and a model of channel activation. Biophys. J. 1994;66:1011–1021. doi: 10.1016/S0006-3495(94)80882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoppa N.E., Sigworth F.J. Activation of Shaker potassium channels. III. An activation gating model for wild-type and V2 mutant channels. J. Gen. Physiol. 1998;111:313–342. doi: 10.1085/jgp.111.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zagotta W.N., Hoshi T., Aldrich R.W. Shaker potassium channel gating. II: Transitions in the activation pathway. J. Gen. Physiol. 1994;103:279–319. doi: 10.1085/jgp.103.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith-Maxwell C.J., Ledwell J.L., Aldrich R.W. Role of the S4 in cooperativity of voltage-dependent potassium channel activation. J. Gen. Physiol. 1998;111:399–420. doi: 10.1085/jgp.111.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.del Camino D., Kanevsky M., Yellen G. Status of the intracellular gate in the activated-not-open state of shaker K+ channels. J. Gen. Physiol. 2005;126:419–428. doi: 10.1085/jgp.200509385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalstrup T., Blunck R. Dynamics of internal pore opening in K(V) channels probed by a fluorescent unnatural amino acid. Proc. Natl. Acad. Sci. USA. 2013;110:8272–8277. doi: 10.1073/pnas.1220398110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ledwell J.L., Aldrich R.W. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 1999;113:389–414. doi: 10.1085/jgp.113.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loboda A., Armstrong C.M. Resolving the gating charge movement associated with late transitions in K channel activation. Biophys. J. 2001;81:905–916. doi: 10.1016/S0006-3495(01)75750-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schoppa N.E., McCormack K., Sigworth F.J. The size of gating charge in wild-type and mutant Shaker potassium channels. Science. 1992;255:1712–1715. doi: 10.1126/science.1553560. [DOI] [PubMed] [Google Scholar]
- 18.Tao X., Lee A., MacKinnon R. A gating charge transfer center in voltage sensors. Science. 2010;328:67–73. doi: 10.1126/science.1185954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lacroix J.J., Bezanilla F. Control of a final gating charge transition by a hydrophobic residue in the S2 segment of a K+ channel voltage sensor. Proc. Natl. Acad. Sci. USA. 2011;108:6444–6449. doi: 10.1073/pnas.1103397108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCormack K., Joiner W.J., Heinemann S.H. A characterization of the activating structural rearrangements in voltage-dependent Shaker K+ channels. Neuron. 1994;12:301–315. doi: 10.1016/0896-6273(94)90273-9. [DOI] [PubMed] [Google Scholar]
- 21.Kirsch G.E., Shieh C.C., Brown A.M. Segmental exchanges define 4-aminopyridine binding and the inner mouth of K+ pores. Neuron. 1993;11:503–512. doi: 10.1016/0896-6273(93)90154-j. [DOI] [PubMed] [Google Scholar]
- 22.Shieh C.C., Kirsch G.E. Mutational analysis of ion conduction and drug binding sites in the inner mouth of voltage-gated K+ channels. Biophys. J. 1994;67:2316–2325. doi: 10.1016/S0006-3495(94)80718-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caballero N.A., Meléndez F.J., Muñoz-Caro C. Molecular docking study of the binding of aminopyridines within the K+ channel. J. Mol. Model. 2007;13:579–586. doi: 10.1007/s00894-007-0184-9. [DOI] [PubMed] [Google Scholar]
- 24.Labro A.J., Grottesi A., Snyders D.J. A Kv channel with an altered activation gate sequence displays both “fast” and “slow” activation kinetics. Am. J. Physiol. Cell Physiol. 2008;294:C1476–C1484. doi: 10.1152/ajpcell.00479.2007. [DOI] [PubMed] [Google Scholar]
- 25.Labro A.J., Raes A.L., Snyders D.J. Gating of shaker-type channels requires the flexibility of S6 caused by prolines. J. Biol. Chem. 2003;278:50724–50731. doi: 10.1074/jbc.M306097200. [DOI] [PubMed] [Google Scholar]
- 26.Hoshi T., Zagotta W.N., Aldrich R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 27.Perozo E., MacKinnon R., Stefani E. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 1993;11:353–358. doi: 10.1016/0896-6273(93)90190-3. [DOI] [PubMed] [Google Scholar]
- 28.Boussif O., Lezoualc’h F., Behr J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labro A.J., Lacroix J.J., Bezanilla F. Molecular mechanism for depolarization-induced modulation of Kv channel closure. J. Gen. Physiol. 2012;140:481–493. doi: 10.1085/jgp.201210817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefani E., Toro L., Bezanilla F. Gating of Shaker K+ channels: I. Ionic and gating currents. Biophys. J. 1994;66:996–1010. doi: 10.1016/S0006-3495(94)80881-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris T., Graber A.R., Covarrubias M. Allosteric modulation of a neuronal K+ channel by 1-alkanols is linked to a key residue in the activation gate. Am. J. Physiol. Cell Physiol. 2003;285:C788–C796. doi: 10.1152/ajpcell.00113.2003. [DOI] [PubMed] [Google Scholar]
- 32.Hackos D.H., Chang T.H., Swartz K.J. Scanning the intracellular S6 activation gate in the shaker K+ channel. J. Gen. Physiol. 2002;119:521–532. doi: 10.1085/jgp.20028569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Batulan Z., Haddad G.A., Blunck R. An intersubunit interaction between S4-S5 linker and S6 is responsible for the slow off-gating component in Shaker K+ channels. J. Biol. Chem. 2010;285:14005–14019. doi: 10.1074/jbc.M109.097717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lacroix J.J., Labro A.J., Bezanilla F. Properties of deactivation gating currents in Shaker channels. Biophys. J. 2011;100:L28–L30. doi: 10.1016/j.bpj.2011.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeola S.W., Rich T.C., Snyders D.J. Molecular analysis of a binding site for quinidine in a human cardiac delayed rectifier K+ channel. Role of S6 in antiarrhythmic drug binding. Circ. Res. 1996;78:1105–1114. doi: 10.1161/01.res.78.6.1105. [DOI] [PubMed] [Google Scholar]
