

Abstract
microRNAs (miRNAs) are small RNAs endogenously expressed in multiple organisms that regulate gene expression largely by decreasing levels of target messenger RNAs (mRNAs). Over the past few years, numerous studies have demonstrated critical roles for miRNAs in the pathogenesis of many cancers, including lung cancer. Cellular miRNA levels can be easily manipulated, showing the promise of developing miRNA-targeted oligos as next-generation therapeutic agents. In a comprehensive effort to identify novel miRNA-based therapeutic agents for lung cancer treatment, we combined a high-throughput screening platform with a library of chemically synthesized miRNA inhibitors to systematically identify miRNA inhibitors that reduce lung cancer cell survival and those that sensitize cells to paclitaxel. By screening three lung cancer cell lines with different genetic backgrounds, we identified miRNA inhibitors that potentially have a universal cytotoxic effect on lung cancer cells and miRNA inhibitors that sensitize cells to paclitaxel treatment, suggesting the potential of developing these miRNA inhibitors as therapeutic agents for lung cancer. We then focused on characterizing the inhibitors of three miRNAs (miR-133a/b, miR-361-3p, and miR-346) that have the most potent effect on cell survival. We demonstrated that two of the miRNA inhibitors (miR-133a/b and miR-361-3p) decrease cell survival by activating caspase-3/7-dependent apoptotic pathways and inducing cell cycle arrest in S phase. Future studies are certainly needed to define the mechanisms by which the identified miRNA inhibitors regulate cell survival and drug response, and to explore the potential of translating the current findings into clinical applications.
Keywords: cell viability, drug response, lung cancer, miRNA, paclitaxel
Introduction
Lung cancer is the leading cause of cancer-related deaths in the United States, with a five-year survival rate that remains less than 15%.1 The high frequency of resistance to currently available therapeutic agents is a key contributor to the poor survival rates. This highlights the need to further elucidate the molecular mechanisms underlying lung cancer tumorigenesis and drug response in order to identify novel therapeutic targets and agents. Dysregulation of microRNAs (miRNAs) has recently been shown to play a critical role in regulating cancer cell survival and drug response in various types of cancers, including lung cancer,2-4 showing the promise of integrating miRNAs into the therapeutic armamentarium.
miRNAs are short, 19 to 23-nucleotide long RNAs found in multiple organisms that regulate gene expression largely by decreasing levels of target messenger RNAs (mRNAs)5,6 through binding to specific target sites in the mRNA 3′ untranslated regions (3′UTRs). miRNAs have been shown to play important roles in regulating a broad range of pathological processes. Over the past few years, many tumor suppressor genes (TSGs) and oncogenes have been demonstrated to be regulated by miRNAs, with these miRNAs therefore acting as oncogenes or TSGs themselves7-9 to regulate cancer cell survival and proliferation. The critical roles of miRNAs in modulating cancer cell response to chemotherapeutic agents have also been documented.3,4,10-12
Since miRNAs are small oligonucleotides (oligos), it is easy to manipulate their intracellular levels, making them attractive agents and targets in cancer therapy.13-16 A chemically stabilized, single-stranded RNA oligonucleotide complementary to a specific miRNA acts as a competitive inhibitor (known as a miRNA inhibitor, anti-miR or antagomir) that binds to the target miRNA with high affinity.16 This prevents the association of the miRNA with the complementary site(s) in its target mRNA(s), blocking its endogenous activity and restoring expression of its target mRNAs. Such molecules have been used to inhibit the activity of oncogenic miRNAs in several studies,13-16 demonstrating the feasibility of using miRNA inhibitors as therapeutic agents.
We are interested in identifying novel miRNA inhibitors that modulate lung cancer cell survival and response to paclitaxel, a microtubule-targeting agent (MTA) that remains a first-line therapeutic agent in lung cancer treatment. High-throughput screening (HTS) approaches have been used to identify novel regulators, including protein coding genes and miRNAs, of both cancer cell survival and drug response.17-19 For example, a screen based on a library of human miRNA mimics (synthetic small, double-stranded RNA oligos that are used to raise the intracellular level of a specific miRNA) in colon cancer cell line HCT-116 identified miRNAs that affect sensitivity to BCL2 inhibitor ABT-263 (navitoclax).18 In another study, Izumiya, et al. applied a miRNA virus library to identify miRNAs that have tumor suppressor function in pancreatic cell line MIA PaCa-2.19 The above studies demonstrate the feasibility and promise of restoring tumor suppressor miRNAs as a therapeutic approach in cancer treatment. However, no studies have directly and systematically investigated the effect of synthetic inactivation of oncogenic miRNAs on cancer cell survival and drug response. Here, we implemented an HTS screen to systematically identify miRNA inhibitors that modulate cell survival and regulate response to paclitaxel in lung cancer cell lines.
Results
HTS identifies multiple miRNA inhibitors that affect cell survival and response to paclitaxel in NSCLC cell lines
In order to identify miRNA inhibitors that affect viability and response to paclitaxel of NSCLC cells, we combined an HTS platform with a library of inhibitors for 747 human miRNAs. The experiment was designed with two arms, one assessing the effect of the miRNA inhibitors on cell viability and the other assessing the degree to which the inhibitors sensitize cells to paclitaxel (Fig. S1). In order to optimally identify miRNA inhibitors that affect response to paclitaxel in both directions—that is, either sensitize or desensitize cells to paclitaxel—we used a drug concentration close to the IC50 (Fig. S2A–C) for each cell line.
In order to identify miRNA inhibitors that potentially have general effects on lung cancer cells, we chose for the screen three NSCLC cell lines that have distinct genetic backgrounds: H1155, H1993, and H358 (Table S3). Figure 1A–C shows the distribution of the cell viabilities (Vcarrier) in the absence of paclitaxel, reflecting the effect of individual miRNA inhibitors on cell survival alone. Figure 1D–F shows the distribution of paclitaxel sensitivity ratios (S), reflecting the effect of the miRNA inhibitors on cellular response to paclitaxel. The results show that the miRNA inhibitors have a wide range of effects on cell survival and response to paclitaxel in the three cell lines. Further statistical analysis indicated that the screens of cell viability and paclitaxel response produced more hits than expected as shown by a dramatic excess of large z-scores (Fig. S3A–F).

Figure 1. HTS identified miRNA inhibitors that affect cell viability or sensitivity to paclitaxel in NSCLC cells. Cells were transiently transfected with 50 nM miRNA inhibitors. After 48 h of incubation and 72 h treatment with paclitaxel or carrier, cell viability was measured by quantification of ATP produced using the Luminescent Cell Viability Assay (Promega). (A–C) The distributions of the cell viability measurements for individual inhibitors. (D–F) The distributions of paclitaxel responses for individual inhibitors, determined by the ratio of cell viability measurements in the presence and absence of paclitaxel.
We further identified miRNA inhibitors that have effects on cell viability or paclitaxel response in more than one cell line. As shown in Figure 2A and Table S1, using a threshold of a 30% decrease or increase in cell viability or paclitaxel response, we identified inhibitors of three miRNAs (miR-133a/b, miR-361-3p, and miR-346) as decreasing cell viability in all three cell lines. With this cutoff, however, no miRNA inhibitors are identified as increasing cell viability or affecting paclitaxel sensitivity in all of the three cell lines. When we relax the threshold to 20%, additional miRNA inhibitors that are cytotoxic to all three cell lines are identified, and miRNA inhibitors that increase cell viability and desensitize cells to paclitaxel are also identified (Fig. 2B and Table S1). Interestingly, even with a modest threshold of 20%, no inhibitors are identified as common paclitaxel sensitizers of the three cell lines.
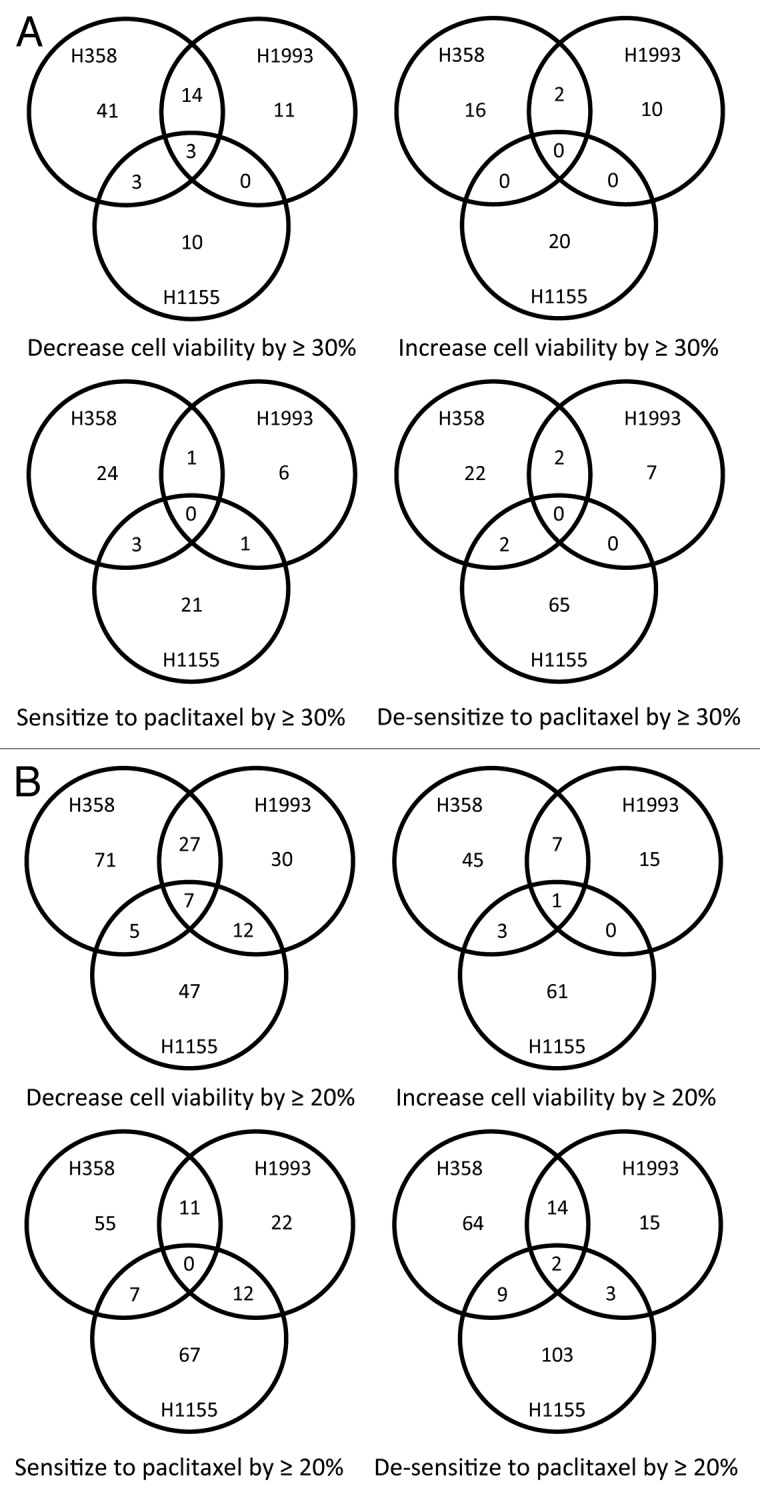
Figure 2. HTS identified miRNA inhibitors that have general effects on lung cancer cell survival and response to paclitaxel. Shown are Venn diagrams of the miRNA inhibitors that affect cell survival and response to paclitaxel of the three lung cancer cell lines, using thresholds of 30% (A) and 20% (B) for the magnitude of their effects on cell survival and response to paclitaxel.
Overall, our screens identified miRNA inhibitors that have common effects on cell survival and paclitaxel response. From the analysis of our results, we see that the miRNA inhibitors have a more potent effect on cell viability alone than on drug response. We therefore focused on characterizing the three miRNA inhibitors that have potent cytotoxic effects on all three lung cancer cell lines.
Inhibitors of miR-133a/b, miR-361-3p, and miR-346 potently decrease viability of NSCLC cells with different genetic backgrounds
In order to validate our screen results, we examined the dose-dependence of the effect of the three inhibitors on cell viability in H1993 cells. As shown in Figure 3A, inhibitors of these three miRNAs decrease cell viability in a dose-dependent manner in H1993 cells. The cytotoxicities of the three inhibitors differ, with miR-133a/b having the lowest IC50 (Table S2). To further examine whether these miRNA inhibitors have a universal effect on lung cancer cell survival, we examined their effect on cell viability in additional lung cancer cell lines with different genetic backgrounds (Table S3). Figure 3B–F indicates that the three inhibitors show cytotoxic effects in all tested cell lines, although the IC50s (Table S2) for these cell lines vary greatly. The variability in the response of lung cancer cell lines to the miRNA inhibitors is likely due to the different endogenous expression levels of the miRNAs. As shown in Figure S4, lung cancer cell lines show great variation in expression of these miRNAs, suggesting differential sensitivity to the corresponding inhibitors. We next examined the effect of the miRNA inhibitors on long-term cell proliferative capacity of H1993 cells using a colony formation assay. As shown in Figure 3G–H, the three miRNA inhibitors significantly inhibited cell growth, leading to reduced numbers of colonies. Overall, the results indicate that the three candidate inhibitors have a universal and potent effect on both short- and long-term survival of lung cancer cells.

Figure 3. miR-133a/b, miR-361-3p, and miR-346 inhibitors show general inhibitory effects on cell viability in lung cancer cell lines. (A–F) Cell viability as a function of the concentration of the miRNA inhibitors. Cells were transfected with different concentrations of the indicated miRNA inhibitors or control oligos. After 120 h, cell viability was measured as described above. (G) Colony formation assay as a function of miRNA inhibitors in H1993 cells. Treatments were conducted in triplicate. (H) Quantification of the number of colonies for the colony formation assay. ** P < 0.01; *** P < 0.001.
Combining the cytotoxic miRNA inhibitors with each other or with chemotherapeutic agents results in enhanced cytotoxicity in lung cancer cells
In order to examine whether the three miRNA inhibitors have synergistic cytotoxic effects on lung cancer cells, we tested the effect of combining the inhibitors on cell survival. As shown in Figure 4A, miR-133ab and miR-361-3p inhibitors together act synergistically to reduce cell viability compared with each miRNA inhibitor alone, as assessed by Bliss independence.20 miR-133ab and miR-361-3p inhibitors were delivered individually and in combination at 12.5 nM each. We further examined whether these miRNA inhibitors potentiate the cytotoxic effect of other chemotherapeutic agents. As show in Figure 4B–G, miR-133ab and miR-361-3p inhibitors significantly potentiate the effects of paclitaxel, vinorelbine, and gemcitabine. This suggests that the identified miRNA inhibitors have the potential to be applied in combination with other anticancer drugs.

Figure 4. Combining the miRNA inhibitors with each other and with other anti-cancer agents enhances their effects on cell viability. (A) Effect of combining the miR-133ab and miR-361-3p inhibitors on cell viability in H1993 cells. (B–G) Effect of combining miR-133ab inhibitor (B–D) and miR-361-3p inhibitor (E–G) with paclitaxel, vinorelbine, and gemcitabine on cell viability in H1993 cells. The red lines indicate predicted thresholds for synergy under the assumption of Bliss independence.