- 36.Chen F.S., Fedida D. On the mechanism by which 4-Aminopyridine occludes quinidine block of the cardiac K+ channel, hKv1.5. J. Gen. Physiol. 1998;111:539–554. doi: 10.1085/jgp.111.4.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H., Zhu B., Tseng G.N. Differential effects of S6 mutations on binding of quinidine and 4-aminopyridine to rat isoform of Kv1.4: common site but different factors in determining blockers’ binding affinity. J. Pharmacol. Exp. Ther. 1998;287:332–343. [PubMed] [Google Scholar]
- 38.Bett G.C., Lis A., Rasmusson R.L. Interaction of the S6 proline hinge with N-type and C-type inactivation in Kv1.4 channels. Biophys. J. 2012;103:1440–1450. doi: 10.1016/j.bpj.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henrion U., Renhorn J., Elinder F. Tracking a complete voltage-sensor cycle with metal-ion bridges. Proc. Natl. Acad. Sci. USA. 2012;109:8552–8557. doi: 10.1073/pnas.1116938109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jensen M.O., Jogini V., Shaw D.E. Mechanism of voltage gating in potassium channels. Science. 2012;336:229–233. doi: 10.1126/science.1216533. [DOI] [PubMed] [Google Scholar]
- 41.Pathak M.M., Yarov-Yarovoy V., Isacoff E.Y. Closing in on the resting state of the Shaker K(+) channel. Neuron. 2007;56:124–140. doi: 10.1016/j.neuron.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 42.Upadhyay S.K., Nagarajan P., Mathew M.K. Potassium channel opening: a subtle two-step. J. Physiol. 2009;587:3851–3868. doi: 10.1113/jphysiol.2009.174730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vargas E., Bezanilla F., Roux B. In search of a consensus model of the resting state of a voltage-sensing domain. Neuron. 2011;72:713–720. doi: 10.1016/j.neuron.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vargas E., Yarov-Yarovoy V., Roux B. An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J. Gen. Physiol. 2012;140:587–594. doi: 10.1085/jgp.201210873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yarov-Yarovoy V., Baker D., Catterall W.A. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K(+) channels. Proc. Natl. Acad. Sci. USA. 2006;103:7292–7297. doi: 10.1073/pnas.0602350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith-Maxwell C.J., Ledwell J.L., Aldrich R.W. Uncharged S4 residues and cooperativity in voltage-dependent potassium channel activation. J. Gen. Physiol. 1998;111:421–439. doi: 10.1085/jgp.111.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Denning E.J., Woolf T.B. Cooperative nature of gating transitions in K(+) channels as seen from dynamic importance sampling calculations. Proteins. 2010;78:1105–1119. doi: 10.1002/prot.22632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Imbrici P., Grottesi A., Pessia M. Contribution of the central hydrophobic residue in the PXP motif of voltage-dependent K+ channels to S6 flexibility and gating properties. Channels (Austin) 2009;3:39–45. doi: 10.4161/chan.3.1.7548. [DOI] [PubMed] [Google Scholar]
- 49.Mashl R.J., Jakobsson E. End-point targeted molecular dynamics: large-scale conformational changes in potassium channels. Biophys. J. 2008;94:4307–4319. doi: 10.1529/biophysj.107.118778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fowler P.W., Sansom M.S. The pore of voltage-gated potassium ion channels is strained when closed. Nat. Commun. 2013;4:1872. doi: 10.1038/ncomms2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding S., Horn R. Effect of S6 tail mutations on charge movement in Shaker potassium channels. Biophys. J. 2003;84:295–305. doi: 10.1016/S0006-3495(03)74850-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haddad G.A., Blunck R. Mode shift of the voltage sensors in Shaker K+ channels is caused by energetic coupling to the pore domain. J. Gen. Physiol. 2011;137:455–472. doi: 10.1085/jgp.201010573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirsch G.E., Drewe J.A. Gating-dependent mechanism of 4-aminopyridine block in two related potassium channels. J. Gen. Physiol. 1993;102:797–816. doi: 10.1085/jgp.102.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kalia J., Swartz K.J. Elucidating the molecular basis of action of a classic drug: guanidine compounds as inhibitors of voltage-gated potassium channels. Mol. Pharmacol. 2011;80:1085–1095. doi: 10.1124/mol.111.074989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bocksteins E., Snyders D.J. Electrically silent Kv subunits: their molecular and functional characteristics. Physiology (Bethesda) 2012;27:73–84. doi: 10.1152/physiol.00023.2011. [DOI] [PubMed] [Google Scholar]
- 56.Yang W.P., Levesque P.C., Blanar M.A. Functional expression of two KvLQT1-related potassium channels responsible for an inherited idiopathic epilepsy. J. Biol. Chem. 1998;273:19419–19423. doi: 10.1074/jbc.273.31.19419. [DOI] [PubMed] [Google Scholar]
- 57.Yang W.P., Levesque P.C., Blanar M.A. KvLQT1, a voltage-gated potassium channel responsible for human cardiac arrhythmias. Proc. Natl. Acad. Sci. USA. 1997;94:4017–4021. doi: 10.1073/pnas.94.8.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.