Inhibitors of miR-133a/b, miR-361-3p, and miR-346 reduce cell survival through different mechanisms
The most common mechanism by which anticancer agents cause cell death is through inducing caspase-dependent apoptotic pathways. In order to further examine whether the cytotoxicity of the three miRNA inhibitors is mediated by their activation of caspase-3/7-dependent apoptotic pathways, we used live cell imaging to monitor caspase3/7 activation as a function of time following transfection of cells with 10 nM oligos. As shown in Figure 5A, miR-133a/b inhibitor dramatically increases apoptotic events relative to control oligo, as measured by the percentage of cells that undergo apoptosis. Compared with miR-133a/b inhibitor, miR-361-3p and miR-346 inhibitors are much less potent in inducing apoptosis, suggesting that additional mechanisms are involved in the cytotoxicity induced by the latter. The corresponding growth curves in Figure 5B show that the proliferative capacity of cells transfected with the three inhibitors is significantly decreased as compared with control oligo. Consistent with the results showing that miR-133a/b is the most potent in inducing apoptosis, miR-133a/b inhibitor has the most dramatic effect on reducing cell growth rate. The representative images in Figure 5C show the staining of apoptotic cells at the end point of the apoptotic assay, consistent with the results shown in Figure 5A. Figure 5D shows the activated caspase-3 levels detected by western blot. Consistent with the results shown in Figure 5A and C, miR-133a/b inhibitor dramatically increases the levels of activated caspase-3 after 3 d of transfection compared with control oligo. The miR-361-3p inhibitor shows a more modest effect on caspase-3 activation, while the miR-346 inhibitor doesn’t show detectable cleaved caspase.

Figure 5. Effect of miR-133a/b, miR-346, and miR-361-3p inhibitors on caspase-3 activation in H1993 cells. (A) Time-dependent effect of the miRNA inhibitors on the induction of cell apoptosis. Cells were transfected with 10 nM of the indicated oligos. Cells undergoing apoptosis were stained using the CellPlayer Caspase-3/7 Reagent (Essen BioScience) and apoptotic events were counted using the IncuCyte live cell imaging system. The percentage of cells induced into apoptosis was calculated by normalizing to total cell numbers quantified by staining for total DNA content. (B) Cell confluence as a function of time was quantified using the IncuCyte live cell imaging system. (C) Representative images at the end point of the apoptotic assay. Apoptotic cells fluoresce green. (D) Western blot analysis of Caspase-3 activation. Cells were transfected with 50 nM of the indicated oligos and incubated for 72 h, after which cell lysates were harvested. Caspase-3 was detected using the specified antibodies, with calnexin levels measured as a loading control. Band intensities were quantified using ImageJ.
Another common mechanism by which anticancer agents inhibit cell growth is through arrest of cells in specific stages of the cell cycle, thereby blocking cell proliferation. We therefore tested the effect of the miRNA inhibitors on cell cycle distribution. As shown in Figure 6A–E, cells transfected with miR-133a/b and miR-361-3p inhibitors have significantly more cells in S phase than cells transfected with control oligos (42.9% and 55.7% vs. 28.9% in control cells), with the cell cycle distribution showing a sharp peak at the beginning of S phase; in contrast, miR-346 inhibitor does not dramatically change the cell cycle distribution. Figure 6F shows that miR-133a/b and miR-361-3p significantly decrease BrdU incorporation into DNA synthesis, while miR-346 inhibitor has much less of an effect. These results suggest that S phase arrest contributes to miR-133a/b and miR-361-3p inhibitor-induced cytotoxicity. We therefore further examined the effect of miR-133a/b and miR-361-3p inhibitors on cell cycle progression in G1/S phase synchronized cells.21-23 As shown in Figure 6G, after release from G1 phase synchronization, cells transfected with miR-133a/b and miR-361-3p inhibitors show a significant accumulation in S phase as compared with control cells, indicating S phase arrest is induced by miR-133a/b and miR-361-3p inhibitors. Quantitation of the fractions of cells in the G1, S, and G2 phases at each time point are given in Table S4. Figure 6H further shows that miR-133a/b and miR-361-3p inhibitors dramatically increase the levels of phosphorylated Chk1 and Chk2 (pChk1 and pChk2), markers of intra-S-phase arrest,24 while miR-346 inhibitor does not dramatically affect pChk1 and pChk2. Together, these results demonstrate that S phase arrest significantly contributes to cytotoxicity induced by miR-133a/b and miR-361-3p inhibitors, but not miR-346 inhibitor.

Figure 6. Effect of miR-133a/b, miR-346, and miR-361-3p inhibitors on cell cycle distribution. Cells were transfected with 50 nM of the indicated oligos. After 48 h, cells were collected and stained with PI for cell cycle analysis. (A–D) Flow cytometry images show the distribution of cell numbers at each cell cycle stage under each treatment condition. (E) The fraction of cells in G1, S, and G2 phases was quantified using the Watson pragmatic model. Similar results were obtained from two independent experiments. (F) Effect of miR-133a/b, miR-346, and miR-361-3p inhibitors on DNA synthesis as measured by BrdU incorporation. Shown are representative results from three replicate experiments. (G) Effect of miR-133a/b and miR-361-3p inhibitors on cell cycle progression in G1 phase-synchronized cells. (H) Effect of miR-133a/b, miR-346, and miR-361-3p inhibitors on total and phosphorylated Chk1 and Chk2 protein levels in H1993 cells.
Overall, the above results indicate that knockdown of the three miRNAs decreases lung cancer cell survival through different mechanisms.
The cytotoxicity of miR-346 inhibitor is likely to be caused by off-target effects of the synthetic oligo
EXIQON miRCURY LNA™ miRNA inhibitors are chemically stabilized, single-stranded RNA oligonucleotides complementary to specific miRNAs. The binding of the miRNA inhibitor to the target miRNA prevents the association of the miRNA with the complementary site(s) in its target mRNA(s), blocking the endogenous activity of the miRNA and restoring protein translation from the mRNA transcript(s). It is possible, however, that these oligos cause off-target cytotoxic effects by binding to other nucleic acid molecules with sequences similar to the targeted miRNAs. In order to preliminarily address this question, we examined whether co-transfection of miRNA mimics with the corresponding miRNA inhibitors blocks the cytotoxic effect of the inhibitors. As shown in Figure 7, mimics of miR-133a/b (Fig. 7A) and miR-361-3p (Fig. 7B) rescue the cytotoxic effect of the inhibitors of these two miRNAs respectively, indicating that the effects of these inhibitors are likely to be mediated through inactivation of their corresponding miRNAs. The mimic of miR-346, however, does not rescue the cytotoxic effect of the miR-346 inhibitor (Fig. 7C). Overall, although our results cannot exclude the possibility that the cytotoxicity of miR-133a/b and miR-361-3p inhibitors is caused by off-target effects, our results indicate that the cytotoxicity of the miR-346 inhibitor is most likely to be caused by off-target effects of the synthetic oligo rather than knocking down the expression of the endogenous miR-346.
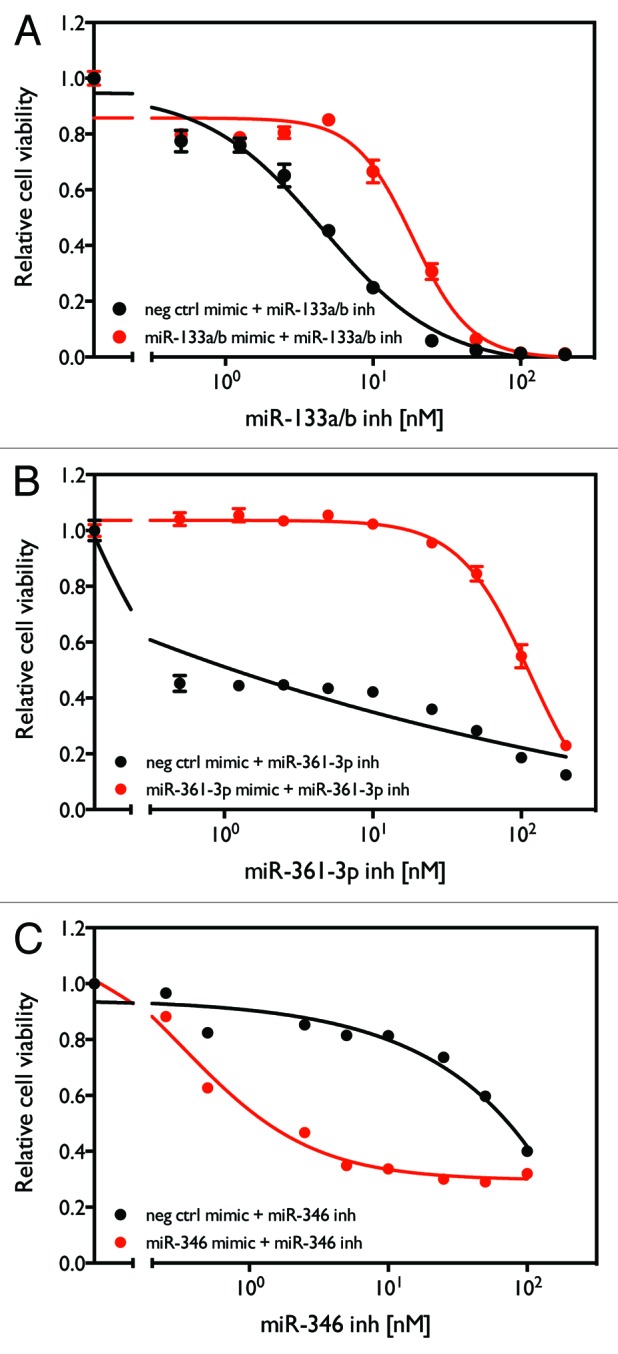
Figure 7. Rescue of cytotoxicity induced by miR-133a/b, miR-361-3p, and miR-346 inhibitors with miRNA mimics. H1993 cells were transfected with combinations of either the indicated miRNA mimic or negative control oligo with different concentrations of the indicated miRNA inhibitor. After 96 h, cell viability was measure as described above.
miRNA:target prediction indicates that the three miRNAs have distinct “targetomes”
Based on the above results, we speculate that miR-361-3p and miR-133a/b function as oncogenes in lung cancer cells by downregulating expression of tumor suppressor genes. It is well known that each miRNA targets hundreds of genes. However, among these genes, we speculate that only a small fraction is important to cell survival and drug response. In this study, in order to identify target genes that are highly likely to mediate the effect of the miRNAs on cell survival, we first combined the miRmate algorithm developed in our lab and the public TargetScan program25 to identify the predicted targets of the two miRNAs. We then annotated these target genes with Gene Ontology terms and performed a comprehensive literature search to identify those that have been suggested to function as tumor suppressor genes that repress cancer cell growth. We consider this group of genes (Table 1) high-confidence candidate targets that mediate the growth-promoting function of these miRNAs. Since the two miRNA inhibitors share common cellular mechanisms that lead to decreased cell survival and growth (as shown in Figs. 5 and 6), we speculate that miR-361-3p and miR-133a/b share direct target(s) regulating lung cancer cell survival. We therefore analyzed whether the two miRNAs share common direct target(s). As shown in Table 1, however, no target was identified in common for the two miRNAs. These results indicate that the effect of the two miRNAs on lung cancer cell survival is mediated by distinct “targetomes.”
Table 1. Predicted direct targets of miR-133a/b and miR-361-3p.
| miR-133a/b | ||||||
|---|---|---|---|---|---|---|
| Gene | miRmate score | TargetScan PCT | Biological process (GO) | Function | Implication | Ref. |
| PPP2CA | -0.49 | regulation of cell death | cell proliferation; negative control of cell growth and division | tumor suppressor gene | 51 | |
| SGMS2 | 83.0 | -0.52 | cell cycle, differentiation, autophagy, apoptosis; required for cell growth | tumor suppressor gene | 52 | |
| SUMO1 | 52.5 | -0.48 | negative regulator of transcription | cell proliferation, tumor growth | tumor suppressor gene | 53 |
| EDEM1 | 58.0 | -0.66 | antitumor immunity | tumor suppressor gene | 54 | |
| LRRFIP1 | 65.0 | -0.48 | regulation of transcription from RNA polymerase II promoter | resistance to chemotherapy | tumor suppressor gene | 55 |
| RBMXL1 | 67.0 | -0.55 | mRNA processing | N/A | tumor suppressor gene | 56 |
| miR-361-3p | ||||||
|---|---|---|---|---|---|---|
| BTG2 | 103.0 | negative regulation of apoptosis, negative regulation of proliferation | proliferation, invasion, and apoptosis | tumor suppressor gene | 57 | |
| C12orf5 | 105.5 | proliferation, cell cycle | tumor suppressor gene | 58 | ||
| CEBPA | 97.0 | -0.75 | regulation of transcription, regulation of proliferation, lung development | carcinogenesis, cellular differentiation | tumor suppressor gene | 59 |
| CPEB1 | 103.0 | regulation of translation | invasion, metastases | tumor suppressor gene | 60 | |
| IKZF1 | 101.0 | regulation of transcription, regulation of cell differentiation | migration and invasion | tumor suppressor gene | 61 | |
| MYT1 | 101.0 | cell differentiation, regulation of transcription | apoptosis | tumor suppressor gene | 62 | |
| NFE2L3 | 103.0 | regulation of transcription | oxidative damage, carcinogen detoxification | tumor suppressor gene | 63 | |
| PEG3 | 101.0 | regulation of transcription, programmed cell death | cancer growth | tumor suppressor gene | 64 | |
| RANBP10 | 106.0 | microtubule cytoskeleton organization | tumor development, progression | tumor suppressor gene | 65 | |
| SLC36A1 | 104.0 | transmembrane transport | androgen receptor | tumor suppressor gene | 66 | |
| YAP1 | 102.0 | regulation of cell proliferation, regulation of transcription | anoikis, metastasis | tumor suppressor gene | 67 | |
| SYNPO | 72.5 | -0.65 | regulation of cytoskeleton organization | cell migration | tumor suppressor gene | 68 |
| MGLL | 70.0 | -0.48 | colony formation, cell growth | tumor suppressor gene | 69 | |
| KDM5C | 63.0 | -0.47 | regulation of transcription, chromosome organization | tumor growth | tumor suppressor gene | 70 |
| CADM4 | 92.5 | -0.70 | cell adhesion | tumor formation | tumor suppressor gene | 71 |
Shown are gene names, TargetScan PCT scores, miRmate interaction scores, biological process terms (derived from GO), cellular functions, their role(s) in cancer, and selected references.
Discussion
Many studies have demonstrated the critical role of miRNAs in regulating cancer cell survival and drug response, and manipulating miRNA levels has shown great promise as a therapeutic tool to treat cancers, including lung cancer.26,27 However, there is still a lack of comprehensive studies of miRNAs in regulating lung cancer cell survival and drug response, which is a major obstacle to developing efficacious miRNA-based therapeutics for lung cancer treatment. Here, we conducted the first comprehensive analysis of miRNA inhibitors regulating lung cancer cell survival and response to paclitaxel. Our study uncovered a small group of novel miRNA inhibitors that are potential universal modulators of lung cancer cell survival and paclitaxel sensitivity, independent of genetic background. These novel inhibitors have direct potential to be developed as therapeutic agents for lung cancer treatment.
Our experimental design was based on the high-throughput synthetic lethal siRNA screen used by Whitehurst, et al. to identify chemosensitizer loci in non-small cell lung cancer cells. In order to identify general sensitizers and regulators of drug response in lung cancer cells, we expanded the screen along the cell line axis and included three NSCLC cell lines—H1155, H1993, and H358—representing common but distinct mutations found in lung cancer as reflected in their KRAS and TP53 mutational status. In general, the HTS approach described here could also be used to identify miRNAs that regulate cell viability or drug resistance in the context of a specific pathway by using several cell lines that share a specific molecular characteristic, such as a KRAS mutation, and comparing them to cell lines that do not share that trait.
The follow-up investigations of the HTS results presented here are focused on the three miRNA inhibitors identified as most potently cytotoxic to all three cell lines used in the screens. Our investigation shows that, among the three, miR-133a/b has the most potent effect. miR-133b has been shown to participate in apoptosis pathways in lung cancer cells28 by targeting the BCL-2 family of anti-apoptotic genes. Crawford, et al. demonstrated that two members of the BCL-2 family of pro-survival molecules (MCL-1 and BCL2L2) are direct targets of miR-133b, with overexpression of miR-133b inducing apoptosis of tumor cells following gemcitabine exposure. These results suggest a tumor suppressor function for miR-133b. Our results, however, indicate that miR-133a/b inhibitor has a potent cytotoxic effect and that miR-133b overexpression rescues the cytotoxicity of miR-133a/b inhibitor, which is inconsistent with previous findings. There are several possible explanations for this inconsistency. One possible reason is that the cellular function of miR-133a/b is context-dependent. Our target prediction analysis showed that miR-133a/b targets both oncogenes and tumor suppressor genes; it is therefore possible that the major effect of miR-133a/b in the lung cancer cell lines that we investigated is to downregulate tumor suppressor genes, thereby functioning as an oncogene and promoting cell survival. Another possibility is that the observed cytotoxicity of the miR-133a/b inhibitor might be caused by off-target effects of the synthetic oligos and not by downregulation of miR-133a/b levels. Although our results show that miR-133a/b mimic rescues the cytotoxicity of the miR-133a/b inhibitor, further study is needed to completely exclude the possibility of off-target effects.
Another miRNA that we identified as regulating cell survival and proliferation is miR-361-3p. Two mature miRNAs, miR-361-3p and miR-361-5p, are produced from the mir-361 precursor. Differential expression of miR-361-5p has been linked to bleomycin-induced pulmonary fibrosis29 and fatty acid-mediated insulin resistance30 in mouse models. The function of miR-361-3p, however, has not been characterized. We are therefore the first to identify the novel oncogenic function of miR-361-3p.
The third synthetic oligo that exhibited cytotoxicity in lung cancer cells is the miR-346 inhibitor. A role has been suggested for miR-346 in several pathological processes including rheumatoid arthritis,31 inflammatory response,32,33 and metabolic processes.34 In cancers, miR-346 has been shown to be significantly overexpressed in follicular thyroid carcinoma (FTC).35 However, the function as well as the targets of miR-346 in the context of FTC were not defined. Our results indicate that knockdown of miR-346 inhibits cell survival and proliferation. Further investigation, however, shows that the cytotoxic effect of the miR-346 inhibitor is not rescued by miR-346 mimic, suggesting that the cytotoxicity of the miR-346 inhibitor is likely due to off-target effects. This conclusion is based on the assumption that, if the cytotoxicity of the miRNA inhibitors is caused by their depletion of endogenous miRNAs, transfection of exogenous miRNA mimics should restore the intracellular expression levels of these miRNAs and therefore rescue the cytotoxic effects of the inhibitors; if the cytotoxicity of the miRNA inhibitors is caused not by their depletion of endogenous miRNAs but rather through targeting other sequences, the miRNA mimics are unlikely to rescue the observed cytotoxicity, since transfection of exogenous miRNA mimics will only restore the expression levels of the miRNAs, and not the expression levels of other sequences targeted by the inhibitor. Theoretically, for a complementary mimic and inhibitor that are delivered together, the mimic and inhibitor will bind to and sequester each other, canceling each other out, even if the mimic doesn't produce a functional miRNA, assuming that the inhibitor has much higher binding affinity with the corresponding miRNA mimic than with other off-target sequence(s). A possible explanation for the failure of the miR-346 mimic to rescue the cytotoxic effect of the miR-346 inhibitor is that the miR-346 inhibitor has higher binding affinity to sequence(s) other than the miR-346 mimic, letting the miR-346 inhibitor escape, at least partially, sequestration by the mimic—strongly supporting off-target effects of the miR-346 inhibitor. Therefore, the function of miR-346 in lung cancer cells is not clearly defined in the present study. Although this observation needs further confirmation, our results do suggest that when using synthetic oligos to manipulate endogenous expression of target genes, off-target effects of the synthetic oligos might be more common than presently appreciated. Therefore, each novel gene or molecular pathway identified using synthetic oligo-based screening, including the miRNAs identified in our current study, needs to be individually evaluated with the potential off-target effects of each synthetic oligo investigated through additional approaches.
In the current study, we also preliminarily investigated the mechanisms underlying the cytotoxicity of the three miRNA inhibitors. We assessed their ability to activate apoptotic pathways and induce cell cycle arrest. We also identified potential direct targets that mediate the cellular functions of miR-133a/b and miR-361-3p. Well-defined mechanisms of anticancer agent action include cell cycle arrest, apoptosis, necrosis, and autophagy,36 among which caspase-dependent apoptosis is recognized as the most common and central mechanism. Our results show that caspase-3 activation plays an important role in miR-133a/b inhibitor- and miR-361-3p inhibitor-induced cytotoxicity. Cell cycle arrest is another critical mechanism in anticancer agent-induced cell death and/or growth arrest. Our analysis of cell cycle distributions shows that miR-133a/b and miR-361-3p inhibitors induce significant S phase arrest, whereas miR-346 does not have a significant effect on cell cycle distribution. S phase arrest has been demonstrated to be one of the most important mechanisms of cell cycle arrest in cancer cells.37 The mechanisms of S phase arrest and the protein machinery involved in regulating the S phase checkpoint have also been well defined.37-41 However, our target prediction shows that miR-133a/b and miR-361-3p do not directly target genes relevant to S phase progression (Table 1), suggesting that the effect of these miRNAs on S phase progression is through indirect regulation. The direct targets and downstream signaling pathways mediating the effect of these miRNAs on the S phase checkpoint need to be defined further.
Our investigation of the predicted targets of the miR-133a/b and miR-361-3p indicate that they have distinct “targetomes,” suggesting that distinct signaling pathways are involved in mediating their effects on lung cancer cell survival. The diversity of the targets of these miRNAs suggests the potential of developing combined therapeutic agent-based inhibitors of these miRNAs. Because miRNAs have multiple regulatory targets and can intervene in multiple oncogenic pathways simultaneously, they may provide more effective therapeutic strategies than agents that are currently in use.42 Recent studies have demonstrated that tumor cells show significant intra-tumoral heterogeneity, with somatic mutations and gene expression profiles indicative of both good and poor prognosis found in different regions of the same tumor and between the primary tumor and distant metastases.42 This heterogeneity may be the primary contribution to tumor adaptation leading to drug resistance and therapeutic failure,43 and the multi-targeting feature of miRNAs might be a way to overcome this obstacle to traditional strategies. The combination of multiple cytotoxic miRNA inhibitors would hit an even broader spectrum of molecular pathways, further addressing the problem of heterogeneity. In addition, the combination would allow smaller doses of each individual miRNA inhibitor while still inducing effective cell death and would therefore decrease any toxic side effects specific to each inhibitor. The therapeutic efficacy of such combinations of miRNA inhibitors, including those previously identified, as well as those identified here, certainly warrants further investigation.
Overall, our HTS identified novel miRNA inhibitors that modulate lung cancer cell survival and response to paclitaxel. Although the mechanisms by which the identified inhibitors regulate cell survival and drug response need to be further investigated, the universal effect of the identified inhibitors on lung cancer cells with different genetic backgrounds highlights the potential of applying these inhibitors as therapeutic agents in lung cancer treatment. Future studies are certainly warranted to explore the possibility of translating these laboratory findings into clinical applications.
Materials and Methods
Reagents and materials
miRNA mimics and siRNA oligos were obtained from Dharmacon. miRNA inhibitors were obtained from Exiqon. Paclitaxel dissolved in Cremophor EL (polyoxyethylated castor oil) and dehydrated ethanol was obtained from Teva Pharmaceuticals. Aphidicolin was obtained from Sigma-Aldrich.
Cell lines
Cell lines beginning with “H” were established at the National Cancer Institute. Cell lines beginning with “HCC” and the immortalized human bronchial epithelial cells (HBECs) were established by the Hamon Center for Therapeutic Oncology Research at UT Southwestern Medical Center. All cancer cell lines were grown in RPMI-1640 medium (Life Technologies) supplemented with 5% fetal bovine serum (Atlanta Biologicals). All cell lines were grown in a humidified atmosphere with 5% CO2 at 37 °C. Total RNA was extracted using TRIzol (Invitrogen).
High-throughput screen (HTS)
The inhibitors, each of which targets one of the 747 known human miRNAs, were arrayed in a one-inhibitor one-well format in the central 60 wells of 96-well micro-titer plates. Transfections of NSCLC cells were performed in quadruplicate (H1155) or triplicate (H358 and H1993). After incubation for 120 h, cell viability was assessed by measuring ATP concentration (CellTiter-Glo Luminescent Cell Viability Assay; Promega). Data from the drug and carrier arms of the screens were collected in parallel, with the same plate and well locations for each miRNA. Each miRNA inhibitor was assigned a relative viability calculated by normalizing replicate means to the mean of the central 60 wells on each plate to assess the effect of each inhibitor on cell viability in the absence (Vcarrier) or presence of paclitaxel (Vpaclitaxel). Each miRNA inhibitor was also assigned a sensitivity ratio (S) calculated as mean viability in the presence of paclitaxel divided by mean viability in the presence of carrier (S = Vpaclitaxel/Vcarrier) to assess the effect of each inhibitor on cell response to paclitaxel.
miRNA qRT-PCR
Total RNA was prepared using the mirVana™ miRNA Isolation Kit (Ambion). mRNA and miRNA levels were assessed by qRT-PCR using an ABI PRISM 7900 Sequence Detection System using miRNA Expression Assay primer and probe sets (Applied Biosystems). RNU19 expression was used as a control for normalization of cDNA loading. Threshold cycle times (Ct) were obtained and relative gene expression was calculated using the comparative cycle time method.44
miRNA expression profiling
Total RNA from lung cancer cell lines was prepared as above. miRNA expression profiling was done using the Agilent miRNA expression platform following standard protocols.
Cell viability assay
Cells were plated in 96-well format and transfected with oligos, and/or with drug added in different concentrations after 72 h, followed by incubation for additional 48–72 h. Cell viability was determined using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega).
Colony formation assay
One thousand cells were seeded on each 10 cm dish. After 14 d, colonies were visualized by staining with 1% crystal violet. Colonies were counted using ImageJ (NIH), and differences were assessed by two-tailed t-test.
Cell cycle analysis
Cells were detached by trypsinizing and collected by centrifuging at 1000 rpm for 5 min. The cells were washed once with 1X PBS and fixed with 1X PBS containing 1 mM EDTA and 85% ethanol at 4 °C. After 1 h, the cells were harvested by centrifugation at 1400 rpm for 5 min at 4 °C. The cells were then re-suspended in 1X PBS, and treated with 50 µg/ml propidium iodide and 100 µg/ml RNase A for 30 min at 37°C. Cell cycle data were collected on a Cytomics FC 500 flow cytometer (Beckman Coulter), with 20 000 events collected per sample. Data were analyzed using FlowJo version 7.6.5 (TreeStar).
Intra-S phase checkpoint activation
Cells were transfected with 10 nM miR-133a/b inhibitor, miR-361-3p inhibitor, or scrambled control oligo for 12 h, followed by serum starvation for 12 h. Cells were then treated with aphidicolin (5 µg/ml) for 12 h for G1/S phase synchronization. To release cells from synchronization, cells were washed with PBS twice and medium was replaced with fresh medium. Cells were then collected at 0, 2, 4, 6, 8, 10, and 12 h for cell cycle analysis as above.
Western blots
Cell lysates were prepared using NP-40 buffer. Protein concentration was determined using the Pierce BCA assay (Thermo Fisher). For electrophoresis, equal amounts of cell lysate were resolved by SDS-PAGE and transferred to Immun-Blot PVDF membranes (Bio-Rad). Membranes were blocked and probed with rabbit anti-Caspase-3 (Cell Signaling Technology), anti-Chk1 (Cell Signaling), anti-Chk2 (Cell Signaling), anti-pChk1 (Cell Signaling), anti-pChk2 (Cell Signaling), or goat anti-calnexin (Santa Cruz Biotechnology). Bound antibodies were detected with secondary antibodies conjugated with horseradish peroxidase (HRP) (Santa Cruz Biotechnology) and visualized by enhanced chemiluminescent (ECL) substrate (Pierce/Thermo Fisher).
miRNA target prediction
We combined the predictions from TargetScan and miRmate to identify high-confidence targets for each miRNA. The miRmate method45,46 rewards complete complementarity of the seed region at positions 2–8 of the miRNA, mismatches and insertions in the central bulge at positions 9–11 of the miRNA, some complementarity at the 3′ end, and specific sequence composition at positions 1 (A) and 9 (A or C) of the miRNA, according to the findings of Lewis, et al.47 Each miRmate predicted miRNA:target interaction is given a predicted interaction score based on the calculations of the above parameters, with higher interaction scores indicating a higher probability of miRNA:target interaction. For miRNA:target interaction identified by TargetScan, we rank the prediction by the probability of conserved targeting (PCT) score, with higher scores considered to indicate a higher probability of miRNA:target interaction.48-50 We consider predicted targets that have either a TargetScan PCT score > 0.1 or a miRmate interaction score > 90 as high-confidence targets of each miRNA.
Cell apoptosis and growth rate assays
Cells were plated in 96-well plates and treated with specified conditions. After 18 h, CellPlayer Caspase 3/7 Reagent (Essen BioScience) was added and apoptotic events were detected with an IncuCyte live-cell imaging system (Essen BioScience). Cell confluence was monitored at the same time. After 54 d, the total number of cells in each well was determined by staining for total DNA content using Vybrant DyeCycle Green DNA stain (Invitrogen). The percentage of cells undergoing apoptosis was determined from the ratio of apoptotic events to the total number of cells, with the latter estimated by combining the DNA content assay at the end point and the cell confluence at each time point. Cell growth curves were derived from the observed confluence at each time point.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported in part by R01 CA129632 (A Pertsemlidis) from the NCI, Academic Excellence Grant EDUD-7824-021007-US (A Pertsemlidis) from Sun Microsystems, and Cancer Center Support Grants P30 CA142543 and P30 CA054174.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/rnabiology/article/26541
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/26541
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Ferracin M, Veronese A, Negrini M. Micromarkers: miRNAs in cancer diagnosis and prognosis. Expert Rev Mol Diagn. 2010;10:297–308. doi: 10.1586/erm.10.11. [DOI] [PubMed] [Google Scholar]
- 3.Garofalo M, Condorelli G, Croce CM. MicroRNAs in diseases and drug response. Curr Opin Pharmacol. 2008;8:661–7. doi: 10.1016/j.coph.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Hummel R, Hussey DJ, Haier J. MicroRNAs: predictors and modifiers of chemo- and radiotherapy in different tumour types. Eur J Cancer. 2010;46:298–311. doi: 10.1016/j.ejca.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 5.Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci U S A. 2006;103:4034–9. doi: 10.1073/pnas.0510928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–40. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hebert C, Norris K, Scheper MA, Nikitakis N, Sauk JJ. High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol Cancer. 2007;6:5. doi: 10.1186/1476-4598-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DY, Deng Z, Wang CH, Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci U S A. 2007;104:20350–5. doi: 10.1073/pnas.0706901104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–43. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 10.Ma J, Dong C, Ji C. MicroRNA and drug resistance. Cancer Gene Ther. 2010;17:523–31. doi: 10.1038/cgt.2010.18. [DOI] [PubMed] [Google Scholar]
- 11.Zheng T, Wang J, Chen X, Liu L. Role of microRNA in anticancer drug resistance. Int J Cancer. 2010;126:2–10. doi: 10.1002/ijc.24782. [DOI] [PubMed] [Google Scholar]
- 12.Enfield KS, Stewart GL, Pikor LA, Alvarez CE, Lam S, Lam WL, Chari R. MicroRNA gene dosage alterations and drug response in lung cancer. J Biomed Biotechnol. 2011;2011:474632. doi: 10.1155/2011/474632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corsten MF, Miranda R, Kasmieh R, Krichevsky AM, Weissleder R, Shah K. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007;67:8994–9000. doi: 10.1158/0008-5472.CAN-07-1045. [DOI] [PubMed] [Google Scholar]
- 14.Matsubara H, Takeuchi T, Nishikawa E, Yanagisawa K, Hayashita Y, Ebi H, Yamada H, Suzuki M, Nagino M, Nimura Y, et al. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene. 2007;26:6099–105. doi: 10.1038/sj.onc.1210425. [DOI] [PubMed] [Google Scholar]
- 15.Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, Forloni M, Boldrini R, Donfrancesco A, Federici V, Giacomini P, et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One. 2008;3:e2236. doi: 10.1371/journal.pone.0002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis S, Lollo B, Freier S, Esau C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 2006;34:2294–304. doi: 10.1093/nar/gkl183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446:815–9. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 18.Lam LT, Lu X, Zhang H, Lesniewski R, Rosenberg S, Semizarov D. A microRNA screen to identify modulators of sensitivity to BCL2 inhibitor ABT-263 (navitoclax) Mol Cancer Ther. 2010;9:2943–50. doi: 10.1158/1535-7163.MCT-10-0427. [DOI] [PubMed] [Google Scholar]
- 19.Izumiya M, Okamoto K, Tsuchiya N, Nakagama H. Functional screening using a microRNA virus library and microarrays: a new high-throughput assay to identify tumor-suppressive microRNAs. Carcinogenesis. 2010;31:1354–9. doi: 10.1093/carcin/bgq112. [DOI] [PubMed] [Google Scholar]
- 20.Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995;47:331–85. [PubMed] [Google Scholar]
- 21.Krokan H, Wist E, Krokan RH. Aphidicolin inhibits DNA synthesis by DNA polymerase alpha and isolated nuclei by a similar mechanism. Nucleic Acids Res. 1981;9:4709–19. doi: 10.1093/nar/9.18.4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kota KP, Benko JG, Mudhasani R, Retterer C, Tran JP, Bavari S, Panchal RG. High content image based analysis identifies cell cycle inhibitors as regulators of Ebola virus infection. Viruses. 2012;4:1865–77. doi: 10.3390/v4101865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Alvarez C, Doll R, Kurata H, Schebye XM, Parry D, Lees E. Intra-S-phase checkpoint activation by direct CDK2 inhibition. Mol Cell Biol. 2004;24:6268–77. doi: 10.1128/MCB.24.14.6268-6277.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 25.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trang P, Wiggins JF, Daige CL, Cho C, Omotola M, Brown D, Weidhaas JB, Bader AG, Slack FJ. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol Ther. 2011;19:1116–22. doi: 10.1038/mt.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiggins JF, Ruffino L, Kelnar K, Omotola M, Patrawala L, Brown D, Bader AG. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010;70:5923–30. doi: 10.1158/0008-5472.CAN-10-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crawford M, Batte K, Yu L, Wu X, Nuovo GJ, Marsh CB, Otterson GA, Nana-Sinkam SP. MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochem Biophys Res Commun. 2009;388:483–9. doi: 10.1016/j.bbrc.2009.07.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie T, Liang J, Guo R, Liu N, Noble PW, Jiang D. Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol Genomics. 2011;43:479–87. doi: 10.1152/physiolgenomics.00222.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li ZY, Na HM, Peng G, Pu J, Liu P. Alteration of microRNA expression correlates to fatty acid-mediated insulin resistance in mouse myoblasts. Mol Biosyst. 2011;7:871–7. doi: 10.1039/c0mb00230e. [DOI] [PubMed] [Google Scholar]
- 31.Semaan N, Frenzel L, Alsaleh G, Suffert G, Gottenberg JE, Sibilia J, Pfeffer S, Wachsmann D. miR-346 controls release of TNF-α protein and stability of its mRNA in rheumatoid arthritis via tristetraprolin stabilization. PLoS One. 2011;6:e19827. doi: 10.1371/journal.pone.0019827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alsaleh G, Suffert G, Semaan N, Juncker T, Frenzel L, Gottenberg JE, Sibilia J, Pfeffer S, Wachsmann D. Bruton’s tyrosine kinase is involved in miR-346-related regulation of IL-18 release by lipopolysaccharide-activated rheumatoid fibroblast-like synoviocytes. J Immunol. 2009;182:5088–97. doi: 10.4049/jimmunol.0801613. [DOI] [PubMed] [Google Scholar]
- 33.Bartoszewski R, Brewer JW, Rab A, Crossman DK, Bartoszewska S, Kapoor N, Fuller C, Collawn JF, Bebok Z. The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J Biol Chem. 2011;286:41862–70. doi: 10.1074/jbc.M111.304956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsai NP, Lin YL, Wei LN. MicroRNA mir-346 targets the 5′-untranslated region of receptor-interacting protein 140 (RIP140) mRNA and up-regulates its protein expression. Biochem J. 2009;424:411–8. doi: 10.1042/BJ20090915. [DOI] [PubMed] [Google Scholar]
- 35.Weber F, Teresi RE, Broelsch CE, Frilling A, Eng C. A limited set of human MicroRNA is deregulated in follicular thyroid carcinoma. J Clin Endocrinol Metab. 2006;91:3584–91. doi: 10.1210/jc.2006-0693. [DOI] [PubMed] [Google Scholar]
- 36.Ricci MS, Zong WX. Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 2006;11:342–57. doi: 10.1634/theoncologist.11-4-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 38.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 39.Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002;16:571–82. doi: 10.1101/gad.970702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D’Andrea AD. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109:459–72. doi: 10.1016/S0092-8674(02)00747-X. [DOI] [PubMed] [Google Scholar]
- 41.Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, Mathew CG, Kastan MB, Weaver DT, D’Andrea AD. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol. 2002;4:913–20. doi: 10.1038/ncb879. [DOI] [PubMed] [Google Scholar]
- 42.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011;71:1858–70. doi: 10.1158/0008-5472.CAN-10-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bookout AL, Mangelsdorf DJ. Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl Recept Signal. 2003;1:e012. doi: 10.1621/nrs.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du L, Schageman JJ, Subauste MC, Saber B, Hammond SM, Prudkin L, Wistuba II, Ji L, Roth JA, Minna JD, et al. miR-93, miR-98, and miR-197 regulate expression of tumor suppressor gene FUS1. Mol Cancer Res. 2009;7:1234–43. doi: 10.1158/1541-7786.MCR-08-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Du L, Subauste MC, DeSevo C, Zhao Z, Baker M, Borkowski R, Schageman JJ, Greer R, Yang CR, Suraokar M, et al. miR-337-3p and its targets STAT3 and RAP1A modulate taxane sensitivity in non-small cell lung cancers. PLoS One. 2012;7:e39167. doi: 10.1371/journal.pone.0039167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 48.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18:1139–46. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bluemn EG, Spencer ES, Mecham B, Gordon RR, Coleman I, Lewinshtein D, et al. PPP2R2C loss promotes castration-resistant prostate cancer growth and is associated with increased prostate cancer-specific mortality. Mol Cancer Res. 2013;11(6):568–78. doi: 10.1158/1541-7786.MCR-12-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barceló-Coblijn G, Martin ML, de Almeida RF, Noguera-Salvà MA, Marcilla-Etxenike A, Guardiola-Serrano F, Lüth A, Kleuser B, Halver JE, Escribá PV. Sphingomyelin and sphingomyelin synthase (SMS) in the malignant transformation of glioma cells and in 2-hydroxyoleic acid therapy. Proc Natl Acad Sci U S A. 2011;108:19569–74. doi: 10.1073/pnas.1115484108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang J, Yan J, Zhang J, Zhu S, Wang Y, Shi T, Zhu C, Chen C, Liu X, Cheng J, et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun. 2012;3:911. doi: 10.1038/ncomms1919. [DOI] [PubMed] [Google Scholar]
- 54.Marin MB, Ghenea S, Spiridon LN, Chiritoiu GN, Petrescu AJ, Petrescu SM. Tyrosinase degradation is prevented when EDEM1 lacks the intrinsically disordered region. PLoS One. 2012;7:e42998. doi: 10.1371/journal.pone.0042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, Li W, Yang Y, Lu Y, He C, Hu G, Liu H, Chen J, He J, Yu H. MicroRNA-21 targets LRRFIP1 and contributes to VM-26 resistance in glioblastoma multiforme. Brain Res. 2009;1286:13–8. doi: 10.1016/j.brainres.2009.06.053. [DOI] [PubMed] [Google Scholar]
- 56.Jiang Z, Guo J, Xiao B, Miao Y, Huang R, Li D, Zhang Y. Increased expression of miR-421 in human gastric carcinoma and its clinical association. J Gastroenterol. 2010;45:17–23. doi: 10.1007/s00535-009-0135-6. [DOI] [PubMed] [Google Scholar]
- 57.Zhang YJ, Wei L, Liu M, Li J, Zheng YQ, Gao Y, Li XR. BTG2 inhibits the proliferation, invasion, and apoptosis of MDA-MB-231 triple-negative breast cancer cells. Tumour Biol. 2013;34:1605–13. doi: 10.1007/s13277-013-0691-5. [DOI] [PubMed] [Google Scholar]
- 58.Madan E, Gogna R, Kuppusamy P, Bhatt M, Pati U, Mahdi AA. TIGAR induces p53-mediated cell-cycle arrest by regulation of RB-E2F1 complex. Br J Cancer. 2012;107:516–26. doi: 10.1038/bjc.2012.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang S, Jiang T, Feng L, Sun J, Lu H, Wang Q, Pan M, Huang D, Wang X, Wang L, et al. Yin Yang-1 suppresses differentiation of hepatocellular carcinoma cells through the downregulation of CCAAT/enhancer-binding protein alpha. J Mol Med (Berl) 2012;90:1069–77. doi: 10.1007/s00109-012-0879-y. [DOI] [PubMed] [Google Scholar]
- 60.Caldeira J, Simões-Correia J, Paredes J, Pinto MT, Sousa S, Corso G, Marrelli D, Roviello F, Pereira PS, Weil D, et al. CPEB1, a novel gene silenced in gastric cancer: a Drosophila approach. Gut. 2012;61:1115–23. doi: 10.1136/gutjnl-2011-300427. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Z, Xu Z, Wang X, Wang H, Yao Z, Mu Y, Ma Z, Liu Z. Ectopic Ikaros expression positively correlates with lung cancer progression. Anat Rec (Hoboken) 2013;296:907–13. doi: 10.1002/ar.22700. [DOI] [PubMed] [Google Scholar]
- 62.Choi HS, Bode AM, Shim JH, Lee SY, Dong Z. c-Jun N-terminal kinase 1 phosphorylates Myt1 to prevent UVA-induced skin cancer. Mol Cell Biol. 2009;29:2168–80. doi: 10.1128/MCB.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.auf dem Keller U, Huber M, Beyer TA, Kümin A, Siemes C, Braun S, Bugnon P, Mitropoulos V, Johnson DA, Johnson JA, et al. Nrf transcription factors in keratinocytes are essential for skin tumor prevention but not for wound healing. Mol Cell Biol. 2006;26:3773–84. doi: 10.1128/MCB.26.10.3773-3784.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feng W, Marquez RT, Lu Z, Liu J, Lu KH, Issa JP, Fishman DM, Yu Y, Bast RC., Jr. Imprinted tumor suppressor genes ARHI and PEG3 are the most frequently down-regulated in human ovarian cancers by loss of heterozygosity and promoter methylation. Cancer. 2008;112:1489–502. doi: 10.1002/cncr.23323. [DOI] [PubMed] [Google Scholar]
- 65.Harada N, Yokoyama T, Yamaji R, Nakano Y, Inui H. RanBP10 acts as a novel coactivator for the androgen receptor. Biochem Biophys Res Commun. 2008;368:121–5. doi: 10.1016/j.bbrc.2008.01.072. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y, Yang Y, Yeh S, Chang C. ARA67/PAT1 functions as a repressor to suppress androgen receptor transactivation. Mol Cell Biol. 2004;24:1044–57. doi: 10.1128/MCB.24.3.1044-1057.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu SJ, Hu JY, Kuang XY, Luo JM, Hou YF, Di GH, Wu J, Shen ZZ, Song HY, Shao ZM. MicroRNA-200a promotes anoikis resistance and metastasis by targeting YAP1 in human breast cancer. Clin Cancer Res. 2013;19:1389–99. doi: 10.1158/1078-0432.CCR-12-1959. [DOI] [PubMed] [Google Scholar]
- 68.Wong JS, Iorns E, Rheault MN, Ward TM, Rashmi P, Weber U, Lippman ME, Faul C, Mlodzik M, Mundel P. Rescue of tropomyosin deficiency in Drosophila and human cancer cells by synaptopodin reveals a role of tropomyosin α in RhoA stabilization. EMBO J. 2012;31:1028–40. doi: 10.1038/emboj.2011.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun H, Jiang L, Luo X, Jin W, He Q, An J, Lui K, Shi J, Rong R, Su W, et al. Potential tumor-suppressive role of monoglyceride lipase in human colorectal cancer. Oncogene. 2013;32:234–41. doi: 10.1038/onc.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Niu X, Zhang T, Liao L, Zhou L, Lindner DJ, Zhou M, Rini B, Yan Q, Yang H. The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene. 2012;31:776–86. doi: 10.1038/onc.2011.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Williams YN, Masuda M, Sakurai-Yageta M, Maruyama T, Shibuya M, Murakami Y. Cell adhesion and prostate tumor-suppressor activity of TSLL2/IGSF4C, an immunoglobulin superfamily molecule homologous to TSLC1/IGSF4. Oncogene. 2006;25:1446–53. doi: 10.1038/sj.onc.1209192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.